

Abstract
Artemis is a member of the β–CASP family of nucleases in the metallo-β-lactamase superfamily of hydrolases. Artemis has been demonstrated to be involved in V(D)J-recombination and in the NHEJ-catalyzed repair of DNA DSBs. In vitro, both DNA-PK independent 5’ to 3’ exonuclease activity and DNA-PK dependent endonuclease activity have been attributed to Artemis, though mutational analysis of the Artemis active site only disrupts endonuclease activity. This suggests that either the enzyme contains two different active sites, or the exonuclease activity is not intrinsic to the Artemis polypeptide. To distinguish between these possibilities, we sought to determine if it was possible to biochemically separate Artemis endonuclease activity from exonuclease activity. Recombinant [His]6–Artemis was expressed in a Baculovirus insect-cell expression system and isolated using a three-column purification methodology. Exonuclease and endonuclease activity, the ability to be phosphorylated by DNA-PK, and Artemis antibody reactivity was monitored throughout the purification and to characterize final pools of protein preparation. Results demonstrated the co-elution of exonuclease and endonuclease activity on a Ni-Agarose affinity column but separation of the two enzymatic activities upon fractionation on a hydroxyapatite column. An exonuclease free fraction of Artemis was obtained that retained DNA-PK dependent endonuclease activity, was phosphorylated by DNA-PK and reacted with an Artemis specific antibody. These data demonstrate that the exonuclease activity thought to be intrinsic to Artemis can be biochemically separated from the Artemis endonuclease.
Keywords: Artemis, DNA end processing, NHEJ, DNA-PK
1. Introduction
In higher eukaryotes ionizing-radiation (IR) induced DNA double strand breaks (DSB) are primarily repaired by the non-homologous end joining (NHEJ) pathway [1]. Ku, a heterodimeric protein with a unique bridge and pillar structure has a very high affinity for DNA termini and binds to the site of the DSB [2]. The DNA-dependent protein kinase catalytic subunit (DNA-PKcs) is then recruited to the site of the break interacting with both the DNA terminus and the Ku heterodimer. The resulting heterotrimeric complex, termed DNA-PK, is active as a serine/threonine protein kinase and can phosphorylate downstream substrates. As IR induced DSBs often contain other DNA structural damage including thymine glycols, ring fragmentation, 3’ phosphoglycolates, 5’ hydroxyl groups and abasic sites, processing of DNA termini is often necessary before ligation of the double strand break by the XRCC4/Ligase IV/XLF complex can occur [1, 3]. A variety of enzymes have been implicated in DNA processing, including but not limited to, FEN-1[4], polynucleotide kinase (PNK) [5], Werner protein [6, 7], MRN [8], DNA polymerase μ and λ [9, 10], and the nuclease Artemis [11].
The results implicating the involvement of Artemis in the NHEJ pathway are based on in vivo data showing that Artemis null cells are more sensitive to IR than wild type counterparts [12]. Artemis has DNA-PK dependent endonuclease activity on DNA hairpin structures, and DNA-PK dependent endonuclease processing of 3’ and 5’ single-strand overhangs, with preferential cleavage at the dsDNA/ssDNA junction [11]. It has been suggested that the stimulation of endonuclease activity of Artemis requires binding and phosphorylation by DNA-PK which causes a conformational change in the C-terminal region of Artemis, resulting in relief of Artemis autoinihibition of the endonuclease active site [13]. Other labs have suggested that autophosophorylation of DNA-PK results in a conformational change in the DNA-bound kinase which in turn alters the conformation of DNA such that it can be easily recognized and cleaved by Artemis [14, 15]. While each model differs slightly in mechanism, both models suggest that Artemis endonuclease activity is DNA-PK and ATP dependent. In addition to DNA-PK-dependent endonuclease activity, Artemis has been suggested to possess an intrinsic 5’ to 3’ DNA-PK-independent exonuclease activity based on in vitro analysis of partially purified preparations of Artemis [11].
Artemis is a member of the β-CASP family, a new group of the metallo-β-lactamase fold superfamily made up of enzymes acting on nucleic acids [16, 17]. Mutational analysis of conserved residues in the catalytic domain disrupt the endonuclease activity of Artemis, although each of these mutants still possess robust exonuclease activity [18, 19]. This could be a result of Artemis having two independent catalytic sites, one for each of its proposed nuclease activities. However, this would make Artemis a unique enzyme within its family, as metallo-β-lactamase fold enzymes have been classified as only having one active site that has been shown to be the functional catalytic site for all activity [20]. Interestingly, the exonuclease activity has not to date been shown to have a role in vivo, whereas the endonuclease activity has been demonstrated both in vitro and in vivo [1, 21]. In vitro characterization of the exonuclease activity has largely relied on partially purified Artemis protein produced in exogenous systems. A variety of protein purification protocols have been used to obtain purified Artemis, and all include a tagged form of Artemis and affinity chromatography [3, 11, 14, 22-24]. Some preparations also include an ionic exchange fractionation step, but all final preparations contain both endonuclease and exonuclease activity. Considering the discrepancy between the existing genetic, biochemical and structural data, we pursued the fractionation of Artemis in a baculovirus expression system to ascertain if the exonuclease and endonuclease activity were biochemically separable. We developed a three-step purification protocol which results in the separation of the exonuclease activity from the intrinsic endonuclease activity of Artemis. Biochemical analyses demonstrate unequivocally that the exonuclease activity associated with Artemis is not intrinsic to the Artemis polypeptide. These results are discussed in the context of in vitro and in vivo processing of DNA termini in the NHEJ pathway.
2. Materials and Methods
2.1. Cloning, protein expression and purification of [His]6-Artemis
The Artemis gene was amplified via PCR from a B-cell cDNA library using primers specifically designed to encompass the entire gene. The PCR product was cloned into a BLUNT-TOPO-II to create the pCR-Blunt-Artemis construct. The Artemis gene was excised with Xba I which was filled in with Sequenase and dNTP’s prior to digestion with KpnI. The fragment was then gel purified and cloned into the pRSETC vector to incorporate an N-terminal His6 tag. The His6-tagged Artemis gene was excised using Xba I and Not I, and cloned into the pBacPAK8 vector to create BacPAK8-Art-His. Sequencing analysis verified the insert sequence and this construct was transfected into SF9 insect cells in conjunction with Bsu36I-digested BacPAK6 viral DNA to generate recombinant baculovirus. The virus was isolated, amplified, and titered as described previously [25].
200 mL of SF9 cells were infected with this recombinant baculovirus, driving expression of the [His]6-Artemis protein for 48 hours. Infected cells were sedimented at 4,000 × g at 4° C for 15 min and washed once in PBS, repelleted and resuspended in buffer P50/10 (50 mM KPi, pH 7.85, 10mM KCl, 10% glycerol, 5 mM imidazole). All buffers used in protein purification were supplemented with protease inhibitors (0.5 mM PMSF and 1 μg/mL each leupeptin and pepstatin). Cells were lysed by Dounce homogenization and sonication, and extract was sedimented at 10,000 × g at 4°C for 30 minutes. The supernatant was retrieved, and KCl was added to adjust the salt concentration to 500 mM KCl. The high-salt extract was batch adsorbed to 10 mL of Phosphocellulose matrix equilibrated in buffer P50/500 (50 mM KPi, pH 7.85, 500 mM KCl, 10% glycerol, 5 mM imidazole) and the flow-through was collected and immediately applied to a 2 mL Nickel-agarose column equilibrated in the same buffer. The flow-through material was re-applied and the column subsequently washed with 20 mL buffer P50/500 followed by a 20 mL wash with P50/10. Bound protein was eluted in P50/10 with 200 mM imidazole. Fractions were collected and screened for protein using Bradford reagent. Fractions containing an absorbance greater than 50% of the maximal absorbance were pooled and dialyzed overnight against buffer P10/200 (10 mM KPi, pH 7.85, 200 mM KCl, 10% glycerol, 2 mM DTT). A portion of protein pooled from the Nickel-agarose column was dialyzed overnight against Buffer A (50 mM Tris, pH 7.5, 200 mM KCl, 20% glycerol, 1 mM DTT), aliquoted and stored directly at -80° C. Following dialysis, the remaining sample was further fractionated by chromatography on a 5mL CHT-Hydroxyapatite (HAP) column (Biorad) equilibrated in buffer P10/200. The column was washed with buffer P10/200 and bound protein was eluted with a 25 mL linear gradient from 10 mM KPi to 500 mM KPi. All fractions were assayed for total protein and screened for exonuclease activity. Pooled fractions were dialyzed overnight against Buffer A and frozen at -80° C.
