

Abstract
Motivation: Genomes in higher eukaryotic organisms contain a substantial amount of repeated sequences. Tandem Repeats (TRs) constitute a large class of repetitive sequences that are originated via phenomena such as replication slippage and are characterized by close spatial contiguity. They play an important role in several molecular regulatory mechanisms, and also in several diseases (e.g. in the group of trinucleotide repeat disorders). While for TRs with a low or medium level of divergence the current methods are rather effective, the problem of detecting TRs with higher divergence (fuzzy TRs) is still open. The detection of fuzzy TRs is propaedeutic to enriching our view of their role in regulatory mechanisms and diseases. Fuzzy TRs are also important as tools to shed light on the evolutionary history of the genome, where higher divergence correlates with more remote duplication events.
Results: We have developed an algorithm (christened TRStalker) with the aim of detecting efficiently TRs that are hard to detect because of their inherent fuzziness, due to high levels of base substitutions, insertions and deletions. To attain this goal, we developed heuristics to solve a Steiner version of the problem for which the fuzziness is measured with respect to a motif string not necessarily present in the input string. This problem is akin to the ‘generalized median string’ that is known to be an NP-hard problem. Experiments with both synthetic and biological sequences demonstrate that our method performs better than current state of the art for fuzzy TRs and that the fuzzy TRs of the type we detect are indeed present in important biological sequences.
Availability: TRStalker will be integrated in the web-based TRs Discovery Service (TReaDS) at bioalgo.iit.cnr.it.
Contact: marco.pellegrini@iit.cnr.it
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Tandem Repeats (TRs) are multiple (two or more) duplications of substrings in the DNA that occur contiguously, and may involve some base mutations (such as substitutions, insertions and deletions). TRs of several forms (satellites, microsatellites, minisatellites and others) have been studied extensively because of their role in several biological processes. In fact, TRs are privileged targets in activities such as fingerprinting or tracing the evolution of populations (Kelkar et al., 2008; Vogler et al., 2006). Several diseases, disorders and addictive behaviors are linked to specific TR loci (Wooster et al., 1994). The role of TRs has been studied also within coding regions (O'Dushlaine et al., 2005) and in relation to gene functions (Legendre et al., 2007). Large scale comparative studies on TRs of the human genome are described in Ames et al. (2008) and Warburton et al. (2008). Data Bases of repetitive elements such as RepBase (Jurka et al., 2005) and Tandem Repeats Database (TRDB) (Gelfand et al., 2007) are now available; and the detection of repetitive elements via library-based similarity matching, for example by using the tool Repeatmasker (Smit et al., 2004), is a popular practice. However, tools for ab initio detection of repetitive elements that are not based on prior knowledge accumulated in data bases are still important in order to extend our comprehension of the role of TRs in biological mechanisms. Existing ab initio tools are successful when the TR exhibits a moderate amount of divergence and when the TR is easily validated. However, there is an emerging need for new tools that are able to cope with higher levels of sequence divergence and/or TR computationally more difficult to validate. For example, Boeva et al. (2006) study so called Fuzzy TRs and their role in gene expression. The technique in Boeva et al. (2006) works well for the Hamming metric (only substitutions and no insertions/deletions allowed) and for short repeat units (from 3 to 24 bp) that are common in micro- and mini-satellite families.
Some of the most successful ab initio tools, such as TRF (Benson, 1999) and ATRHunter (Wexler et al., 2005), are based on a multi-stage filtering approach [see also (Peterlongo et al., 2009)]. In the first stage the input sequence is analyzed to detect, via statistical criteria, likely position and length of candidate subsequences. The final stage is the validation one in which a more expensive test is applied to candidate substrings passing the first stages, so to determine an output that matches the implicit definition of TR and the user-defined filtering parameters.
1.1 Our contribution
Our contribution is a novel multi-stage filtering algorithm, called TRStalker, for finding long fuzzy TRs under the edit distance, that introduces new techniques (w.r.t. previous TR finding algorithms) in all stages. For the first stage, where over-represented distances between probes are sought, we employ gapped q-grams (Burkhardt and Kärkkäinen, 2003) in place of the standard ungapped q-grams in order to collect evidence on the candidate substrings. Gapped q-grams have been used before in the context of textual and biological database searching, but less so in the area of TRs detection [with the exception of the system TEIRESIAS (Stolovitzky et al., 1999)]. Because of errors due to insertion/deletions, the period of a TR is subject to fluctuations, thus we employ a weighting scheme with exponential decay so to reinforce the signal even in presence of this smearing effect. Finally, we use ranking instead of thresholds when deciding the substrings to pass to the next phases, in order to concentrate the computational effort on the zones with candidates with higher weight. For the final validation stage we employ an NP-complete definition of TR involving the concept of generalized median string under edit distance (de la Higuera and Casacuberta, 2000; Sim and Park, 2003), together with an efficient heuristic for computing an approximation of such median string (Jiang et al., 2003) previously not used in a biological context.
By extensive experimental comparisons of TRStalker with two state-of-the-art tools, namely TRF and ATRHunter, we did find out that TRStalker has consistently better performance for a large range of error and length parameters for the class of fuzzy TRs under edit distance, with a recall ranging from 100 to 60%. Thus TRStalker improves the capability of TR detection for classes of TRs for which existing methods do not perform well. Tests performed on standard evolutionary TRs definitions (verifiable in polynomial time) also show recall performance close to 100%. Incidentally, this result confirms of the power of the new techniques developed for the initial filtering phase.
1.2 State of the art
We will briefly survey the state of the art in finding tandem repeats. First we will describe methods that for a given definition of TR are able to find all maximal substrings in the input that match the definition (exhaustive algorithms). Often exhaustive algorithms may not be available, or when available they may be too slow in practice. Thus, several heuristic algorithms have been developed which are shown experimentally to be able to detect a large fraction of TRs efficiently. Note that the time/precision trade-off is severely influenced by the allowed error thresholds. Performance often degrades quickly with increasing error levels.
1.2.1 Exhaustive algorithms
When we allow no error, it is possible to find all maximal exact TRs in a string of length n in time O(n) (Gusfield and Stoye, 2004; Kolpakov and Kucherov, 1999). When we allow two consecutive repeats to differ by an amount at most k (either in Hamming or in edit distance) Landau et al. (2001) give exhaustive algorithms running in time O(nklog(n/k)) for Hamming distance, and O(nklog klog(n/k)) for edit distance. A simpler algorithm with the same asymptotic complexity for the edit distance is proposed by Sokol et al. (2007). Kolpakov and Kucherov (2003) improved the bound for the Hamming distance to O(nklogk + s) where s is the number of TRs found. For the Hamming distance, Krishnan and Tang (2004) give an exhaustive method running sequentially in time O(n3), that can be easily implemented onto a parallel architecture, since every possible pattern length is searched independently.
1.2.2 Heuristic algorithms
The algorithmic techniques in Kolpakov and Kucherov (1999, 2003) have been extended in the tool mreps (Kolpakov et al., 2003) so to be able to handle approximate TRs (ATRs) under edit distance, with some additional heuristic filtering steps.
