

Abstract
The ability to discriminate between different cations efficiently is essential for the proper physiological functioning of many membrane transport proteins. One obvious mechanism of ion selectivity is when a binding site is structurally constrained by the protein architecture and its geometry is precisely adapted to fit an ion of a given size. This mechanism is not effective in the case of flexible protein binding sites that are able to deform structurally or to adapt to a bound ion. In this study, the concept of nontrivial ion selectivity arising in a highly flexible protein binding site conceptually represented as a microdroplet of ligands confined to a small volume is explored. The environment imposed by the spatial confinement is a critical feature of the reduced models. A large number of reduced binding site models (1077) comprising typical ion-coordinating ligands (carbonyl, hydroxyl, carboxylate, water) are constructed and characterized for Na+/K+ and Ca2+/Ba2+ size selectivity using free energy perturbation molecular dynamics simulations. Free energies are highly correlated with the sum of ion-ligand and ligand-ligand mean interactions, but the relative balance of those two contributions is different for K+-selective and Na+-selective binding sites. The analysis indicates that both the number and the type of ligands are important factors in ion selectivity.
Introduction
The mechanism of ion selectivity in biology is a subject that has fascinated scientists for a long time. K+ channels, in particular, have always been intriguing due to their remarkable ability to conduct K+ ions near the diffusion limit while remaining very selective for K+ over Na+. When the atomic structure of the KcsA K+ channel was resolved using x-ray crystallography (1,2), it seemed to offer direct support for an appealing structural explanation of ion selectivity in close correspondence with the snug-fit mechanism proposed in the early 1970s (3), which thereafter, became almost universally adopted (4). The idea, familiar in the field of host-guest chemistry (5), is that the narrow pore was perfectly suited (at the sub-ångström level) to provide a cavity of the appropriate size to fit K+, but unable for structural reasons to adapt and cradle the slightly smaller Na+. However, very early on, the picture arising from molecular dynamics (MD) simulations carried out by several independent groups suggested that the selectivity filter of the KcsA channel might be inherently too dynamical and flexible to satisfy the requirement of a strict snug-fit mechanism (6–8). K+ and Na+ are very similar, differing only slightly in their atomic radius by ∼0.38 Å. At room temperature, the channel can easily distort locally with little energetic cost to cradle one Na+ (6,8), a view that is inconsistent with the picture of a rigid site of the proper size to fit a K+ ion.
The issue came into sharper focus when free energy perturbation MD simulations (FEP/MD) showed that the flexible/fluctuating pore in its conductive conformation displayed nonetheless a substantial selectivity for K+ over Na+ (9–11). Those FEP/MD computations assume that ion binding selectivity can be addressed from a thermodynamic viewpoint. Accordingly, the key quantity is the relative free energy of Na+ and K+ in the biding site and in bulk solution,
| (1) |
(A binding site is K+-selective when ΔΔGNa,K is positive, and is Na+-selective when it is negative.) The suggestion of a flexible pore undergoing ångström-scale fluctuations able to yield a robust ΔΔGNa,K favoring K+ over Na+ seemed very surprising. Our analysis of ion selectivity in the KcsA channel, using quantum mechanical ab initio calculations as well as a series experiments based on free energy MD simulations led us to a surprising and somewhat provocative conclusion (12). For the KcsA channel in MD simulations, the free energy differences controlling selectivity arises principally from the number and properties of ion-coordinating ligands, without the need to enforce the position of those ligands to sub-ångström precision.
To further illustrate and explore the concept of size selectivity in the absence of any precise structural geometry on a sub-ångström level, we introduced simple reduced toy models, comprising only one ion and a small number of freely fluctuating ligands confined to a spherical region of 3.5 Å radius (12). By construction, no restraint forces prevent the collapse of the ligands onto the smaller Na+ ion. Yet, using free energy simulations, a reduced model with eight carbonyl-like ligands was shown to be as selective for K+ over Na+ than the binding site S2 in KcsA. That such an exceedingly simple model would display any size selectivity at all was unanticipated and quite surprising. Following this work, several theoretical studies have examined the properties of analogous reduced toy models (13–19), trying to explain the underlying cause of this observation. Although there are differences in interpretation, the studies have broadly confirmed that reduced models can display nontrivial size selectivity on the basis of local interactions, in the absence of a geometry enforced at sub-ångström precision (12–19).
