

Abstract
Most mechanistic studies of repair of DNA double-strand breaks (DSBs) produced by in vivo expression of endonucleases have utilized enzymes that produce cohesive-ended DSBs such as HO, I-SceI and EcoRI. We have developed systems for expression of PvuII and EcoRV, nucleases that produce DSBs containing blunt ends, using a modified GAL1 promoter that has reduced basal activity. Expression of PvuII and EcoRV caused growth inhibition and strong cell killing in both haploid and diploid yeast cells. Surprisingly, there was little difference in sensitivities of wildtype cells and mutants defective in homologous recombination, nonhomologous end-joining (NHEJ), or both pathways. Physical analysis using standard and pulsed field gel electrophoresis demonstrated time-dependent breakage of chromosomal DNA within cells. Although ionizing radiation-induced DSBs were largely repaired within 4 hrs, no repair of PvuII-induced breaks could be detected in diploid cells, even after arrest in G2/M. Rare survivors of PvuII expression had an increased frequency of chromosome XII deletions, an indication that a fraction of the induced DSBs could be repaired by an error-prone process. These results indicate that, unlike DSBs with complementary single-stranded DNA overhangs, blunt-ended DSBs in yeast chromosomes are poor substrates for repair by either NHEJ or recombination.
Keywords: endonuclease, DNA repair, end-joining, homologous recombination, double-strand break
1. Introduction
Chromosomal DNA within eukaryotic cells is subject to many types of damage that cause alterations to the bases or to the sugar-phosphate backbone. Damage that leads to breakage of phosphodiester linkages that are in close proximity on opposite strands results in formation of double-strand breaks (DSBs). This type of lesion can be caused endogenously, e.g., by free radical attack via reactive oxygen species, by endonucleases, or indirectly by stalling of DNA replication fork progression [1-3]. Exogenous causes of DSBs include ionizing radiation as well as chemicals such as the anti-tumor drug bleomycin and various other chemical clastogens.
Repair of DSBs occurs via two major pathways in eukaryotic organisms, homologous recombination and nonhomologous end-joining (NHEJ). Repair of DSBs by homologous recombination requires several proteins, including the Rad51, Rad52, Rad54, Rad55, Rad57 and Rad59 proteins, the Mrx complex (Mre11, Rad50 and Xrs2), the Rpa single-stranded DNA binding protein, and other proteins associated with end-processing, strand exchange, and downstream steps of the pathway [4-7]. DSB ends are initially resected to produce single-stranded 3’ overhangs by the actions of the Mrx nuclease working in conjunction with several other proteins, including Exo1, Sae2, Sgs1, and Dna2 [8,9]. The long 3’ tails generated by these enzymes then serve as substrates for binding of Rpa and Rad51 and recruitment of other proteins involved in repair, cell cycle checkpoint signaling, nucleosome remodeling and sister chromatid cohesion [6, 10-14].
Repair by the second pathway, NHEJ, requires Mrx, the DNA end-binding Yku70/Yku80 complex and DNA Ligase IV, which is composed of Dnl4, Lif1, and Nej1 [15,16]. Proteins such as Pol4, Rad27 (Fen1), the Rsc complex, Smc cohesins and checkpoint response complexes may also affect efficiency of the pathway [7,14-16].
A great deal has been learned about the repair of DSBs and their consequences through the use of radiation and DNA damaging chemicals as inducers. However, a confounding factor in such studies is that these clastogens also produce many other lesions within DNA. For example, ionizing radiation generates more SSBs than DSBs, and also produces many different types of altered bases and sugars via oxidation [1,3]. The ends of many radiation-induced DSBs do not retain canonical chemical groups, with a large fraction of broken ends containing 3’ phosphate or 3’ phosphoglycolate plus damaged sugars or bases. To circumvent this lack of specificity, DNA sequence-targeting endonucleases have been employed to generate DSBs. In many experiments the nuclease proteins were added directly to cells under conditions in which the enzymes pass inside through the cell envelope and remain active [1,17,18]. Such studies have demonstrated the ability of endonucleases to induce DNA strand breaks, reduce cell viability, and stimulate chromosome aberrations and gene mutations. When restriction enzymes were utilized, the endonucleases that create DSBs with blunt (or flush) ends were frequently found to induce aberrations more efficiently than those retaining single-stranded DNA overhangs, though this was not always the case [1,19-21].
Studies involving in vivo expression of genes encoding site-specific endonucleases have been especially informative in understanding DSB repair mechanisms. In the yeast S. cerevisiae many investigations have analyzed the effects of expression of the frequent-cutting endonuclease EcoRI [22-26] or the rare-cutting nucleases HO and I-SceI [24,27,28]. Each of these nucleases induces DSBs that require genes from both major DSB repair pathways to be functional for efficient repair. For example, mutants deficient in NHEJ or recombination frequently exhibit checkpoint responses, chromosome structure changes, growth inhibition and reduced survival phenotypes after expression of EcoRI or HO that are not seen in WT cells [22,24,26,29]. Each of these three nucleases produces breaks with 4 nt single-stranded DNA (ssDNA) overhangs, though the end structures are not identical. EcoRI cuts a palindromic 6 bp sequence (GˆAATTC) to create 5’ ssDNA overhangs. In contrast, I-SceI cuts a non-palindromic 18 bp sequence, producing 3’ overhangs that are 4 nt long. Similar to I-SceI, HO cuts a large (≥ 24 bp) asymmetric target sequence and creates 3’ ssDNA overhangs that are also 4 nt long. Although the repair of DSBs containing 5’ or 3’ overhangs such as these has been extensively studied, the impact of expression of nucleases producing blunt ends in vivo has not been investigated. In this study we demonstrate that DSBs caused by controlled expression of the blunt end-producing nucleases PvuII (recognition sequence CAGˆCTG) or EcoRV (GATˆATC) are strongly toxic to yeast cells and are poor substrates for repair by either of the major DSB repair pathways.
2. Materials and methods
2.1. Strains and plasmids
Yeast strains used in the study were derivatives of T344 (MATα ura3-52 leu2-3,112 prb1-1122 trp1Δ∷hisG reg1-501 gal1 pep4-3) [30]. This strain contains the reg1-501 mutation that permits regulation of GAL promoter activity with galactose while cells are grown using glucose as carbon source. Expression of EcoRI utilized strain YLKL350 (his3Δ∷[GAL1p∷EcoRI TRP1]). For expression of PvuII using the mutant GAL1-V10 promoter that has reduced basal activity, the PvuII expression cassette from plasmid pV10 [27] was integrated into the HIS3 locus of T334 by PCR fragment-mediated gene disruption to create JW1689 (his3Δ∷[GAL1-V10p∷PvuII URA3]). The PvuII gene within pV10 was derived from plasmid pPvuRM3.4 [31]. Strain JW1689 was subsequently used to create all repair-deficient haploid and diploid mutants. DNA repair-deficient haploid JW1689 derivatives included rad50Δ∷G418r (JW1714), rad52Δ∷G418r (JW1708), dnl4Δ∷G418r (JW1775), and rad9Δ∷TRP1 (JW1716).