2.2. Protein purification of DNA-PK
Cell free extracts were prepared from 12 L of HeLa cells and DNA-PK was purified as described previously [26].
2.3. SDS-PAGE and western blot analysis
Protein samples were separated by SDS-PAGE and either stained with Coomassie Blue or transferred to Immobilon-FL membranes (Millipore, Bedford, MA), probed and then visualized using chemiluminescent detection, and the LAS-3000 imaging system (FujiFilm) was used to document and quantify blots. [His]6-Artemis was detected in immunoblots using either a monoclonal antibody against the N-terminal [His]6 fusion tag, anti-xpress, (Invitrogen) or a polyclonal antibody directed against the full length of Artemis (Bethyl Laboratories, Montgomery, TX).
2.4. In vitro DNA-PK kinase assays
Kinase assays were performed at 37°C in a final volume of 20 μL containing 50 mM HEPES, pH 7.5, 100 mM KCl, 10 mM MgCl2, 0.2 mM EGTA, 0.1 mM EDTA, 1 mM DTT, 125 μM ATP, [γ-32P]-ATP (1.0 μCi), 100 nM 30-bp full duplex DNA, 60 nM DNA-PK and varying concentrations of [His6]-Artemis, as indicated. DNA-PK was added to buffer and DNA and incubated on ice for 5 minutes, followed by addition of Artemis. Reactions were initiated with the addition of ATP, incubated at 37°C for 30 minutes, and terminated by addition of SDS loading dye. Reactions were heated at 95°C for 5 minutes and separated by SDS-PAGE. Gels were dried and phosphorylated products were visualized by PhosphorImager analysis [26].
2.5. In vitro exonuclease assays
The 5’ radiolabeled DNA substrate (CCCCTATCCTTTCCGCGTCCTTACTTCCCC) used for single-strand nuclease assays was radiolabeled with T4 polynucleotide kinase and [γ-32 P] ATP [27]. To generate the 3’ radiolabeled DNA substrate [14] for single-strand nuclease assays, complementary oligonucleotide were, extended and labeled with [α-32P] dCTP and Klenow (exo-) fragment. The extension reactions were performed for 30 minutes at 37°C, followed by a chase reaction containing 1mM dCTP to ensure full extension. The DNA was denatured at 95°C in formamide buffer and separated on a 12% polyacrylamide/urea denaturing gel. The radiolabeled band was visualized using film, excised, eluted from the gel piece, ethanol precipitated and resuspended in water. Single-strand nuclease assays were carried out in a final volume of 15 μL in nuclease buffer (25 mM Tris, pH 8.0, 10 mM KCl, 10 mM MgCl2, 1 mM DTT) with 50 fmol of radiolabeled DNA and varying amounts of Artemis (as indicated) at 37°C for 30 minutes. Reactions were terminated by the addition of formamide loading dye, heated at 95°C for 5 min, loaded onto a 12% polyacrylamide/urea denaturing gel, and products were visualized by PhoshorImager analysis.
2.6. In vitro endonuclease assays
The 5’ radiolabeled hairpin substrate with a 6 base single-strand overhang and the 3’ overhang DNA substrates were prepared as previously described [11]. The 3’ radiolabeled substrate with a 5’ single-strand overhang was prepared as described above (2.4), except following ethanol precipitation the substrate was re-annealed to its complement. Endonuclease assays were carried out in a final volume of 10 μL containing nuclease buffer with 50ng/ul of BSA, varying amounts of Artemis, 50 nM of DNA-PK, 250 fmol of radiolabeled DNA and 250 μM ATP (unless otherwise indicated). Reactions were incubated, terminated and visualized as described above for single-strand nuclease assays.
3. Results
Artemis has been reported to possess 5’ to 3’ exonuclease activity in vitro on ssDNA, as well as DNA-PK-dependent endonuclease activity on single-strand overhang and hairpin DNA structures [11]. However, enzymes within the metallo-β-lactamase family typically contain only one active site that has been shown to be the functional catalytic site for all substrates [20]. Possessing two different nuclease activities that are located within two different active sites would make Artemis unique in the metallo-β-lactamase family. We sought to determine biochemically if in fact the reported 5’-3’ exonuclease activity of Artemis is an intrinsic activity of the Artemis polypeptide. To accomplish this, following cloning and overexpression of Artemis, we undertook the process of fractionating the [His]6-Artemis fusion protein via column chromatography and monitoring exonuclease activity. A three-step protein purification procedure was developed including anionic exchange, Nickel-affinity and hydroxyapatite column chromatography (Fig. 1A).
Fig. 1.
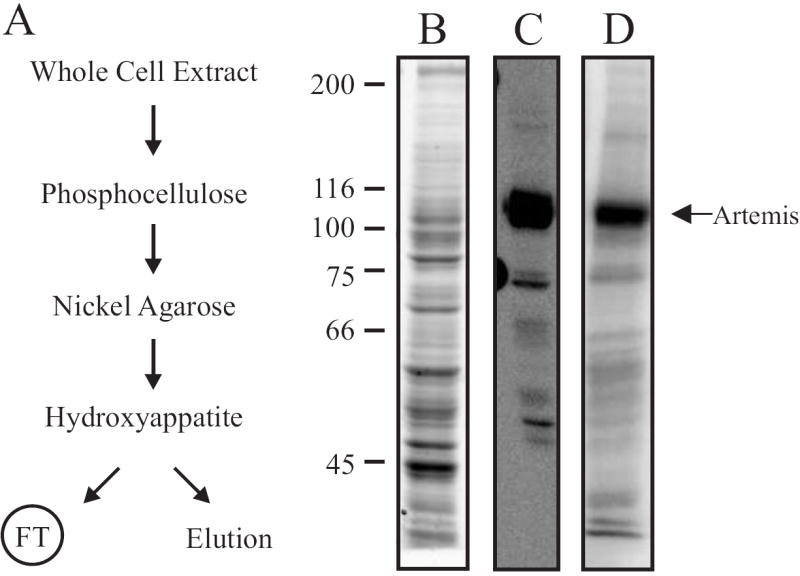
(A) Purification scheme for [His]6-Artemis. A whole cell extract preparation from SF9 insect cells was fractionated over a phosphocellulose column. The flow-through was applied over a 2 mL Nickel-Agarose column, and [His]6-Artemis was eluted with 200 mM imidazole. Pooled fractions were further fractionated over a 5mL hydroxyappatite column. The FT containing [His]6-Artemis and elution containing exonuclease activity were collected from the HAP column. (B) Analysis of whole cell extract from SF9 insect cells containing over-expressed Artemis-[His]6. Coomassie-stained SDS-PAGE gel of the whole cell extract (2.5 μg) made from insect cells infected with recombinant [His]6-Artemis baculovirus. (C) Western blot analysis of whole cell extract (2.5 μg) using the anti-Xpress antibody, as described in Materials and Methods. (D) Phosphorylation of Artemis by DNA-PK was measured by incubating whole cell extract containing [His]6-Artemis (5 μg) with 60 nM DNA-PK in presence of 100 nM 30- mer full duplex DNA, 125 μM ATP and 1.0 μCi [γ-32P]-ATP as described in Materials and Methods. Reactions were terminated by addition of SDS, separated by 8% SDS-PAGE, and the gel was dried and exposed to a PhosphorImager. Arrow depicts phosphorylated Artemis or DNA-PKcs product on each gel.