Despite their apparent simplicity, the reduced models can give rise to a wide range of selectivity. In the context of a flexible binding site or a reduced toy model, the relative free energy of K+ and Na+ reflects a complicated interplay between the favorable ion-ligand interaction and the unfavorable ligand-ligand repulsion. However, only a small number of reduced models comprising a few carbonyl groups or water molecules have been explored until now, and the full range of possible behaviors remains unknown. In the absence of a more complete characterization, our conclusions regarding the selectivities arising from the ion-ligand and ligand-ligand interactions in a flexible binding site have remained somewhat controversial. For instance, a number of studies have put forth the notion that selectivity is governed primarily by the number of coordinating ligands and that their chemical type plays a minor role (14–16,19). Partly to address this controversy and partly to flesh out our previous conclusions, ion selectivity was characterized using FEP/MD simulations for an extensive collection of ∼1077 toy models comprising four to nine coordinating oxygen ligands of different types. The body of data accumulated from large set of calculations provides an unprecedented illustration of the broad variability of free energies in these reduced models.
Methods
Realistic chemical moieties were used to avoid ambiguities that arise when representing coordinating ligands by fragments extracted from molecular mechanical force fields. N-methylacetamide and diglycines were used to model the backbone carbonyl. Using ab initio calculations as shown previously (20), backbone carbonyl models based on formaldehyde such as used in some theoretical studies (15) do not provide a realistic model of ion-backbone interactions. All the toy models comprise four to nine coordinating oxygen ligands, with zero to eight N-methylacetamide, methanol (CH3OH), or water, zero to four diglycines (each with two backbone carbonyls), and zero to two neutral or ionized acetic acid (CH3COOH or CH3COO−, each with two oxygen ligands).
All the computations were carried out using the program CHARMM (21) with the force field PARAM22 (22). The water model is TIP3P (23). The FEP/MD simulations were carried out as described previously (12,24); they include a total of ∼2.8 ns of sampling for each model. A steep half-harmonic spherical restriction was imposed such that the oxygen ligands remain within 3.5 Å from the ion. The size of the confinement sphere was chosen as previously to mimic the amount of freedom observed in all-atom MD of the KcsA channel (12). The number of toy models with 4, 5, 6, 7, 8, and 9 ligands is 39, 69, 117, 183, 276, and 393, respectively, for a total of 1077 models. Simulations were generated at 300 K with a time-step of 2 fs and a Langevin friction of 5 ps−1 on each nonhydrogen atom. Average interaction energies were calculated from 5 ns of Langevin dynamics. The initial configuration of each reduced model was assembled by randomly placing the constituting molecules within a large sphere with the ion fixed at the center. This was followed by successive energy minimization and MD simulations during which a radial restraint on the oxygen ligands was increased progressively. One concern is whether the small crowded systems are kinetically trapped (locked) into their starting configuration, implying that MD is only sampling the small local atomic fluctuations. To address this issue, tests simulations with some of the most crowded models comprising nine ligands were carried out. The results indicate that there is exchange of configurations leading to a mixing of the molecules on the ns timescale. Further tests with FEP/MD simulations starting from different configurations yielded variations in relative free energies of ∼0.6 kcal/mol or less, suggesting that the sampling is sufficiently converged for the purpose of this analysis. The Lennard-Jones parameters of K+, Na+, Ca2+, and Ba2+ were adjusted to yield the proper difference in solvation free energy in bulk water based on experimental data. The free energy difference between K+ and Na+ is and Ba2+ and Ca2+ are ∼17.5 kcal/mol and 59 kcal/mol, respectively.