For analysis of PvuII expression in diploid strains, JW1689 was crossed with the MATa strain JW1698. To create rad52-/- diploids, JW1708 was crossed with JW1706 (a rad52Δ∷G418r derivative of JW1698). rad9-/- diploids were constructed by crossing JW1716 with JW1718 (a rad9Δ∷TRP1 derivative of JW1698). An EcoRV-expressing strain was created by replacing the PvuII and URA3 genes on chromosome XV in JW1689 with a DNA fragment containing the EcoRV coding sequence. The EcoRV gene was PCR amplified from plasmid pLB1, provided by Stephen Halford using primers v10.erv.dw (TCAATATGCCTCTATACTTAAACGTCAAGGAGAAAAAACCCCGGATCCTCTCAGTTAAAAATGAGTCTTCGTTCTGATTT) and hisA.erv.up (CCTAGTAAAGCGTATTACAAATGAAACCAAGATCAGAGCAGATTGTA CTGAGAGTGCACCTTATTTTCTTCCTCGGTATATCCAG) and transformed into JW1689 followed by selection for Ura- cells on 5-fluoroorotic acid (5-FOA) plates. Colonies were subsequently tested for low Ura- --> Ura+ revertability, non-growth on galactose plates and by sequencing of the complete integrated EcoRV gene. The resulting strain was called JW1752 (his3Δ∷[GAL1-V10p∷EcoRV]). Diploid strains containing GAL1-V10p∷EcoRV were created by crossing JW1752 with strain JW1698.
2.2. Molecular biology reagents
PCR fragment-mediated gene disruptions employed ExTaq DNA polymerase (Takara Mirus Bio) or Vent DNA polymerase (New England Biolabs) and the lithium acetate method for transformation of yeast cells [32]. Geneticin/G418 (Invitrogen) and Hygromycin B (Sigma) were added to plates for selection of resistant strains at concentrations of 200 and 300 ug/ml, respectively.
2.3. Dilution pronging survival assays
For the pronging experiments depicted in Figure 1 and Figure 8, cells grown on YPDA plates (1% bacto yeast extract, 2% bacto peptone, 2% glucose, 2% bacto agar, 0.001% adenine) were harvested into water, counted by hemacytometer, and 2 × 107 cells in a total volume of 220 ul were placed into a microtiter dish well and serially diluted five-fold. Cells were then pronged to YPDA plates supplemented with or without various concentrations of galactose.
Fig. 1.

PvuII expression from the mutant GAL1-V10 promoter that has reduced activity is more toxic than expression of EcoRI from the WT GAL1 promoter. T334, YLKL350, and JW1689 cells were diluted 5-fold and pronged to YPDA plates with or without supplemental galactose.
Fig. 8.
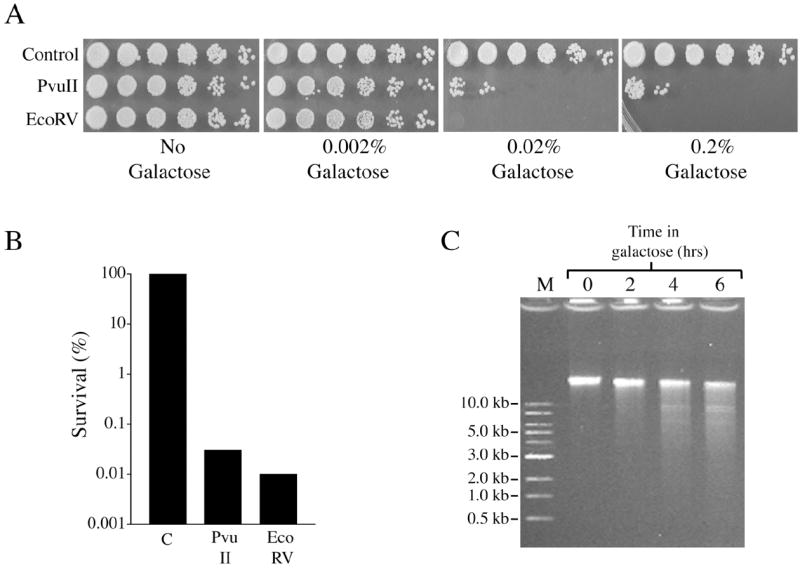
Another blunt-end producing endonuclease, EcoRV, has toxicity similar to that of PvuII. Induction of EcoRV expression from an integrated GAL1-V10p∷EcoRV promoter fusion reduces cell survival in (A) haploid cells and (B) diploid cells. (C) Induction of EcoRV expression causes time-dependent breakage of chromosomal DNA in logarithmically growing haploid cells.
2.4. Analysis of DNA breakage using standard and pulsed field gel electrophoresis
Pulsed field gel analysis of yeast chromosomes was performed using a Geneline II apparatus (Beckman Instruments) run for 24 hrs at 14 °C using 1% agarose and 0.25 X TBE running buffer. Gel bands were visualized using ethidium bromide. Molecular weight standards employed were 1 Kb Ladder, HindIII-cut Lambda DNA Ladder, and PFG Lambda Ladder (New England Biolabs). To analyze PvuII-induced DSBs using standard agarose gels, DNA was purified using a MasterPure yeast DNA purification kit (Epicentre) except that the initial lysis at elevated temperatures was conducted at 75 °C rather than 65 °C to promote inactivation of the enzymes. For Southern blots, the DNA within each gel was transferred to a Hybond N+ membrane using a Stratagene PosiBlot pressure blotter system. Hybridization was performed using a 32P-labelled DNA fragment prepared by random priming (using the Stratagene Prime-It RmT Random Primer Labeling Kit) of a gel-purified PCR product containing part of the LYS2 gene using primers o-4819L2 and s-2870L2 (gifts from Kirill Lobachev). Blots were visualized using X-ray film.
2.5 Cell growth, cell cycling and use of liquid holding to assess DSB repair proficiency
Growth of cell cultures was monitored by counting cells using a hemacytometer. Survival after induction of endonuclease expression in YPDA medium containing 2% glucose supplemented with galactose was calculated as the number of viable cells per ml observed from colonies on YPDA plates divided by the number of cells per ml in the culture determined by hemacytometer. For survival studies in Figure 2, overnight cultures were diluted to 1 × 106 cells per ml into YPDA media, grown for 2 hrs to obtain log phase cultures, and galactose was added to begin induction. Cell cycle progression was monitored as described [33]. Large-budded G2/M cells were defined as cells in which the bud was greater than or equal to 50% of the size of the mother cell.
Fig. 2.
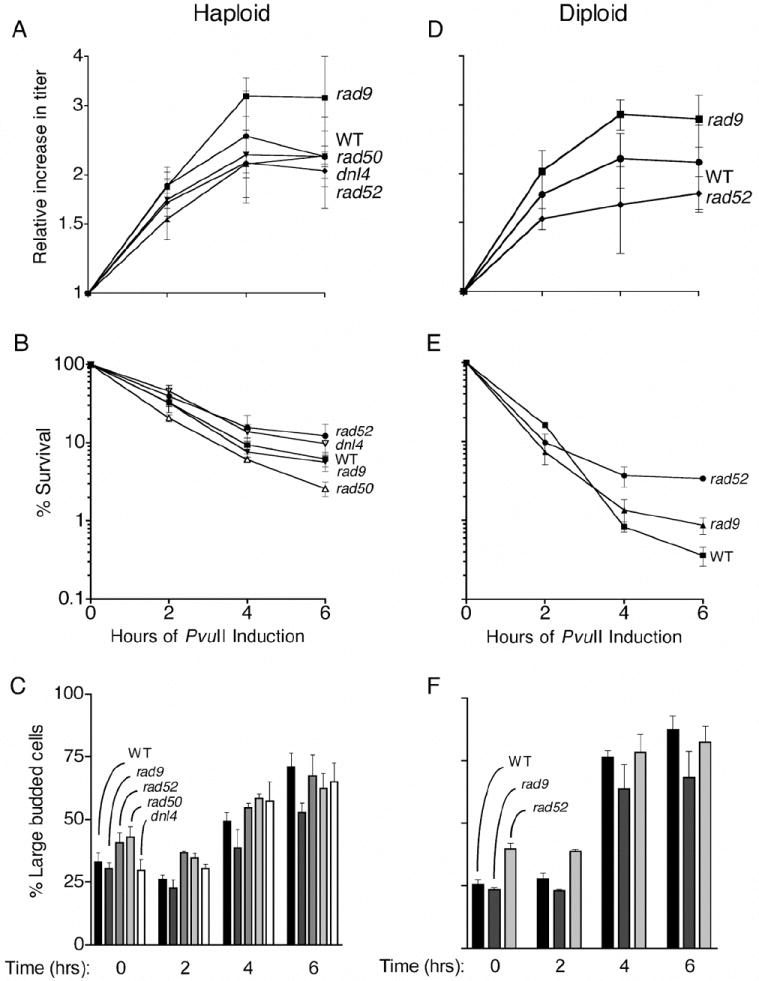
Impact of PvuII expression on cell growth rates, survival and cell cycle progression. Logarithmically growing haploid or diploid cells were induced with galactose at time zero. Growth and cell cycling were assessed microscopically and viability was determined after spreading cells to YPDA plates.