Following expression of the human recombinant [His]6-Artemis protein in insect cells using a baculovirus expression system, a cell-free extract was prepared and Artemis expression analyzed. Analysis of Artemis nuclease enzymatic activity is not possible in crude extracts due to the abundance of endogenous nucleases (data not shown). To detect Artemis expression, we analyzed cell free extracts prepared from infected cells by SDS-PAGE, western blot and phosphorylation by DNA-PK. Coomassie Blue staining of an SDS denaturing gel revealed expression of the [His]6-Artemis protein (Fig. 1B) that was confirmed by western blot analysis. Which revealed a dominant band at the expected molecular mass for the recombinant His-tagged fusion protein using both a monoclonal antibody against the N-terminal [His]6 fusion tag (Fig. 1C) and a polyclonal antibody against the full length Artemis polypeptide (data not shown). Artemis has been shown to possess eleven serine/threonine residues that are phosphorylated by DNA-PK in vitro [22]. To confirm that the baculovirus expressed [His]6-Artemis can be phosphorylated by DNA-PK, we performed an in vitro DNA-PK phosphorylation reaction. The cell-free extract was incubated with purified DNA-PK, DNA and [γ-32P]ATP and the reaction products were separated by SDS-PAGE and radioactive incorporation of the phosphate into protein was detected by PhosphorImager analysis. Again, a prominent band appeared at the same size as that seen in the western blot of Artemis (Fig. 1D), indicative of DNA-PK-dependent phosphorylation of [His]6-Artemis in the cell-free extract.
Following preparation of the whole cell extract from insect cells infected with recombinant [His]6-Artemis baculovirus, the concentration of potassium chloride was increased to 0.5 M and mixed with phosphocellulose chromatography media. The high salt concentration allowed for the majority of the protein contained in the extract (including [His]6-Artemis) to flow through the matrix. The flow-through protein was collected and loaded directly onto a 2 mL Nickel-NTA agarose column. The column was washed extensively with high ionic strength buffer followed by low ionic strength buffer, both containing 5mM imidazole. The bound protein was eluted with 200 mM imidazole and collected in 1 mL fractions. Analyses of these pools revealed that the majority of [His]6-Artemis bound to the Ni-NTA agarose matrix and was eluted with 200 mM imidazole. The load, flow-through and eluate from the nickel column were analyzed by Coomassie Blue staining of SDS-PAGE (Fig. 2A). The presence of [His]6-Artemis in these fractions was assessed by western blotting (Figure 2B), and phosphorylation by DNA-PK (Fig. 2C). In each case the majority of the Artemis applied to the column (lane 1) was retained in the imidazole elution (lane 3). We found that retention of Artemis on the Ni- matrix was very sensitive to chromatographic conditions and the presence of reducing agents, which should be avoided to maximize yield (data not shown). While these results indicate a degree of purity of [His]6-Artemis can be achieved by Nickel-affinity chromatography, the Coomassie Blue stained gel and phosphorylation assay reveal significant impurities in the eluate, indicating the need for further fractionation to achieve a higher degree of purity.
Fig. 2.
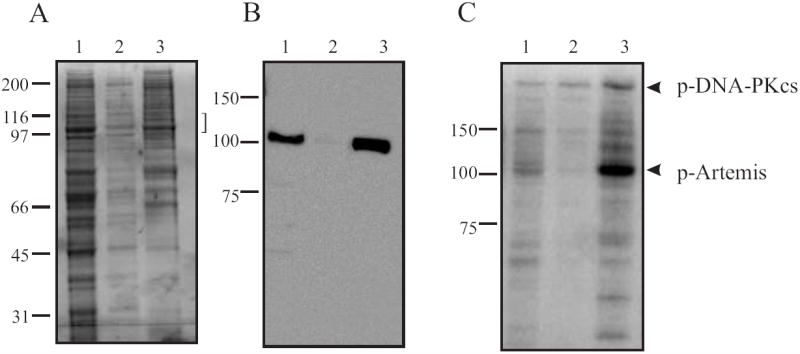
Analysis of fractionation on a Nickel-Agarose column. (A) Coomassie-Blue stained 8% SDS-PAGE of pools from Nickel-Agarose column. The bracket indicates anticipated position of [His]6-Artemis migration. (B) Western blot analysis using the anti-Xpress antibody. (C) Phosphorylation of Nickel-Agarose pools by DNA-PK. For A, B and C, Lane 1, Nickel-Agarose load, Lane 2, Nickel-Agarose flow-through, Lane 3, Nickel-Agarose pool. All assays were conducted as described in legend for Fig. 1 and arrows indicate migration the position of Artemis.
In attempts to further purify [His]6-Artemis, numerous matrices were assessed including anion and cation exchange, and hydrophobic interaction chromatography. The results from these matrices were universally poor (data not shown). Fractionation via adsorption chromatography on a hydroxyapatite (HAP) matrix at a relatively high pH of 7.8 did however result in substantial purification of Artemis. The majority of total protein applied (greater than 90%) was retained on the HAP column while the majority of [His]6-Artemis was not retained on a HAP column under these conditions but was identified in the flow-through fractions. Western blot analysis of the flow-through fractions (Fig 3A) and elution fractions (Fig.3B) demonstrates the vast majority of Artemis was recovered in the flow-through fraction. Coomassie Blue stained SDS-PAGE of the HAP load and flow-through fractions confirmed the decrease in complexity of the HAP FT pool (Fig. 3C), though definitive determination of the Artemis polypeptide remains. The Artemis containing flow-through fractions were pooled and Artemis levels in each were determined by the more quantitative in vitro phosphorylation analysis which confirmed that the vast majority of the Artemis protein was present in the flow-through pool and was capable of being phosphorylated by DNA-PK (Fig. 3D, lane 1) compared to control reactions without the HAP FT (lane 2).
Fig. 3.
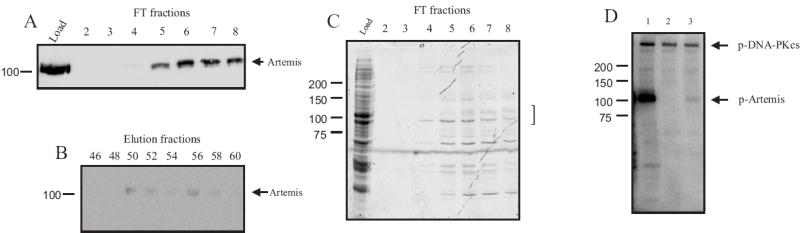
Analysis of hydroxyapatite (HAP) column fractionation of Artemis. (A) Western blot analysis of load and flow through fractions and (B) fractions obtained from the gradient elution. Aliquots of each fraction were separated by SDS-PAGE, transferred, probed with anti-Xpress antibody and detected as described in “Methods”. (C) Coomassie-Blue stained SDS-PAGE analysis of the load and flow-through fractions. Samples were processed as described in “Methods”. The bracket indicates anticipated position of [His]6-Artemis migration. (D) Phosphorylation of [His]6-Artemis by DNA-PK. Reactions were performed as described in the legend for Figure 2. Lane 1, HAP flow-through pool (140 ng), Lane 2, DNA-PK control without Artemis. Lane 3, HAP elution pool (190 ng). Arrows indicate phosporylated [His]6-Artemis and DNA-PKcs.
