

Abstract
We recently developed a polarizable atomic multipole refinement method assisted by the AMOEBA force field for macromolecular crystallography. Compared to standard refinement procedures, the method uses a more rigorous treatment of x-ray scattering and electrostatics that can significantly improve the resultant information contained in an atomic model. We applied this method to high-resolution lysozyme and trypsin data sets, and validated its utility for precisely describing biomolecular electron density, as indicated by a 0.4–0.6% decrease in the R- and Rfree-values, and a corresponding decrease in the relative energy of 0.4–0.8 Kcal/mol/residue. The re-refinements illustrate the ability of force-field electrostatics to orient water networks and catalytically relevant hydrogens, which can be used to make predictions regarding active site function, activity, and protein-ligand interaction energies. Re-refinement of a DNA crystal structure generates the zigzag spine pattern of hydrogen bonding in the minor groove without manual intervention. The polarizable atomic multipole electrostatics model implemented in the AMOEBA force field is applicable and informative for crystal structures solved at any resolution.
Introduction
The x-ray crystallographic structure of a molecule typically yields atomic resolution information based on the density of electrons within the crystal. To generate calculated structure factors from an atomic model, assumptions must be made regarding the distribution of electrons around atoms and their disorder. Typically, atoms are assumed to scatter as isolated Gaussian spheres with some thermal vibration. This formalism has the benefit of simplicity; however, a recently developed scattering model based on Cartesian Gaussian multipoles offers an advantage in that it captures the features of the electron density due to chemical bonding exhibited by macromolecules (1). Whereas the benefits of nonspherical scattering models are most pronounced at very high resolution, the inclusion of a polarizable atomic multipole force field (based on the atomic multipole optimized energetics for biomolecular applications (AMOEBA) force field (2,3)) that replaces the standard geometric force field (4) can provide improvements at any resolution. Furthermore, our new refinement method uses a more descriptive and physically transferable model of molecular energetics.
The limitations of the isolated atom model (IAM) were first made clear when Bragg described the appearance of a 222 reflection in diffraction patterns of diamond, which can only be explained by a tetrahedral description of the electron density about each atom (5). This result was expanded on by Renninger (6,7) and later modeled by Dawson (8,9) using an aspherical harmonic expansion of the electron density. This improved the scattering model and led to many developments using similar approaches; however, it has not been widely employed, primarily because of the computational cost—up until recently, there were no fast Fourier transform (FFT)-based methods (10,11) available for aspherical scattering models. As a result, aspherical expansions have only been employed for a few cases in which high-resolution (<0.8 Å) data were available and the deviations from the IAM were most apparent. Previous aspherical treatments of the electron density, such as the Hansen and Coppens (12) formalism, describe the angular dependence of the electron density around an atom using spherical harmonics combined with radial Slater-type orbitals. In contrast, our method uses Cartesian Gaussian multipoles. We have shown that this approach is readily amenable to FFT methods to compute structure factors, and as a result, aspherical and anisotropic electron density can be computed rapidly even for large systems (1).
As a replacement for the commonly used simple geometric force field (4), we use the AMOEBA force field, which includes polarization (2,3). Permanent electrostatics represents the electron density of a group of atoms in the absence of interactions with the environment, whereas the induced dipoles model the polarization response of the electron density to the local electric field (13,14). The inclusion of polarization in the AMOEBA force field allows transferability between gas and condensed phases, and therefore quantum calculations can be used directly to parameterize the force field. In principle, AMOEBA can accurately model molecular energetics across biological environments of different polarities (15). Improved modeling of the electrostatic potential via polarization effects provides more informative/accurate descriptions of protein interaction energies (such as in a β-sheet or between protein and ligand (16)) and charge density (17) to enrich crystallographic model interpretation.
Water plays a widely appreciated central role in dictating biomolecular structure and function: the hydrophobic force drives folding, hydrogen bonding stabilizes nucleic acid and protein structures, and water itself has a high nucleophilicity that must either be shielded or harnessed by enzymes (18). Crystallography is one of only a few techniques that can directly analyze water structure and bonding patterns; however, the high experimental demands for neutron diffraction and the lack of an Ewald electrostatics (19) treatment in crystallographic refinement have been limiting factors in the detailed analysis of water structure in crystal structures (20). The AMOEBA force field thus described includes a model of water that has been validated against vacuum and condensed phase experimental data in addition to precise electronic structure calculations (2). Long-range electrostatics are rigorously calculated using a particle mesh Ewald (PME) approach that avoids artifacts and instability caused by using cutoffs (21–23). The explicit inclusion of polarization effects facilitates a water structure that matches experimental observations in varied environments (24). The resultant water model provides a reliable means of determining the orientation and energetics of water hydrogen-bonding networks, and obtaining information from crystallographic models that has not been available in the past.
Here we present re-refinements of high-resolution crystal structures of lysozyme at 0.65 Å resolution (25) and trypsin at 0.84 Å resolution (26) using AMOEBA force-field-assisted multipolar refinement. The re-refinements significantly lower the Rfree-values, suggesting that the method improves agreement between the model and data. The polarizable Ewald treatment of electrostatics yields information that suggests a mechanism of pKa coupling in lysozyme and provides additional evidence for the charge-relay mechanism of trypsin. To validate the AMOEBA electrostatic model, we re-refined a nine-basepair DNA duplex at 0.89 Å (27), which resulted in a complete hydrogen-bond network in the minor groove that agrees precisely with the Dickerson model of hydration (28,29). Our results illustrate the power of energy-function-assisted refinement when a state-of-the-art force field is used, and provide a method to orient and refine hydrogen positions (including solvent)—a crucial component in obtaining structural insights into enzymatic mechanisms, ligand-binding specificity, and protein-ligand design that we propose as a generally applicable method for macromolecular crystallography.