For analysis of repair of DSBs formed by ionizing radiation or PvuII, aliquots of cells were centrifuged for 0.5 min, washed with water, and resuspended and incubated in YPDA medium. To arrest cell cycling with nocodazole, aliquots of a 10 mg/ml stock solution dissolved in DMSO were added to a final concentration of 15 ug/ml and G2 arrest was confirmed microscopically. For the EcoRV survival experiments depicted in Figure 8B, cells were diluted into YPDA medium for 1 hr as described above, induced with 2% galactose for 2 hr and spread to YPDA plates to assess survival. Two cultures were analyzed for each strain and results were averaged.
3. Results
3.1. In vivo expression of PvuII is highly toxic in both WT and repair-deficient haploid and diploid cells
In order to investigate the consequences and repair of blunt-ended DSBs, systems were developed for expression of PvuII and EcoRV within yeast cells. The PvuII gene was successfully cloned downstream of the GAL1 promoter in the CEN/ARS yeast plasmid vector pRS316 [27]. However, when GAL1p∷PvuII plasmids cloned in E. coli and confirmed by DNA sequencing were subsequently transformed into yeast cells, few colonies were observed on glucose media plates (conditions where the GAL1 promoter should be repressed). Sequencing of plasmids within these yeast colonies revealed that all contained mutations in the GAL1p∷PvuII cassette. These results indicated that even the low basal levels of PvuII expression occurring in non-inducing media (glucose) were toxic to yeast cells. To overcome this problem, the GAL1 promoter region of the plasmid pJW8 (GAL1p∷PvuII URA3 CEN/ARS) was mutagenized by PCR in the presence of Mn2+. The promoter fragments were then subcloned upstream of the PvuII gene in place of the original promoter and new plasmids were identified (a) that could transform yeast cells and form colonies with normal growth rates on standard glucose plates, (b) that did not grow when transferred to plates containing 2% galactose (inducing conditions), and (c) which were confirmed by sequencing to have no mutations in the PvuII coding region. This work was described in our previous paper [27], and resulted in the identification of four new mutant GAL1 promoters that have reduced basal activity in glucose media but are still strongly inducible in galactose. Luciferase assays and other tests indicated that one of the new promoters, GAL1-V10, had the lowest levels of activity under non-inducing conditions. For the current work, a new strain called JW1689 (his3Δ∷[GAL1-V10p∷PvuII URA3]) was created that contained the PvuII gene integrated into chromosome XV. This strain, a derivative of T334, has the reg1-501 mutation that permits expression from GAL promoters to be modulated with galactose while cells grow on glucose [22,30].
The impact of PvuII expression on growth of haploid cells is compared to that of the widely studied nuclease EcoRI in Figure 1. The EcoRI expression strain, called YLKL350, contains a his3Δ∷[GAL1p∷EcoRI TRP1] insertion [22]. Inhibition of cell growth by PvuII was observed using ten times less galactose (0.02% vs. 0.2%) than that required for inhibition by EcoRI, even though EcoRI is more strongly induced because it is controlled by the WT GAL1 promoter (Figure 1). This experiment demonstrated the strong toxicity of PvuII and revealed that the threshold for lethal induction of the nuclease from the GAL1-V10 promoter occurs between 0.002% and 0.02% galactose.
Effects of PvuII expression on growth rates, survival, and cell cycling were analyzed in WT cells and in mutants defective in DNA repair (Figure 2). For these experiments overnight cultures were diluted into YPDA medium, grown to log phase, and then induced with 0.02% galactose for 0, 2, 4 and 6 hrs as described in Materials and Methods. Surprisingly, the kinetics of growth inhibition and cell killing (Figures 2A and 2B) were similar in WT haploid cells and in mutants defective in recombination (rad52), NHEJ (dnl4), or both pathways (rad50). Survival was reduced to approximately 10% in each of the mutants, with only rad50 cells showing a small, statistically significant decrease in survival (non-overlapping standard deviation bars). In similar tests using diploid cells, rad52-/- mutants were not more sensitive than WT cells and actually exhibited greater survival at longer expression times (Figures 2D and 2E). Although these survival tests might be influenced by variations in the steady-state levels of PvuII achieved in each mutant, the overall results are striking because they differ strongly from those obtained with EcoRI and HO [22-24,29] where repair mutants are much more affected than WT cells.
DSBs produced by radiation, chemicals, or nucleases such as HO and EcoRI are potent inducers of DNA damage checkpoint responses. As shown in Figures 2C and 2D, expression of PvuII caused a large increase in G2/M cells after 4 and 6 hrs, many of which were enlarged in size, an indication of cellular stress. Unlike other well-studied nucleases and clastogenic chemicals, PvuII expression did not produce a stronger G2 arrest response in the DNA repair mutants than in WT cells, which is consistent with the survival responses.
Rad9 is an important component of the checkpoint system [34,35] and rad9 cells display a diminished G2 arrest response to conventional clastogens. Haploid rad9 cells stopped dividing after 4 hrs, similar to WT cells, and exhibited survival responses that were also similar to WT (Figures 2A and 2B). PvuII induction in haploids caused increases in large-budded cells from ~30% to 65% in WT cells and from 30% to 55% in rad9 mutants (Figure 2C). In diploids, the corresponding increases were from 25% to 65% for WT and 20% to 60% in rad9-/- mutants (Figure 2F). Thus, PvuII expression caused at least a doubling of G2 cells in both WT and rad9 strains. Expression of PvuII in rad9-/- diploids produced growth and survival phenotypes analogous to those seen in haploids (Figures 2D and 2E). These results suggest that PvuII-induced DNA damage is being recognized in the cells and that some level of processing and signaling leading to G2 arrest is involved. However, the arrest and survival response to PvuII-damaged DNA is largely RAD9-independent.
3.2. PvuII expression causes extensive fragmentation of chromosomal DNA within haploid and diploid cells
The ability of PvuII to generate DSBs in cellular DNA was investigated next under conditions where PvuII was either fully induced (2% galactose) or only partially induced (0.02% galactose). Fragmentation of haploid cell DNA was readily detectable on standard agarose gels after 2 hrs induction with 2% galactose (Figure 3A). The fraction of the original uncut chromosomal DNA band that was degraded was small, however. Analysis using pulsed field gel electrophoresis (PFGE) revealed that most whole chromosome bands were broken within the first hr in both haploids and diploids, with somewhat faster kinetics in the latter cells (Figure 3B). Interestingly, most of the broken DNA fragments formed a broad band at 40-50 kb within 1 hr that persisted throughout the time-course, suggesting that many sites were not cut. The target sequence of PvuII (CAGCTG) occurs 2,149 times in the published sequences of all 16 yeast chromosomes (excluding mitochondrial sites; from the Saccharomyces Genome Database at yeastgenome.org). Since the size of the haploid yeast genome is ~ 14,000,000 bp, with some variation caused by size differences in the rDNA repeats on chromosome XII, PvuII recognition sites occur approximately once every 6,500 bp. The accumulation of fragments that are much larger than 6500 bp (~ 40,000 - 50,000) is likely due in part to interference with digestion at some sites by proteins bound to the DNA.