The tool TRF (Tandem Repeat Finder) developed by Benson (1998, 1999), based on statistical filtering of zones of DNA likely to contain TRs, is currently one of the standard heuristic methods. ATRHunter by Wexler et al. (2004) is also based on a statistical filtering approach, placing greater emphasis in techniques for designing thresholds for the quantities of interest. Other proposed heuristics for finding TRs are REPuter (Kurtz and Schleiermacher, 1999; Kurtz et al., 2001), STRING (Parisi et al., 2003), TEIRESIAS (Stolovitzky et al., 1999) and TandemSWAN (Boeva et al., 2006). A class of papers (see e.g. Brodzik, 2007; Buchner and Janjarasjitt, 2003; Gupta et al., 2007; Sharma et al., 2004) tackle the problem of finding TRs as a problem in signal processing theory and usually map the input string into a time-signal in a suitable numerical domain for which several spectral techniques can be used, such as the Periodicity Transform or the Fourier Transform. Other methods use data compression techniques to detect repetitive elements (Rivals et al., 1997).
The methods cited above are rather general since they aim at treating efficiently TRs in a wide range of length values. There is also a large class of methods that are aimed at handling particular or special classes of TRs such as: microsatellites [e.g. IMEx (Mudunuri and Nagarajaram, 2007)], palindromic repeats [e.g. CRISPFinder (Grissa et al., 2007)], Variable Length TRs (VLTR) and Multi-period TRs (MPTR) (Hauth and Joseph, 2002) and Variable Number TRs (VNTR) (Sammeth and Stoye, 2006). Since the focus of our research on TRs at present is on the more classical forms of TRs, we do not dwell longer on them. However, we just note that often methods for MPTR, VNTR, VLTR use standard TR finding as a subroutine, thus our proposed algorithm can increase also the ability to detect such higher order structures.
Systematic comparison among TR finding tools and algorithms operating ab initio, that is without support of specific biological data bases has been tackled in recent years (Leclercq et al., 2007; Saha et al., 2008). A survey of problems on TRs in the context of evolutionary mechanisms, such as the construction of TR Evolutionary Trees, is proposed in Rivals (2004); see also Elemento and Gascuel (2002).
1.3 Organization of the article
The article is organized as follows: in Section 2 we describe at a high level the principles guiding the different phases of the TRStalker algorithm. Section 3 gives a more technical description of key ingredients of TRStalker and discusses the formal definition of fuzzy TR employed. Section 4 describes the experiments devised to demonstrate the capacity of TRStalker in detecting fuzzy TRs, and a few interesting fuzzy TRs found in sequences of biological significance.
2 APPROACH
An example: To focus on the main ideas, let us consider the very simple case of Exact TR. Consider an alphabet Σ = {A, C, G, T} of four symbols, and a string X = x1x2 …xt formed by the concatenation of t strings xi, embedded in a random string Y, where xi = x1 for all i and |x1| = k, thus all replicas of x1 are of the same length. An ungapped q-gram is a string of q symbols from Σ that appears as a consecutive sequence of q symbols in Y. We aim at discovering k just by looking at the distances between occurrences of homologous (i.e. identical) q-grams in Y. For q-grams in X, the period k will appear at least (k − q + 1)(t − 1) times as the distance between homologous probes. More generally the distance hk, an integer multiple of k, will appear at least (k − q + 1)(t − h) times for each value h = 1 …t − 1. A gapped q-gram is a sequence of q characters from Σ with additional ‘don't care’ symbols, also called ‘gaps’, that appears as a consecutive sequence in Y. For gapped q-grams similar formulae hold. For values of k and t large enough, the period k and its integer multiples will occur more frequently than the expected number of occurrences of any distance of homologous q-grams in a random string, thus the empirical number of occurrences of the value k and its multiples will tend to be in the higher part of a ranking by frequency. This observation holds true as long as the length of the super-string Y is sufficiently limited so that the frequencies generated by the random portion of Y do not overrun the frequencies generated by X. An exact characterization of such a distribution in terms of the parameters k, t, q and |Y| is complex since it can be characterized as the sum of non-independent random variables each with a negative binomial distribution. However we avoid the issue of characterizing exactly such a distribution by: (i) splitting the input string into blocks of predefined length and limiting the analysis to each block separately, providing mechanisms to deal with TRs stranded across the block boundaries; (ii) ranking the periods by weighted frequency and exploring only the top L positions (for L = 50 in our experiments). Note that in most cases the top ranking periods not corresponding to TRs will be discarded quickly when the positional density is considered, thus we can be very slack in choosing L without incurring in a computational burden. The choice of block length could be critical too, but experimental results showed that blocks of length within a factor of up to 40 of the length of the TR do work well. For long input, string occurrences of the same q-gram that are too distant are unlikely to be related to a TR, thus we limit the number of pairs of homologous q-grams considered. While scanning each block of the input Y we record for each occurrence of a gapped q-gram in Y its distance to the five preceding and the five following homologous occurrences (10 in total). The high-level pseudocode of TRStalker is shown in the Supplementary Materials while we expose next the key algorithmic choices.
Gapped q-grams: The presence of substitutions/insertions/deletions in X has the effect that many instances of q-grams will be affected by error and a match will be missed, thus reducing the frequency counts for the period k. To cope with this effect, we use gapped q-grams (Burkhardt and Kärkkäinen, 2002, 2003) that are more resilient to the presence of substitutions/insertions/deletions. As suggested by experiments in Burkhardt and Kärkkäinen (2002), just few gaps are sufficient to be effective, thus we will use the family of all gapped q-grams with three alphabet symbols and at most two gaps.
Anti-smear weighting: If q1 and q2 are occurrences of homologous q-grams in X at distance k, before the implant of mutations, the effect of insertion and deletions on the positions of the string X between q1 and q2 is to alter their distance so that a different period k′ is detected. The difference k − k′ is equal to the algebraic sum of number of insertions and deletions in the positions between q1 and q2. Assuming that any such position can be an insertion or a deletion independently with the same probability, the random variable k − k′ is distributed as a sum of independent random variables with values in {+1, −1, 0} with mean value 0, thus, by a Chernoff bound argument, its tail distribution decays exponentially (Motwani and Raghavan, 1995; Mulmuley, 1993). Also near-by probes in X have small variations in the value of the shift k − k′. Inspired by the above observation, we devise a weighting scheme that increments the total weight of period k if another period of value 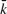 is discovered in a near-by position, with weights that decay exponentially with
is discovered in a near-by position, with weights that decay exponentially with 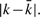 . The final weight w0(k) for a given period k is the sum of the individual anti-smear weights computed above for probes at distance k.
. The final weight w0(k) for a given period k is the sum of the individual anti-smear weights computed above for probes at distance k.
Multiplicity weighting: Let w0(k) be the weight of the period k as assigned by the anti-smear weighting procedure. As observed before, for a TR with a large number of copies we will find also integer multiples of k with a relatively high frequency. We take advantage of this fact and compute new weights:
The candidate periods are then sorted by the weight w1(.), and processed in decreasing order.