To determine the retention of exonuclease activity upon HAP fractionation, we assayed the column fractions for 5’-3’ exonuclease activity using a 34–base single-strand oligonucleotide with a 5’-[32P]-label. The release of the 5’ radiolabeled nucleoside monophosphate is a direct measure of 5’ to 3’ exonuclease activity. The majority of exonuclease activity was resolved in six fractions that bound to the column and were subsequently eluted during the phosphate gradient. Minimal exonuclease activity was observed in the flow-through fractions, which contain the majority of Artemis protein (Fig. 4A and B). The exonuclease containing fractions in the HAP phosphate elution were pooled. These data demonstrate a small portion of the overall exonuclease activity loaded onto the HAP column was identified in the flow-through, while the majority was located in the eluate (Fig 4B). Importantly, measurement of the bound exonuclease activity is potentially an underestimation, as all of the substrate was completely degraded in the peak elution fractions (fractions 48-50, Fig. 4A and B). These results suggest that at specific conditions of this fractionation, [His]6-Artemis does not bind to a HAP column while the majority of exonuclease activity remains bound under the same conditions. Further confirmation of the fractionation of Artemis into the HAP FT separated from the majority of exonuclease is revealed by the presence of DNA products consistent with a sequence specific, single-stranded endonuclease activity recently attributed to Artemis in the HAP FT fractions [28].
Fig. 4.
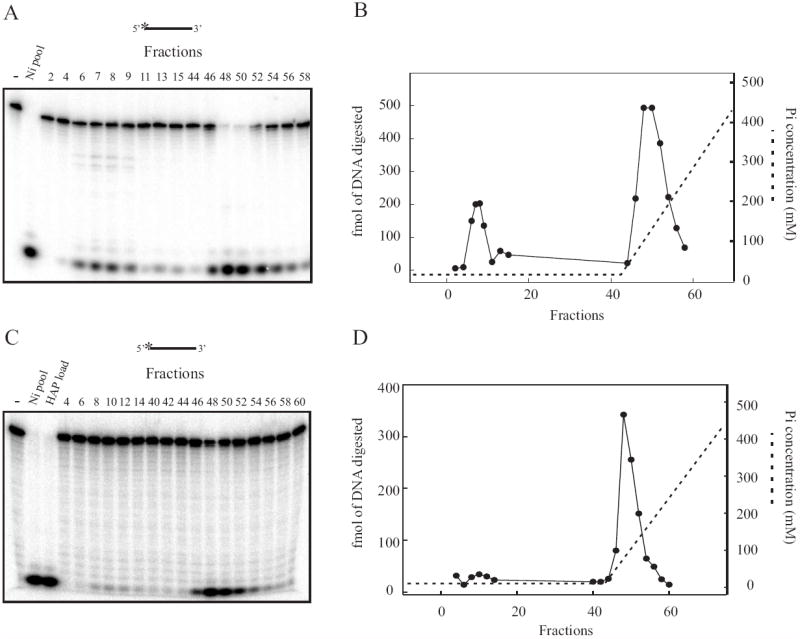
Quantitative assessment of exonuclease activity from HAP fractionation of [His]6- Artemis and [His]6-XPA. (A) A 2 μL aliquot of each 1mL HAP fraction from a [His]6-Artemis purification was assayed for exonuclease activity using a 5’[32P] labeled single-strand DNA 30 bases in length. Reaction products were separated and visualized as described in Materials and Methods. (B) Quantification of exonuclease activity from (A). (C) A 2 μL aliquot of each 1mL HAP fraction from a [His]6-XPA purification was assayed as described above. (D) Quantification of exonuclease activity from (C).
In an effort to determine if the minimal exonuclease activity found in the HAP FT and the major exonuclease in the elution was specific to the [His]6-Artemis purification, we designed an experiment to determine whether the exonuclease activity is a contaminating nuclease that has a high affinity for a Nickel-agarose column. Using recombinant baculovirus, we overexpressed another His-tagged DNA repair protein, XPA, with no intrinsic nuclease activities and minimal protein interaction domains. A whole cell extract was prepared in the same fashion as extracts of overexpressed [His]6-Artemis, and the identical purification protocol was followed. Following fractionation over the Nickel-agarose column and HAP column, fractions were assayed for exonuclease activity. Robust exonuclease activity was seen in the whole cell extract, as well as in protein pooled from the Nickel column (data not shown). Fractions assayed from the HAP column again revealed a minimal amount of exonuclease activity flowing through the column (Fig. 4C and 4D). Furthermore, the peak of exonuclease activity eluted from the HAP column with the phosphate gradient coincides exactly with the peak of exonuclease activity eluted from the HAP column in the [His]6-Artemis prep. Interestingly, the low level of exonuclease activity flowing through the HAP column coincides with the low level of exonuclease activity seen flowing through the HAP column from the [His]6-Artemis prep. Finally, pools of protein from the Nickel-agarose elution, HAP flow-through and HAP elution were examined for DNA-PK dependent endonuclease activity, and all pools were completely devoid of this activity, as expected (Supplemental Fig. 1). Analysis of the HAP elution pool for DNA-PK phosphorylation of Artemis reveals an extremely low level of Artemis in this pool of protein (Figure 3D lane 3), consistent with the spreading out of the Artemis proteins over the entire gradient as assessed by western blot analysis of the fractions (Fig. 3B).