Results and Discussion
The concept of the reduced binding site and its relation to a realistic system is depicted schematically in Fig. 1. By construction, the simplified reduced models are designed to mimic flexible protein binding sites with N flexible ligands. Some amount of spatial limitation on the extent of the fluctuations of the ligands is a critical component of the reduced models. Even in the case of highly flexible protein binding sites, the ligands are never free to make arbitrarily large displacements. Protein ligands are always, by virtue of the three-dimensional architecture of the folded protein, restricted to remain within some small spatial region (Fig. 1 A). This suggests a simplified representation of a flexible protein binding site as a crowded microdroplet of ligands confined to a small spatial region (Fig. 1 B). In the reduced models, the physical confinement arising from the structure is approximated by a spherical half-harmonic restraining potential to keep the oxygen of each ligand within a distance of 3.5 Å away from the ion. In proteins binding sites, the ion-coordinating ligands are typically surrounded by nonpolar side chains (25), which confers a low dielectric to the surrounding of a binding site. In the reduced binding site models, this low dielectric nonpolar is simply represented by vacuum. One important consequence of representing protein binding sites as confined clusters surrounded by vacuum is that there is no accounting for long-ranged interactions with more distant ligands or solvent. Although such long-ranged contributions certainly amount to a high fraction of the total binding free energy of the ions, this does not actually pose a problem for a characterization of the selectivity regarding ions of same valence (e.g., Na+ and K+), which requires only relative free energies (see Eq. 1). It is important to note that the number of ligands N present in the confined reduced models as depicted in Fig. 1 B is not meant to impose a constraint on the canonical coordination number n, i.e., the number of ligands strictly within the inner coordination shell of the ion. Within the small crowded volume, the ion and the N ligands are free to interact and self-organize, and the actual coordination number n may differ from N. Whereas the ligands in the reduced binding site models are restricted to a small spherical region (3.5 Å), the latter is meant to incorporate the generic physical confinement arising from the surrounding protein. In contrast, some studies of ion selectivity have considered toy models constrained in such a way to impose specific values of the canonical coordination number n (14–16,19). In fact, strong architectural protein forces dictating a specific binding site geometry must be present to strictly impose a given value of n. Therefore, those reduced models are implicitly designed to mimic fairly rigid binding sites by construction, and for this reason, may differ in an essential way from the flexible reduced models considered in this analysis (see below).
Figure 1.
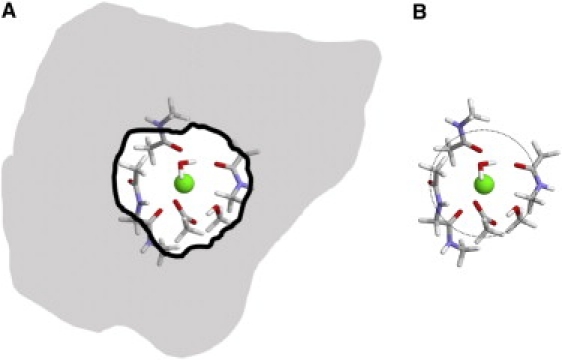
Schematic illustration of the concept of a confined microdroplet of ligands as a representation of a flexible ion binding site in a protein. (A) Typical flexible protein binding site (25). (B) Reduced binding site model of N ligands restricted by a spherical flat-bottom potential surrounded by vacuum. The 1077 reduced model were from diglycine, N-methylacetamide, methanol, water, acetate, and acetic acid.
As a word of caution, it is emphasized that the concept of the confined microdroplet depicted in Fig. 1 is not intended to be a valid representation of all existing ion binding sites in proteins. Some binding sites do indeed present a rigid geometrical arrangement of coordinating ligands suitable for an ion of a given size and their selectivity is straightforwardly understood on the basis of a snug-fit mechanism (12,26–29). The two Na+-selective binding sites in LeuT (Na1 and Na2) present nice examples of each of these two mechanisms (30). Analysis of LeuT extended to the structural database showed that binding sites with high local molecular stiffness rely on the local covalent connectivity to provide ion coordination via backbone and side-chain atoms of near-neighbor residues along the sequence (i, i + 1, i − 1) (30). Furthermore, the dynamical fluidity of the coordination structure within the reduced models such as depicted in Fig. 1 B should be properly understood: the radial root mean-square fluctuations of the ligands around the ion are typically on the order of 0.2–0.4 Å, indicating that the ion remains well-coordinated at all time despite the lack of precise structural geometry.