Positional density: We further exploit the property of TRs that the same period is detected by probes in near-by positions. We define a notion of positional k-density, that is the density of probes that contribute to the counter for the candidate period k. We search for position in Y of high k-density as candidates for the starting point of a TR.
Validation: In the third phase we take each candidate pair (p, i) and we test explicitly whether there is a TR of period p starting in position i according to the definition (Section 3). In particular when using the definition of a Steiner-STR (Section 3.2) we use a double filtering. The fist filter uses a wraparound dynamic programming technique (WDP; Fischetti et al., 1993). The second filter computes an approximation to the generalized media string [inspired by an algorithm proposed in Jiang et al. (2003)]. In this phase, besides validating the TRs, we discover the (fractional) repetition number of the TRs eventually extracted.
Post-processing: As a post-processing, we check for inclusion the TRs found and we filter out those TRs completely enclosed in another one. For TRs in the same position and length but different period we report the TR with shorter period. Finally we align the approximate generalized median string with the TR units so to give a graphical compact output of the TR.
3 METHODS
3.1 Basic definitions
A TR in a DNA sequence is the repetition of two or more contiguous exact or approximate copies of a substring (called the motif) of the TR.
3.1.1 Exact TR
Formally, given an alphabet Σ, and a set of strings xi ∈ Σ*, consider the concatenation X = x1x2 …xt. The string X is an exact TR (ETR) of period k and repeat number t, when |xi| = k and xi = x1, for each i ∈ [1 …t]. In general we may suppose there is a longer string Y of which X is a substring. The string x1 that is repeated exactly is called the motif of the ETR. A TR X is called maximal if it cannot be extended in Y while still being a TR.
3.1.2 ATR
ETRs are sometimes found in biological sequences, but they tell us only part of the story, thus several notions of an ATR have been developed. Denote with DH(a, b) the hamming distance of two strings with equal length. If the length of a and b is different, we consider the smallest possible mismatch in an alignment of the two strings without gaps. Denote with DE(a, b) the edit distance of the two strings a and b.
3.2 Our definitions of TR
We used two different definitions of TRs:
Neighboring TR (NTR): a string X, so that for each i ∈ [1 …t − 1], DE(xi, xi+1)≤μ|xi|, for a user defined parameter 0≤μ≤1
- Steiner-STR with sum: a string X = x1x2 …xt for which two conditions hold for a user defined error parameter 0≤μ≤1, and constant c with 1≤c≤2:
- for each i ∈ [1 …t − 1], DE(xi, xi+1)≤cμ|xi|.
- there exists a Steiner string
 so that
so that 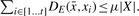
Intuitively, in a Steiner-STR the TR consists of t duplications of a single Steiner consensus string  with
with 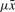 mutations on average in each copy, such that consecutive copies do not diverge too much w.r.t. the average. Note that condition (a) is vacuous for μ ≥ 1/c. The choice for the constant c depends also on the level of divergence. For low divergence c = 2 is a sensible choice since two copies at distance
mutations on average in each copy, such that consecutive copies do not diverge too much w.r.t. the average. Note that condition (a) is vacuous for μ ≥ 1/c. The choice for the constant c depends also on the level of divergence. For low divergence c = 2 is a sensible choice since two copies at distance 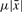 from
from 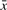 are also at distance at most
are also at distance at most 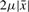 from each other by the triangular inequality. Thus (a) is a necessary condition for (b). For higher level of divergence above 30%, the value c = 2 is too loose and we use a lower value c = 1.5, so as to maintain a good filtering ability of condition (a) and to avoid having as a possible solution a TR where the consecutive pairs may have a very irregular divergence.
from each other by the triangular inequality. Thus (a) is a necessary condition for (b). For higher level of divergence above 30%, the value c = 2 is too loose and we use a lower value c = 1.5, so as to maintain a good filtering ability of condition (a) and to avoid having as a possible solution a TR where the consecutive pairs may have a very irregular divergence.
3.3 Output of TRStalker
The aim of TRStalker is to produce a ranked list of all maximal tandem repeat sub-sequences present in the input string that satisfy the definition above (NTR or Steiner-STR), where maximality means that it is not possible to extend the TR (as a substring of the input) to the left or to the right without violating the definition within the given user-defined parameters. We avoid producing meaningless TRs by imposing also a lower bound on the TRs length.
3.4 Other definitions
In Sokol et al. (2007) it is used the following definition: X is called a k-edit ATR when ∑i=1t−1 DE(xi, xi+1)≤k, where the last repeat xt might be incomplete so DE(xt−1, xt) is computed as the minimum edit distance of xt and the prefixes of xt−1. This definition is inspired by the evolutionary model of TRs in which it is assumed that TRs are generated by duplicating the last copy of a previous TR, possibly with duplication errors that truncate it. A k-edit repeat is maximal if it cannot be extended either to the left or to the right without violating its definition.
In Wexler et al. (2004), for a similarity function ϕ that measures the alignment score of two sequences, it is defined a η-Simple ATR (η-SATR) a string X = x1 …xt such that: there exists a motif  so that for every i ∈ [1, …, t],
so that for every i ∈ [1, …, t], 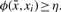 . In other words, the TR consists of t duplications of a single consensus string
. In other words, the TR consists of t duplications of a single consensus string  with mutations. Such string
with mutations. Such string  is also called a Steiner motif if
is also called a Steiner motif if  is not constrained to be equal to some repeat xj. Often in practice
is not constrained to be equal to some repeat xj. Often in practice 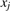 is chosen as the repeat xj that minimizes the error function, and is called a pivot motif. The distinction is critical since, as mentioned before, Steiner motifs lead to NP-complete recognition problems, while pivot motifs do not.
is chosen as the repeat xj that minimizes the error function, and is called a pivot motif. The distinction is critical since, as mentioned before, Steiner motifs lead to NP-complete recognition problems, while pivot motifs do not.
The η-Neighboring ATR (η-NATR) is a string X, so that for each i ∈ [1,.., t − 1], ϕ(xi, xi+)≥η (Wexler et al., 2004). The Pairwise ATR (PATR) is a string X, such that for every pair of indices i, j∈[1,.. t]2 with i ≠ j we have ϕ(xi, xj)≥ηij, where ηij is set to be a monotonically decreasing function of |i − j|, thus allowing more slackness when comparing distant copies of the basic motif.
In Krishnan and Tang (2004) it is used a definition similar to that of the NATR, except that the Hamming distance is used and that the threshold is not absolute but relative to the length. A γ-Hamming ATR (γ-HATR) is a string X such that: for each i ∈ [1, …t − 1], DH(xi, xi+1)≤|xi|γ.
In Stolovitzky et al. (1999), a more complex definition is given that takes into account the substring alignment score density function for pairs of random substrings of a given length. Here, the definition of a TR X depends on the properties of the longer string Y into which X is embedded. In particular, a (μ, p)-TR must comply to two conditions: (i) ∑i=0t−1 ϕ(xi, xi+1)≥(t − 1)μ, that imposes an average high similarity score for adjacent repeats, and (ii) define α(p, k) as the value of similarity such that there is probability p that two random substrings of length k in Y have similarity above α(p, k). There must be an index q ∈ [1, …t] such that ϕ(xq, xj)≥α(p, k) for all j ∈ [1, …t]. Note that this condition limits the dispersion of the similarity with respect to one of the copies (called the pivot).