Materials and Methods
Details of the polarizable multipole refinement methodology have been described elsewhere (1). Briefly, the expression used to describe the core and valence electron density for atom j at position r is given by
| (1) |
Where is the integer number of core electrons (carbon has 2), is the integer number of valence electrons (carbon has 4), is one component of the del operator, and Θ represents the traceless quadrupole moment for the atom type. The Greek subscripts (α,β) represent the use of the Einstein summation over tensor elements where . The superscripts on the anisotropic Gaussian form factors (, where ρ is described in Eq. 2) denote the number of Gaussians n that the core, valence, dipole, and quadrupole densities each utilize, and κ is a fixed (in the case of core scattering) or refined width component, respectively. The same sets of six ai and bi scattering parameters are used for the core and valence electron densities, and the dipole and quadrupole densities use a single Gaussian with their ai and bi parameters set to unity. The widths (κν, κd, κΘ) are optimized against the diffraction data for each AMOEBA multipole type. The multipole moments are held fixed based on the AMOEBA force field, requiring only a coordinate transformation into the global frame. Each atomic dipole is a sum of permanent and induced (d and u, respectively) components to account for polarization, where the latter is determined using a self-consistent field calculation. The Gaussian form factor ρ includes anisotropic displacement parameters (ADPs) in all of the cases presented (except hydrogens) following the form:
| (2) |
The ADP formalism includes a Uadd parameter as described by Ten Eyck (11):
| (3) |
where I3 is a 3 × 3 identity matrix, and κ is the width parameter described above. The full details of this model, including its derivation and associated derivatives, are available in Schnieders et al. (1).
All refinements were carried out in a modified version of CNS 1.2 (30) using customized calls to TINKER (15) to compute the chemical terms and gradients. All refinements took advantage of the real space equation for electron density given in Eq. 1 to allow for FFT-based computation of structure factors. The target function for minimization,
| (4) |
utilized a weight (wA) of 1.0 for all refinements, as determined by trial and error using the R- and Rfree-values (data not shown). It is worthwhile to emphasize that not only does the AMOEBA force field contribute to the force-field energy term in Eq. 4, the induced dipoles and multipole coefficients also contribute to the scattering equation used to compute Ex-ray (also see the discussion above), and therefore the force field contributes to both energy terms.
The general refinement scheme follows a protocol similar to that used for the IAM and the AMOEBA with interatomic scatterering (AMOEBA-IAS) model described by Schnieders et al. (1) unless noted otherwise. The AMOEBA-IAS model differs from the IAM model only in the inclusion of the multipolar coefficients and interatomic scatterers in the x-ray scattering terms as given in Eq. 1 (and therefore differ in Ex-ray in Eq. 4); both use the full force field for the chemical term (EForceField in Eq. 4). Modifications to this protocol included an initial round of slow-cooling simulated annealing refinement using Cartesian molecular dynamics to optimize hydrogen positions (which were initially assigned using purely geometric criteria for protein atoms, and randomly placed in the case of solvent waters). Ionizable residues were considered on a case-by-case basis using previous experimental data to determine the protonation states. In the case of alternate conformers, a complete AMOEBA energy evaluation per conformation is carried out because the potential is many-body and not pairwise. Fortunately, computation of the AMOEBA potential energy remains less expensive than evaluation of structure factors. The use of a polarizable force field substitutes for fitting multipole coefficients for the scattering term, and offers the unique ability to orient waters via a rigorous PME-based electrostatic term. Other electrostatic models have been included in the geometry term of x-ray refinements, but they tended to neglect periodic boundary conditions (31) or implement conditionally convergent truncation schemes (typically using a minimum image convention (32)). Our treatment adds hydrogen positions to the refined parameters as part of the model, increasing the parameter count by three times the number of hydrogen atoms. Timings for the force field relative to the x-ray term (both with and without PME) are presented in the Supporting Material. After refinement was completed, the models were inspected with Coot (33) and O (34), and further rounds of refinement were carried out as necessary. All resulting models, data, and AMOEBA force-field parameters are available as Supporting Material.
Results
Lysozyme
The triclinic hen egg white lysozyme (HEWL) diffraction data collected by Wang et al. (25) at 100 K extend to 0.65 Å resolution and were originally refined to an R-value of 8.39% and an Rfree-value of 9.52%. Beginning from the deposited structure (PDB ID: 2VB1) and using the same reflections reserved for calculation of Rfree (provided courtesy of Z. Dauter, Argonne National Labs, personal communication, 2009), we re-refined the model using AMOEBA-assisted multipole refinement. The occupancies and definitions of alternate conformations were not altered from the original work. Using the AMOEBA-IAS scattering model and AMOEBA force field energetics, refinement converged to a final R-value of 7.87% and Rfree of 8.60% (Table 1). In this model, all solvent molecules included explicit hydrogen atoms. The AMOEBA electrostatic treatment, calculated rigorously via PME, is largely responsible for orienting water molecules into hydrogen-bond networks that are consistent with the observed density. The inclusion of PME increases the total energy evaluation time by an order of magnitude (Table S1), although the majority of the evaluation time (>98%) is spent on the electron density calculation and subsequent FFT to compute structure factors. Therefore, the overall change in time spent on each refinement step is negligible compared to that required by current methods. However, we are developing parallelization methods for the PME calculation to further reduce the time required for electrostatic calculations.
Table 1.
Refinement statistics
| Molecule | dmin (Å) | Scattering model | Npar | Nhkl |
Rwork / Rfree (%) |
Relative energy (Kcal/mol) | |
|---|---|---|---|---|---|---|---|
| Fobs/σ(Fobs)>0 | Fobs/σ(Fobs)>3 | ||||||
| Lysozyme | 0.65 | IAM | 20681 | 187165 | 8.40 / 9.05 | 8.21 / 8.87 | 109.3 |
| AMOEBA-IAS | 21887 | 187165 | 7.87 / 8.60 | 7.66 / 8.38 | 0 | ||
| Trypsin | 0.84 | IAM | 29523 | 138150 | 10.90 / 11.62 | 10.60 / 11.28 | 93.4 |
| AMOEBA-IAS | 30597 | 138150 | 10.45 / 11.11 | 10.16 / 10.77 | 0 | ||
| DNA∗ | 0.89 | IAM | 7786 | 30475 | 14.21 / 16.59 | 14.10 / 16.37 | NA |
For the DNA case, only the IAM model with AMOEBA chemical forces was used, because the AMOEBA-IAS model did not lead to a significant statistical improvement (data not shown).