To ascertain separation of the distinct enzymatic activities found in this protein preparation following separation on the HAP column, a rigorous quantitative analysis was conducted on the Nickel-agarose elution and HAP-FT pools of protein. For these analyses, each pool of protein was dialyzed in the identical buffer to minimize any difference in reactions conditions that may stimulate or inhibit at eh enzymatic activities of the protein. Comparison of the Ni pool and HAP FT via western blot analysis (Fig. 5A) revealed the presence of significant level of Artemis in each pool as expected. This result was confirmed in analysis of DNA-PK phosphorylation of each pool (Supplemental Fig. 2). As mentioned previously, the flow-through fractions from the HAP column retained minimal exonuclease activity on single-strand DNA, an activity previously attributed to the Artemis polypeptide. In an effort to directly compare the exonuclease activity collected from each pool of protein, increasing amounts of the nickel pool of protein and the HAP flow-through pool were incubated with a 5’ radiolabeled oligonucleotide and the release of the 5’ deoxynucleoside monophosphate (dNMP) monitored. Increasing concentrations of Ni pool containing Artemis resulted in increased 5’ to 3’ exonuclease activity. However, the Artemis containing HAP flow-through pool revealed an NMP product barely above the detection limit of our assay (1% of the input DNA) demonstrating that this protein pool does not contain 5’ to 3’ exonuclease activity (Fig. 5B). Importantly, there are preparation specific variations of exonuclease activity and differences observed in the analysis of column fraction versus pools. In the absence of 5’ to 3’ exonuclease activity in vitro, the more relevant activity of Artemis, found both in vitro and in vivo, is DNA-PK dependent hairpin-opening activity and the 5’ and 3’ overhang endonuclease activity. In an effort to ensure that the HAP FT pool of [His]6-Artemis devoid of exonuclease activity still retained its endonuclease catalytic activity, we assayed each of the two pools of protein for hairpin opening activity. In agreement with results demonstrating the importance of Ku in forming the DNA-PK-Artemis complex, we conducted all of our endonuclease assays with purified heterotrimeric DNA-PK [14]. DNA-PK-dependent Artemis catalyzed endonuclease activity was assessed on a hairpin substrate containing a 6 base 5’ overhang and yields an endonuclease product of approximately 28 bases in length as previously reported [11]. Both the Nickel pool of protein and the HAP FT pool catalyzed hairpin opening activity in the presence of ATP on this substrate (Fig. 5C lanes 6 and 7, and Fig. 5C lanes 9 and 10 respectively). Importantly, there is slightly more hairpin opening activity in the HAP FT pool of protein, as evidenced by the more prominent cleavage products (Fig. 5C, lanes 9 and10). The intensity of these products could also result from the lack of exonuclease activity in the HAP FT, which would necessarily result in less exonucleolytic removal of the 5’ label on the cleavage product. Also evident, though of reduced intensity compared to the hairpin opening activity, are products that result from 5’ overhang cleavage activity of Artemis which is anticipated to release a short 5-7 base product. Interestingly, the 5’ dNMP product produced by exonuclease activity cleaving the 5’ label on the hairpin substrate is evident in reactions performed with the Nickel pool, but is substantially reduced in reactions performed using the HAP flow-through fraction. This is evident upon reduced exposure of the gel to minimize bleed-over from the Ni-fractions (Fig. 5C bottom panel).
Fig. 5.
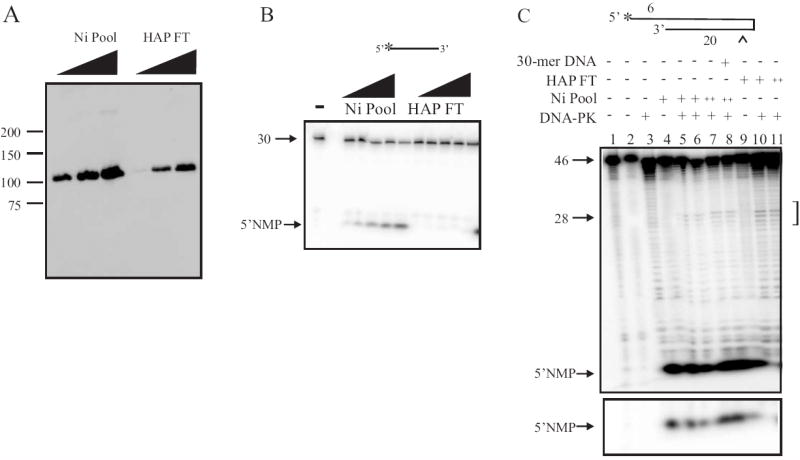
Biochemical characterization of Nickel-Agarose and HAP flow-through pools of [His]6- Artemis. (A)Western blot analysis. Nickel-Agarose pool (200 ng, 1 μg and 2 μg) and HAP flow-through (10 ng, 20 ng and 50 ng) were analyzed by Western blot using an anti-Artemis antibody. (B) Analysis of [His]6-Artemis Ni pool and HAP FT exonuclease activity on 30 base 5’ radiolabed single-strand DNA. Increasing amounts of Ni pool (100 ng to 1 μg) or HAP FT (5 ng to 50 ng) pool was assayed for exonuclease activity using 50 fmol of 5’ [32P] single-strand DNA substrate as described in Materials and Methods. (C) Analysis of hairpin opening activity. Ni pool (400 ng and 1 μg) or HAP FT (20 ng and 50 ng) was incubated with 250 fmol of 5’ radiolabeled hairpin DNA substrate, 200 nM double-strand cold DNA (30-mer), and 50 nM of DNA-PK as indicated. Following endonuclease cleavage, reaction products were separated and visualized as described in Materials and Methods.
Despite rigorous experimental precautions taken, the potential exists that our interpretation of the dNMP observed in Figure 5B is incorrect. In order to ensure that the radiolabeled nucleotide products observed in Figure 5B are in fact the result of exonuclease activity and not a dinucleotide product produced by an endonuclease activity, we assessed nuclease activity on a single-strand DNA substrate with a 3’ radiolabeled terminus. Increasing amounts of the Nickel pool of protein resulted in a ladder of products consistent with sequential single nucleoside monophosphate removal from the 5’ terminus of the oligonucleotide (Figure 6A). The HAP flow-through pool of protein again, contained barely detectable 5’ to 3’ single strand exonuclease activity. Overexposure of these gels (Fig. 5B and 6A) however, reveals products indicative of low level exonuclease activity which comprises less than 1% of those observed in the Ni-pool. These experiments confirm that while [His]6-Artemis fractionated over a Nickel column retains significant 5’ to 3’ exonuclease activity, fractionation of this protein over a HAP column results in near quantitative separation of the exonuclease from the Artemis protein.
Fig. 6.
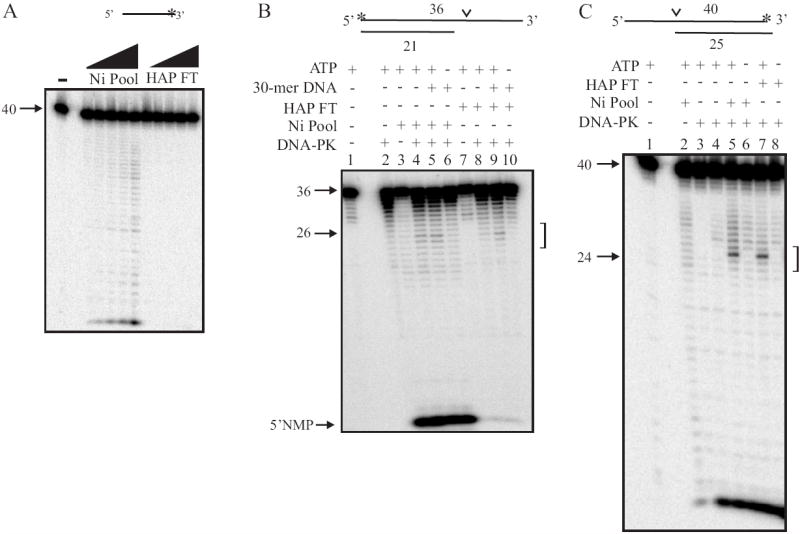
Characterization of Artemis nuclease activity. (A) Analysis of Ni pool and HAP FT exonuclease activity on single-strand DNA with a 3’ 32P label. Assays were conducted as described in the legend to figure 4B), except the single-strand oligonucleotide was radiolabeled by incorporation of [α-32P] on the 3’ termini. (B) Analysis of endonuclease activity on a 5’ radiolabeled DNA substrate with a 3’ single-strand overhang. Reactions were conducted as described in 4C, except Ni pool, HAP FT, 250 μM of ATP and 200 nM of cold duplex DNA (30-mer) were added as indicated. (C) Analysis of endonuclease activity on a 3’ radiolabeled DNA substrate with a 5’ single-strand overhang. Reaction conditions were conducted as described in “ Materials and Methods”. Each DNA substrate is depicted above the gel and the position of the [32P] label is denoted by the asterisk and the position of Artemis endonuclease activity is denoted by the carat. DNA strand lengths of each DNA substrate are also noted in the figure. The 5’ nucleoside monophosphate product (5’ dNMP) is indicated by the arrows in panel B.