TRF (Benson, 1999) uses as final validation algorithm the WDP that tests efficiently the alignments of a given candidate motif with the surrounding portions of the input sequence, so as to determine the maximum number of adjacent repetitions within a user-defined score bound. This implies a notion of TR akin to that of SATRs with pivot motif. Classical results on string alignments (Gusfield, 1997, p. 351) ensures that, for the metric score given by the sum of motif-repeats distances, the solution found using the optimal pivot motif has a score within a factor (2 − 1/t) of the score induced by the optimal Steiner motif. For low levels of errors one could use a pivot-SATR definition doubling the error threshold to capture a Steiner-SATR, however for higher error levels (say, above 25%), doubling the error threshold forces the existing systems to work in a range of values (say, above 50%) where most methods do not perform well.
3.5 Gapped q-grams
Let I be a finite subset of non-negative integers. We call I an index set. The span of I is span(I) = max{i − j|i, j ∈ I}, the position of I is pos(I) = mini ∈ I and the shape of I is shape(I) = {i − pos(I)|i ∈ I}. When set I has |I| = q and span(I) = s, its shape belongs to the class of (q, s)-shapes. Any set of non-negative integers Q containing 0 is a shape. For an alphabet Σ={A, C, G, T}, a string S ∈ Σ* of length n can be seen as a function defined over [0, …, n − 1] with values in Σ, and for any subset I ⊂ [0, …, n − 1] the restriction of S to I, denoted by S[I] a substring of S.
Given any shape Q in the class of (q, s)-shapes, all sets I ⊂ [0, …n − 1] such that shape(I) = Q, form the set of Indexes(Q, n). We can use elements from the Indexes(Q, n) to generate restrictions for the string S. Given two index sets I1, I2 ∈ Indexes(Q, n), we call them matching (or homologous in S, if S[I1] = S[I2]. The value |pos(I1) − pos(I2)| is called the period of the match.
An index set I with |I| = q and span(I) = q − 1 is called an ungapped q-gram since its shape is shape(I) = [0, …q − 1]. If we have an index set J with |J| = q and span(J) = s≥q we have a gapped q-gram since its shape is formed of non-consecutive integers. In order to generate a population of candidate periods we consider now all possible (q, s)-shapes with q = 3 and s = 4, 3 ,2. Denoting with – the gaps and with # symbols from Σ, (the first and last positions must be always #), we have the (3, 4)-shapes ##− −#, #−#−# and #−−##; the (3, 3)-shapes #−##, ##−#; and the (3, 2)-shape ###.
As noted in Burkhardt and Kärkkäinen (2003) and Burkhardt and Kärkkäinen (2002), if we fix an ungapped shape and an error level in Hamming distance, there are error patterns for which every corresponding ungapped q-gram is affected by error. In contrast with the same Hamming error level, for some gapped shapes, there are always some gapped q-grams unaffected by the injected error. Thus using a small complete family of gapped q-grams we can detect the correct period in situations where ungapped q-grams cannot.1
3.6 Anti-smear weighting
Let P be a q-gram in the input string Y at position i. Let j1,…, jh be the next h occurrences of P in Y following the occurrence at position i. The h corresponding detected distances are xg = jg − i, for g ∈ [1, …h]. For the period xg, we increment its weight:
where Q is a queue holding the last H detected distances in the sequential scan of the input string Y. After the weight update, we enqueue all h values xg in the queue Q, and we dequeue an equal number h of items. In line with other constants fixed in TRStalker, we have chosen h = 5 and H = 20 since they do work well in our synthetic experiments for a large range of TR error and length values. A fine tuning of these parameters as a function of the characteristics of the TR sought is possible, but beyond the focus of this article.
3.7 Positional density
Let k be the period under investigation. Consider the set Kk of the positions of those q-grams (i.e. substrings of Y) that contribute to the weighting of k through the multiplicity weighting. In order to avoid double counting, we always take the position of the first of the two matching probes. Note that, if a position is shared by several pairs of probes it will be counted only once. Let f :[1, …, |Y|]→{0, 1} the characteristic function that for each position in Y denote the membership of that position to Kk. Consider the k-window smoothing of f: F(i) = ∑j=ii+k f(j) that computes the k-smoothed density of the function f, for i ∈ [1, …, |Y| − k]. Finally, we define a threshold t(k) proportional to the average k-density by a user-defined constant, and we consider as a candidate position set CP(Y, k) = {i ∈ [1, …, |Y| − k]|F(i)≥t(k)}. The output of this positional density computation is a sequence of pairs (k, i) where k is a candidate period and i a candidate position.
3.8 Validation
The definition of Steiner-STR is composed of two conditions that will be tested in cascade starting from the one less computationally demanding.
3.8.1 Testing condition (a)
The WDP technique in Fischetti et al. (1993) solves the following problem. Given a string P of length m and a text T of length n, with m ≪ n, find the best alignment of Pn (concatenation of n copies of P in T), in time and storage O(nm). Note that a naive application of the standard dynamic programming based optimal alignment of two strings would require O(n2m) time/storage. We modify the WDP approach in order to (i) work with edit distance instead of similarity matrices, (ii) take as pattern the candidate initial tandem copy in positions [i, i + k − 1] and as text an adjacent portion of the input string of size O(m), (iii) we iteratively expand the the text length till the termination condition is met and (iv) we stop the matching as soon as the next adjacent copy of the TR differ from the previous one by more than cμm in edit distance.
3.8.2 Testing condition (b)
Let x1, …xt be the candidate TR to test for property (b) that passed the test for property (a). We incrementally compute an approximate generalized median 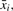 , using xi and the previously computed approximate generalized median string
, using xi and the previously computed approximate generalized median string 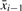 . Initially
. Initially 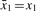 . Let k and h be two positive integers and K = {j/k|j ∈ [0, k]} be the set formed by k + 1 equally spaced real values between 0 and 1. For each value α ∈ K , we determine up to h median strings between xi and
. Let k and h be two positive integers and K = {j/k|j ∈ [0, k]} be the set formed by k + 1 equally spaced real values between 0 and 1. For each value α ∈ K , we determine up to h median strings between xi and 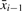 with weight α. This set of at most hk candidates is then searched for the string a that minimizes the function ∑j=1i DE(a, xj). So we set
with weight α. This set of at most hk candidates is then searched for the string a that minimizes the function ∑j=1i DE(a, xj). So we set  and start the next iteration.
and start the next iteration.
A median string of weight α ∈ [0, …, 1] of two strings a and b is obtained as follows. Compute the edit distance e=DE(a, b) and record the set A(a, b) of edit operations that transform a into b. Pick any subset of size ⌊αe⌋ in A(a, b). The median weighted string c is obtained by applying those operations to the string a. It is not difficult to show that it holds that DE(a, c)=αDE(a, b) and DE(b, c) = (1 − α)DE(a, b). Note that depending on the value of e we have 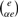 different subsets of A(a, b) we can choose. In our algorithm we randomly select
different subsets of A(a, b) we can choose. In our algorithm we randomly select 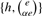 of them.
of them.
