

Abstract
We recently described a set of drug-like molecules obtained from an in silico screen that stabilize mutant superoxide dismutase-1 (SOD-1) linked to familial amyotrophic lateral sclerosis (ALS) against unfolding and aggregation but exhibited poor binding specificity towards SOD-1 in presence of blood plasma. A reasonable, but not a conclusive model for the binding of these molecules, was proposed based on restricted docking calculations and site-directed mutagenesis of key residues at the dimer interface. A set of hydrogen bonding constraints obtained from these experiments were used to guide docking calculations with compound library around the dimer interface. A series of chemically unrelated hits were predicted, which were experimentally tested for their ability to block aggregation. At least six of the new molecules exhibited high specificity of binding towards SOD-1 in presence of blood plasma. These molecules represent a new class of molecules for further development into clinical candidates.
Introduction
Protein misfolding diseases such as Alzheimer's disease, Parkinson's disease, Huntington disease, Familial polyneuropathy, Gaucher's disease and amyotrophic lateral sclerosis (ALS) are related to aggregation of proteins often caused by loss of protein stability 1-5. Point mutations - the most common cause of human genetic diseases - can inhibit correct folding and assembly of the protein, cause mistrafficking in the endoplasmic reticulum and in the majority of cases aggregation into toxic structures 1-5. These diseases can occur with the wild-type proteins, as mutations are not absolutely required, although mutations can often accelerate the onset of diseases. Traditional approaches of ‘function based screening’ and drug development are not generally applicable to these protein misfolding diseases since such approaches are designed to screen for antagonistic ligands6-8. Although small molecule enzyme inhibitors in some cases have been shown to stabilize proteins against misfolding, they also interfere in enzyme function 9,10. Pharmacological chaperones are small molecules that can bind to their protein target and function to either inhibit aggregation, block mistrafficking and/or protect cells from death without interfering with normal protein function1,9,11-16. Though osmolytes have been shown to function as general chemical chaperones12-14, there is also precedence for small molecules as specific pharmacological chaperones in the case of familial amyloid polyneuropathy (FAP), caused by mutations in the gene encoding transthyretin (TTR) and Gaucher's disease, caused by misfolding of glucocerebrosidase 6,8-10,17-23. Many FAP mutations destabilize the native TTR tetramer, facilitating its dissociation, partial unfolding, and aggregation. The natural ligand of TTR, thyroxine, stabilizes the tetramer and prevents its aggregation in vitro. Drug-like molecules that are thyroxine analogs bind and stabilize the native TTR tetramer, preventing its aggregation in vitro 6,8,17-21. These compounds could potentially be used for the treatment of FAP. Similarly, mutations in glucocerebrosidase lead to misfolding and mislocalization of the protein in vivo. Inhibitors of this enzyme have been shown to significantly stabilize the protein and lead to correct subcellular localization 23. In this manuscript, we describe a methodology for improving binding specificity of pharmacological chaperones for targeting novel binding sites on superoxide dismutase-1 (SOD-1), which has been linked to familial ALS.
Amyotrophic lateral sclerosis (ALS), or Lou Gehrig's disease, is a fatal motor neuron disease that affects > 50,000 Americans and many more individuals worldwide. Though most ALS cases are sporadic with no genetic disposition, a small percentage of patients carry point mutations in the gene-encoding SOD-1, a β-sheet-rich dimeric metalloenzyme that is normally responsible for scavenging superoxide ions24. Familial ALS (FALS) does not appear to arise from a loss of this important activity but rather from a “gain of toxic function” due to aggregation of the mutant form of SOD-1. There are 114 known SOD-1 FALS mutations, which are distributed throughout the primary sequence and tertiary structure25. It is proposed that these mutations affect the structural stability of SOD-1. One or more FALS SOD-1 mutations have been linked to decreased metal binding, decreased formation of a stabilizing intramolecular disulfide, decreased structural stability, and increased propensity to monomerize and aggregate26-38. Though the consequences of the mutations may differ, the common underlying factor in all cases is the loss of thermodynamic stability29,34,39-43. Analyzing a number of mutants in simulation studies revealed long range effects of widely distributed mutations on the stability of the dimer interface of SOD-140,44,45.
Though increasing metallation as a strategy to preserve stability has been suggested as a therapeutic approach46-48, we have pursued an alternate strategy to inhibit SOD-1 aggregation with small drug-like molecules, which act as pharmacological chaperones 47,49. Although no natural ligands are known for SOD-1, we have recently described a number of drug-like molecules identified from an in silico screen (docking calculations), which bind at the dimer interface and stabilize SOD-1 against aggregation and unfolding 50. Potent inhibition of SOD-1 aggregation, though necessary, is not sufficient for efficacy in humans. An ideal drug molecule should be specific for SOD-1 and have low binding specificity towards other human proteins in order to reduce toxic side-effects51. In this manuscript, we use a series of structurally related compounds and mutagenesis of the protein to arrive at a model for compound binding, and use structural features (docking constraints) derived from this model to identify novel compounds from a database with improved ADME properties. A general outline for the strategy used in this manuscript to identify new compounds based on existing hits is shown in supplemental figure 1. The preliminary structure-activity relationship from this investigation can be utilized for further optimization of the pharmacological chaperones.
Materials and Method
Chemicals and reagents
All solvents, chemicals and protein purification reagents, unless otherwise stated, were commercially available, were of the best grade, and were used without further purification. Synthesis and characterization of 14, 13 and 14 is described in the supporting information section. All compounds used in this study were homogeneous (purity of not less than 98%), as determined by high-performance liquid chromatography (HPLC).
Cloning, purification and enzymatic activity
Genes encoding human SOD-1, WT and A4V, were cloned by a PCR strategy into pGex6P1 (Amersham) using the BamH1 and Xho1 restriction sites of the vector. SOD-1 was expressed as a glutathione-S-transferase (GST) fusion in the Rosetta E.coli strain (Novagen). Purification of SOD-1 and its mutants was carried out as described previously52. The superoxide dismutase activity of purified SOD-1 and the mutants was measured by a coupled enzyme assay using xanthine oxidase to generate superoxide. SOD-1 activity was measured colorimetrically with (2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide).
Preparation of apo-protein and ICP-MS analysis
Metal-free SOD was generated by repeated dialysis (5 times) against 100mM acetate buffer pH 3.8 and 10mM EDTA. Following this dialysis, the sample was dialyzed against 100mM acetate buffer pH 5.5 containing 100mM NaCl 5 times to remove EDTA from the sample. All buffers used in this experiment except the ones containing EDTA were passed over 250ml of chelex 100 resin to ensure complete removal of any trace copper or zinc in the buffer. Aggregation assay was carried out in plastic tubes rather than glass vials to prevent introduction of stray copper or zinc into the sample. The compounds used for this analysis were diluted from their stock solutions into chelex 100-treated water prior to their addition into the aggregation assay.