Ion selectivity was characterized using free energy simulations for 1077 toy models comprising four to nine coordinating oxygen ligands of different types. The main results are shown in Fig. 2. The coordinating ligands in the toy models are formed by methanol, water, N-methylacetamide, diglycine, acetic acid, and acetate ligands. Chemically realistic species were used to avoid the ambiguities associated with extracting generic chemical groups from molecular mechanical force fields. As shown in Fig. 2 A, the toy models can display size selectivity for either Na+ or K+, with relative free energies spanning from about −10 kcal/mol to +10 kcal/mol. A large fraction of the toy models yield marginal ion selectivity, with relative free energies between −1.5 and +1.5 kcal/mol, but some are quite selective for either Na+ or K+. Almost any type of ligand can be involved in a Na+-, or K+-selective model, although there is clearly an increase of carboxylates (acetic acid or acetate) in Na+-selective sites, and an increase of backbone carbonyls (N-methylacetamide or diglycine) in K+-selective sites (Fig. 2 C). The free energy of the models comprising eight identical ligands is: 7.6 kcal/mol (eight N-methylacetamide), 4.8 kcal/mol (four diglycines), 2.7 kcal/mol (eight methanol), and 1.4 kcal/mol (eight water). These results based on a large number of toy models further clarify previous observations that selectivity in the binding site S2 of KcsA was determined primarily by the intrinsic physical properties of the carbonyl ligands coordinating the cation (12). To be more precise, it is important to keep in mind that the crowded environment of the flexible ligands imposed by the confinement is an important feature that underlies selectivity. For example, completely free and unrestricted carbonyls, as well as liquid amides, do not display any significant selectivity.
Figure 2.
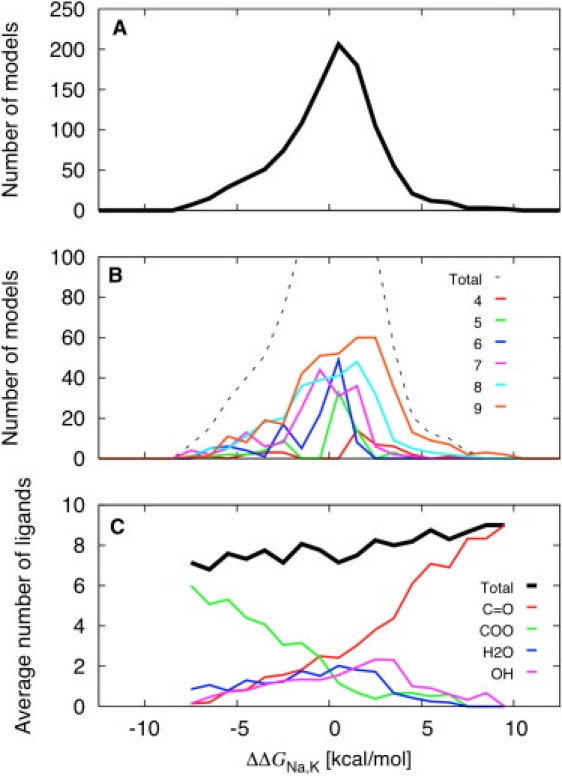
Results from free energy simulations carried out on 1077 reduced toy models of ion binding sites. (A) Distribution of the calculated free energy for all the toy models. (B) Distribution of the calculated free energy for all the toy models is broken down. (C) Average number of ligands as a function of free energy.