3.9 Evaluation of recall in synthetic sequences
In order to measure the quality of the TRs reported by TRSTalker and by other benchmark algorithms in our synthetic experiments, we need to give a score to a pair of TRs. The higher the similarity of the two TRs, the higher should be the score. Since perfect equality is rare we need a more flexible score function. A TR can be characterized by the triple: (b, p, r), where b is the initial position, p the period, r the repetition number. Also, the same TR covers the positions in Y from index b to b+rp − 1. We identify the TR with the set of positions Seg(TR)=[b, b + rp − 1]. Given two TRs TR1 and TR2 represented as sets of positions, the classical Jaccard coefficient measure of set similarity JC is:
3.9.1 Modified Jaccard coefficient
Let t0 be a TR embedded in Y. Even if t0 is a TR according to the definition, when we embed t0 in a string Y, it is well possible that t0 is not maximal in Y, thus if an algorithm reports correctly t′ ⊃ t0 there will be a slight penalization in the JC measure. This phenomenon arose a number of times, thus we decided to use a modified version of the Jaccard coefficient, called JC2, where the denominator is changed. The resulting measure is thus more robust w.r.t. this penalization:
Given a TR t0 and a set of TRs: T = {t1 …ts} we define the best-match BM(t0, T):
and the best-match-score (BMS):
In our controlled experiments, the evaluation module knows the embedded TR t0 and receives the output of an algorithm T, giving back the BMS. For a series of experiments, we will report the average of the BMS. Note that BMS has values in the range [0, …, 1], and higher values correspond to better quality. At first sight one might consider this metric as overly generous. However, since we cannot rule out the existence of other TRs in Y besides the embedded ones, we do not want to penalize the presence in T of valid TRs different from t0. Also, the set T will not contain nested TRs.
3.10 Evaluation of recall on biological sequences
The evaluation has been carried out according to the following procedure. Let TTRS, TTRF, TATR be the set of TRs found by TRStalker, TRF and ATRHunter respectively. First, we removed from every set all the TRs that have a Jaccard coefficient greater than a threshold J when compared with another TR in the same set. In other words, we removed TR duplicates from every set of results, where two TRs are considered as duplicates when they cover the same region with an approximation J. Since TRF and ATRHunter have been executed with options that discard all TRs having a score lower than a given threshold, we filtered TTRS by removing all the TRs with a score under such value (this has been done to not penalize TRF and ATRHunter with respect to TRStalker). More in detail, TRF has been executed with match, mismatch and indel score equal to 2, 3 and 3, respectively, maximum motif length equal to 2000bp2, and threshold equal to 30. ATRHunter has been executed with match, mismatch, gap and terminal gap score equal to 1 0 -1 0, maximum motif length equal to 500bp3 and threshold equal to 30. For the TRs found by TRStalker, the score is computed by using the same weights used by TRF and ATRHunter then we filtered the results using the same threshold. After the filtering phase, we computed the union of the TRs found by all algorithms, 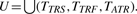 . The removal of duplicates with threshold J is also applied to U. Naturally the higher the value of J less filtering will be performed.
. The removal of duplicates with threshold J is also applied to U. Naturally the higher the value of J less filtering will be performed.
4 DISCUSSION
We have performed comparative experiments both with synthetic and with biological sequences. Here we describe the experimental set up, how the synthetic sequences are generated and the outcome of the comparison. For biological data, we briefly indicate the reason why that sequence has been selected, and the new TRs found by the application of TRStalker.
4.1 Synthetic data
4.1.1 Generation of synthetic data
We carried out a first set of experiments by using synthetic data. This allows a fine grained control on the amount of mutations introduced within the regions covered by the TRs. The sequences we gave as input to the programs have been built according to the following steps:
the background sequence is generated by selecting the four bases A,C,G and T with equal probability;
a perfect TR is embedded within the previous sequence, the TR is generated as r repetitions of a motif with length l;
the region covered by the TR is mutated according to substitution, insertion and deletion probabilities (ps, pi and pd); the number of substitutions, insertions and deletion for every repetition of the motif is exactly equal to lps, lpi and lpd; and
if the TR is a Steiner-STR, mutations are introduced in every repeat with respect to the consensus motif; if the TR is a NTR, mutations are introduced with respect to the previous repeat.
The experiments have been carried out running ATRHunter with these parameters: match, mismatch, gap and terminal gap score equal to 1 0 -1 0 (the most permissive setting on the website); maximum motif length equal to 500 bp (the maximum allowed by the tool). In order to select the definition of TRs among those allowed by ATRHunter, we performed a preliminary set of experiments: the definition that gave the best results was the third one (minimum alignment score). In this case, ATRHunter reports only the TRs that have a score higher than a given threshold. The value of the threshold has been set to 30.
For the web-based version of TRF, all the experiments have been carried out with these parameters: match, mismatch and indel score equal to 2, 3 and 5, respectively; maximum period equal to 500; minimum score equal to 30. For the binary version, we used the following ones: match, mismatch and indel score equal to 2, 3 and 3, respectively; match and indel probability equal to 0.75 and 0.20; maximum period equal to 500; minimum score equal to 30. The parameters of the experiments have been set so as to make sure that the minimum allowed score for all the tools tested is attained on the input data. TRStalker is run with the error parameter μ = 0.3 and the constant c = 1.5.
4.1.2 Discussion of the comparative experiments
For the experiments on NTR (Fig. 1), we tested TRs with motifs of length from 60 to 300, and a number of repeats from 2 to 8. TRStalker has recall always above 95%. TRF (binary) has always a recall above 80% except for TR with repeat number 2 for which the recall drops to 60%. ATRHunter has recall of a about 60%. These experiments confirm the effectiveness of the new techniques for the initial filtering steps.
Fig. 1.

BMS as a function of copy number for NTR. Motif lengths 60 (a), 100 (b), 100 (c) and 300 (d). The total length of the input sequence is 10 000 bp; the amount of substitutions, insertions and deletions are equal to 10% of the motif length each (thus with total error allowed of 30%). Every point is the average of 30 measurements and the 95% confidence intervals are shown.
Results on Steiner-STR with motifs of length from 60 to 300, and a number of repeats from 2 to 8 are shown in Figure 2. Here we notice that all methods have degraded performance for longer motifs (>200 bases) while TRStalker still manages to have recall above 60%. For shorter motifs (of <100 bases) TRF (binary) is able to match TRStalker only when the repeat number is above 6. Thus for a large range of values, TRStalker attains the best performance in recall, or a matching one, always above 80%.
Fig. 2.

BMS as a function of copy number for Steiner-STR. Motif lengths 60 (a), 100 (b), 200 (c) and 300 (d). The total length of the input sequence is 10 000 bp; the amount of substitutions, insertions and deletions are equal to 10% of the motif length each (thus with total error allowed of 30%). Every point is the average of 30 measurements and the 95% confidence intervals are shown.
The time performance of TRStalker has not been yet optimized. At the moment it is within an order of magnitude of TRF and ATRHunter. More details on the running time are in the Supplementary Materials.