Quantitative metal analyses of apo-protein samples were conducted using a Perkin Elmer 6100DRC ICP-MS. Sample aspiration into the ICP-MS was through a quartz Meinhard concentric nebulizer and a cyclonic spray chamber. Gas flow was set at 1.08, 1.225 and 18.5L·min-1 for the nebulizer, auxiliary and plasma flows respectively and the incident R.F. power was set at 1250 Watts. The instrument was tuned using a 10ng·mL-1 solution of Li, Mg, Ce, Co, In, Ba and U to minimize oxide adduct formation and doubly charged species without unduly compromising sensitivity. Count rates were typically better than 600× 103 cps for 115In with a RSD of <0.5%. The Ce:CeO ratio was typically < 0.02 under these operating conditions.
Protein solution extracts together with appropriate buffer and dialysis reagent blanks were diluted 3 × using 2% HNO3 prepared in 18μ Ohm water containing 10 μg·L-1 of gallium. The gallium was used as an internal standard to correct for changes in instrumental sensitivity, sample viscosity and aspiration rate during ICP-MS analysis. Acid blanks containing gallium were prepared in a similar manner. Data acquisition was conducted in the peak-hopping mode. Five readings were taken per sample with a dwell time of 100msec·amu-1 and 20 sweeps per reading. The Zn and Cu content of each sample was quantified from response curves generated for 64Zn, 66Zn and 68Zn, 63Cu and 65Cu from a dilution series of certified metal standard solutions of known concentration. Linear responses (r2 > 0.999) were observed over a dynamic range of four orders of magnitude of metal concentration. Individual analyses of the standards, acid blanks and samples were corrected for variations in the response for the internal standard, and the content of metal in each sample was corrected for dilution and normalized for protein concentration after subtraction of the acid blank. Highly comparable values were obtained for the other isotopes analyzed, indicating minimal interference effects from the matrix (data from 64Zn and 65Cu responses shown). Overall, accuracy was confirmed by the analysis of five independent replicate samples of certified standards and was better than ± 3%. Detection limits, as determined by the values obtained from the analysis of multiple acid blanks, were better than 0.2 μg·L-1 for Cu and Zn.
Database preparation and Docking
All computations were carried out on an 16-node Beowulf Cluster (each node∼ 3.0Ghz quad-core Xeon processor) running RedHat Linux (http://www.redhat.com). Structure data files for small molecules from 11 commercially available databases were consolidated and filtered to remove duplicates. These molecules are from updated versions of the same databases that we used in our previous publication50. 2.2 million molecules consolidated from these databases were treated with the ligprep module of Schrödinger First Discovery Tool (Schrödinger Inc.) to remove counter ions, adjust charge states and generate tautomers wherever applicable. The final database was stored in maestro (Schrödinger Inc. proprietary format). The protein was prepared for docking using the protein prep utility of GLIDE. Following removal of water molecule coordinates from the first dimer in the asymmetric unit SOD-1A4V structure (1UXM.pdb), hydrogen atoms were added using the applyhtreat suite of GLIDE and energy minimized using the OPSLAA forcefield. Positions of hydrogen bonds in the structure were refined using the Schrödinger protassign utility. The protassign utility simply uses a set of rules to generate an optimal hydrogen bonding network in the structure in order to resolve ambiguity in protonation of residues such as histidine. Residue 148 was chosen as the center of mass for generation of a 20Å grid for docking calculations. The program Glide (part of the Schrödinger First Discovery Suite) was used for docking. The first main docking job was carried out using the default Glide parameters. The default glide parameters allow for flexible ligand docking but not flexible protein. In case of constrained docking, hydrogen constraint parameters were placed on Val 7 and Asn 53 prior to grid calculations. The desired number of hydrogen bonds was selected at the time of actual docking. To facilitate computation, the main job was split into subjobs using the paraglide utility to allow for parallel processing on the linux cluster. The results obtained from the first round of docking were docked again using the Glide extra precision mode which exacts severe penalties for complexes that violate established principles, including ligand stereochemistry, and solvent exposure of charged groups. The results of docking were analyzed using the pose viewer utility built into Glide. Shape comparison was carried out, which evaluates integral overlap between the compounds.
Gel filtration and aggregation assay
Dimeric SOD-1 was purified on a superdex 75 (16/60) gel filtration column (Pharmacia) to produce starting material for each aggregation experiment. Incubation was performed at 37°C and aliquots were periodically removed and analyzed by gel filtration on a superdex 200 (3.2/30) gel filtration column (Pharmacia). All chromatography was performed in TBS (20 mM Tris pH 7.4, 150 mM NaCl) on a Waters 2695 Alliance HPLC, and monitored at 220 nm and 276 nm. 5mM EDTA was added to induce aggregation prior to incubation at 37°C. In order to test the effect of compounds on aggregation, 75μM of compound was added to 25μM of apoprotein and incubated at 37°C for 15 min. Following this incubation period, 5mM EDTA was added to initiate aggregation. To test the effect of compound concentration, samples were prepared as above with increasing concentrations of compounds and aggregation was allowed to proceed for 4 hrs prior to analysis. Samples were analyzed using size exclusion chromatography and amount of dimer was estimated from area under the peak. Percentage inhibition was calculated using the differences between the untreated A4V samples and samples treated with various compound concentrations.
Guanidinium chloride unfolding
Equilibrium unfolding transitions, as a function of GdnCl concentration, were monitored by circular dichroism spectroscopy. SOD-1 samples with compounds (50μM) at a final concentration of 12μM in 20mM Tris (pH 7.4) / 150mM NaCl were equilibrated over a range of GdnCl concentrations from 0 - 5M at 0.2M intervals. Far UV CD spectra were recorded to measure changes in secondary structure content as a function of GdnCl concentration.
The slit width was 5 nm for both monochromators. Each measurement was an average of five readings. The data were analyzed directly for a two-state (N← →U) transition as follows: the raw data for the GdnCl-induced denaturation studies were converted to fractions of the protein in the unfolded state (fu) as a function of GdnCl concentration using the equation:
where Y0 is the observed spectroscopic property, Yf and mf are the slope and intercept of the folded state baseline and Yu and mu represent the respective values of the unfolded baseline. The folded fraction was calculated as (fn = 1-fu) and the equilibrium constant was determined by Keq = fu/fn. The free energy of unfolding was determined using the equation ΔG = -RT ln(Keq) where T is the temperature in Kelvin and R is the universal gas constant (1.987 cal mol-1 K-1).
CSF and plasma selectivity assay
GST-SOD fusion protein or GST (tag) at a final concentration of 75μM was added to 1 ml of human blood plasma or CSF and incubated with 100μM of the various compounds. This mixture was incubated on an orbital shaker at 60 rpm and 4°C for 24 hrs. A 1:1 gel/slurry (250 μl) of glutathione-sepharose (GE bioscience) was added to the solution and incubated on a gentle rocker at 20 rpm and 4°C for 1 h. The solution was centrifuged (16,000 × g) and the supernatant was discarded. 150μl of pre-chilled 5% TFA solution was added to the beads and incubated on a gentle rocker for 2 hrs. The samples were centrifuged (16,000 × g) and the supernatant was removed and split into batches of 50μl. The supernatant elution samples from the beads were directly loaded onto a Waters Alliance 2690 HPLC using an autosampler. The entire sample was injected for a single analysis and separated on a C18 reverse-phase column using a 40-100% gradient of solution B over 20 min (solution A: 94.8% water/5% acetonitrile/0.2% trifluoroacetic acid; solution B: 94.8% acetonitrile/5% water/0.2% trifluoroacetic acid). Pure compound was injected at a concentration of 25μM to obtain retention time in each case. A buffer control without blood plasma was treated the same way with the compounds and evaluated for compound binding.