While apparent that [His]6-Artemis fractionated over a HAP column is devoid of 5’ to 3’ exonuclease activity but still retains DNA-PK dependent hairpin opening activity, we sought to further assess DNA-PK dependent Artemis overhang cleavage activity to ensure all the in vivo, intrinsic enzymatic activities are retained in this purified preparation of protein. We therefore prepared a 5’-radiolabeled DNA substrate with a 3’ single-strand overhang [11]. This substrate was efficiently cleaved in close proximity to the single-strand/double-strand junction by both the Nickel and HAP flow-through pools of protein when incubated with DNA-PK and ATP (Fig. 6B, lanes 4, 5 and 6 and lanes 7, 8 and 9 respectively). As expected, the endonucleolytic product is completely dependent on DNA-PK and ATP. Although the presence of duplex DNA has been reported to stimulate DNA-PK dependent Artemis endonuclease activity [11], the addition of a 30-mer double-stranded cold DNA substrate did not increase endonuclease cleavage on the hairpin radiolabeled substrate (Fig. 6A, lane 7) or the 3’ overhang substrate (Fig. 6B, lane 5 and 9). Again, the 5’ NMP is apparent in reactions performed with the Nickel pool of protein but is reduced to background levels in reactions containing the HAP flow-through fraction. Finally, a DNA substrate with a 5’ single-strand overhang and 3’ [α-32P] dCMP label was used to assay endonuclease activity on a 5’ overhang [14]. DNA-PK dependent Artemis-catalyzed endonuclease activity on this substrate is anticipated to result in a product of approximately 26 nucleotides following cleavage at the single-strand/double-strand junction. In the presence of DNA-PK and ATP, both the Nickel pool of protein and the HAP flow-through endonucleolytically cleaved the 3’ overhang to generate a 26 nucleotide product (Fig. 6C, lanes 5 and 7). An exonuclease-like product appears at the bottom of the gel, but as it only appears in lanes that have DNA-PK, including but not limited to the lane that contains DNA-PK alone (Fig. 6C, lane 4), it is attributed to a contaminating 3’ exonuclease in our preparation of DNA-PK from Hela cells. The data presented in Figure 5 and 6 demonstrate that both pools of protein, from the Nickel and the HAP FT column, maintain hairpin opening and endonuclease activity that has previously been reported to be intrinsic to Artemis. Importantly, the HAP flow-through pool of protein, containing [His]6-Artemis, no longer exhibits single-strand exonuclease activity but retains DNA-PK dependent endonucleolytic activities. This data demonstrates that the exonuclease activity of Artemis previously reported is not an intrinsic component of the Artemis polypeptide. It is possible that the exonuclease activity is another enzyme that is associated with Artemis and plays a physiological role with Artemis in the cell, but it is equally possible that the exonuclease activity is simply a contaminating protein that is difficult to separate from the pool of Artemis protein during a purification procedure.
To finally ensure that there are no other alternatives to the separation of exonuclease activity from Artemis we quantified the endonuclease and exonuclease activity throughout the purification and the results are presented in Table 1. These results clearly demonstrate that following fractionation on a HAP column, 100 % of the endonuclease activity is recovered in the HAP FT resulting in nearly a 30-fold purification while only 2.5% of the exonuclease activity is retained in that fraction with no increase in specific activity despite the loss of 96% of the total protein. Consistent with this data, analysis of the Artemis polypeptide as determined by western blotting in conjunction with analysis of exonuclease activity was performed on the HAP-FT and elution pools of protein from a separate preparation. The percent of total antibody reactivity or exonuclease activity resolved in the two pools of protein was determined and the results demonstrate that greater than 90% of Artemis protein loaded onto the HAP column was recovered in the HAP flow-through material, while less than 10% was recovered in the gradient elution pool while greater than 90% of the exonuclease was identified in the elution pool and less than 10% in the FT pool of protein (Supplemental Fig. 3). These results indicate that the exonuclease found to co-purify with [His]6-Artemis under certain conditions can be separated away from [His]6-Artemis under other conditions (as described above), and therefore is probably a prominent exonuclease that has a similar affinity as Artemis for certain column conditions, but is not intrinsic to the Artemis polypeptide.
Table 1.
| Endonuclease activity | Exonuclease activity | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Fraction | Protein (mg) | Specific activity (units/mg) | total activity (units)* | Purification-fold | yield % | Specific activity (units/mg) | total activity (units)* | purification-fold | yield % |
| Ni pool | 1.200 | 1.57 | 1883.45 | 1.00 | 100.00 | 27.00 | 32400.00 | 1.00 | 100.00 |
| HAP FT | 0.04 | 47.01 | 1880.30 | 29.95 | 99.83 | 20.00 | 800.00 | 0.74 | 2.47 |
1 unit equal 1 fmol of product generated in a 30 min. reaction at 37°C.
4. Discussion
The biochemical separation of exonuclease activity from DNA-PK dependent endonuclease activity reported in this manuscript is consistent with genetic separation of Artemis enzymatic activity. Mutations in the protein which result in disruption of endonuclease activity have no effect on exonuclease activity, consistent with our interpretation that Artemis does not contain intrinsic exonuclease activity [13, 14, 18, 22, 29, 30]. Many of these mutational studies were conducted to determine how Artemis and DNA-PKcs interact, and what role this interaction and DNA-PK mediated phosphorylation of Artemis play in endonucleolytic cleavage activity [13, 14, 22, 30]. Differing results from these studies have left the mechanism of Artemis endonuclease activation an open question. Analysis of Artemis phosphorylation site mutants and large scale deletion of sites of phosphorylation have led to the conclusion that Artemis must be phosphorylated by DNA-PKcs to gain endonuclease activity [13, 22]. Analysis of DNA-PKcs phosphorylation mutants led others to conclude that autophosphorylation of DNA-PKcs is required to facilitate Artemis catalyzed endonuclease activity [14, 15]. Importantly, the analysis of an extensive collection of N-terminal Artemis mutations located in the enzymatically important metallo-β-lactamase and β-CASP domains resulted in identification of a sub-set of mutants which functionally abrogated endonuclease activity via disrupting metal coordination [18]. Analysis of these mutants resulted in no loss of exonuclease activity. A recent paper generated two additional mutations, also in the N-terminus domain, which have reduced and inactive endonuclease activity, respectively and these endonuclease deficient mutants also retained exonuclease activity [29] . An additional phosphorylation mutant, associated with partial immunodeficiency in a mouse model, exhibits reduced endonuclease activity but nearly complete retention of exonuclease activity [30]. While the potential exists that the exonuclease activity could be located in another active site other than those identified by generating mutants, this seems unlikely, as it is thought that metallo-β-lactamase fold enzymes have one active site that is responsible for all enzymatic processing [20]. This is further supported by data published regarding SNM1, a 5’ to 3’ mammalian exonuclease classified in the metallo-β-lactamase superfamily. SNM1 is characterized by having only exonuclease activity on single-strand DNA, with no accompanying endonuclease activity, and a mutation of a conserved aspartate (D736) in the β-lactamase domain functionally disrupts the exonuclease activity [31, 32, 32]. Paradoxically, mutagenesis of the conserved aspartate in Artemis (D37) eliminated endonuclease activity, but the exonuclease activity remained [18]. This indicates that the exonuclease activity is not located within the same active site as the endonuclease activity, and the extensive mutational analysis performed to date has yet to locate an exonuclease active site within Artemis. The combination of these genetic studies coupled with our biochemical analysis indicates that not only is the exonuclease activity separate from the endonucleolytic active site, but is not part of the Artemis polypeptide at all.