Providing simple intuitive rules to explain of ion selectivity in these small and crowded systems proves to be challenging. For instance, there is an induced fit on the binding of K+ or Na+, and the difference in the mean ion-ligand interaction between Na+ and K+ 〈ΔEi-L〉 is not sufficient to quantify selectivity (13,17). This is confirmed with the 1077 models. As shown in Fig. 3 bottom, 〈ΔEi-L〉 is not a good indicator of the relative free energy (the calculated correlation coefficient is only 0.56). In contrast, the difference between Na+ and K+ in the total sum of ion-ligand and ligand-ligand interactions (Fig. 3 top) correlates much better with the calculated free energies, though not perfectly (the calculated correlation coefficient is 0.88). Strongly coupled fluctuations mark the ion-ligand and ligand-ligand interactions (17); the two contributions are thermodynamically important and one should not try to explain ion selectivity by one to the exclusion of the other. The relative free energy of K+ and Na+ in each of the 1077 model reflects a complicated interplay between the favorable ion-ligand interaction 〈ΔEi-L〉, and the unfavorable ligand-ligand repulsion 〈ΔEL-L〉. Although the free energy differences between Na+ and K+ are dominated by enthalpic contributions (24,28), thermal fluctuations are important (13). For this reason, it is important to consider thermally averaged energies rather than simply the energy minimum from optimized geometries (31). Two key physical factors seem to control the relative free energy in these small confined systems: 1), the number of available ligands N; and 2), their physical properties (12,17,28). The physical properties of the coordinating ligand include both the electrostatic (dipole, charge, polarizability) as well as nonelectrostatic aspects (shape, radius, size).
Figure 3.
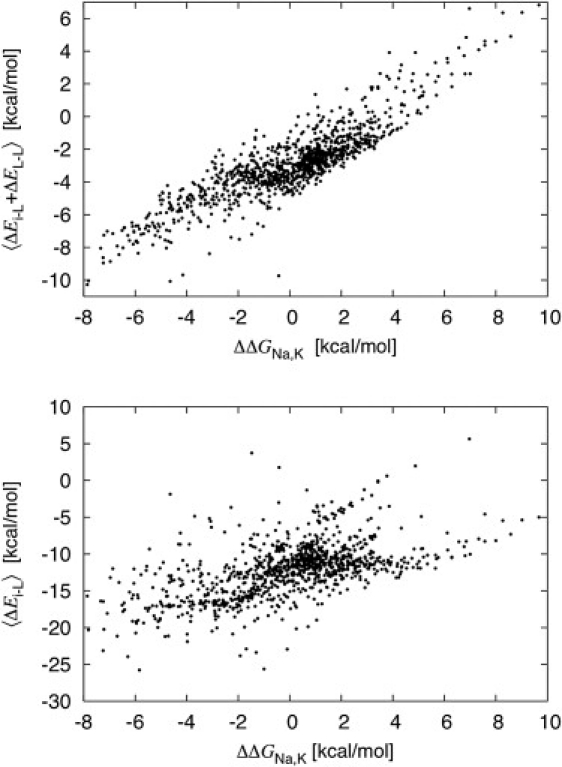
Correlation between the Na+/K+ relative free energy and the mean ion-ligand interaction (bottom) and the total ion-ligand and ligand-ligand mean energy (top) calculated for the 1077 reduced models. The calculated correlation coefficient for bottom is 0.56, and 0.88 for the top plot.
The mean ligand-ligand interaction becomes progressively more unfavorable for a small cation. The ligands are strongly oriented with their electronegative moieties pointing toward the cation that they coordinate. Their electronegative atoms are getting closer to one another when they coordinate a smaller cation than a larger cation, thus giving rise to an unfavorable size-dependent energy. The mean ligand-ligand interaction act much like the familiar concept of the strain energy, which plays an important role in the induced fit picture (32). The concept of strain energy is normally associated with structural distortions of the host (32). In the flexible binding site, an important component of strain energy is realized via through-space electrostatic interactions between the ligands coordinating the cation. The notion that ligand-ligand interactions in a flexible binding site contribute indirectly to the thermodynamics of ion selectivity is, to our knowledge, a novel concept that contrasts sharply with the classical treatments (3,33), which reasoned exclusively in terms of the ion-ligand interaction. It should be noted, however, that the concept of indirect solvent-solvent energy contributing to the thermodynamics of ion solvation has long been recognized (34).