To illustrate the improvement gained by re-refinement, the active site residues and their surrounding waters are shown in Fig. 1. For the deposited structure, a riding model was used to place the hydrogens; for example, in the case of a serine hydroxyl group, the hydrogen is added using an idealized torsion angle and bond length that depend only on the coordinates of the nonhydrogen atoms. During refinement, the hydrogen positions are updated based on these criteria, and thus they ride on the heavy atoms to which they are bonded, even though the energetic barrier for rotation by the serine hydroxyl group about the Cβ-Oγ axis may be low, and alternative rotations may be more favorable in the surrounding protein environment. Also, this model assigns protonation states assuming a pH (typically 7.0) that may be significantly different from the crystallographic conditions. Therefore, we reevaluated the absence or presence of protons in the lysozyme model using crystallographic bond lengths (35), NMR proton chemical shift measurements of pKa values (36), and neutron diffraction data of crystals grown under conditions similar to those used for the x-ray structure (37), the results of which are summarized in Table 2. Based on a protonated (neutral) Glu-35 and deprotonated (charged) Asp-52 species, we utilized two methods to assign the water network. The first method employs the popular PDB2PQR program (38–40), based on an inexpensive, local ad hoc hydrogen potential without the diffraction data (Fig. S1). The second implements both the diffraction data and AMOEBA chemical forces as described (Fig. 1 B). The results contrast with the deposited model (Fig. 1 A), in which the hydrogen bonding can only be inferred from the positions of the hydrogen-bond donors and acceptors. The AMOEBA-assisted refinement model shows an explicit and extensive network of hydrogen bonds that carries from the protonated Glu-35 to Asp-52, forming a complete and stable hydrogen-bonding network. Our interpretation of this result is that the rigorous treatment of electrostatics and x-ray data together provides a powerful method that will augment and improve currently available tools, such as PDB2PQR. The highly organized nature of the water network from the AMOEBA refinement suggests that the protonation states of both residues may be coupled. Early work on lysozyme suggested that the local hydrophobic environment around Glu-35 causes the residue to remain neutral (41), although the hydrogen-bond network may offer an alternative explanation for the elevated pKa (36). The view that hydrogen-bond networks are conformationally coupled and may be involved in signal transduction/activity is not without precedent (42).
Figure 1.
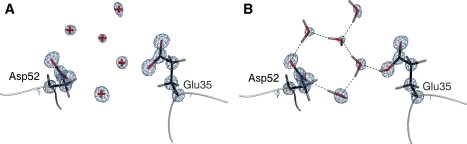
Final model of the lysozyme (PDB ID: 2VB1) active site for the deposited structure (A) and after the addition of hydrogens and re-refinement with AMOEBA forces and the x-ray data (B). Shown are the nucleophile (Asp-52), general acid (Glu-35), and surrounding water molecules. Water molecules without hydrogens are depicted as red crosses. Hydrogen bonds are drawn as dashed lines, and electron density represents 2Fo-FcσA-weighted maps contoured at 3.0 σ. Glu-35 was modeled as protonated based on bond lengths, available data, and crystallization conditions. All figures were generated using POVScript+ (64) and rendered using POVRay.
Table 2.
Protonation state assignment for HEWL
| Residue | pKa∗ | Neutron assigment† | C-O bond lengths‡ | C-O bond lengths§ | Assignment |
|---|---|---|---|---|---|
| Glu-7 | 2.85 ± 0.25 | neutral | 1.28, 1.25 | 1.28, 1.24 | charged |
| Asp-18 | 2.66 ± 0.08 | charged | 1.25, 1.24 | 1.25, 1.25 | charged |
| Glu-35 | 6.20 ± 0.10 | neutral | 1.33, 1.23 | 1.32, 1.23 | neutral |
| Asp-48 | < 2.5 | charged | 1.27, 1.22 | 1.27, 1.22 | charged |
| Asp-52 | 3.68 ± 0.08 | charged | 1.27, 1.22 | 1.27, 1.22 | charged |
| Asp-66 | < 2.0 | charged | 1.27, 1.26 | 1.27, 1.25 | charged |
| Asp-87 | 2.07 ± 0.15 | charged | 1.27, 1.24 | 1.27, 1.24 | charged |
| Asp-101 | 4.09 ± 0.07 | charged | 1.30, 1.20 | 1.31, 1.20 | neutral |
| Asp-119 | 3.20 ± 0.09 | charged | 1.27, 1.24 | 1.26, 1.24 | charged |
| Leu-129 | 2.75 ± 0.12 | charged | 1.26, 1.24 | 1.26, 1.24 | charged |
| His-15 | 5.36 ± 0.07 | charged | - | - | charged |
pKa standard deviation from Bartik et al. (36) measured by monitoring proton chemical shifts by NMR during titration at 35°C and 100 mM salt.
Assignments from a 1.7 Å neutron diffraction study at pH 4.7 by Bon et al. (37).
Bond lengths from the re-refined lysozyme structure reported here. For protonated carboxylic acids, the equilibrium C=O and C-OH bond lengths are 1.21 and 1.31 Å, respectively. This assumes that that the proton is not shared between the oxygen atoms. For a charged carboxylic acid, the equilibrium C-O bond lengths are both 1.26 Å (35).
Bond lengths from re-refinement of the lysozyme structure with the force field turned off (i.e., refined against the x-ray diffraction data only).
The effect of the improved scattering model on the refinement of the HEWL data is shown in Fig. 2. Scattering at bond centers and lone pairs is primarily affected, as shown by the loss of difference density at these sites (green mesh) relative to the electron density calculated from the deposited structure (purple mesh). Further, the X-H bond lengths are relaxed (note the difference in red versus blue hydrogen atoms) as the density along the bond is captured by the multipolar scattering coefficients, as opposed to the deposited model, which centers density at the hydrogen nucleus.
Figure 2.
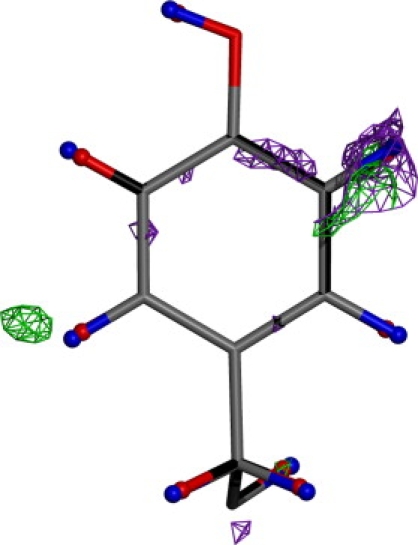
Tyr-53 from the lysozyme model with electron density (σA-weighted Fo-Fc maps, contoured at 1.8 σ) obtained before (purple) and after (green) introduction of the aspherical and anisotropic scattering model. Also highlighted are the hydrogen positions before (red) and after (blue) the same procedure. Note the average lengthening of X-H bonds and the disappearance of difference density at bond centers.