Separation of the nuclease activities, as presented in this paper, was achieved with multiple protein purification preparations. However, it is important to note that the separation of exonuclease activity from Artemis did vary between protein preparations. As we continued to improve our purification procedures, specific changes, albeit small, in the protocol resulted in subtle differences in separation of exonuclease activity from endonuclease activity. In separating [His]6-Artemis over the HAP column, we found that greater separation of activity was achieved on a 5 mL HAP column compared to a 2 mL HAP column, despite more than enough protein-binding capacity on the 2 mL column. However, the residual exonuclease activity that flowed through the 2 mL column could be separated from Artemis by re-running the flow-through on a second HAP column (data not shown). This suggests that saturation of the hydroxyapatite column with exonuclease activity, at least to a certain level, can occur, leading to sub-optimal separation. These variations are largely a result of the specifics of our protocol, and can be impacted by numerous factors, including specificity of the matrix used, MacroPrep Ceramic Hydroxyapatite (Biorad), or relatively high pH (7.85) of the buffer used in the purification. Interestingly, we did observe a nominal amount of Artemis in many of the fractions collected from the HAP column, including the wash and elution. The diminutive levels of [His]6-Artemis were often only observed by Western blot analysis. This phenomenon was also observed during fractionation over the Nickel-agarose column, indicating a certain degree of spreading of the fusion protein during all fractionation steps.
Our data supports the conclusion that the exonuclease activity thought to be intrinsic to Artemis is not a component of the Artemis polypeptide. Many possibilities exist to explain the presence of the exonuclease activity in less-pure fractions of Artemis. It is possible that the exonuclease is simply a contaminating enzyme that is endogenously expressed in the insect cells and co-purifies with Artemis independent of any biologically relevant interaction. This possibility is supported by data presented where single-strand 5’-3’ exonuclease activity co-purifies with another DNA repair protein, [His]6-XPA, overexpressed in insect cells. Not only do these results support the hypothesis that the exonuclease is from SF9 cells, but it also raises the possibility that this exonuclease has an affinity for Nickel-agarose resin, as both the XPA and Artemis were fractionated over this column first. This is not an unlikely scenario, as insect cells contain many endogenous nucleases and the 5’-3’ exonuclease SNM1 was also identified in D. Melanogaster [33]. It is also possible that the exonuclease is endogenously associated with Artemis, and is a biologically relevant interaction. This intriguing possibility has yet to be investigated. The results presented here demonstrate that the exonuclease activity previously thought to be intrinsic to Artemis is separable from the DNA-PK and ATP dependent endonuclease activity of Artemis prompting further studies to clarify a role for exonuclease processing in NHEJ and the protein responsible for that activity.
Supplementary Material
Analysis of endonuclease activity on a 3’ radiolabeled DNA substrate with a 5’ single-strand overhang from [His]6-XPA preparation. Reaction conditions were conducted as described in “Materials and Methods” and the legend to Figure 6. The DNA substrate is depicted above the gel and the position of the [32P] label is denoted by the asterisk and the position of Artemis endonuclease activity is denoted by the carat. DNA strand lengths of each DNA substrate are also noted in the figure. A positive control demonstrating ATP dependent endonuclease activity in the HAP FT fraction from a [His]6 Artemis preparation is presented in lanes 1 and 2, while no endonuclease activity is observed in any fractions from the XPA preparation 3-8.
DNA-PK phosphorylation of Nickel (A) and HAP FT (B) pool of protein. In vitro phosphorylation reactions were conducted as described in Materials and Methods and contained DNA-PK, [His]6-Artemis and DNA as indicated. Reaction products were separated via SDS PAGE and products visualized by phosphorImager analysis.
Quantified antibody reactivity and exonuclease activity from the HAP flow-through and HAP elution pools. Total intensity of antibody reactivity was obtained from MultiGauge analysis (Fuji) of representative western blots and total exonuclease activity was quantified using ImageQuant (Amersham) analysis of product released.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Mahaney BL, Meek K, Lees-Miller SP. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem J. 2009;417:639–650. doi: 10.1042/BJ20080413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walker JR, Corpina RA, Goldberg J. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature. 2001;412:607–614. doi: 10.1038/35088000. [DOI] [PubMed] [Google Scholar]
- 3.Povirk LF, Zhou T, Zhou R, Cowan MJ, Yannone SM. Processing of 3’-phosphoglycolate-terminated DNA double strand breaks by Artemis nuclease. J Biol Chem. 2007;282:3547–3558. doi: 10.1074/jbc.M607745200. [DOI] [PubMed] [Google Scholar]
- 4.Wu X, Wilson T, Lieber M. A role for FEN-1 in nonhomologous DNA end joining: the order of strand annealing and nucleolytic processing events. Proc Natl Acad Sci USA. 1999;96:1303–1308. doi: 10.1073/pnas.96.4.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karimi-Busheri F, Rasouli-Nia A, lalunis-Turner J, Weinfeld M. Human polynucleotide kinase participates in repair of DNA double-strand breaks by nonhomologous end joining but not homologous recombination. Cancer Res. 2007;67:6619–6625. doi: 10.1158/0008-5472.CAN-07-0480. [DOI] [PubMed] [Google Scholar]
- 6.Perry JJ, Yannone SM, Holden LG, Hitomi C, Asaithamby A, Han S, Cooper PK, Chen DJ, Tainer JA. WRN exonuclease structure and molecular mechanism imply an editing role in DNA end processing. Nat Struct Mol Biol. 2006;13:414–422. doi: 10.1038/nsmb1088. [DOI] [PubMed] [Google Scholar]
- 7.Kusumoto R, Dawut L, Marchetti C, Wan LJ, Vindigni A, Ramsden D, Bohr VA. Werner protein cooperates with the XRCC4-DNA ligase IV complex in end-processing. Biochemistry. 2008;47:7548–7556. doi: 10.1021/bi702325t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee JH, Paull TT. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science. 2004;304:93–96. doi: 10.1126/science.1091496. [DOI] [PubMed] [Google Scholar]
- 9.Capp JP, Boudsocq F, Bertrand P, Laroche-Clary A, Pourquier P, Lopez BS, Cazaux C, Hoffmann JS, Canitrot Y. The DNA polymerase lambda is required for the repair of non-compatible DNA double strand breaks by NHEJ in mammalian cells. Nucleic Acids Res. 2006;34:2998–3007. doi: 10.1093/nar/gkl380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davis BJ, Havener JM, Ramsden DA. End-bridging is required for pol mu to efficiently promote repair of noncomplementary ends by nonhomologous end joining. Nucleic Acids Res. 2008;36:3085–3094. doi: 10.1093/nar/gkn164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ma YM, Pannicke U, Schwarz K, Lieber MR. Hairpin opening and overhang processing by an Artemis/DNA- dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell. 2002;108:781–794. doi: 10.1016/s0092-8674(02)00671-2. [DOI] [PubMed] [Google Scholar]