Fig. 4, showing the mean ion-ligand and ligand-ligand energies 〈ΔEi-L〉 and 〈ΔEL-L〉 as a function of the free energy ΔΔGNa,K, sheds some new light on the energetic balance supporting K+ or Na+ selectivity in flexible sites. There is a clear linear anticorrelation between 〈ΔEi-L〉 and 〈ΔEL-L〉, and in all cases, the mean ligand-ligand repulsion increases as the mean ion-ligand interactions becomes more negative. This is consistent with the analysis of Dixit et al. (17) on the S2 binding site in KcsA. The data follow roughly the linear pattern,
| (2) |
where C is a constant that varies with the chemical composition of the reduced binding site. The slope of −1 suggests a direct response of the mean ligand-ligand repulsion to changes in the mean ion-ligand attraction. The relationship differs from the mean-field response governed by Gaussian fluctuations, which typically gives rise to a factor of 1/2 (e.g., the Born model of ion solvation (35,36)). Because ΔΔGNa,K ≈ 〈ΔEi-L〉 + 〈ΔEL-L〉, the constant C is related to the relative free energy of the ions in the binding site (although this relationship is not strictly exact; see Fig. 3). The value of C can be fitted empirically from the data in Fig. 4. For the binding sites that are highly selective for Na+ over K+, C is about −24 kcal/mol, whereas for the binding sites that are highly selective for K+ over Na+, C is about −15 kcal/mol. Interestingly, it seems that the mean ion-ligand attraction reaches larger negative values in binding sites that are highly Na+-selective, resulting in a relatively more important mean ligand-ligand repulsion for the binding sites that are highly K+-selective. This underlying cause can be traced back to the presence of small number (1,2) high-field ligands in the Na+-selective sites (e.g., CH3COOH or CH3COO−), shifting the ion-ligand attraction toward large negative values without raising considerably the ligand-ligand repulsion. In contrast, the K+-selective sites typically comprise neutral ligands and ligand-ligand repulsion becomes, relative to ion-ligand attraction, more important. This is consistent with previous analyses of the S2 binding site in the selectivity filter of the KcsA channel, which have noted the importance of the ligand-ligand repulsion (12,17,28).
Figure 4.
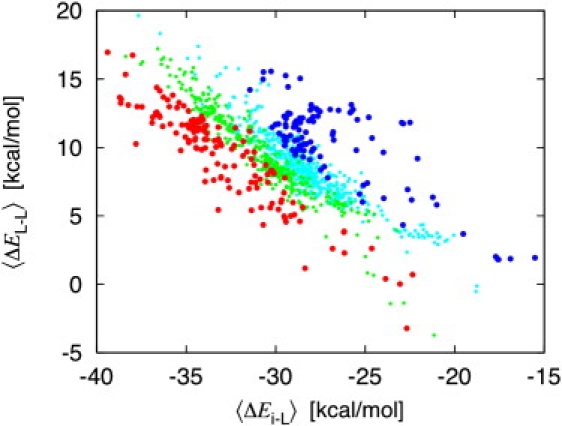
Correlation between the mean ion-ligand interaction (bottom) and the total ion-ligand and ligand-ligand mean energy (top) calculated for the 1077 reduced models for different range of the Na+/K+ relative free energies: highly selective for K+ over Na+ (ΔΔGNa,K > +3 kcal/mol, blue circles), weakly selective for K+ over Na+ (0 kcal/mol < ΔΔGNa,K < 3 kcal/mol, magenta circles), weakly selective for Na+ over K+ (−3 kcal/mol < ΔΔGNa,K < 0 kcal/mol, green circles), and highly selective for Na+ over K+ (ΔΔGNa,K < −3 kcal/mol, red circles).