Fig. 3.
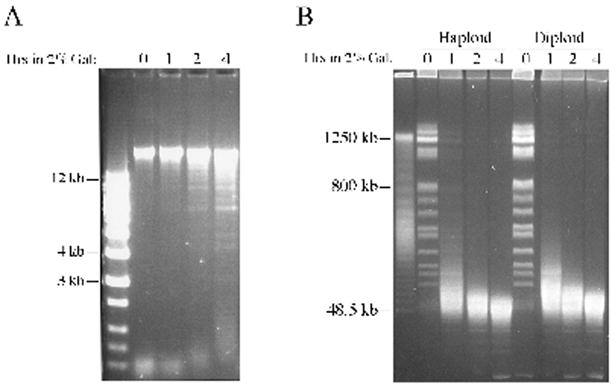
Expression of PvuII in vivo causes chromosomal DNA breakage. DNA from logarithmically growing cells propagated in YPDA media supplemented with 2% galactose was analyzed using (A) a conventional 0.7% agarose gel or (B) a 1% agarose pulsed field gel. 1 Kb ladder was used as molecular weight standard in (A) and PFG Lambda ladder DNA served as control in (B).
We next tested whether DNA breakage could be detected under sub-inducing conditions (0.02% galactose). As shown in Figure 4, modest breakage was observed in haploids after 4-6 hrs. In contrast, chromosome bands were largely abolished in diploids within 4 hrs, in agreement with the stronger effect seen in diploids when 2% galactose was used (Figure 3). There was once again an accumulation of ~ 50 kb fragments (Figure 4B). To investigate this phenomenon further, a Southern blot was performed with the diploid DNA gel using a LYS2 gene-specific probe. In agreement with the stained gels, this more sensitive hybridization experiment demonstrated few fragments below 40-50 kb. Cell survival was also determined after induction with 0.02% galactose and percentages are indicated at the bottom of each lane in Figures 4A and 4B. After 6 hrs, haploid cell cultures contained 6% viable cells and diploid cell survival was reduced to 0.4%. The decreased survival of diploids at each time point is qualitatively consistent with the increased level of chromosome breakage visible in DNA from the diploid cells.
Fig. 4.
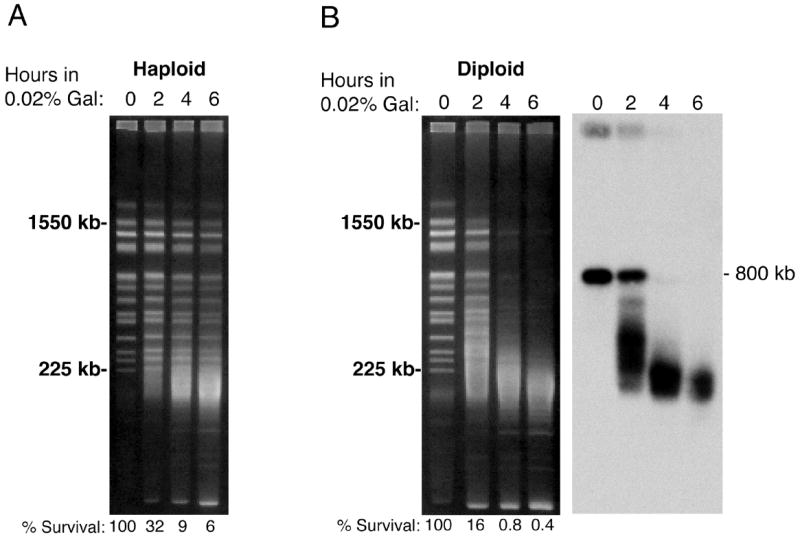
Analysis of PvuII-induced DNA breakage and cell survival in haploid and diploid cells after sub-induction with 0.02% galactose. (A) Time-course demonstrating DSB formation and reduction of survival in haploid cells. (B) Decreased survival and faster DNA breakage kinetics in diploid cells determined using standard pulsed field gel analysis and Southern blotting.
3.3. PvuII-induced DSBs, unlike radiation-induced DSBs, are poorly repaired in diploid cells
We next addressed the question of whether PvuII-induced DSBs can be rejoined if cells are given time to repair the damage prior to DNA extraction and analysis. As a control, log phase diploid cells were treated with either 20 or 150 krad, washed with water and incubated in YPDA media to allow repair prior to PFGE analysis. After exposure of cells to 20 krad, repair of broken fragments (seen as a smear of staining between the chromosome bands) was essentially complete after 4 hrs (Figure 5A). Partial repair was also readily detected after the extensive damage caused by 150 krad, with most chromosome bands becoming visible again during the repair period.
Fig. 5.
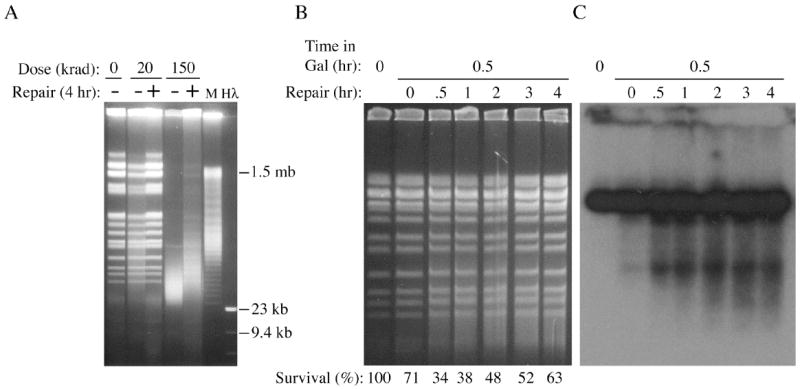
Use of liquid holding experiments to monitor repair of DSBs in diploid cells. (A) Reassembly of broken chromosomes after exposure to 20 or 150 krad gamma radiation followed by a 4 hr liquid holding period. (B and C) Lack of apparent repair of DSBs produced by induction of PvuII for 0.5 hr using 0.2% galactose. Southern blotting was performed as for Figure 4.
To determine if PvuII damage can be repaired, diploid cells containing the integrated GAL1-V10p∷PvuII cassette were grown to log phase and induced for 0.5 hr with 0.2% galactose. Cells were then washed and suspended in YPDA media, which caused glucose repression of the GAL1-V10∷PvuII fusion and provided time for repair of the induced DSBs. Because of the time required for transcription, translation, nuclear import, etc., of the endonuclease at this low dose of galactose, breakage of DNA was not readily apparent until one hour after induction, or 0.5 hr after washing out the galactose (Figure 5B and 5C). A strong smear of broken fragments became visible at this time in ethidium bromide stained gels and also in Southern blots. The strong breakage observed 0.5 hr after galactose removal showed no indication of repair in the stained gel over the course of the next 4 hrs (Figure 5B). Similarly, no repair was detectable between 0.5 hr and 4 hrs using the more sensitive blot hybridization technique, and indeed a slight increase in low molecular weight fragments was observed after 2 hrs that remained essentially unchanged for the rest of the time-course (Figure 5C).