- 12.Rooney S, Alt FW, Lombard D, Whitlow S, Eckersdorff M, Fleming J, Fugmann S, Ferguson DO, Schatz DG, Sekiguchi J. Defective DNA repair and increased genomic instability in artemis-deficient murine cells. J Exp Med. 2003;197:553–565. doi: 10.1084/jem.20021891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Niewolik D, Pannicke U, Lu H, Ma Y, Wang LC, Kulesza P, Zandi E, Lieber MR, Schwarz K. DNA-PKcs dependence of Artemis endonucleolytic activity, differences between hairpins and 5’ or 3’ overhangs. J Biol Chem. 2006;281:33900–33909. doi: 10.1074/jbc.M606023200. [DOI] [PubMed] [Google Scholar]
- 14.Goodarzi AA, Yu Y, Riballo E, Douglas P, Walker SA, Ye R, Harer C, Marchetti C, Morrice N, Jeggo PA, Lees-Miller SP. DNA-PK autophosphorylation facilitates Artemis endonuclease activity. EMBO J. 2006;25:3880–3889. doi: 10.1038/sj.emboj.7601255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yannone SM, Khan IS, Zhou RZ, Zhou T, Valerie K, Povirk LF. Coordinate 5’ and 3’ endonucleolytic trimming of terminally blocked blunt DNA double-strand break ends by Artemis nuclease and DNA-dependent protein kinase. Nucleic Acids Res. 2008;36:3354–3365. doi: 10.1093/nar/gkn205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moshous D, Callebaut I, de Chasseval R, Poinsignon C, Villey I, Fischer A, de Villartay JP. The V(D)J recombination/DNA repair factor artemis belongs to the metallo-beta-lactamase family and constitutes a critical developmental checkpoint of the lymphoid system. Immune Mechanisms and Disease. 2003;987:150–157. doi: 10.1111/j.1749-6632.2003.tb06043.x. [DOI] [PubMed] [Google Scholar]
- 17.Callebaut I, Moshous D, Mornon JP, de Villartay JP. Metallo-beta-lactamase fold within nucleic acids processing enzymes: the beta-CASP family. Nucleic Acids Res. 2002;30:3592–3601. doi: 10.1093/nar/gkf470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pannicke U, Ma YM, Hopfner KP, Niewolik D, Lieber MR, Schwarz K. Functional and biochemical dissection of the structure-specific nuclease ARTEMIS. EMBO J. 2004;23:1987–1997. doi: 10.1038/sj.emboj.7600206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Villartay JP, Shimazaki N, Charbonnier JB, Fischer A, Mornon JP, Lieber MR, Callebaut I. A histidine in the beta-CASP domain of Artemis is critical for its full in vitro and in vivo functions. DNA Repair (Amst) 2009;8:202–208. doi: 10.1016/j.dnarep.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 20.Dominski Z. Nucleases of the metallo-beta-lactamase family and their role in DNA and RNA metabolism. Crit Rev Biochem Mol Biol. 2007;42:67–93. doi: 10.1080/10409230701279118. [DOI] [PubMed] [Google Scholar]
- 21.Dahm K. Functions and regulation of human artemis in double strand break repair. J Cell Biochem. 2007;100:1346–1351. doi: 10.1002/jcb.21226. [DOI] [PubMed] [Google Scholar]
- 22.Ma YM, Pannicke U, Lu HH, Niewolik D, Schwarz K, Lieber MR. The DNA-dependent protein kinase catalytic subunit phosphorylation sites in human artemis. J Biol Chem. 2005;280:33839–33846. doi: 10.1074/jbc.M507113200. [DOI] [PubMed] [Google Scholar]
- 23.Weterings E, Verkaik NS, Keijzers G, Florea BI, Wang SY, Ortega LG, Uematsu N, Chen DJ, van G. The Ku80 carboxy terminus stimulates joining and artemis-mediated processing of DNA ends. Mol Cell Biol. 2009;29:1134–1142. doi: 10.1128/MCB.00971-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu H, Shimazaki N, Raval P, Gu J, Watanabe G, Schwarz K, Swanson PC, Lieber MR. A biochemically defined system for coding joint formation in V(D)J recombination. Mol Cell. 2008;31:485–497. doi: 10.1016/j.molcel.2008.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hermanson IL, Turchi JJ. Overexpression and purification of human XPA using a Baculovirus expression system. Protein Expression and Purification. 2000;19:1–11. doi: 10.1006/prep.2000.1224. [DOI] [PubMed] [Google Scholar]
- 26.Pawelczak KS, Turchi JJ. A mechanism for DNA-PK activation requiring unique contributions from each strand of a DNA terminus and implications for microhomology-mediated nonhomologous DNA end joining. Nucleic Acids Res. 2008;36:4022–4031. doi: 10.1093/nar/gkn344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pawelczak KS, Andrews BJ, Turchi JJ. Differential activation of DNA-PK based on DNA strand orientation and sequence bias. Nucleic Acids Res. 2005;33:152–161. doi: 10.1093/nar/gki157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gu J, Li S, Zhang X, Wang LC, Niewolik D, Schwarz K, Legerski RJ, Zandi E, Lieber MR. DNA-PKcs regulates a single-stranded DNA endonuclease activity of Artemis. DNA Repair (Amst) 2010 doi: 10.1016/j.dnarep.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Villartay JP, Shimazaki N, Charbonnier JB, Fischer A, Mornon JP, Lieber MR, Callebaut I. A histidine in the beta-CASP domain of Artemis is critical for its full in vitro and in vivo functions. DNA Repair (Amst) 2009;8:202–208. doi: 10.1016/j.dnarep.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 30.Huang Y, Giblin W, Kubec M, Westfield G, St CJ, Chadde L, Kraftson S, Sekiguchi J. Impact of a hypomorphic Artemis disease allele on lymphocyte development, DNA end processing, and genome stability. J Exp Med. 2009;206:893–908. doi: 10.1084/jem.20082396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hazrati A, Ramis-Castelltort M, Sarkar S, Barber LJ, Schofield CJ, Hartley JA, McHugh PJ. Human SNM1A suppresses the DNA repair defects of yeast pso2 mutants. DNA Repair (Amst) 2008;7:230–238. doi: 10.1016/j.dnarep.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 32.Hejna J, Philip S, Ott J, Faulkner C, Moses R. The hSNM1 protein is a DNA 5’- exonuclease. Nucleic Acids Res. 2007;35:6115–6123. doi: 10.1093/nar/gkm530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laurencon A, Orme CM, Peters HK, Boulton CL, Vladar EK, Langley SA, Bakis EP, Harris DT, Harris NJ, Wayson SM, Hawley RS, Burtis KC. A large-scale screen for mutagen-sensitive loci in Drosophila. Genetics. 2004;167:217–231. doi: 10.1534/genetics.167.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Analysis of endonuclease activity on a 3’ radiolabeled DNA substrate with a 5’ single-strand overhang from [His]6-XPA preparation. Reaction conditions were conducted as described in “Materials and Methods” and the legend to Figure 6. The DNA substrate is depicted above the gel and the position of the [32P] label is denoted by the asterisk and the position of Artemis endonuclease activity is denoted by the carat. DNA strand lengths of each DNA substrate are also noted in the figure. A positive control demonstrating ATP dependent endonuclease activity in the HAP FT fraction from a [His]6 Artemis preparation is presented in lanes 1 and 2, while no endonuclease activity is observed in any fractions from the XPA preparation 3-8.
DNA-PK phosphorylation of Nickel (A) and HAP FT (B) pool of protein. In vitro phosphorylation reactions were conducted as described in Materials and Methods and contained DNA-PK, [His]6-Artemis and DNA as indicated. Reaction products were separated via SDS PAGE and products visualized by phosphorImager analysis.
Quantified antibody reactivity and exonuclease activity from the HAP flow-through and HAP elution pools. Total intensity of antibody reactivity was obtained from MultiGauge analysis (Fuji) of representative western blots and total exonuclease activity was quantified using ImageQuant (Amersham) analysis of product released.