Some similarities with the classic field strength theory of Eisenman (33) are worth noting. For instance, the magnitude of the ion-ligand interaction varies linearly with the number of ligands and their dipoles, and becomes more negative as the radius of the ion gets smaller. It one or a few ligands possess a very large dipole (or even a net negative charge), the reduced model then behaves as a classic Eisenman high field-strength binding site, favoring the smaller ion. If only a single ligand has a large dipole (e.g., on carboxylate group), the ligand-ligand repulsion may remain relatively small whereas the ion-ligand dominates (Fig. 4). However, the similarities with field-strength theory can fall short and should not be overstated to avoid confusion. For instance, it was pointed out early on that traditional ion-ligand field-strength arguments do not explain why a flexible binding site of eight carbonyls remains robustly selective for K+ because the interaction between a cation and a single backbone carbonyl is significantly larger than with a water molecule, and that this difference is even more prominent in the case of Na+ than for K+ (28). The dipole of the backbone amide carbonyl is about twice as large as the dipole of a water molecule. In other words, carbonyls are high field ligands according to the traditional Eisenman picture (33,37). They would favor Na+ over K+ if it were not for the counter-effect of ligand-ligand interactions (17,24). There are also circumstances where selectivity is set by trivial factors. For example, reduced models with very small number of neutral ligands often favor K+ over Na+, because of their inability to compensate for the loss of hydration free energy between K+ and Na+. One may note that selectivity for K+ over Na+ is also maintained if the carbonyl dipole is artificially scaled down. This is illustrated in Fig. 5 using the 4-diglycine model (8 carbonyl ligands). As the carbonyl dipole is scaled down, the reduced model starts to behave as a classic Eisenman low field-strength binding site, favoring the larger ion. One reason is that the mean ligand-ligand contribution (that grows quadratically with the magnitude of the ligand dipole) becomes less and less important as the dipoles are scaled down, and selectivity is dominated by the ion-ligand interactions. Achieving selectivity by scaling down the dipoles can become meaningless, however, because there is a point beyond which the total affinity is so small that no cation would bind. For example reduced models comprising eight methanol or eight water molecules can also display some preference for K+ over Na+ (Fig. 5). However, the mean binding energy produced by those models is significantly smaller than that with the model comprising eight carbonyls. Discussing the role of absolute affinities much further in the context of reduced models is difficult because long-range interactions that are not accounted for could have a large impact. Nevertheless, general considerations suggest that reduced models in which the mean ion-ligand interaction becomes weaker than ∼50% of an ion's enthalpy are unlikely to provide an effective binding site. For example, in the case of the S2 binding site of KcsA, the mean potential energy of K+ arising from four diglycines is about −130 kcal/mol, representing considerably >50% of the total enthalpy of K+ ion (about −160 kcal/mol). By comparison, the nearest hydration shell contributes ∼40–50% of the total hydration free energy of a K+ in bulk water (38). In general, it seems likely that a larger fraction of the total stabilization enthalpy than occurring in bulk water must arise from the nearest ligands in the case of protein binding sites, as the latter are generally surrounded by nonpolar residues (25) (Fig. 1), although there certainly are exceptions. What is remarkable with the flexible site of eight carbonyls is that selectivity for K+ over Na+ is robustly maintained whereas the large ligand dipoles confer high binding affinity. In a previous study, selectivity was maintained for dipoles varying up to 4.2 Debye in a toy model with eight carbonyl-like ligands (12).
Figure 5.
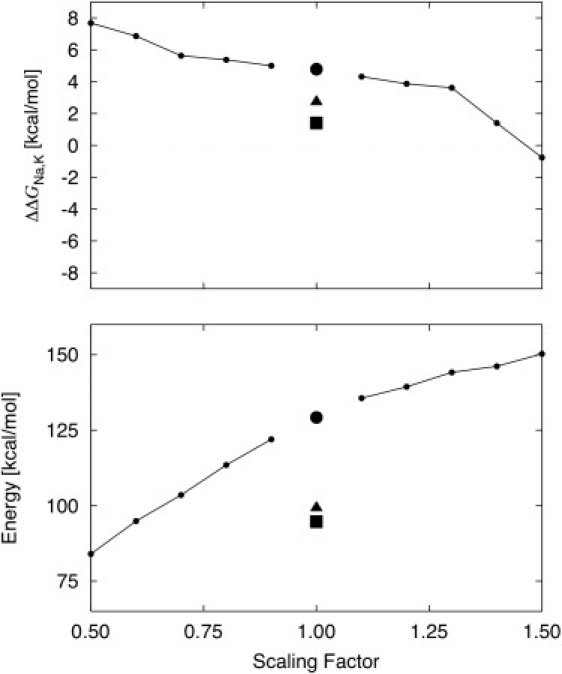
Results for the reduced model comprising four diglycine ligands. The C=O partial charges were multiplied by a scaling factor between 0.5 and 1.5 and FEP/MD calculations were generated. The results for the reduced model of eight methanol (triangle) or eight water molecules (square) are also shown.