To increase the opportunity for cells to repair PvuII-generated damage by homologous recombination, cells were arrested in G2/M with nocodazole, induced with galactose as before, and then allowed to repair in the continued presence of nocodazole. Control nocodazole-arrested cells maintained in glucose (non-inducing conditions) for up to 6 hrs displayed no changes in chromosome integrity (Figure 6A). After induction with galactose in the presence of nocodazole, DNA from the G2-arrested cells contained an abundance of broken fragments that were not repaired even after 6 hrs incubation in YPDA liquid containing nocodazole. We note that most molecules making up the top DNA band in Figure 6B (chromosome XII) were broken after induction of PvuII and did not reappear by the end of the experiment.
Fig. 6.
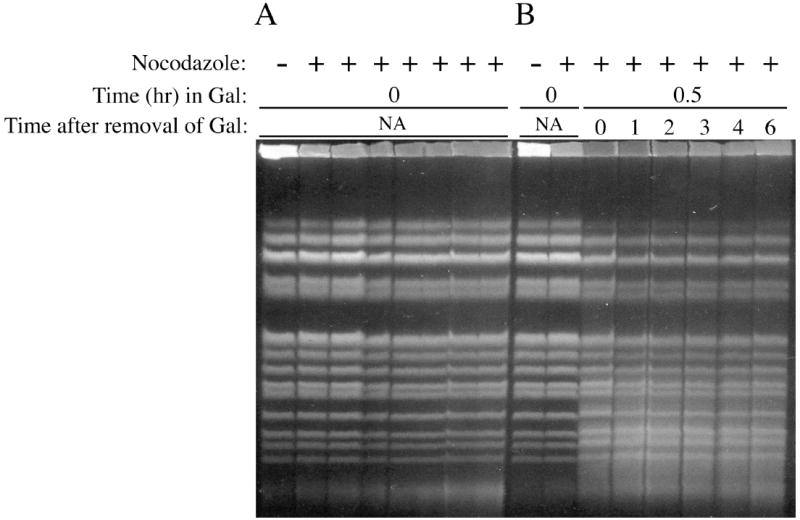
Analysis of changes in PvuII-induced DSBs in diploid cells arrested in G2 phase, conditions that maximize opportunities for recombinational repair. (A) Control experiment demonstrating that incubation of cells in glucose media (non-inducing conditions) in the presence of nocodazole does not affect chromosome structure. Lanes labelled as ‘+’ correspond to 0, 1, 2, 3, 4, 5 and 6 hrs. (B) PvuII-induced DSBs produced after 0.5 hr in 0.2% galactose media supplemented with nocodazole are not repaired after transfer to glucose media containing nocodazole.
3.4. Cells that survive brief induction of PvuII expression exhibit increased frequencies of chromosome XII aberrations
To examine the consequences of PvuII expression on chromosome integrity, we purified DNA from colonies arising from rare cells that survived 6 hr PvuII expression (2% galactose) and assessed chromosome abnormalities using PFGE. Chromosome bands observed for 30 haploid survivors are displayed in Figure 7A. Interestingly, most chromosomes within haploid and diploid survivors of PvuII expression appeared normal, but a high frequency of chromosome XII bands with reduced sizes was observed. Only 4% of colonies from uninduced haploid or diploid cells (grown in glucose only) contained visible changes in chromosome XII (Figure 7B). In contrast, 23% of haploid survivors of PvuII expression and 21% of diploid survivors exhibited aberrations. This 5-fold increase in deletions suggests that repair at a fraction of induced DSBs within the largest yeast chromosome was initiated but could not be completed accurately.
Fig. 7.
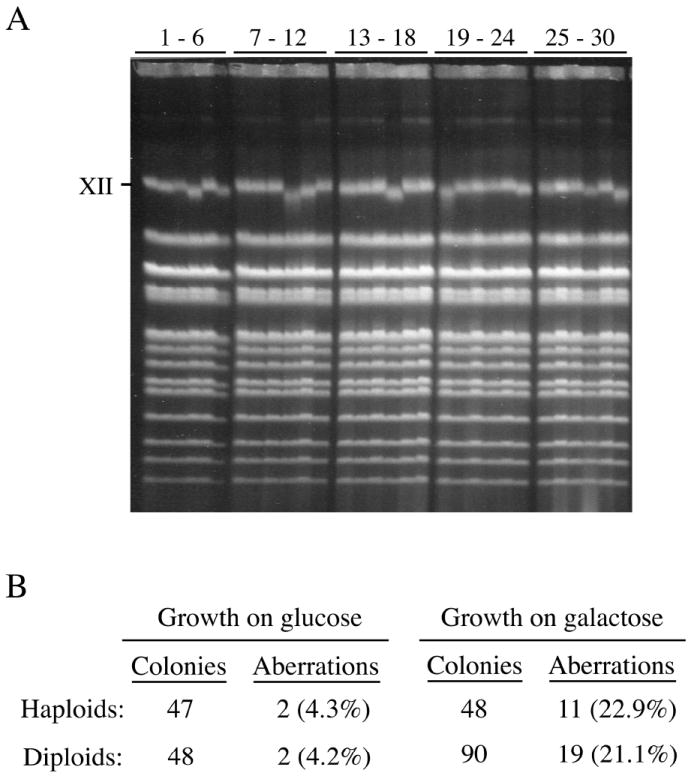
Cells that survive induction of PvuII expression contain a high frequency of chromosome XII aberrations. (A) Pulsed field gel analysis of chromosomes from 30 haploid colonies formed on glucose plates by surviving cells after 6 hrs induction. (B) Chromosome XII deletions are increased among haploid and diploid cells surviving expression of PvuII.
3.5. Blunt-ended DSBs produced by EcoRV are also toxic to haploid and diploid yeast cells
The similar lethality of PvuII expression in both WT cells and DSB repair mutants, combined with the absence of detectable repair, suggests that blunt-ended breaks are not subject to the same repair processes as the sticky-ended DSBs produced by EcoRI, HO and I-SceI. However, it is formally possible that the extreme toxicity of PvuII might be a consequence of a unique characteristic of this enzyme’s mechanism of interaction with DNA or other proteins, rather than from poor repair of blunt DSBs. We therefore tested another blunt end restriction enzyme, EcoRV. New strains were constructed that contained an expression cassette for EcoRV integrated into the same locus used for PvuII expression (his3Δ∷[GAL1-V10p∷EcoRV]). In side-by-side comparisons using haploid cells (Figure 8A), EcoRV toxicity was observed at galactose concentrations similar to those seen with PvuII and distinctly different than EcoRI (Figure 1). In addition, after induction for 2 hrs in 2% galactose, survival of diploid cells was reduced to similarly low levels by EcoRV and PvuII (0.008% and 0.030%, respectively) (Figure 8B). To confirm that the lethality of EcoRV was associated with DSBs, log phase haploid cells grown in YPDA media were induced with galactose and cellular DNA was analyzed by gel electrophoresis after 2, 4 and 6 hrs. Chromosomal DNA breakage was evident at 2 hrs and increased in a time-dependent fashion thereafter (Figure 8C). These results imply that blunt end-producing nucleases are generally more toxic than nucleases that create sticky ends and reinforce the conclusion that blunt-ended DSBs are poor substrates for repair in yeast cells.