Trypsin
The serine protease family is one of the best-characterized systems in terms of both biochemistry and structure, and is also one of the largest (roughly one-third of all proteases belong to the serine protease class). The classical catalytic triad mechanism of serine proteases utilizes a charge relay system to generate the nucleophilic serine, which attacks the carbonyl group of a given peptide substrate (for a more complete overview of serine proteases, see the excellent review by Hedstrom (43) and references therein). The electrostatics predominates the charge relay mechanism, as the catalytic serine (Ser-195) must donate a proton to the general base (His-57) to form a nucleophile (44–46). The charge that forms on His-57 is stabilized through the Nδ1 position by the carboxylate of the third residue in the catalytic triad, Asp-102 (44,47). Further, the tetrahedral intermediate that forms during the reaction is stabilized by electrostatic interactions provided by the protein (47–50). Given the importance of the electrostatics for catalysis in serine proteases, we were interested in applying the proposed refinement method to this system.
A 0.84 Å resolution apo crystal structure of trypsin solved at pH 6.0 (26) was used as a starting point with a deposited R-value of 10.8% (PDB ID: 1XVO). The Rfree-value for this model was not available; however, a CNS-based simulated annealing refinement on the deposited structure yields an R-value of 11.71% and an Rfree of 12.38%. We re-refined this structure using our AMOEBA-assisted multipole refinement method. In the case of lysozyme, the occupancies and definitions of alternate conformations were not altered from the original work. Re-refinement using the aspherical and anisotropic scattering model (AMOEBA-IAS) reduced R and Rfree to 10.45% and 11.11%, respectively (Table 1), suggesting that the AMOEBA force field is a significant improvement over the Engh and Huber (4) model.
A notable aspect of the re-refinement is presented in Fig. 3. The riding model used in the deposited structure places hydrogen atoms as described above. The addition of the AMOEBA electrostatic model allows the hydrogens to independently refine against both the electric field and the crystallographic data (as well as contribute to the scattering term Ex-ray; see Materials and Methods). In the case of trypsin, this orients the serine Hγ slightly away from His-56 (as per the Fusarium oxysporum numbering) and toward a sulfate in the oxyanion hole (note the difference in purple and red serine hydroxyl positions in Fig. 3). This is further evidenced by the colinear polarization vector denoted by the green arrow: the polarization vectors point in the direction of the self-consistent electric field and away from induced increases in electron density. The polarization vector lies along the Oγ-Hγ bond, consistent with the strong anionic character of the bound sulfate, and causing the Hγ to rotate out toward the solvent. The polarization model also indicates that Asp-99 is functioning to build a partial negative charge character at its carboxylate as a mechanism to withdraw the Nδ1 proton from His-56, as expected for the charge transfer mechanism. The polarization vectors on the amide backbone are directed toward the oxyanion hole, lending electrostatic support to the concept of tetrahedral intermediate stabilization by these residues.
Figure 3.
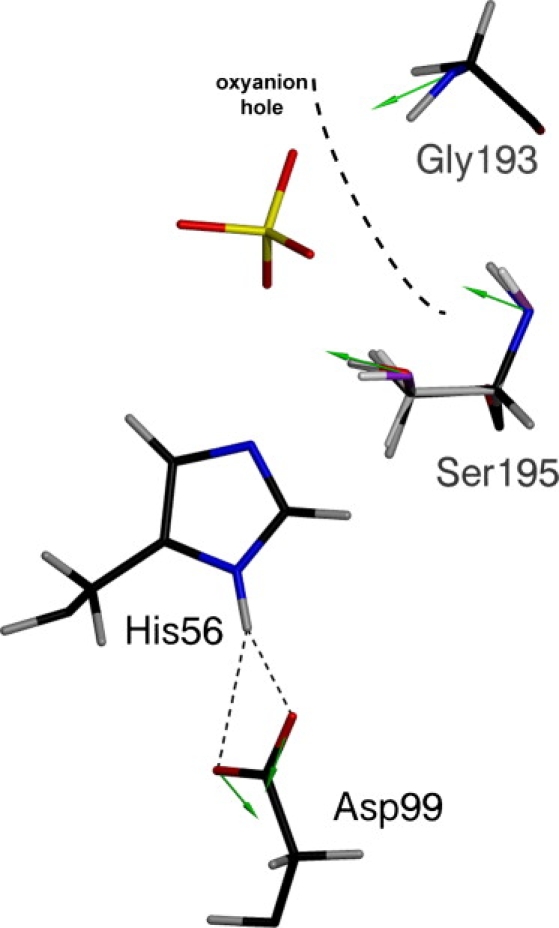
Trypsin catalytic triad prior (purple hydroxyl group on Ser-195) and after (red hydroxyl group on Ser-195) introduction of the electrostatic model. The oxyanion hole is depicted with the thick black dashed line. Residue numbering corresponds to trypsin from Fusarium oxysporum. Green arrows represent polarization vectors at the displayed atomic positions. A 3.0 Å vector length corresponds to 1 D.
B-form DNA
In early fiber diffraction studies of DNA, it was found that drying of the sample leads to loss of helical diffraction, suggesting that water is integral to DNA stability and structure (51). Later work suggested that an extensive and stable water network is required to maintain the B-form of DNA (52), which led to the use of a CGCGAATTCGCG dodecamer and the development of the current zigzag spine of hydration theory in the minor groove (28,29).
The crystal structure of a DNA 9-mer (GCGAATTCG) at 0.89 Å resolution represents the core sequence of the Dickerson dodecamer, and thus allowed us to revisit the geometry of the water network and the importance of hydration to nucleic acid structure. The deposited model (PDB ID: 1ENN) contained several lone oxygen atoms that were modeled as a proxy for phosphate backbone disorder (27). Since this was impermissible with the AMOEBA force field (or any physical model), parts of the phosphate backbone were split into discrete alternate conformers to fully model the observed disorder. Also, two magnesium ions (of seven total) and one chloride ion were removed from the model because the density and coordination in some cases were weak or insufficient. Only the IAM model with AMOEBA chemical forces was used, because the AMOEBA-IAS model did not lead to a significant statistical improvement (data not shown).