α-synuclein aggregation assay
Samples of α-synuclein were dissolved in 50mM Tris-HCl (pH 8.0) and filtered through a Millipore Microcon 100K MWCO filter. Equimolar amounts of DJ-1 samples and α-synuclein were diluted to a final concentration of 20μM in 50mM Tris-HCl, pH 8.0 / 3.1% 1,1,1,3,3,3-hexafluoroisopropanol (HFIP). Aggregation was initiated by incubating the samples at room temperature with roller agitation. 1mM stock solution of thioflavin T (thio T, Sigma) was filtered through a 0.2-μm polyether sulfone filter and added to the sample wells at a final concentration of 20μM and incubated for 1 min prior to recording of fluorescence. Fluorescence was monitored at 490nm by an Analyst 96-384 plate reader (LJL Biosystems). Fluorescence readings were obtained at 5 min intervals for 1 h. Data points from three independent experiments were used for averaging and calculation of error bars. For testing compounds, samples of α-synuclein were dissolved in 50mM Tris-HCl and incubated with the small molecule at a final concentration of 100μM and pre-incubated on ice for 30 min prior to the addition of HFIP.
Results
Binding of small molecule to SOD-1 bearing aza-uracil and uracil sub-structures in buffer and blood plasma
Analysis of the hits obtained from the initial in silico screen 50 revealed four molecules that bear either a uracil or an aza-uracil substructure. Eleven new commercially available molecules (Supplementary figure 2) bearing both sub-structures and linkers were tested for their ability to block mutant SOD-1 aggregation (Figure 1a). The familial mutant, SOD-1A4V, was chosen for the aggregation assay since it is known to aggregate rapidly compared to other SOD-1 mutants at low protein concentrations. 25μM metal deficient SOD-1A4V (prepared using a method described previously) was allowed to aggregate in the presence of 75μM of small molecules and analyzed over size exclusion chromatography for aggregation kinetics. 5mM EDTA was also added to accelerate aggregation. SOD-1A4V aggregates rapidly, with greater than 50% of dimer lost within the first four hours of incubation at 37°C. Exceptionally good inhibitors led to less than 20% loss of dimeric SOD-1A4V over a period of 48 hrs, whereas poor inhibitors (or non-inhibitors) had no effect on accelerated aggregation. Seven molecules (1, 2, 3, 7, 8, 9 and 10) out of the eleven were found to be strong inhibitors of SOD-1 aggregation at a concentration of 75μM (Figure 1a). In general, it was observed that large molecules such as 11, which has greater solvent exposure and lacks conformational flexibility, were poor inhibitors of aggregation.
Figure 1.

(a) Aggregation of SOD-1A4V monitored using size-exclusion chromatography in presence of a series of compounds having either uracil or aza-uracil like substructures. The amount of dimer observed after 48hrs of incubation is plotted after normalization of the aggregation curves. Two molecules 4, 5 bear methylation at different positions on the aza-uracil ring and are indicated by (*) on top of the bar chart. (b) Unfolding of SOD-1A4V measured as a function of GdnCl. in presence of the small molecules tested in the left panel. The data have been fitted to a two-state model to facilitate the calculation of thermodynamics parameters shown in the table below (left). (c) Measurement of compound specificity towards a GST-SOD-1 fusion protein (green bars) in presence of buffer and blood plasma. Control experiments include binding in buffer (black bars) and GST (tag) alone (red bars).
The aggregation assay is sensitive enough to distinguish non-stabilizers from stabilizers, but it cannot accurately measure the differences between the seven stabilizers. We measured the effects of these compounds on the chaotrope-induced (GdnCl) denaturation of A4V. This appeared to be a valid approach in absence of a direct assay for measuring dimerization of A4V or binding of ligand. A4V (12 μM) was incubated with compound (50 μM) and increasing concentrations of GdnCl. Far-UV circular dichroism signal was measured as function of [GdnCl]. Typical transition curves for GdnCl-induced unfolding are shown in Figure 1b. In these curves, the straight lines indicate the pre-transition and post-transition regions of unfolding. The data were analyzed directly, assuming a two-state (N→U) transition, and thermodynamic properties were measured by fitting the data to a linear extrapolation model 53. The stabilization of A4V in the presence of the compounds was expressed as ΔG values, listed in Table 1.
Table-1.
| Protein/protein + compound | Free Energy ΔG Kcal Mol-1 | Free Energy of stabilization ΔΔG Kcal Mol-1 | m value Kcal mol-1 M-1 |
|---|---|---|---|
| A4V | 5.25 | 0 | 4.0 |
| WT | 12.45 | 7.2 | 3.9 |
| A4V + 1 | 8.56 | 3.31 | 4.2 |
| A4V + 7 | 8.89 | 3.64 | 3.8 |
| A4V + 3 | 10.99 | 5.74 | 4.1 |
| A4V + 2 | 9.78 | 4.53 | 4.2 |
| A4V + 9 | 11.09 | 5.84 | 3.8 |
| A4V + 10 | 11.77 | 6.52 | 4.1 |
| A4V + 8 | 10.99 | 5.74 | 3.9 |
| A4V + 12 | 12.01 | 6.76 | 4.2 |
| A4V + 14 | 11.99 | 6.74 | 3.8 |
| A4V + 13 | 11.98 | 6.73 | 4.1 |
The strongest SOD-1 aggregation inhibitors in buffer may not necessarily function well in biological fluids such as blood plasma or cerebrosipinal fluid (CSF), primarily due to competition from large amounts of plasma proteins such as blood plasma albumin. The top seven SOD-1 stabilizers were evaluated for their ability to bind a GST-SOD-1 fusion protein relative to the rest of the proteins in blood plasma. Similar approaches have been used by Kelly and co-workers to obtain chemical chaperones for blocking transthyretin aggregation (involved in FAP) in human plasma. While their assays used a specific antibody towards TTR, we used a fusion protein in our investigation. Both assays work on very similar principles 54-56. SOD-1 dimer (Kd in nM range) is a substantially stronger dimer compared to GST (Kd in μM range) and therefore the solution dimer will be dictated by SOD-157,58. It is reasonable to assume that the dimer interface cavity of SOD-1 will be intact and not affected by the GST tag. Briefly, GST-SOD fusion protein was added ex vivo to blood plasma and incubated with the various compounds (see materials and methods for details). The GST-SOD-drug complex was isolated using glutathione beads and analyzed using reverse phase HPLC after liberating compound from the complex using 5% TFA. A buffer control (no blood plasma) treated in an identical fashion was taken as the theoretical maximum in each case for calculating relative binding. For blood plasma binding, any molecules having greater than 50% binding specificity were categorized as a highly specific binder. Control experiments were also carried out where compound binding to GST (tag) in buffer was measured in an identical fashion. None of the molecules exhibited any specific binding affinity towards GST (Figure 1c). This ruled out the possibility of interference or competition towards compound binding to GST-SOD-1 from the tag. All seven molecules tested by this assay showed relatively poor binding selectivity towards the GST-SOD-1 fusion protein as shown in Figure 1c.