4. Discussion
We have demonstrated that expression of the blunt end-producing endonucleases PvuII and EcoRV is highly toxic in haploid and diploid yeast cells. This toxicity was initially identified because single-copy plasmids containing a GAL1p∷PvuII promoter fusion did not yield colonies after transformation into haploid or diploid cells, even under conditions where the GAL1 promoter was repressed. After cloning PvuII under the control of a new promoter, GAL1-V10, that has reduced basal expression in glucose media, subsequent experiments revealed that PvuII expression was more toxic than that of EcoRI, an endonuclease that generates DSBs with cohesive ends.
Cell survival after brief expression of PvuII was not decreased in mutants deficient in homologous recombination, NHEJ or both pathways. These results are strikingly different from observations made in previous work with EcoRI and HO [22,24,26,28,29], which demonstrated that breaks with 4 nucleotide overhangs could be acted upon by both repair pathways. Although PvuII stimulated accumulation of distended (stressed) G2/M cells in the induced cultures, cell cycle arrest and survival were largely independent of the checkpoint gene RAD9. In contrast, cohesive-ended DSBs were potent inducers of cell cycle arrest that was dependent on RAD9 and other damage response checkpoint genes [26,28,33].
Structural analysis of chromosomal DNA isolated from haploid and diploid strains containing the GAL1-V10p∷PvuII fusion demonstrated breakage that was both galactose concentration-dependent and time-dependent. Interestingly, PFGE analysis revealed that PvuII cleavage products became progressively shorter with time until they formed a broad band at approximately 40 – 50 kb. This is considerably larger than the average size of 6,500 bp predicted if digestion were to go to completion (based on 2,149 PvuII sites in the published S. cerevisiae sequence and typical genome size of 14,000,000 bp). The data suggest that some recognition sites remain unavailable in vivo, possibly because they are protected by proteins bound to the DNA.
When gamma radiation-treated diploid cells were incubated in glucose media prior to plating, allowing time for repair of induced DSBs, broken chromosomes were restored. In contrast, no repair of PvuII-induced fragments could be detected. This was also true in diploid cells arrested in G2/M with nocodazole, indicating that DSBs were not repaired even under conditions that maximize opportunities for repair by homologous recombination.
Evidence that at least some breaks can be repaired came from analysis of rare survivors of PvuII expression. DNA purified from colonies formed by surviving cells displayed a 5-fold increase in chromosome XII deletions relative to uninduced cells, with no other chromosome aberrations detected at the level of resolution attainable with PFGE. The induction of chromosome aberrations suggests strongly that DSBs were created but were subsequently repaired by an error-prone process. It is possible that the abundance of chromosome XII deletions is an indication that breaks occurred in the 100 – 200 tandem repeats of 9.1 kb rDNA on this chromosome (each repeat has 3 potential PvuII recognition sites) (36). We speculate that the DSBs were repaired by a non-conservative, homology-dependent mechanism such as single-strand annealing [5,6]. It remains unclear why, if most DSBs induced by PvuII are not repaired, these cells survived the incubation in galactose media. A possibility is that galactose induction within cell cultures is a stochastic process, whereby GAL promoters in most cells are activated efficiently but the process is delayed or reduced in a small fraction of cells. These rare cells would experience few or no breaks and be capable of forming colonies on plates after PvuII expression was repressed.
It was necessary to address the possibility that the toxicity of PvuII might be due to some unique characteristic of the enzyme rather than from induction of blunt-ended DSBs. For example, the enzyme might cut both DNA strands and then remain bound to a broken end, interfering with repair enzymes and increasing lethality, though such a mechanism has not been proposed for this endonuclease. New tests using strains containing the unrelated blunt end-producing restriction enzyme EcoRV under the control of the GAL1-V10 promoter demonstrated that this nuclease was fully as toxic as PvuII. The recognition sequence for EcoRV (GATˆATC) is of similar size to that of PvuII (CTGˆCAG). However, the S. cerevisiae genome sequence has 4203 predicted chromosomal EcoRV sites versus only 2149 for PvuII. Both enzymes reduced survival strongly, but EcoRV expression produced four-fold higher killing than PvuII in diploid cells (Figure 8). It is possible that this result is a consequence of the larger number of EcoRV target sites within the cells.
Our general conclusion that blunt ends are repaired extremely poorly in yeast cells is consistent with previous reports of inefficient repair in eukaryotic cells. Experiments involving direct transfer of restriction enzymes into cells of higher eukaryotes have frequently found that blunt end-producing endonucleases induce aberrations more efficiently than those that generate ssDNA overhangs [1,19-21]. In yeast cells, polyethylene glycol-mediated transfer of active AluI restriction enzyme (inducing blunt ended DSBs at the sequence AGˆCT) produced a dose-dependent killing effect, though this effect was not compared to that of nucleases producing sticky ends [37]. Recircularization of linear plasmids with blunt ends by NHEJ after transformation into yeast cells is greatly decreased compared to plasmids with complementary overhangs [38-42]. In two such recircularization studies essentially all rejoined plasmids had undergone deletions during repair [38,39], though another report found that over 50% of the blunt ended plasmid DNAs were repaired accurately [41]. In a more comprehensive study, Daley and Wilson [42] observed that blunt-ended linear plasmids were poorly repaired, but ends with short (≤ 4 nt) overhangs were rejoined effectively by NHEJ. Ends with even longer overhangs (> 4 nt) were rejoined most efficiently, being repaired by both NHEJ and by a separate, RAD52-dependent process [42].
Studies utilizing cell extracts or purified DNA repair proteins have been less consistent. For example, human cell extracts were found to rejoin purified DNA fragments containing cohesive ends more efficiently than fragments with blunt ends [43]. However, several proteins associated with DNA repair and replication in yeast and human cells (e.g., Ku70/Ku80, Mre11) are able to act on blunt-ended substrates in vitro, while others, including several DNA helicase enzymes, cannot (e.g., see references [4,44-47]. In a review of studies analyzing recruitment of GFP-tagged repair proteins to DSBs, Harrison and Haber suggested a model in which the Mrx complex and checkpoint proteins such as Tel1 associate with blunt ends in vivo, but in which many more proteins are recruited to DSBs containing ssDNA overhangs [35].
Enzymatic and structural properties of the restriction endonucleases EcoRI, PvuII and EcoRV have been extensively investigated. Each of the enzymes forms Mg2+-dependent, dimeric complexes, and protein:DNA co-crystal structures have been determined [48]. Similar to several other sequence-specific DNA binding proteins, EcoRI and EcoRV induce bending of the DNA helix when bound, but PvuII does not. EcoRI (and BamHI, another widely studied cohesive end-producing endonuclease) binds primarily to the major groove of the helix, whereas both PvuII and EcoRV associate primarily with nucleotides in the minor groove [48]. At present it is unclear if characteristics such as these or another shared property might in some way contribute to the greater toxicity of the two blunt end-producing enzymes.
An important question that arises from this work is why ionizing radiation-induced DSBs are repaired so much more efficiently than blunt-ended DSBs. Ends produced by radiation essentially always contain damaged bases and sugars and are frequently missing a terminal nucleotide [1,3]. In addition, these ends often retain 3’ phosphate or phosphoglycolate moieties. In principle, it would appear that blunt ends could be repaired by simple ligation, while radiation-induced DSBs require several steps to remove the damage, restore all sequence information to its original state, and rejoin the ends. However, experiments have demonstrated that chromosomal DSBs induced by radiation are resected rapidly and repaired accurately most of the time [51,52].