4.2 Biological sequences
Testing of TRStalker on biological sequences has confirmed the potential of our method for finding very fuzzy TRs not detected by TRF and ATRHunter, and, to the best of our knowledge, not reported in literature. We tested the following sequences:
U43748 Homo sapiens frataxin gene, promoter region and exon—2465 bp long (FRDA).
L3609 Homo sapiens germline T-cell receptor βchain, complete gene—684 973 bp long (HSBT).
NC_001133.8 Saccharomyces cerevisiae Chromosome I—230 208 bp long (YCh1).
4.2.1 Experimental settings
The three algorithms have been run with the setting used in the synthetic experiments4 (thus with a very permissive acceptance policy). In general, none of the three algorithms generates all TRs found by the two others, and in Table 1 we show the percentage of the TRs found by each algorithm with respect to the union of the TRs found. In Table 2, we report some very long TRs that were detected by TRStalker but missed by the other two methods. We check the motif/repeat alignments using the tool jaligner (http://jaligner.sourceforge.net/) using the BLOSUM62 score matrix, that confirms the good quality of the motifs found (Table 3).
Table 1.
Evaluation of recall for the three methods under evaluation
| Algorithm | Filter 90% | Filter 70% |
|---|---|---|
| Frataxin | ||
| TRStalker (TRF filter) | 59 (56.2) | 43 (56.5) |
| TRStalker (ATR filter) | 43 (41.0) | 30 (39.4) |
| TRF | 24 (22.9) | 18 (23.6) |
| ATRHunter | 24 (22.9) | 23 (30.2) |
| Union | 105 (100.0) | 76 (100.0) |
| Homo sapiens T-cell receptor β chain | ||
| TRStalker (TRF Filter) | 22 557 (59.1) | 14 137 (60.2) |
| TRStalker (ATR Filter) | 18 124 (47.5) | 11 427 (48.7) |
| TRF | 9977 (26.1) | 8521 (36.0) |
| ATRHunter | 7392 (19.3) | 7034 (29.6) |
| Union | 38218 (100.0) | 23743 (100.0) |
| Saccharomyces cerevisiae chromosome I | ||
| TRStalker (TRF Filter) | 7168 (61.8) | 4656 (63.5) |
| TRStalker (ATR Filter) | 5621 (48.4) | 3655 (49.9) |
| TRF | 2892 (24.9) | 2518 (34.1) |
| ATRHunter | 2037 (17.6) | 1958 (26.4) |
| Union | 11 616 (100.0) | 7407 (100.0) |
Each entry in the table gives the absolute number of unique TR found, and in the percentage of unique TR w.r.t the union of the three methods. For TRSTalker, we used both a TRF-like and an ATRHunter-like filtering (more restrictive) on the TRs found.
Table 2.
Examples of TRs found by TRStalker and missed by TRF and ATRHunter
| No. | Sequence | Seq. length | TR start | TR end | TR length | Consensus | Repetitions | Score | Norm. score |
|---|---|---|---|---|---|---|---|---|---|
| 1 | HSBT | 684 973 | 411 000 | 413127 | 2127 | 1061 | 2.00 | 2868 | 1.384 |
| 2 | HSBT | 684 973 | 448 001 | 449687 | 1686 | 842 | 2.00 | 2310 | 1.370 |
| 3 | HSBT | 684 973 | 636 116 | 638622 | 2506 | 1253 | 2.00 | 3323 | 1.326 |
| 4 | YCh1 | 230 208 | 186 168 | 188347 | 2179 | 1089 | 2.00 | 3053 | 1.401 |
| 5 | FRDA | 2465 | 2029 | 2407 | 378 | 188 | 2.011 | 501 | 1.325 |
We report the original sequence name and length, the TR starting and ending positions, the TR length and the TR repeating unit length and copy number. The score is computed by assigning +2 to matches and −1 to mismatches and gaps w.r.t the consensus string. The normalized score is the score divided the TR length.
Table 3.
Motif/repeats alignment scores computed by jaligner using the BLOSUM62 score matrix with gap open penalty set to 10.0 and gap extend penalty set to 0.5 for the TRs reported in Table 2
| Seq. | No. | Repeat | Length, N | Identity, n(%) | Gaps, n(%) | Score |
|---|---|---|---|---|---|---|
| HSBT | 1 | 1 | 1107 | 805(72.72) | 91 (8.22) | 3657.00 |
| - | 1 | 2 | 1093 | 895(81.88) | 70 (6.40) | 4291.00 |
| HSBT | 2 | 1 | 878 | 638(72.67) | 85 (9.68) | 3045.50 |
| - | 2 | 2 | 866 | 716(82.68) | 52 (6.00) | 3568.00 |
| HSBT | 3 | 1 | 1300 | 1000(76.92) | 94 (7.23) | 5206.00 |
| - | 3 | 2 | 1313 | 1004(76.47) | 120 (9.14) | 5176.50 |
| YCh1 | 4 | 1 | 1130 | 895(79.20) | 83 (7.35) | 4280.50 |
| - | 4 | 2 | 1123 | 901(80.23) | 77 (6.86) | 4345.50 |
| FRDA | 5 | 1 | 193 | 149(77.20) | 10 (5.18) | 723.50 |
| - | 5 | 2 | 191 | 146(76.44) | 5 (2.62) | 765.00 |
4.2.2 Frederich's ataxia
Frederich's ataxia is an autosomal recessive degenerative disease involving the central and peripheral nervous system and the heart, that roughly affects one person in 50 000 (Wells, 2008). In 1996, it was shown (Campuzano et al., 1996) that in 98% of the cases this disease was caused by an abnormal expansion in the copy number of a triplet TR in the first intron of the Frataxin coding sequence. It belongs to the family of trinucleotide repeat disorders. Very recently (Vissers et al., 2009) it has been shown that the local repetitive structure of DNA may play a role in variable copy number genomic disorders. Applying TRStalker to the frataxin sequence we detected a divergent TR in positions [2036–2414], of period 188 and copy number 2, to the best of our knowledge not previously reported, that includes the breakpoint region of the repeat disorder (Table 2). Experimental data reported in Brodzik (2007) on the Frataxin sequence did find a number of short TRs (of period up to 10/13) that are completely covered by the longer fuzzy TR reported by TRStalker.
4.2.3 Human beta T-cell receptor locus
The cellular immune system detects the presence of pathogens largely through the activation of T-cell receptor proteins (TCR) (Glusman et al., 2001), which come in four different families α, β, γ and δ. The complete DNA sequence of the human β T-cell receptor locus has been determined (Rowen et al., 1996) and it has been found that a large fraction of the locus sequence (about 47%) is formed by locus-specific repeats (Rowen et al., 1996). This sequence was selected as a test case for TRStalker because of its richness in repeating elements with the aim of highlighting the ability of TRStalker in finding repeats with high divergence among adjacent copies. Here, (Table 2) we could find a few such repeats apparently not recorded in the GenBank: L36092.2 record, nor found by TRF and ATRHunter (still set with very loose parameters).