Modeling of aza-uracil and uracil based inhibitors and mutational analysis of the binding site
Non-specific binding to human proteins other than SOD-1 in blood plasma and/or CSF may lead to off-pathway toxicity; therefore it is necessary to obtain molecules with minimal binding to blood plasma and CSF proteins in order to be a viable lead compound. In order to improve the selectivity of the molecules, it is necessary to understand the interactions of these molecules with the protein in detail. The top seven aggregation inhibitors from the assays above were docked into the dimer interface cavity around residue 148 (20Å grid) using the program GLIDE v2.5. Control calculation was also carried out using a much larger grid (38Å), which included almost 90% of the protein. The molecules still preferentially bound at the dimer interface despite the bigger grid. A second set of control docking calculations were carried out where the molecules were “forced docked” using a 20Å grid around residue F45 (supplementary material 5). The scores obtained from these control calculations were significantly lower than those obtained from docking at the dimer interface (supplementary material 6). This docking calculation revealed that the molecules preferentially bound at the dimer interface cavity. The modeling results show that the aza-uracil substructure appears to occupy roughly the same position within the protein dimer interface with r.m.s deviation between the poses being <1.3Å (Figure 2a). The side-chains however do not appear to have any preferred orientation (Figure 2a). The dimer interface cavity of SOD-1 is composed of hydrophobic residues such as Val 148 and Val 7 with a small number of peripheral charged or polar residues such as Lys 9 and Asn 53. The pocket is symmetric in nature, with similar side chain orientation for residues on either side. The model indicates the possibility of a hydrogen bond between N3 of either uracil or aza-uracil substructure with the backbone carbonyl oxygen of Val 7 in the majority of the cases (Figure 2a). Similarly, a hydrogen bond was observed between N5 and the side chain carbonyl Asn 53 (Figure 2a). The Val 148 residue from either subunit provides a “hydrophobic base” for proper orientation of the aromatic rings. In order to test the importance of these residues to compound binding, Val 148 and Asn 53 were mutated to Ala using site-directed mutagenesis. Each of these mutations independent of each other led to complete loss of compound binding (Figure 2b), indicating that the presence of both these residues at the dimer interface is critical for compound binding. The mutant SOD-1A4V/N53A and SOD-1A4V/V148A showed CD spectra and secondary structure content similar to showed SOD-1A4V. Unfolding of the double mutants using GdnCl. showed only a slight change in stability when compared to SOD-1A4V (Figure 1b). This is not surprising since effects of mutations are often not additive in terms of free energy of destabilization59.
Figure 2.
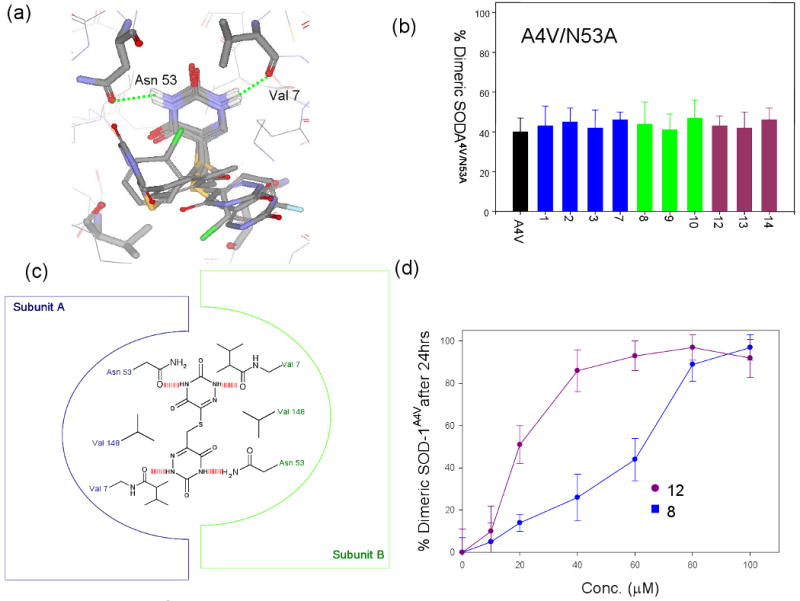
(a) Docked poses for 7 of the molecules that were found to be strong inhibitors of SOD-1 aggregation described in figure 1. The dimer interface region of SOD-1 around residues 148 is shown indicating that uracil or aza-uracil substructures occupy very similar positions in all cases, and makes hydrogen bonds with Asn 53 and Val 7. The rest of the molecules, however, do not appear to have any preferred orientation in the dimer interface. (b) Effect of compounds on aggregation of SOD-1A4V/N53A measured in a fashion similar to the one described in figure 1a. The mutant was generated with the rationale that N53→A should lead to the loss of a hydrogen bond with the small molecules. The experimental data indicates minimal or no effect on aggregation of the double mutant by the small molecules which normally stabilizes SOD-1A4V. (c) Schematic representation of a symmetric molecule with two aza-uracil groups bound at the dimer interface of SOD-1. The schematic representation indicates the possibility of two pairs of hydrogen bonds between the protein and the small molecule. (d) Effect of various concentrations of 8 or 12 on the aggregation of SOD-1A4V. 12 makes two pairs of hydrogen and is more effective at lower concentrations compared to 8.
In conjunction with the mutagenesis experiments, two compounds (4 and 5) were tested for their ability to block aggregation (Figure 1a). These two molecules are methylated either at N3 position of the ring nitrogen (4) or at the N5 ring nitrogen (5), which would disrupt the hydrogen bonds with Asn 53 and Val 7. Neither compound blocked aggregation of SOD-1A4V under demetallating conditions as described above (Figure 1a). This further supports that the nitrogen rings may be involved in critical hydrogen bonds with the protein.
Three new compounds (12, 13 and 14) were synthesized bearing two aza-uracil or uracil substructures but different linker lengths. The purity of these molecules were determined to be not less than 98% using reverse phase HPLC analysis. It was reasoned from the modeling of 12 that having two identical pharmacophores may improve binding since symmetric compounds may be able to make two pairs of hydrogen bonds on opposite subunits (Figure 2c). Three symmetric versions of the inhibitors were synthesized where two aza-uracil pharmacophoric groups were joined using linkers of various lengths. The molecules were analyzed for their chemical identity using NMR and for purity on reverse-phase HPLC analysis (see supporting information). The three new molecules (12, 13 and 14) were all found to be strong inhibitors of SOD-1A4V aggregation (Figure 1a). A dose-response study comparing 8 and 12 revealed that 12 was significantly more effective in blocking aggregation compared to 8 (Figure 2d), supporting the proposed model for compound binding. It was expected that this class of molecules might have better binding specificity towards SOD-1, however they still exhibited poor binding selectivity towards GST-SOD-1 in blood plasma (Figure 1c). These results suggest that it might be hard to proceed towards drug development with the aza-uracil candidates since only minimal modifications can be made to the base pharmacophore without compromising binding. However, it was reasoned that the structural information derived from the experiment above could be used as input into docking calculations to identify better lead molecules.