A possible explanation for the more efficient repair of radiation-induced DSBs is that the damaged ends, or structures created by initial damage processing, act as high affinity binding sites for DNA damage recognition proteins. For example, several radiation-induced oxidized base structures are recognized by specific proteins of the base excision repair pathway. In addition, 3’ phosphates are targeted by a 3’ phosphatase, Tpp1, as well as the Rad1/Rad10 complex and the endonucleases Apn1 and Apn2 [49,50]. Such proteins may create new structures, e.g., short ssDNA regions, that are binding sites for proteins such as Rpa (single stranded DNA binding protein), which may then attract other repair proteins. Alternatively, the proteins binding to the initial lesions may actively recruit other protein complexes including those involved in end resection and DSB repair. A similar binding and recruiting mechanism might also explain why DSBs containing 4 nt ssDNA overhangs are repaired so much more efficiently than blunt ends, though in this case there is the added possibility of simple annealing of the complementary overhangs to restore duplex structure followed by ligation.
In summary, the results presented here provide strong evidence that blunt-ended chromosomal DSBs such as those induced by PvuII and EcoRV are recognized and repaired distinctly differently than DSBs containing either complementary cohesive ends or ends produced by ionizing radiation. Elucidating these differences is important because DSBs with many types of end structures are generated in eukaryotic organisms, e.g., as part of programmed events such as immunoglobulin gene switching, crossing over in meiosis, and mating type switching, and after exposure to physical and chemical DNA damaging agents.
Acknowledgments
The authors wish to thank Robert Blumenthal and Stephen Halford for gifts of PvuII and EcoRV gene plasmids. LKL was supported in part by National Institutes of Health grant 1R15AG028520-01A1 and a departmental grant from the Welch Foundation. MAR and JWW were supported by the Intramural Research Program of the NIEHS (NIH, DHHS) under project 1Z01ES065073.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Obe G, Johannes C, Schulte-Frohlinde D. DNA double-strand breaks induced by sparsely ionizing radiation and endonucleases as critical lesions for cell death, chromosomal aberrations, mutations and oncogenic transformation. Mutagenesis. 1992;7:3–12. doi: 10.1093/mutage/7.1.3. [DOI] [PubMed] [Google Scholar]
- 2.Branzei D, Foiani M. Interplay of replication checkpoints and repair proteins at stalled replication forks. DNA Repair (Amst) 2007;6:994–1003. doi: 10.1016/j.dnarep.2007.02.018. [DOI] [PubMed] [Google Scholar]
- 3.Shikazono N, Noguchi M, Fujii K, Urushibara A, Yokoya A. The yield, processing, and biological consequences of clustered DNA damage induced by ionizing radiation. J Radiat Res (Tokyo) 2009;50:27–36. doi: 10.1269/jrr.08086. [DOI] [PubMed] [Google Scholar]
- 4.Krogh BO, Symington LS. Recombination proteins in yeast. Annu Rev Genet. 2004;38:233–271. doi: 10.1146/annurev.genet.38.072902.091500. [DOI] [PubMed] [Google Scholar]
- 5.Li X, Heyer WD. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008;18:99–113. doi: 10.1038/cr.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu Rev Biochem. 2008;77:229–257. doi: 10.1146/annurev.biochem.77.061306.125255. [DOI] [PubMed] [Google Scholar]
- 7.Shrivastav M, De Haro LP, Nickoloff JA. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008;18:134–147. doi: 10.1038/cr.2007.111. [DOI] [PubMed] [Google Scholar]
- 8.Zhu Z, Chung WH, Shim EY, Lee SE, Ira G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell. 2008;134:981–994. doi: 10.1016/j.cell.2008.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mimitou EP, Symington LS. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature. 2008;455:770–774. doi: 10.1038/nature07312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ira G, Pellicioli A, Balijja A, Wang X, Fiorani S, Carotenuto W, Liberi G, Bressan D, Wan L, Hollingsworth NM, Haber JE, Foiani M. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature. 2004;431:1011–1017. doi: 10.1038/nature02964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lewis LK, Karthikeyan G, Cassiano J, Resnick MA. Reduction of nucleosome assembly during new DNA synthesis impairs both major pathways of double-strand break repair. Nucl Acids Res. 2005;33:4928–4939. doi: 10.1093/nar/gki806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sonoda E, Hochegger H, Saberi A, Taniguchi Y, Takeda S. Differential usage of non-homologous end-joining and homologous recombination in double strand break repair. DNA Repair (Amst) 2006;5:1021–1029. doi: 10.1016/j.dnarep.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 13.Wyman C, Kanaar R. DNA double-strand break repair: all’s well that ends well. Annu Rev Genet. 2006;40:363–383. doi: 10.1146/annurev.genet.40.110405.090451. [DOI] [PubMed] [Google Scholar]
- 14.Shim EY, Hong SJ, Oum JH, Yanez Y, Zhang Y, Lee SE. RSC mobilizes nucleosomes to improve accessibility of repair machinery to the damaged chromatin. Mol Cell Biol. 2007;27:1602–1613. doi: 10.1128/MCB.01956-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Daley JM, Palmbos PL, Wu D, Wilson TE. Nonhomologous end joining in yeast. Annu Rev Genet. 2005;39:431–451. doi: 10.1146/annurev.genet.39.073003.113340. [DOI] [PubMed] [Google Scholar]
- 16.Hefferin ML, Tomkinson AE. Mechanism of DNA double-strand break repair by non-homologous end joining. DNA Repair. 2005;4:639–648. doi: 10.1016/j.dnarep.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 17.Rogers-Bald M, Sargent RG, Bryant PE. Production of chromatid breaks by a single DSB: evidence supporting the signal model. Int J Radiat Biol. 2000;76:23–29. doi: 10.1080/095530000138970. [DOI] [PubMed] [Google Scholar]
- 18.Johannes C, Horstmann M, Durante M, Chudoba I, Obe G. Chromosome intrachanges and interchanges detected by multicolor banding in lymphocytes: searching for clastogen signatures in the human genome. Radiat Res. 2004;161:540–548. doi: 10.1667/rr3157. [DOI] [PubMed] [Google Scholar]
- 19.Bryant PE. Use of restriction endonucleases to study relationships between DNA double-strand breaks, chromosomal aberrations and other end-points in mammalian cells. Int J Radiat Biol. 1988;54:869–890. doi: 10.1080/09553008814552291. [DOI] [PubMed] [Google Scholar]
- 20.Costa ND, Bryant PE. Differences in accumulation of blunt- and cohesive-ended double-strand breaks generated by restriction endonucleases in electroporated CHO cells. Mutat Res. 1991;254:239–246. doi: 10.1016/0921-8777(91)90062-t. [DOI] [PubMed] [Google Scholar]
- 21.Venditti S, Camilloni G. In vivo analysis of chromatin following nystatin-mediated import of active enzymes into Saccharomyces cerevisiae. Mol Genet Genom. 1994;242:100–104. doi: 10.1007/BF00277353. [DOI] [PubMed] [Google Scholar]
- 22.Lewis LK, Westmoreland JW, Resnick MA. Repair of endonuclease-induced double-strand breaks in Saccharomyces cerevisiae: essential role for genes associated with nonhomologous end-joining. Genetics. 1999;152:1513–1529. doi: 10.1093/genetics/152.4.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mills KD, Sinclair DA, Guarente L. MEC1-dependent redistribution of the Sir3 silencing protein from telomeres to DNA double-strand breaks. Cell. 1999;97:609–620. doi: 10.1016/s0092-8674(00)80772-2. [DOI] [PubMed] [Google Scholar]