The results of the re-refinement for the water spine are shown in Fig. 4. The AMOEBA-assisted re-refinement explicitly recapitulates the first-order, hydrogen-bonding network formed by water to the individual bases (Fig. 4 B), including the hydrogen-bonding pattern described by the bridge between a base and the following base on the partner strand (in the 5′→3′ direction) in the shown AATT substructure. Also, secondary waters bridge the nucleotide interacting water layer together, completing the zigzag spine over several base steps. The polarization vectors on the water oxygens (represented as green arrows) align on average with their respective permanent molecular dipoles, as expected. It is important to point out that no manual modeling of the hydrogen positions was performed to achieve this model. Again, these results can only be inferred from the deposited structure (Fig. 4 A), in which waters are modeled only as lone oxygen atoms without hydrogens. The use of the PDB2PQR engine to determine hydrogen positions yields results that do not agree with the Drew and Dickerson (28,29) model (Fig. S2), further suggesting the improved utility of rigorous electrostatics combined with x-ray data compared to currently available tools. The results obtained by AMOEBA-assisted refinement augment the Drew and Dickerson model and reinforce the value added to structures refined with the proposed scattering and energetic engine.
Figure 4.
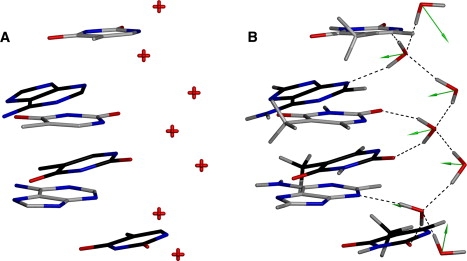
Zigzag spine of hydration in the DNA minor groove. Bases in gray are from the 3′→5′ strand, and bases in black are derived from the 5′→3′ strand. Shown is the AATT subsequence of the deposited structure (A) and after AMOEBA-assisted refinement with the x-ray data (B), with the primary and secondary layers of water forming the zigzag pattern. Green arrows represent polarization vectors originating from the water oxygens, and a 3.0 Å vector length corresponds to 1 D (average vector length: 1.5 Å).
The AMOEBA electrostatic model is able to accurately describe highly charged centers, such as cations (53), as illustrated in Fig. 5. The presence of a divalent cationic species possessing a strong electric field (one of the five structural Mg2+ ions) orients the hydrating water dipoles away from the ion. Further, the Mg2+ coordinates an intricate hydrogen-bonding network between opposing strands and strands in crystallographically related molecules. This agrees with a recent crystallographic analysis of B-DNA that suggested that the presence of ions serves to stabilize lateral contacts and end-to-end overlaps in the crystal lattice (54).
Figure 5.
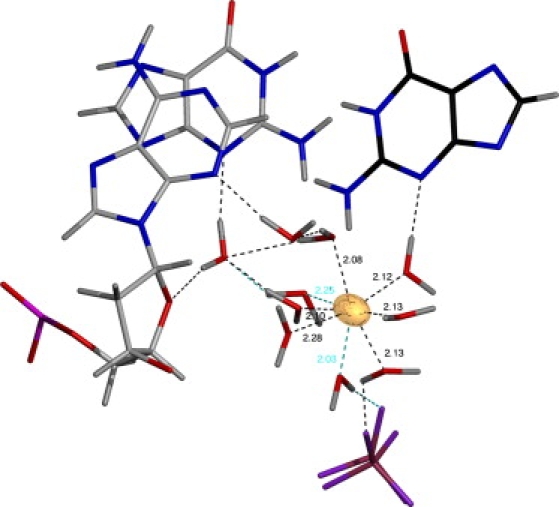
Hydration shell around one of the magnesium ions (shown in gold) in the re-refined crystal structure of the DNA 9mer (GCGAATTCG). Extensive hydrogen bonding is present with both DNA strands and a phosphate from a crystallographically related molecule (shown in purple and maroon at the bottom of the figure). Discretely disordered waters are indicated with cyan hydrogen bonds for clarity. All distances shown are given in angstroms. The magnesium is rendered according to thermal displacement parameters at the 20% isoprobability level.
Discussion
Improvements to the electrostatics of force fields, such as those provided by the AMOEBA model, allow for increasingly accurate and rapid simulations of structures at a level of chemical detail approaching the Born-Oppenheimer approximation. Coupled with Cartesian Gaussian multipoles to facilitate structure factor calculations, the force field can be used in macromolecular protein refinement in place of the commonly used geometric force field by Engh and Huber (4) without greatly affecting the speed of the refinement process. We validated this approach by re-refining several high-resolution x-ray crystal structures, which yielded a concomitant improvement in crystallographic refinement statistics and overall potential energy (Table 1), the latter of which represents an improvement of ∼1 kcal/residue for lysozyme. The resultant models produced additional information regarding pKa values at enzymatic active sites, hydrogen-bonding structure, and molecular stabilization.
The hydrogen-bond network of the lysozyme active site shown in Fig. 1 illustrates the information content that can be obtained by using a modern force field in crystallographic data analysis. Increasing experimental support for hydrogen-bond coupling in proteins suggests that these networks are crucial for propagating electronic and conformational signals (42,55–57). The use of a detailed electrostatic model that includes the crystallographic data suggests the possibility of hydrogen-bond coupling between Glu-35 and Asp-52, which may partly explain the perturbed pKa values for these residues (36). For example, our results suggest that additional experiments designed to probe the pKa/hydrogen-bonding distances between Glu-35 and Asp-52 (perhaps using NMR and isosteric mutations such as a Glu-35-Asp and/or Asp-52-Glu) may show correlated changes between the pKas of the two residues.