Docking of ligand and virtual screening of a chemical library using an improved scoring function from experimentally derived constraints
Though a detailed description of the GLIDE methodology (Grid-based Ligand Docking with Energetics) is beyond the scope of this manuscript, briefly, the GLIDE algorithm approximates a systematic search of positions, orientations and conformations of the ligand in the receptor binding site using a series of hierarchical filters. The shape and properties of the receptor are represented on grids that are computed prior to docking. In absence of any experimental input, the grid is considered unconstrained and the ligands are placed and scored based on shape, size and chemical features of the molecules. In cases such as SOD-1, where no x-ray structure of bound ligand is available for a starting model, GLIDE leads to database enrichment by eliminating molecules that are too big or small (shape driven). However, it can lead to inaccurate ranking of molecules and often enriches false positives. This was observed in our original docking calculations where only 15 out of 100 molecules that were active did not rank order well by docking score 50. However, if experimentally derived constraints such as hydrogen bonds and electrostatic interactions are included as part of the docking grid calculations, that should, in principle, improve the performance of the scoring function for ranking active ligands. In order to optimize the docking parameters, the experimentally verified SOD-1 stabilizer, 12, was combined with a set of 20 “false positive” molecules to develop a “training set”. The “false positives” are defined as molecules that bear a very similar shape to the active molecules but do not necessarily share the same capability to bind (e.g. can not form hydrogen bonds) and found to be poor inhibitors of SOD-1 aggregation experimentally (supplemental figure 3). The objective of this exercise was to fine tune docking parameters in the program to identify 3001 preferentially over the false positive molecules. Two independent trials were conducted as follows:
Trial 1: Unconstrained docking
Trial 2: Hydrogen bond constraints on the same subunit (δO of Asn 53 and backbone carbonyl of Val 7).
The ranking of the first 20 top hits in each case is shown in Figure 3. Trial 1, in absence of any constraints, did not lead to significant enrichment with only the experimentally verified inhibitor (12) ranking below the false positive molecules (Figure 3a). Trial 2, where two sets of independent hydrogen bond constraints on each subunit were used, led to a better score for 12 over all the other false positive molecules (Figure 3b). It is interesting to note that GLIDE rejected six out of the 20 molecules when hydrogen bond constraints were used, because the initial poses did not satisfy even weak hydrogen bonds.
Figure 3.

(a,b) Glidescores obtained after docking calculations were carried out using GLIDE with a set of “false positive” molecules mixed with experimentally verified aggregation inhibitors. The top panel uses standard docking parameters of the GLIDE program. The bottom panel describes docking calculations carried out after two hydrogen bond constraints were placed on C=O of Val 7 and the side-chain of Asn53. The use of the hydrogen bonding constraints leads to substantial enrichment of the experimentally verified inhibitors over the false positives. (c) Aggregation of SOD-1A4V monitored using size-exclusion chromatography in presence of a series of compounds that were obtained after screening of a database of 2.2 million molecules using the experimentally derived hydrogen bond constraints on Val 7 and Asn 53. The amount of dimer observed after 48hrs of incubation is plotted after normalization of the aggregation curves. (d) Binding of the new molecules obtained after docking and verified by aggregation assay as inhibitors, tested in the plasma binding assay as described above. At least six molecules show significantly improved binding specificity towards SOD-1 over blood plasma proteins. Control experiments include binding in presence of buffer and GST (tag only). The compounds show specificity towards binding to the GST-SOD-1 fusion protein but not towards GST (tag).
It is conceivable that if docking was carried out using the model derived constraints, it is likely to enrich a database for molecules that would satisfy the hydrogen bonding constraints and thereby yield molecules that are likely to function as strong pharmacological chaperones for SOD-1. The optimized symmetric scoring function using four hydrogen bond (δO of Asn 53, Asn 53′ and carbonyl O of Val 7 and Val 7′) constraints on both subunits was used to screen a database of 2.2 million small drug-like molecules (details of database preparation in materials and method section). The top 20 molecules obtained were tested for their ability to block SOD-1 aggregation. At least 14 out of these 20 molecules were found to be good inhibitors of SOD-1 aggregation carried out under experimental conditions described previously (Figure 3c). In a control experiment, these molecules failed to block aggregation of α-synuclein, a structurally unrelated protein, suggesting that these pharmacological chaperones are specific to SOD-1. Most of these molecules do not bear any structural resemblance to the 12, however, they satisfy the hydrogen bond constraints on the receptor (Figure 4 and Figure 4 supplemental).
Figure 4.

Chemical structures for the molecules that were found to have high docking scores, and satisfy the hydrogen bonding constraints imposed by the docking program.
The 14 new inhibitors of aggregation were tested for their ability to bind GST-SOD-1 in blood plasma using the assay method described before. Control experiments were also carried out where compound binding to GST (tag) in buffer was measured in an identical fashion. None of the molecules exhibited any specific binding affinity towards GST. At least six out the 14 molecules exhibited significantly high specificity of binding towards SOD-1 over blood plasma components (Figure 3d).
Discussion
We recently described a set of molecules that can bind at the SOD-1 dimer interface as pharmacological chaperones, and block aggregation of the mutant form of the protein. Pharmacological chaperones, unlike enzyme inhibitors, work by increasing the free energy of unfolding of a mutant protein, and may not bind their target with high affinity. This allows more than chemical scaffolding to satisfy the constraints necessary for a good pharmacological chaperone. In this manuscript, we extend our previous study to develop a more generalized method for predicting molecules that are likely to serve as good inhibitors of mutant SOD-1. Furthermore, we use this algorithm to predict molecules that have improved binding specificity towards SOD-1 over protein components of blood plasma.
The 11 new molecules chosen for this study typically possess either an aza-uracil or uracil group (base pharmacophore) linked to various aromatic or aliphatic sidechains by either an imino or thioether linker. Seven out of 14 molecules tested were found to be excellent pharmacological chaperones effective in blocking aggregation. The highest affinity SOD-1 pharmacological chaperones in buffer may not necessarily be the ideal candidates for administration as therapeutics in human since these molecules may bind other protein targets in the human body. This would lead to off-pathway toxicity, as well as poor delivery to the intended target. There is precedence for early medicinal chemistry studies to reduce off pathway toxicity by Kelly and co-workers on pharmacological chaperones that block aggregation of TTR54. These seven active compounds were tested for their ability to bind SOD-1 in blood plasma. Almost all the compounds bound strongly to protein components in blood plasma, and showed poor binding specificity towards SOD-1.