- 24.Schär P, Fäsi M, Jessberger R. SMC1 coordinates DNA double-strand break repair pathways. Nucl Acids Res. 2004;32:3921–3929. doi: 10.1093/nar/gkh716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamana Y, Maeda T, Ohba H, Usui T, Ogawa HI, Kusano K. Regulation of homologous integration in yeast by the DNA repair proteins Ku70 and RecQ. Mol Genet Genom. 2005;273:167–176. doi: 10.1007/s00438-005-1108-y. [DOI] [PubMed] [Google Scholar]
- 26.Grenon M, Magill CP, Lowndes NF, Jackson SP. Double-strand breaks trigger MRX- and Mec1-dependent, but Tel1-independent, checkpoint activation. FEMS Yeast Res. 2006;6:836–847. doi: 10.1111/j.1567-1364.2006.00076.x. [DOI] [PubMed] [Google Scholar]
- 27.Lewis LK, Lobachev K, Westmoreland JW, Karthikeyan G, Williamson KM, Jordan JJ, Resnick MA. Use of a restriction endonuclease cytotoxicity assay to identify inducible GAL1 promoter variants with reduced basal activity. Gene. 2005;363:183–192. doi: 10.1016/j.gene.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 28.Sugawara N, Haber JE. Repair of DNA double strand breaks: in vivo biochemistry. Methods Enzymol. 2006;408:416–429. doi: 10.1016/S0076-6879(06)08026-8. [DOI] [PubMed] [Google Scholar]
- 29.Signon L, Malkova A, Naylor ML, Klein H, Haber JE. Genetic requirements for RAD51- and RAD54-independent break-induced replication repair of a chromosomal double-strand break. Mol Cell Biol. 2001;21:2048–2056. doi: 10.1128/MCB.21.6.2048-2056.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hovland P, Flick J, Johnston M, Sclafani RA. Galactose as a gratuitous inducer of GAL gene expression in yeasts growing on glucose. Gene. 1989;83:57–64. doi: 10.1016/0378-1119(89)90403-4. [DOI] [PubMed] [Google Scholar]
- 31.Blumenthal RM, Gregory SA, Cooperider JS. Cloning of a restriction-modification system from Proteus vulgaris and its use in analyzing a methylase-sensitive phenotype in Escherichia coli. J Bacteriol. 1985;164:501–9. doi: 10.1128/jb.164.2.501-509.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gietz RD, Woods RA. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 2002;350:87–96. doi: 10.1016/s0076-6879(02)50957-5. [DOI] [PubMed] [Google Scholar]
- 33.Lewis LK, Kirchner JM, Resnick MA. Requirement for end-joining and checkpoint functions, but not RAD52-mediated recombination, after EcoRI endonuclease cleavage of Saccharomyces cerevisiae DNA. Mol Cell Biol. 1998;18:1891–1902. doi: 10.1128/mcb.18.4.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Toh GW, Lowndes NF. Role of the Saccharomyces cerevisiae Rad9 protein in sensing and responding to DNA damage. Biochem Soc Trans. 2003;31(Pt 1):242–246. doi: 10.1042/bst0310242. [DOI] [PubMed] [Google Scholar]
- 35.Harrison JC, Haber JE. Surviving the breakup: the DNA damage checkpoint. Annu Rev Genet. 2006;40:209–235. doi: 10.1146/annurev.genet.40.051206.105231. [DOI] [PubMed] [Google Scholar]
- 36.Kim YH, Ishikawa D, Ha HP, Sugiyama M, Kaneko Y, Harashima S. Chromosome XII context is important for rDNA function in yeast. Nucl Acids Res. 2006;34:2914–2924. doi: 10.1093/nar/gkl293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Winckler K, Bach B, Obe G. Survival of Saccharomyces cerevisiae after treatment with the restriction endonuclease Alu I. Int J Radiat Biol. 1988;54:563–566. doi: 10.1080/09553008814552001. [DOI] [PubMed] [Google Scholar]
- 38.Boulton SJ, Jackson SP. Saccharomyces cerevisiae Ku70 potentiates illegitimate DNA double-strand break repair and serves as a barrier to error-prone DNA repair pathways. EMBO J. 1996;15:5093–5103. [PMC free article] [PubMed] [Google Scholar]
- 39.Boulton SJ, Jackson SP. Identification of a Saccharomyces cerevisiae Ku80 homologue: roles in DNA double strand break rejoining and in telomeric maintenance. Nucl Acids Res. 1996;24:4639–4648. doi: 10.1093/nar/24.23.4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsukamoto Y, Kato J, Ikeda H. Silencing factors participate in DNA repair and recombination in Saccharomyces cerevisiae. Nature. 1997;388:900–903. doi: 10.1038/42288. [DOI] [PubMed] [Google Scholar]
- 41.Herrmann G, Lindahl T, Schär P. Saccharomyces cerevisiae LIF1: a function involved in DNA double-strand break repair related to mammalian XRCC4. EMBO J. 1998;17:4188–4198. doi: 10.1093/emboj/17.14.4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Daley JM, Wilson TE. Rejoining of DNA double-strand breaks as a function of overhang length. Mol Cell Biol. 2005;25:896–906. doi: 10.1128/MCB.25.3.896-906.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ganesh A, North P, Thacker J. Repair and misrepair of site-specific DNA double-strand breaks by human cell extracts. Mutat Res. 1993;299:251–259. doi: 10.1016/0165-1218(93)90101-i. [DOI] [PubMed] [Google Scholar]
- 44.de Jager M, Dronkert ML, Modesti M, Beerens CE, Kanaar R, van Gent DC. DNA-binding and strand-annealing activities of human Mre11: implications for its roles in DNA double-strand break repair pathways. Nucl Acids Res. 2001;29:1317–1325. doi: 10.1093/nar/29.6.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fisher TS, Zakian VA. Ku: a multifunctional protein involved in telomere maintenance. DNA Repair (Amst) 2005;4:1215–1226. doi: 10.1016/j.dnarep.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 46.Boulé JB, Zakian VA. Roles of Pif1-like helicases in the maintenance of genomic stability. Nucl Acids Res. 2006;34:4147–4153. doi: 10.1093/nar/gkl561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu Y, Siino JS, Sugiyama T, Kowalczykowski SC. The DNA binding preference of RAD52 and RAD59 proteins: implications for RAD52 and RAD59 protein function in homologous recombination. J Biol Chem. 2006;281:40001–40009. doi: 10.1074/jbc.M608071200. [DOI] [PubMed] [Google Scholar]
- 48.Pingoud A, Fuxreiter M, Pingoud V, Wende W. Type II restriction endonucleases: structure and mechanism. Cell Mol Life Sci. 2005;62:685–707. doi: 10.1007/s00018-004-4513-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gros L, Saparbaev MK. Enzymology of the repair of free radicals-induced DNA damage. Oncogene. 2002;21:8905–8925. doi: 10.1038/sj.onc.1206005. [DOI] [PubMed] [Google Scholar]
- 50.Karumbati AS, Deshpande RA, Jilani A, Vance JR, Ramotar D, Wilson TE. The role of yeast DNA 3’-phosphatase Tpp1 and Rad1/Rad10 endonuclease in processing spontaneous and induced base lesions. J Biol Chem. 2003;278:31434–31443. doi: 10.1074/jbc.M304586200. [DOI] [PubMed] [Google Scholar]
- 51.Argueso JL, Westmoreland J, Mieczkowski PA, Gawel M, Petes TD, Resnick MA. Double-strand breaks associated with repetitive DNA can reshape the genome. Proc Natl Acad Sci USA. 2008;105:11845–11850. doi: 10.1073/pnas.0804529105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Westmoreland J, Ma W, Yan Y, Van Hulle K, Malkova A, Resnick MA. RAD50 is required for efficient initiation of resection and recombinational repair at random, gamma-induced double-strand break ends. PLoS Genet. 2009;5 doi: 10.1371/journal.pgen.1000656. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]