4.2.4 Yeast chromosome I
Saccharomyces cerevisiae (baker's yeast) has been the focus of intensive study as the first eukaryotic organism whose genome was completely sequenced (Dujon, 1996), and serves as a model organism in basic genomic investigations. Chromosome I (Bussey et al., 1995) is the smallest of the 16 chromosomes present in yeast. It has been noticed that the yeast genome is remarkably poor in repeated elements (Dujon, 1996), thus finding new TRs in such organism is a challenging task for any algorithm. In Table 2 we report a TR in position [186168, 188347] of copy number 2 and motif length 1089. This TR is not reported in the TRDB database, while ATRHunter in the same region finds 15 shorter TR of length ranging from 50 to 180. This region, according to the NCBI record, is rich in genes of the DUP240 gene family (encoding membrane proteins). The presence of a fuzzy repeat in this region thus suggests a possible remote gene duplication event.
4.2.5 Performance on biological sequences
Reporting interesting single new TRs, as in Table 2, is useful to demonstrate that biological relevant TRs are still unknown. We give also an evaluation of the overall behavior of the three different methods on biological sequences. Thus, we compared TRStalker, TRF and ATRHunter by estimating their recall on the three biological sequences with the methodology described in Section 3.10. Table 1 reports (i) the number of unique TRs found by the different algorithms and (ii) the percentage of the union reported by a given algorithm, with two filtering thresholds at J = 90% and J = 70%. For all the three sequences, TRStalker is able to find a large number of TRs that are not discovered by using the other methods. In practice, a better overall coverage can be attained by using all three methods and merging their results. Although lower J values imply a more aggressive filtering, the percentage of the union attained by TRStalker is almost constant.
5 CONCLUSION
TRStalker is a novel efficient heuristic algorithm for finding Fuzzy TRs in biological sequences. TRStalker aims at improving the capability of TR detection for a class of fuzzy TRs for which existing methods do not perform well. Initial testing on biological data show that fuzzy TRs not previously reported are present in biologically relevant sequences. In the case of the Frataxin sequence, the fuzzy TR reported is associated with the known variable copy number breakpoint of Frederich's ataxia. Future work will involve testing TRStalker on relevant families of repetitive elements such as centromeric α-satellites. An extension of TRStalker to handle amino acid sequences is under development.
Funding: European Community's Seventh Framework Programme FP7/2007-2013 under grant agreement N 223920 (Virtual Physiological Human Network of Excellence) partially.
Conflict of Interest: none declared.
Supplementary Material
Footnotes
1A precise characterization of the relative gain under different error models would be theoretically interesting but is now beyond the focus of this article. Selecting larger values of q and s, as a function of the period to be detected and the error level, may increase the filtering ability of the method at the cost of slower computations. Exploring these connections is left for future research.
2Maximum possible value for TRF.
3Maximum possible value for ATRHunter.
4For TRF the maximum motif length has been raised to 2000 bp.
REFERENCES
- Ames D, et al. Comparative analyses of human single- and multilocus tandem repeats. Genetics. 2008;179:1693–1704. doi: 10.1534/genetics.108.087882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson G. An algorithm for finding tandem repeats of unspecified pattern size. In: Istrail S, Pevzner P, Waterman M, editors. Proceedings of the Second Annual international Conference on Computational Molecular Biology (New York, New York, United States, March 22–25, 1998) New York, NY: RECOMB '98. ACM; 1998. pp. 20–29. [Google Scholar]
- Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–580. doi: 10.1093/nar/27.2.573. Available at http://tandem.bu.edu/trf/trf.html (last accessed date April 27, 2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeva V, et al. Short fuzzy tandem repeats in genomic sequences, identification, and possible role in regulation of gene expression. Bioinformatics. 2006;22:676–684. doi: 10.1093/bioinformatics/btk032. [DOI] [PubMed] [Google Scholar]
- Brodzik AK. Quaternionic periodicity transform: an algebraic solution to the tandem repeat detection problem. Bioinformatics. 2007;23:694–700. doi: 10.1093/bioinformatics/btl674. [DOI] [PubMed] [Google Scholar]
- Buchner M, Janjarasjitt S. Detection and visualization of tandem repeats in DNA sequences. IEEE Trans. Signal Process. 2003;51:2280–2287. [Google Scholar]
- Burkhardt S, Kärkkäinen J. One-gapped q-grams filters for levenshtein distance. In: Apostolico A, Takeda M, editors. Combinatorial Pattern Matching, 13th Annual Symposium, CPM 2002, Fukuoka, Japan, July 3–5, 2002, Proceedings. Vol. 2373. Lecture Notes in Computer Science, Springer; 2002. pp. 225–234. [Google Scholar]
- Burkhardt S, Kärkkäinen J. Better filtering with gapped q-grams. Fundam. Inform. 2003;56:51–70. [Google Scholar]
- Bussey H, et al. The nucleotide sequence of chromosome I from Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA. 1995;92:3809–3813. doi: 10.1073/pnas.92.9.3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campuzano V, et al. Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423–1427. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- de la Higuera C, Casacuberta F. Topology of strings: median string is np-complete. Theor. Comput. Sci. 2000;230:39–48. [Google Scholar]
- Dujon B. The yeast genome project: what did we learn? Trends Genet. 1996;12:263–270. doi: 10.1016/0168-9525(96)10027-5. [DOI] [PubMed] [Google Scholar]
- Elemento O, Gascuel O. Proceedings of the European Conference on Computational Biology (ECCB 2002) Germany: Saarbrücken; 2002. An efficient and accurate distance based algorithm to reconstruct tandem duplication trees; pp. 92–99. October 6-9, 2002 (Supplement of Bioinformatics) [DOI] [PubMed] [Google Scholar]
- Fischetti VA, et al. Identifying periodic occurrences of a template with applications to protein structure. Inf. Process. Lett. 1993;45:11–18. [Google Scholar]
- Gelfand Y, et al. TRDB - the tandem repeats database. Nucleic Acids Res. 2007;35(Database Issue):80–87. doi: 10.1093/nar/gkl1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glusman G, et al. Comparative genomics of the human and mouse T cell receptor loci. Immunity. 2001;15:337–349. doi: 10.1016/s1074-7613(01)00200-x. [DOI] [PubMed] [Google Scholar]
- Grissa I, et al. The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinformatics. 2007;8:172. doi: 10.1186/1471-2105-8-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R, et al. A novel signal processing measure to identify exact and inexact tandem repeat patterns in DNA sequences. EURASIP. J. Bioinform. Syst. Biol. 2007 doi: 10.1155/2007/43596. [Epub ahead of print, doi: 10.1155/2007/43596, March 13, 2007] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusfield D. Algorithms on Strings, Trees, and Sequences - Computer Science and Computational Biology. Cambridge, U.K.: Cambridge University Press; 1997. [Google Scholar]