In order to understand and improve the compounds binding specificity, structural information was necessary to identify new sites for modification on the original molecules in order to reduce off pathway binding to blood plasma components. Docking calculations were performed to model the aggregation inhibitors at the dimer interface binding pocket. The SOD-1 binding pocket is largely hydrophobic in nature, composed of Val 148 and Val 7 contributed by opposite subunits. The docked structures reveal that the aza-uracil/uracil imino groups make electrostatic and hydrogen bonds with the backbone carbonyl of Val 7 and side chain carbonyl of Arg 53. The proposed model for binding of this compound was found to be consistent with site directed mutagenesis studies and modifications of the base pharmacophore.
We carried out docking calculations on a library of ∼2.2 million molecules with four hydrogen bond constraints (2 on Asn 53 and 2 on Val 7). The docking calculations yielded at least 20 molecules that satisfied the docking constraints (hydrogen bonds). Surprisingly, most of these molecules had no resemblance to the original molecules (aza-uracil based), but could still make hydrogen bonds with Asn 53 and Val 7 (see Figure 4 and Figure 4 supplemental). These molecules were tested for their ability to block aggregation of SOD-1A4V and specifically bind SOD-1 over blood plasma components. At least 14 out of the 20 molecules tested here were highly effective in blocking aggregation of SOD-1A4V. We found at least six molecules that had high specificity towards SOD-1 as well, and performed significantly better than the original aza-uracil based molecules when tested in blood plasma.
These molecules represent a good starting point for drug development. In addition, some optimization of their biological properties (ADME, reduced toxicity) will be required before our working hypothesis can be tested by administering compounds in mouse models of FALS. Nevertheless, the studies proposed here represent a critical first step in the search for an effective ALS therapeutic. Finally, we hope that pharmaceutical companies will use the approach described here to screen their own proprietary libraries for potential lead compounds.
Supplementary Material
Acknowledgments
We wish to thank Mark Duggan (Link Medicine Corporation) for insightful discussions on the medicinal chemistry of the SOD-1 aggregation inhibitors and for providing the raw compound libraries. We thank Robert Brown Jr. (UMass, Worcester) and (Larry Hayward, UMass, Worcester) for providing us with cDNA for G85R mutant of SOD. We also thank Dr. A.Z. Mason (California State University, Long Beach) for performing ICP-MS analysis of the SOD samples. The ICP-MS facility at Long Beach is supported by NSF grant OCE-9977564. We also wish to thank Kistine Vernon for help with manuscript preparation.
‡This work was supported by the National Institutes of Health (AG08476 to P.T.L), ALSA (SSR) and a generous gift from the Tow family (NY).
List of abbreviations
- SOD-1
superoxide dismutase-1
- ALS
Amyotrophic lateral sclerosis
- FALS
Familial ALS
- FAP
Amyloid polyneuropathy
- TTR
Transthyretin
- ADME
Absorption, distribution, metabolism, and excretion
- GST
glutathione-S-transferase
- GdnCl
Guanidine hydrochloride
- GLIDE
Grid-based Ligand Docking with Energetics
Footnotes
Availability of supporting information: Electronic supporting information for this manuscript is available immediately from the ACS website
References
- 1.Cohen FE, Kelly JW. Therapeutic approaches to protein-misfolding diseases. Nature. 2003;426:905–909. doi: 10.1038/nature02265. [DOI] [PubMed] [Google Scholar]
- 2.Dobson CM. Protein aggregation and its consequences for human disease. Protein Pept Lett. 2006;13:219–227. doi: 10.2174/092986606775338362. [DOI] [PubMed] [Google Scholar]
- 3.Dobson CM. Principles of protein folding, misfolding and aggregation. Semin Cell Dev Biol. 2004;15:3–16. doi: 10.1016/j.semcdb.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 4.Fink AL. Protein aggregation: folding aggregates, inclusion bodies and amyloid. Fold Des. 1998;3:R9–23. doi: 10.1016/S1359-0278(98)00002-9. [DOI] [PubMed] [Google Scholar]
- 5.Fowler DM, Kelly JW. Aggregating knowledge about prions and amyloid. Cell. 2009;137:20–22. doi: 10.1016/j.cell.2009.03.030. [DOI] [PubMed] [Google Scholar]
- 6.Miroy GJ, Lai Z, Lashuel HA, Peterson SA, Strang C, et al. Inhibiting transthyretin amyloid fibril formation via protein stabilization. Proc Natl Acad Sci U S A. 1996;93:15051–15056. doi: 10.1073/pnas.93.26.15051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baures PW, Peterson SA, Kelly JW. Discovering transthyretin amyloid fibril inhibitors by limited screening. Bioorg Med Chem. 1998;6:1389–1401. doi: 10.1016/s0968-0896(98)00130-8. [DOI] [PubMed] [Google Scholar]
- 8.Adamski-Werner SL, Palaninathan SK, Sacchettini JC, Kelly JW. Diflunisal analogues stabilize the native state of transthyretin. Potent inhibition of amyloidogenesis. J Med Chem. 2004;47:355–374. doi: 10.1021/jm030347n. [DOI] [PubMed] [Google Scholar]
- 9.Sawkar AR, Adamski-Werner SL, Cheng WC, Wong CH, Beutler E, et al. Gaucher disease-associated glucocerebrosidases show mutation-dependent chemical chaperoning profiles. Chem Biol. 2005;12:1235–1244. doi: 10.1016/j.chembiol.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 10.Sawkar AR, Schmitz M, Zimmer KP, Reczek D, Edmunds T, et al. Chemical chaperones and permissive temperatures alter localization of Gaucher disease associated glucocerebrosidase variants. ACS Chem Biol. 2006;1:235–251. doi: 10.1021/cb600187q. [DOI] [PubMed] [Google Scholar]
- 11.Arakawa T, Ejima D, Kita Y, Tsumoto K. Small molecule pharmacological chaperones: From thermodynamic stabilization to pharmaceutical drugs. Biochim Biophys Acta. 2006;1764:1677–1687. doi: 10.1016/j.bbapap.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 12.Jia LY, Gong B, Pang CP, Huang Y, Lam DS, et al. A natural osmolyte corrects the disease phenotype of mutant myocilin causing glaucoma. Invest Ophthalmol Vis Sci. 2009 doi: 10.1167/iovs.08-3151. [DOI] [PubMed] [Google Scholar]