The use of more-accurate internuclear X-H bond lengths, as illustrated in Fig. 2, facilitates accurate measurement of hydrogen-bonding distances, an important consideration in light of the number of enzymes that involve hydrogen in enzymatic reactions, and situations where hydrogen bonding is an important factor in drug or ligand binding. For example, a more detailed hydrogen-bonding network of cyclic nucleotide phosphodiesterases bound to their respective substrates may help explain the differences in preference for individual nucleotides (e.g., AMP versus GMP) and the energetic factors involved in the glutamine switch mechanism of selectivity, thereby informing drug design and binding energy studies (58).
The inclusion of polarization effects in protein force fields serves a dual role in both properly characterizing the energetics of biomolecules, even in the presence of highly charged species (15), and yielding improved chemical information about the charge interactions of a system. This is supported by the trypsin structure (see Fig. 3), as the backbone amides of Gly-193 and Ser-195 orient their induced dipoles to stabilize anionic ligands/intermediates in the oxyanion hole. The carboxylate of Asp-99 functions as expected by withdrawing the positive charge from His-56, which would stabilize positive charge buildup upon deprotonation of the nucleophilic serine. These results agree with the current canon regarding serine proteases (43), validating both the polarization model and the catalytic triad mechanism. Finally, bound substrates serve to influence the orientation of freely rotatable X-H bonds, as indicated by the Hγ atom on the catalytic serine, an important consideration for protein-ligand energetics and chemistry. This has implications for virtual screening and fragment-based drug design studies of proteases, which require accurate structural models as a starting point to facilitate drug development (59).
Information regarding the electrostatics of the molecular system is generally applicable (as shown for all examples presented) and only adds the hydrogen positional terms as additional fit parameters. This is best done when the solvent model is as complete as possible, as it avoids gaps in the water network that can lead to erroneous hydrogen/water orientations due to the lack of nearby hydrogen-bonding partners. Further work in this area will explore the use of explicit bulk solvent models that can be accurately incorporated using a polarizable force field. It is worthwhile to also point out the utility of water networks in protein/enzyme function, and that the analysis of water structure in various systems, such as inhibitor resistance in β-lactamase, should prove informative in determining the role of water in catalysis and drug design (60).
Conclusions
Biological interpretations of crystal structures stand to be significantly improved by information gained through the use of modern force fields. The availability of more precise atomic positions and the explicit inclusion of hydrogens has implications for drug design, interpretation of enzymatic mechanisms, and structure-function analysis overall, particularly at atomic resolution. Medium- to low-resolution structure refinements also stand to benefit from an improved electrostatics treatment, as it affects nonhydrogen atom positions by playing a key role in forming and maintaining α-helix and β-sheet structure. This is also true of intraprotein hydrogen bonds in general, which can occupy up to 82% of a protein (61,62). Further, although neutron diffraction methods can be used to model hydrogen atoms, they typically hold them fixed during refinement due to the lack of an electrostatics treatment; it should be straightforward to adapt our method for this purpose. The models generated are also more physical compared to previous crystallographic treatments, and thus are transferable to other means of analysis, such as energy-related measures (e.g., protein-ligand binding energies, and Poisson-Boltzmann and free-energy calculations). This is also useful for pKa calculations at ionizable sites in macromolecules, which require accurate hydrogen placement for the Poisson-Boltzmann calculation (38–40,63). Finally, our method provides the chemical detail necessary to obtain information-rich descriptions of protein and nucleic acid functions, without significantly changing the number of parameters involved or the time required to refine them.
Supporting Material
Two tables and two figures are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(10)00406-6.
Supporting Material
Acknowledgments
The authors thank Paul Sigala for comments on the manuscript. We also thank Chuanjie Wu, Pengyu Ren, and Jay Ponder for their latest force-field parameters. V.P.S. acknowledges support from a National Science Foundation (NSF) grant for Cyberinfrastructure (NSF CHE-0535616) and Simbios (National Institutes of Health, U54 GM072970).
Footnotes
This is an Open Access article distributed under the terms of the Creative Commons-Attribution Noncommercial License (http://creativecommons.org/licenses/by-nc/2.0/), which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
References
- 1.Schnieders M.J., Fenn T.D., Brunger A.T. Polarizable atomic multipole X-ray refinement: application to peptide crystals. Acta Crystallogr. D Biol. Crystallogr. 2009;65:952–965. doi: 10.1107/S0907444909022707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ren P., Ponder J.W. Polarizable atomic multipole water model for molecular mechanics simulation. J. Phys. Chem. B. 2003;107:5933–5947. [Google Scholar]
- 3.Ren P., Ponder J.W. Temperature and pressure dependence of the AMOEBA water model. J. Phys. Chem. B. 2004;108:13427–13437. [Google Scholar]
- 4.Engh R.A., Huber R. Accurate bond and angle parameters for X-ray protein structure refinement. Acta Crystallogr. A. 1991;47:392–400. [Google Scholar]