- Gusfield D, Stoye J. Linear time algorithms for finding and representing all the tandem repeats in a string. J. Comput. Syst. Sci. 2004;69:525–546. [Google Scholar]
- Hauth AM, Joseph D. Proceedings of the Tenth International Conference on Intelligent Systems for Molecular Biology. Alberta, Canada: Edmondon; 2002. Beyond tandem repeats: complex pattern structures and distant regions of similarity; pp. 31–37. August 3-7, 2002. [DOI] [PubMed] [Google Scholar]
- Jiang X, et al. Dynamic computation of generalised median strings. Pattern Anal. Appl. 2003;6:185–193. [Google Scholar]
- Jurka J, et al. Repbase update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 2005;110:462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- Kelkar Y.DD, et al. The genome-wide determinants of human and chimpanzee microsatellite evolution. Genome Res. 2008;18:30–38. doi: 10.1101/gr.7113408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolpakov R, Kucherov G. Finding approximate repetitions under Hamming distance. Theor. Comput. Sci. 2003;303:135–156. [Google Scholar]
- Kolpakov R, et al. mreps: efficient and flexible detection of tandem repeats in DNA. Nucleic Acids Res. 2003;31:3672–3678. doi: 10.1093/nar/gkg617. http://bioinfo.lifl.fr/mreps/(last accessed date April 27, 2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolpakov RM, Kucherov G. Proceedings of the 40th Annual Symposium on Foundations of Computer Science (October 17–18, 1999). FOCS. Washington, DC: IEEE Computer Society; 1999. Finding maximal repetitions in a word in linear time; p. 596. [Google Scholar]
- Krishnan A, Tang F. Exhaustive whole-genome tandem repeats search. Bioinformatics. 2004;20:2702–2710. doi: 10.1093/bioinformatics/bth311. [DOI] [PubMed] [Google Scholar]
- Kurtz S, Schleiermacher C. Reputer: fast computation of maximal repeats in complete genomes. Bioinformatics. 1999;15:426–427. doi: 10.1093/bioinformatics/15.5.426. [DOI] [PubMed] [Google Scholar]
- Kurtz S, et al. Reputer: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001;29:4633–4642. doi: 10.1093/nar/29.22.4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau GM, et al. An algorithm for approximate tandem repeats. J. Comput. Biol. 2001;8:1–18. doi: 10.1089/106652701300099038. [DOI] [PubMed] [Google Scholar]
- Leclercq S, et al. Detecting microsatellites within genomes: significant variation among algorithms. BMC Bioinformatics. 2007;8:1–125. doi: 10.1186/1471-2105-8-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legendre M, et al. Sequence-based estimation of minisatellite and microsatellite repeat variability. Genome Res. 2007;17:1787–1796. doi: 10.1101/gr.6554007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motwani R, Raghavan P. Randomized Algorithms. Cambridge, UK: Cambridge University Press; 1995. [Google Scholar]
- Mudunuri SB, Nagarajaram HA. Imex: Imperfect microsatellite extractor. Bioinformatics. 2007;23:1181–1187. doi: 10.1093/bioinformatics/btm097. [DOI] [PubMed] [Google Scholar]
- Mulmuley K. Computational Geometry, an Introduction through Randomized Algorithms. Englewood Cliffs, New Jersey: Prentice Hall; 1993. [Google Scholar]
- O'Dushlaine C, et al. Tandem repeat copy-number variation in protein-coding regions of human genes. Genome Biology. 2005;6:R69. doi: 10.1186/gb-2005-6-8-r69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi V, et al. String: finding tandem repeats in DNA sequences. Bioinformatics. 2003;19:1733–1738. doi: 10.1093/bioinformatics/btg268. [DOI] [PubMed] [Google Scholar]
- Peterlongo P, et al. Lossless filter for multiple repeats with bounded edit distance. Algorithms Mol. Biol. 2009;4:1–3. doi: 10.1186/1748-7188-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivals E. A survey on algorithmic aspects of tandem repeats evolution. Int. J. Found. Comput. Sci. 2004;15:225–257. [Google Scholar]
- Rivals E, et al. Detection of significant patterns by compression algorithms: the case of approximate tandem repeats in DNA sequences. Comput. Appl. Biosci. 1997;13:131–136. doi: 10.1093/bioinformatics/13.2.131. [DOI] [PubMed] [Google Scholar]
- Rowen L, et al. The complete 685-kilobase DNA sequence of the human beta T Cell Receptor Locus. Science. 1996;272:1755–1762. doi: 10.1126/science.272.5269.1755. [DOI] [PubMed] [Google Scholar]
- Saha S, et al. Empirical comparison of ab initio repeat finding programs. Nucleic Acids Res. 2008;36:2284–2294. doi: 10.1093/nar/gkn064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sammeth M, Stoye J. Comparing tandem repeats with duplications and excisions of variable degree. IEEE/ACM Trans. Comput. Biology Bioinform. 2006;3:395–407. doi: 10.1109/TCBB.2006.46. [DOI] [PubMed] [Google Scholar]
- Sharma D, et al. Spectral repeat finder (SRF): identification of repetitive sequences using fourier transformation. Bioinformatics. 2004;20:1405–1412. doi: 10.1093/bioinformatics/bth103. [DOI] [PubMed] [Google Scholar]
- Sim JS, Park K. The consensus string problem for a metric is np-complete. J. Discrete Algorithms. 2003;1:111–117. [Google Scholar]
- Smit A.F.A, et al. (1996–2004). [last access date April 27, 2010]. Repeatmasker open-3.0. http://www.repeatmasker.org/ [Google Scholar]
- Sokol D, et al. Tandem repeats over the edit distance. Bioinformatics. 2007;23:30–35. doi: 10.1093/bioinformatics/btl309. [DOI] [PubMed] [Google Scholar]
- Stolovitzky G, et al. Technical Report RC21508. IBM T.J. Watson Research Labs; 1999. Tandem repeat detection using pattern discovery with applications to the identification of yeast satellites. [Google Scholar]
- Vissers LE, et al. Rare pathogenic microdeletions and tandem duplications are microhomology-mediated and stimulated by local genomic architecture. Hum. Mol. Genet. 2009;18:3579–3593. doi: 10.1093/hmg/ddp306. [DOI] [PubMed] [Google Scholar]
- Vogler A, et al. Effect of repeat copy number on variable-number tandem repeat mutations in Escherichia coli O157:H7. J. Bacteriol. 2006;188:4253–4563. doi: 10.1128/JB.00001-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburton P, et al. Analysis of the largest tandemly repeated DNA families in the human genome. BMC Genomics. 2008;9:533. doi: 10.1186/1471-2164-9-533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells RD. DNA triplexes and Friedreich ataxia. FASEB J. 2008;22:1625–1634. doi: 10.1096/fj.07-097857. [DOI] [PubMed] [Google Scholar]
- Wexler Y, et al. Proceedings of the Eighth Annual International Conference on Resaerch in Computational Molecular Biology (RECOMB 2004) New York, NY, USA: ACM; 2004. Finding approximate tandem repeats in genomic sequences; pp. 223–232. [DOI] [PubMed] [Google Scholar]
- Wexler Y, et al. Finding approximate tandem repeats in genomic sequences. J. Comput. Biol. 2005;12:928–942. doi: 10.1089/cmb.2005.12.928. [DOI] [PubMed] [Google Scholar]
- Wooster R, et al. Instability of short tandem repeats (microsatellites) in human cancers. Nat. Genet. 1994;6:152–156. doi: 10.1038/ng0294-152. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.