- 13.Lee D, Santos D, Al-Rawi H, McNeill AM, Rugg EL. The chemical chaperone trimethylamine N-oxide ameliorates the effects of mutant keratins in cultured cells. Br J Dermatol. 2008;159:252–255. doi: 10.1111/j.1365-2133.2008.08596.x. [DOI] [PubMed] [Google Scholar]
- 14.Mehta AD, Seidler NW. Beta-alanine suppresses heat inactivation of lactate dehydrogenase. J Enzyme Inhib Med Chem. 2005;20:199–203. doi: 10.1080/14756360400020538. [DOI] [PubMed] [Google Scholar]
- 15.Kanki K, Kawamura T, Watanabe Y. Control of ER stress by a chemical chaperone counteracts apoptotic signals in IFN-gamma-treated murine hepatocytes. Apoptosis. 2009;14:309–319. doi: 10.1007/s10495-009-0318-x. [DOI] [PubMed] [Google Scholar]
- 16.Park JY, Kim GH, Kim SS, Ko JM, Lee JJ, et al. Effects of a chemical chaperone on genetic mutations in alpha-galactosidase A in Korean patients with Fabry disease. Exp Mol Med. 2009;41:1–7. doi: 10.3858/emm.2009.41.1.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sekijima Y, Dendle MA, Kelly JW. Orally administered diflunisal stabilizes transthyretin against dissociation required for amyloidogenesis. Amyloid. 2006;13:236–249. doi: 10.1080/13506120600960882. [DOI] [PubMed] [Google Scholar]
- 18.Reixach N, Adamski-Werner SL, Kelly JW, Koziol J, Buxbaum JN. Cell based screening of inhibitors of transthyretin aggregation. Biochem Biophys Res Commun. 2006;348:889–897. doi: 10.1016/j.bbrc.2006.07.109. [DOI] [PubMed] [Google Scholar]
- 19.Klabunde T, Petrassi HM, Oza VB, Raman P, Kelly JW, et al. Rational design of potent human transthyretin amyloid disease inhibitors. Nat Struct Biol. 2000;7:312–321. doi: 10.1038/74082. [DOI] [PubMed] [Google Scholar]
- 20.Oza VB, Petrassi HM, Purkey HE, Kelly JW. Synthesis and evaluation of anthranilic acid-based transthyretin amyloid fibril inhibitors. Bioorg Med Chem Lett. 1999;9:1–6. doi: 10.1016/s0960-894x(98)00696-9. [DOI] [PubMed] [Google Scholar]
- 21.Baures PW, Oza VB, Peterson SA, Kelly JW. Synthesis and evaluation of inhibitors of transthyretin amyloid formation based on the non-steroidal anti-inflammatory drug, flufenamic acid. Bioorg Med Chem. 1999;7:1339–1347. doi: 10.1016/s0968-0896(99)00066-8. [DOI] [PubMed] [Google Scholar]
- 22.Sawkar AR, D'Haeze W, Kelly JW. Therapeutic strategies to ameliorate lysosomal storage disorders--a focus on Gaucher disease. Cell Mol Life Sci. 2006;63:1179–1192. doi: 10.1007/s00018-005-5437-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sawkar AR, Cheng WC, Beutler E, Wong CH, Balch WE, et al. Chemical chaperones increase the cellular activity of N370S beta -glucosidase: a therapeutic strategy for Gaucher disease. Proc Natl Acad Sci U S A. 2002;99:15428–15433. doi: 10.1073/pnas.192582899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rosen DR, Sapp P, O'Regan J, McKenna-Yasek D, Schlumpf KS, et al. Genetic linkage analysis of familial amyotrophic lateral sclerosis using human chromosome 21 microsatellite DNA markers. Am J Med Genet. 1994;51:61–69. doi: 10.1002/ajmg.1320510114. [DOI] [PubMed] [Google Scholar]
- 25.Radunovic A, Leigh PN. ALSODatabase: database of SOD1 (and other) gene mutations in ALS on the Internet. European FALS Group and ALSOD Consortium. Amyotroph Lateral Scler Other Motor Neuron Disord. 1999;1:45–49. doi: 10.1080/146608299300080040. [DOI] [PubMed] [Google Scholar]
- 26.Hayward LJ, Rodriguez JA, Kim JW, Tiwari A, Goto JJ, et al. Decreased metallation and activity in subsets of mutant superoxide dismutases associated with familial amyotrophic lateral sclerosis. J Biol Chem. 2002;277:15923–15931. doi: 10.1074/jbc.M112087200. [DOI] [PubMed] [Google Scholar]
- 27.Hough MA, Grossmann JG, Antonyuk SV, Strange RW, Doucette PA, et al. Dimer destabilization in superoxide dismutase may result in disease-causing properties: structures of motor neuron disease mutants. Proc Natl Acad Sci U S A. 2004;101:5976–5981. doi: 10.1073/pnas.0305143101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tiwari A, Hayward LJ. Mutant SOD1 instability: implications for toxicity in amyotrophic lateral sclerosis. Neurodegener Dis. 2005;2:115–127. doi: 10.1159/000089616. [DOI] [PubMed] [Google Scholar]
- 29.Rodriguez JA, Shaw BF, Durazo A, Sohn SH, Doucette PA, et al. Destabilization of apoprotein is insufficient to explain Cu,Zn-superoxide dismutase-linked ALS pathogenesis. Proc Natl Acad Sci U S A. 2005;102:10516–10521. doi: 10.1073/pnas.0502515102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Strange RW, Antonyuk S, Hough MA, Doucette PA, Rodriguez JA, et al. The structure of holo and metal-deficient wild-type human Cu, Zn superoxide dismutase and its relevance to familial amyotrophic lateral sclerosis. J Mol Biol. 2003;328:877–891. doi: 10.1016/s0022-2836(03)00355-3. [DOI] [PubMed] [Google Scholar]
- 31.Rodriguez JA, Valentine JS, Eggers DK, Roe JA, Tiwari A, et al. Familial amyotrophic lateral sclerosis-associated mutations decrease the thermal stability of distinctly metallated species of human copper/zinc superoxide dismutase. J Biol Chem. 2002;277:15932–15937. doi: 10.1074/jbc.M112088200. [DOI] [PubMed] [Google Scholar]
- 32.Elam JS, Malek K, Rodriguez JA, Doucette PA, Taylor AB, et al. An alternative mechanism of bicarbonate-mediated peroxidation by copper-zinc superoxide dismutase: rates enhanced via proposed enzyme-associated peroxycarbonate intermediate. J Biol Chem. 2003;278:21032–21039. doi: 10.1074/jbc.M300484200. [DOI] [PubMed] [Google Scholar]
- 33.Elam JS, Taylor AB, Strange R, Antonyuk S, Doucette PA, et al. Amyloid-like filaments and water-filled nanotubes formed by SOD1 mutant proteins linked to familial ALS. Nat Struct Biol. 2003;10:461–467. doi: 10.1038/nsb935. [DOI] [PubMed] [Google Scholar]
- 34.Antonyuk S, Elam JS, Hough MA, Strange RW, Doucette PA, et al. Structural consequences of the familial amyotrophic lateral sclerosis SOD1 mutant His46Arg. Protein Sci. 2005;14:1201–1213. doi: 10.1110/ps.041256705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tiwari A, Xu Z, Hayward LJ. Aberrantly increased hydrophobicity shared by mutants of Cu,Zn-superoxide dismutase in familial amyotrophic lateral sclerosis. J Biol Chem. 2005;280:29771–29779. doi: 10.1074/jbc.M504039200. [DOI] [PubMed] [Google Scholar]