- 5.Bragg S.W.H. The intensity of X-ray reflection by diamond. Proc. Phys. Soc. Lond. 1920;33:304–311. [Google Scholar]
- 6.Renninger M. “Umweganregung”, eine bisher unbeachtete Wechselwirkungserscheinung bei Raumgitterinterferenzen. Z. Phys. A. 1937;106:141–176. [Google Scholar]
- 7.Renninger M. Beitrag zur Kenntnis der röntgenographischen Unterschiede zwischen den beiden diamant-typen. Acta Crystallogr. 1955;8:606–610. [Google Scholar]
- 8.Dawson B. A general structure factor formalism for interpreting accurate X-ray and neutron diffraction data. Proc. R. Soc. A. 1967;298:255–263. [Google Scholar]
- 9.Dawson B. The covalent bond in diamond. Proc. R. Soc. A. 1967;298:264–288. [Google Scholar]
- 10.Ten Eyck L.F. Crystallographic fast Fourier transforms. Acta Crystallogr. A. 1973;29:183–191. [Google Scholar]
- 11.Ten Eyck L.F. Efficient structure-factor calculation for large molecules by the fast Fourier transform. Acta Crystallogr. A. 1977;33:486–492. [Google Scholar]
- 12.Hansen N.K., Coppens P. Testing aspherical atom refinements on small-molecule data sets. Acta Crystallogr. A. 1978;34:909–921. [Google Scholar]
- 13.Schnieders M.J., Ponder J.W. Polarizable atomic multipole solutes in a generalized Kirkwood continuum. J. Chem. Theory Comput. 2007;3:2083–2097. doi: 10.1021/ct7001336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schnieders M.J., Baker N.A., Ponder J.W. Polarizable atomic multipole solutes in a Poisson-Boltzmann continuum. J. Chem. Phys. 2007;126:124114–124121. doi: 10.1063/1.2714528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ponder J.W., Case D.A. Force fields for protein simulations. Adv. Protein Chem. 2003;66:27–85. doi: 10.1016/s0065-3233(03)66002-x. [DOI] [PubMed] [Google Scholar]
- 16.Beachy M.D., Chasman D., Friesner R.A. Accurate ab initio quantum chemical determination of the relative energetics of peptide conformations and assessment of empirical force fields. J. Am. Chem. Soc. 1997;119:5908–5920. [Google Scholar]
- 17.Williams D.E. Representation of the molecular electrostatic potential by atomic multipole and bond dipole models. J. Comput. Chem. 1988;9:745–763. [Google Scholar]
- 18.Levy Y., Onuchic J.N. Water mediation in protein folding and molecular recognition. Annu. Rev. Biophys. Biomol. Struct. 2006;35:389–415. doi: 10.1146/annurev.biophys.35.040405.102134. [DOI] [PubMed] [Google Scholar]
- 19.Ewald P.P. Die Berechnung optischer und elektrostatischer Gitterpotentiale. Annalen Physik. 1921;369:253–287. [Google Scholar]
- 20.Teeter M.M. Water-protein interactions: theory and experiment. Annu. Rev. Biophys. Biophys. Chem. 1991;20:577–600. doi: 10.1146/annurev.bb.20.060191.003045. [DOI] [PubMed] [Google Scholar]
- 21.Darden T., York D., Pedersen L. Particle mesh Ewald: an N • log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 22.Sagui C., Darden T.A. Molecular dynamics simulations of biomolecules: long-range electrostatic effects. Annu. Rev. Biophys. Biomol. Struct. 1999;28:155–179. doi: 10.1146/annurev.biophys.28.1.155. [DOI] [PubMed] [Google Scholar]
- 23.Schreiber H., Steinhauser O. Cutoff size does strongly influence molecular dynamics results on solvated polypeptides. Biochemistry. 1992;31:5856–5860. doi: 10.1021/bi00140a022. [DOI] [PubMed] [Google Scholar]
- 24.Gregory J.K., Clary D.C., Saykally R.J. The water dipole moment in water clusters. Science. 1997;275:814–817. doi: 10.1126/science.275.5301.814. [DOI] [PubMed] [Google Scholar]
- 25.Wang J., Dauter M., Dauter Z. Triclinic lysozyme at 0.65 A resolution. Acta Crystallogr. D Biol. Crystallogr. 2007;63:1254–1268. doi: 10.1107/S0907444907054224. [DOI] [PubMed] [Google Scholar]
- 26.Schmidt A., Lamzin V.S. Extraction of functional motion in trypsin crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2005;61:1132–1139. doi: 10.1107/S0907444905016732. [DOI] [PubMed] [Google Scholar]
- 27.Soler-Lopez M., Malinina L., Subirana J.A. Solvent organization in an oligonucleotide crystal. The structure of d(GCGAATTCG)2 at atomic resolution. J. Biol. Chem. 2000;275:23034–23044. doi: 10.1074/jbc.M002119200. [DOI] [PubMed] [Google Scholar]
- 28.Wing R., Drew H., Dickerson R.E. Crystal structure analysis of a complete turn of B-DNA. Nature. 1980;287:755–758. doi: 10.1038/287755a0. [DOI] [PubMed] [Google Scholar]
- 29.Drew H.R., Dickerson R.E. Structure of a B-DNA dodecamer: III. Geometry of hydration. J. Mol. Biol. 1981;151:535–556. doi: 10.1016/0022-2836(81)90009-7. [DOI] [PubMed] [Google Scholar]
- 30.Brünger A.T., Adams P.D., Warren G.L. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 31.Yu N., Yennawar H.P., Merz K.M. Refinement of protein crystal structures using energy restraints derived from linear-scaling quantum mechanics. Acta Crystallogr. D Biol. Crystallogr. 2005;61:322–332. doi: 10.1107/S0907444904033669. [DOI] [PubMed] [Google Scholar]
- 32.Moulinier L., Case D.A., Simonson T. Reintroducing electrostatics into protein X-ray structure refinement: bulk solvent treated as a dielectric continuum. Acta Crystallogr. D Biol. Crystallogr. 2003;59:2094–2103. doi: 10.1107/s090744490301833x. [DOI] [PubMed] [Google Scholar]
- 33.Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 34.Jones T.A., Zou J.Y., Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 35.Ahmed H.U., Blakeley M.P., Helliwell J.R. The determination of protonation states in proteins. Acta Crystallogr. D Biol. Crystallogr. 2007;63:906–922. doi: 10.1107/S0907444907029976. [DOI] [PubMed] [Google Scholar]
- 36.Bartik K., Redfield C., Dobson C.M. Measurement of the individual pKa values of acidic residues of hen and turkey lysozymes by two-dimensional 1H NMR. Biophys. J. 1994;66:1180–1184. doi: 10.1016/S0006-3495(94)80900-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bon C., Lehmann M.S., Wilkinson C. Quasi-Laue neutron-diffraction study of the water arrangement in crystals of triclinic hen egg-white lysozyme. Acta Crystallogr. D Biol. Crystallogr. 1999;55:978–987. doi: 10.1107/s0907444998018514. [DOI] [PubMed] [Google Scholar]