- 36.Cao X, Antonyuk SV, Seetharaman SV, Whitson LJ, Taylor AB, et al. Structures of the G85R variant of SOD1 in familial amyotrophic lateral sclerosis. J Biol Chem. 2008;283:16169–16177. doi: 10.1074/jbc.M801522200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chattopadhyay M, Durazo A, Sohn SH, Strong CD, Gralla EB, et al. Initiation and elongation in fibrillation of ALS-linked superoxide dismutase. Proc Natl Acad Sci U S A. 2008;105:18663–18668. doi: 10.1073/pnas.0807058105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Banci L, Bertini I, Boca M, Girotto S, Martinelli M, et al. SOD1 and amyotrophic lateral sclerosis: mutations and oligomerization. PLoS One. 2008;3:e1677. doi: 10.1371/journal.pone.0001677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Valentine JS. Do oxidatively modified proteins cause ALS? Free Radic Biol Med. 2002;33:1314–1320. doi: 10.1016/s0891-5849(02)01080-8. [DOI] [PubMed] [Google Scholar]
- 40.Strange RW, Antonyuk SV, Hough MA, Doucette PA, Valentine JS, et al. Variable metallation of human superoxide dismutase: atomic resolution crystal structures of Cu-Zn, Zn-Zn and as-isolated wild-type enzymes. J Mol Biol. 2006;356:1152–1162. doi: 10.1016/j.jmb.2005.11.081. [DOI] [PubMed] [Google Scholar]
- 41.Doucette PA, Whitson LJ, Cao X, Schirf V, Demeler B, et al. Dissociation of human copper-zinc superoxide dismutase dimers using chaotrope and reductant. Insights into the molecular basis for dimer stability. J Biol Chem. 2004;279:54558–54566. doi: 10.1074/jbc.M409744200. [DOI] [PubMed] [Google Scholar]
- 42.Goto JJ, Zhu H, Sanchez RJ, Nersissian A, Gralla EB, et al. Loss of in vitro metal ion binding specificity in mutant copper-zinc superoxide dismutases associated with familial amyotrophic lateral sclerosis. J Biol Chem. 2000;275:1007–1014. doi: 10.1074/jbc.275.2.1007. [DOI] [PubMed] [Google Scholar]
- 43.Lyons TJ, Nersissian A, Huang H, Yeom H, Nishida CR, et al. The metal binding properties of the zinc site of yeast copper-zinc superoxide dismutase: implications for amyotrophic lateral sclerosis. J Biol Inorg Chem. 2000;5:189–203. doi: 10.1007/s007750050363. [DOI] [PubMed] [Google Scholar]
- 44.Khare SD, Dokholyan NV. Common dynamical signatures of familial amyotrophic lateral sclerosis-associated structurally diverse Cu, Zn superoxide dismutase mutants. Proc Natl Acad Sci U S A. 2006;103:3147–3152. doi: 10.1073/pnas.0511266103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shaw BF, Durazo A, Nersissian AM, Whitelegge JP, Faull KF, et al. Local unfolding in a destabilized, pathogenic variant of superoxide dismutase 1 observed with H/D exchange and mass spectrometry. J Biol Chem. 2006;281:18167–18176. doi: 10.1074/jbc.M600623200. [DOI] [PubMed] [Google Scholar]
- 46.Valentine JS, Hart PJ. Misfolded CuZnSOD and amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2003;100:3617–3622. doi: 10.1073/pnas.0730423100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shaw BF, Valentine JS. How do ALS-associated mutations in superoxide dismutase 1 promote aggregation of the protein? Trends Biochem Sci. 2007;32:78–85. doi: 10.1016/j.tibs.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 48.Banci L, Bertini I, D'Amelio N, Libralesso E, Turano P, et al. Metalation of the amyotrophic lateral sclerosis mutant glycine 37 to arginine superoxide dismutase (SOD1) apoprotein restores its structural and dynamical properties in solution to those of metalated wild-type SOD1. Biochemistry. 2007;46:9953–9962. doi: 10.1021/bi700620r. [DOI] [PubMed] [Google Scholar]
- 49.Ray SS, Lansbury PT., Jr A possible therapeutic target for Lou Gehrig's disease. Proc Natl Acad Sci U S A. 2004;101:5701–5702. doi: 10.1073/pnas.0401934101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ray SS, Nowak RJ, Brown RH, Jr, Lansbury PT., Jr Small-molecule-mediated stabilization of familial amyotrophic lateral sclerosis-linked superoxide dismutase mutants against unfolding and aggregation. Proc Natl Acad Sci U S A. 2005;102:3639–3644. doi: 10.1073/pnas.0408277102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grossman I. ADME pharmacogenetics: current practices and future outlook. Expert Opin Drug Metab Toxicol. 2009;5:449–462. doi: 10.1517/17425250902902322. [DOI] [PubMed] [Google Scholar]
- 52.Ray SS, Nowak RJ, Strokovich K, Brown RH, Jr, Walz T, et al. An intersubunit disulfide bond prevents in vitro aggregation of a superoxide dismutase-1 mutant linked to familial amytrophic lateral sclerosis. Biochemistry. 2004;43:4899–4905. doi: 10.1021/bi030246r. [DOI] [PubMed] [Google Scholar]
- 53.Santoro MM, Bolen DW. A test of the linear extrapolation of unfolding free energy changes over an extended denaturant concentration range. Biochemistry. 1992;31:4901–4907. doi: 10.1021/bi00135a022. [DOI] [PubMed] [Google Scholar]
- 54.Johnson SM, Connelly S, Wilson IA, Kelly JW. Toward optimization of the linker substructure common to transthyretin amyloidogenesis inhibitors using biochemical and structural studies. J Med Chem. 2008;51:6348–6358. doi: 10.1021/jm800435s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Johnson SM, Connelly S, Wilson IA, Kelly JW. Toward Optimization of the Second Aryl Substructure Common to Transthyretin Amyloidogenesis Inhibitors Using Biochemical and Structural Studies (dagger) J Med Chem. 2009 doi: 10.1021/jm801347s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Purkey HE, Dorrell MI, Kelly JW. Evaluating the binding selectivity of transthyretin amyloid fibril inhibitors in blood plasma. Proc Natl Acad Sci U S A. 2001;98:5566–5571. doi: 10.1073/pnas.091431798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wilcox KC, Zhou L, Jordon JK, Huang Y, Yu Y, et al. Modifications of superoxide dismutase (SOD1) in human erythrocytes: a possible role in amyotrophic lateral sclerosis. J Biol Chem. 2009;284:13940–13947. doi: 10.1074/jbc.M809687200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fabrini R, De Luca A, Stella L, Mei G, Orioni B, et al. Monomer-dimer equilibrium in glutathione transferases: a critical re-examination. Biochemistry. 2009;48:10473–10482. doi: 10.1021/bi901238t. [DOI] [PubMed] [Google Scholar]
- 59.Islam MM, Sohya S, Noguchi K, Kidokoro S, Yohda M, et al. Thermodynamic and structural analysis of highly stabilized BPTIs by single and double mutations. Proteins. 2009;77:962–970. doi: 10.1002/prot.22522. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.