- 38.Dolinsky T.J., Nielsen J.E., Baker N.A. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32:W665–W667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dolinsky T.J., Czodrowski P., H, Baker N.A. PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007;35:W522–W525. doi: 10.1093/nar/gkm276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hooft R.W., Sander C., Vriend G. Positioning hydrogen atoms by optimizing hydrogen-bond networks in protein structures. Proteins. 1996;26:363–376. doi: 10.1002/(SICI)1097-0134(199612)26:4<363::AID-PROT1>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 41.Blake C.C.F., Johnson L.N., Sarma V.R. Crystallographic studies of the activity of hen egg-white lysozyme. Proc. R. Soc. Lond. B Biol. Sci. 1967;167:378–388. doi: 10.1098/rspb.1967.0035. [DOI] [PubMed] [Google Scholar]
- 42.Sigala P.A., Tsuchida M.A., Herschlag D. Hydrogen bond dynamics in the active site of photoactive yellow protein. Proc. Natl. Acad. Sci. USA. 2009;106:9232–9237. doi: 10.1073/pnas.0900168106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hedstrom L. Serine protease mechanism and specificity. Chem. Rev. 2002;102:4501–4524. doi: 10.1021/cr000033x. [DOI] [PubMed] [Google Scholar]
- 44.Blow D.M., Birktoft J.J., Hartley B.S. Role of a buried acid group in the mechanism of action of chymotrypsin. Nature. 1969;221:337–340. doi: 10.1038/221337a0. [DOI] [PubMed] [Google Scholar]
- 45.Dixon M.M., Matthews B.W. Is γ-chymotrypsin a tetrapeptide acyl-enzyme adduct of α-chymotrypsin? Biochemistry. 1989;28:7033–7038. doi: 10.1021/bi00443a038. [DOI] [PubMed] [Google Scholar]
- 46.Harel M., Su C.T., Sussman J.L. γ-Chymotrypsin is a complex of α-chymotrypsin with its own autolysis products. Biochemistry. 1991;30:5217–5225. doi: 10.1021/bi00235a015. [DOI] [PubMed] [Google Scholar]
- 47.Warshel A., Naray-Szabo G., Hwang J.K. How do serine proteases really work? Biochemistry. 1989;28:3629–3637. doi: 10.1021/bi00435a001. [DOI] [PubMed] [Google Scholar]
- 48.Henderson R. Structure of crystalline [α]-chymotrypsin: IV. The structure of indoleacryloyl-[α]-chymotrypsin and its relevance to the hydrolytic mechanism of the enzyme. J. Mol. Biol. 1970;54:341–354. doi: 10.1016/0022-2836(70)90434-1. [DOI] [PubMed] [Google Scholar]
- 49.Robertus J.D., Kraut J., Birktoft J.J. Subtilisin. Stereochemical mechanism involving transition-state stabilization. Biochemistry. 1972;11:4293–4303. doi: 10.1021/bi00773a016. [DOI] [PubMed] [Google Scholar]
- 50.Rao S.N., Singh U.C., Kollman P.A. Free energy perturbation calculations on binding and catalysis after mutating Asn 155 in subtilisin. Nature. 1987;328:551–554. doi: 10.1038/328551a0. [DOI] [PubMed] [Google Scholar]
- 51.Franklin R.E., Gosling R.G. The structure of sodium thymonucleate fibres. I. The influence of water content. Acta Crystallogr. 1953;6:673–677. [Google Scholar]
- 52.Wolf B., Hanlon S. Structural transitions of deoxyribonucleic acid in aqueous electrolyte solutions. II. Role of hydration. Biochemistry. 1975;14:1661–1670. doi: 10.1021/bi00679a018. [DOI] [PubMed] [Google Scholar]
- 53.Jiao D., King C., Ren P. Simulation of Ca2+ and Mg2+ solvation using polarizable atomic multipole potential. J. Phys. Chem. B. 2006;110:18553–18559. doi: 10.1021/jp062230r. [DOI] [PubMed] [Google Scholar]
- 54.Minasov G., Tereshko V., Egli M. Atomic-resolution crystal structures of B-DNA reveal specific influences of divalent metal ions on conformation and packing. J. Mol. Biol. 1999;291:83–99. doi: 10.1006/jmbi.1999.2934. [DOI] [PubMed] [Google Scholar]
- 55.Bouvignies G., Bernadó P., Blackledge M. Identification of slow correlated motions in proteins using residual dipolar and hydrogen-bond scalar couplings. Proc. Natl. Acad. Sci. USA. 2005;102:13885–13890. doi: 10.1073/pnas.0505129102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Perálvarez-Marín A., Lorenz-Fonfria V.A., Padros E. Inter-helical hydrogen bonds are essential elements for intra-protein signal transduction: the role of Asp115 in bacteriorhodopsin transport function. J. Mol. Biol. 2007;368:666–676. doi: 10.1016/j.jmb.2007.02.021. [DOI] [PubMed] [Google Scholar]
- 57.Rodríguez J.C., Zeng Y., Rivera M. The hydrogen-bonding network in heme oxygenase also functions as a modulator of enzyme dynamics: chaotic motions upon disrupting the h-bond network in heme oxygenase from Pseudomonas aeruginosa. J. Am. Chem. Soc. 2007;129:11730–11742. doi: 10.1021/ja072405q. [DOI] [PubMed] [Google Scholar]
- 58.Zhang K.Y., Card G.L., Bollag G. A glutamine switch mechanism for nucleotide selectivity by phosphodiesterases. Mol. Cell. 2004;15:279–286. doi: 10.1016/j.molcel.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 59.Turk B. Targeting proteases: successes, failures and future prospects. Nat. Rev. Drug Discov. 2006;5:785–799. doi: 10.1038/nrd2092. [DOI] [PubMed] [Google Scholar]
- 60.Thomas V.L., Golemi-Kotra D., Shoichet B.K. Structural consequences of the inhibitor-resistant Ser130Gly substitution in TEM β-lactamase. Biochemistry. 2005;44:9330–9338. doi: 10.1021/bi0502700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Baker E.N., Hubbard R.E. Hydrogen bonding in globular proteins. Prog. Biophys. Mol. Biol. 1984;44:97–179. doi: 10.1016/0079-6107(84)90007-5. [DOI] [PubMed] [Google Scholar]
- 62.Sticke D., Presta L., Rose G. Hydrogen bonding in globular proteins. J. Mol. Biol. 1992;226:1143–1159. doi: 10.1016/0022-2836(92)91058-w. [DOI] [PubMed] [Google Scholar]
- 63.Nielsen J.E., Vriend G. Optimizing the hydrogen-bond network in Poisson-Boltzmann equation-based pK(a) calculations. Proteins. 2001;43:403–412. doi: 10.1002/prot.1053. [DOI] [PubMed] [Google Scholar]
- 64.Fenn T.D., Ringe D., Petsko G.A. POVScript+: a program for model and data visualization using persistence of vision ray-tracing. J. Appl. Cryst. 2003;36:944–947. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.