The set of calculations illustrates the importance of both the number and type of ligands for ion selectivity in these reduced confined systems meant to represent flexible protein binding sites. This observation seems to be in contrast with the conclusion of a number of studies asserting that selectivity is primarily controlled by the number of coordinating ligands (14–16). According to the concept of over-coordination (14), the cation in the binding site is coordinated by more ligands than in bulk water and altering the number of ligands rather than their type is the way to control selectivity. A similar argument is elaborated in the topological control hypothesis (16). However, this analysis shows that the type of ligands leads to important quantitative differences (24,28,30). A straightforward way to address this issue is to compare free energies for two specific cases. The selectivity for K+ over Na+ in a toy model of eight backbone carbonyl groups is on the order of 4–6 kcal/mol, whereas the selectivity of eight water molecules is on the order of 1–2 kcal/mol under the same conditions. Previous results by Bostick and Brooks (16) showing that the toy model of eight waters could be as selective as that of eight carbonyls were simulated under different conditions (39) and have been subsequently revised by the authors (18). Irrespective of the details of the confining potential in reduced toy models, carbonyl ligands yield larger K+ selectivity than water molecules, a fact that is confirmed by several independent computational studies (12,17,18,24,28) including the original proponents of the topological control hypothesis (18).
In part, it is possible that these diverging views are further compounded by the lack of strict equivalence between the number N of ligands in the flexible reduced models considered here, and the canonical coordination number n considered in other studies (14–16,19). To examine this possibility in more detail, the selectivity free energy displayed by a given reduced model was correlated with the mean canonical coordination number n of Na+ and K+ within that model. The results are shown in Fig. 6. To define the coordination number n, distances of 2.8 Å and 3.2 Å were used for Na+ and K+, respectively (the color code is the same as used in Fig. 4; red for the most Na+-selective models and blue for most K+-selective models). From the scatter plot, it is apparent that the mean coordination number n is typically a little smaller for Na+ than K+, which is expected in a flexible system. The most K+-selective models (Fig. 6 blue) are found on the high-end of the mean coordination number n, approximately five to seven for K+ and approximately four to six for Na+, where they overlap with the weakly K+-selective models (Fig. 6 cyan). However, no obvious correlation is observed for the strongly Na+-selective models, as the mean coordination numbers n from those models are spread over the entire range. This indicates that considerations limited to coordination numbers are important but not sufficient to explain ion selectivity. For example, although the concept of overcoordination sheds an interesting light on the properties of a binding site of eight carbonyls, it plainly cannot account for the properties of the toy models comprising eight ligands with a strong preference for Na+ over K+ (Fig. 2). Likewise, the magnitude of the free energies achieved via topological control cannot account for the considerable selectivity of the S2 binding site in KcsA (39). In view of the above results with 1077 toy models, the proposition that both the number and the type of ligands control ion selectivity in a flexible binding site is unambiguously confirmed (12).
Figure 6.
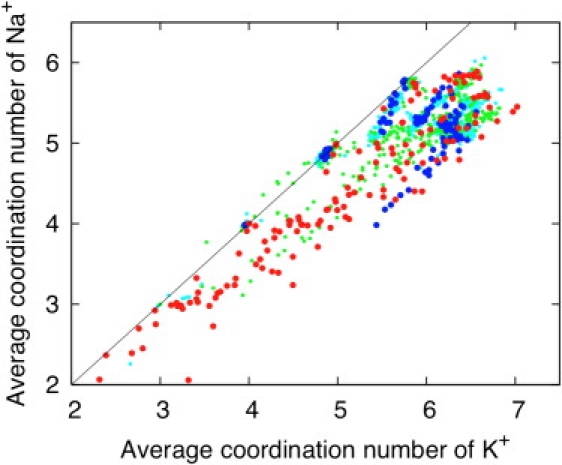
Correlation between the selectivity displayed by a reduced model and the mean canonical coordination number n of Na+ or K+ (calculated by counting the average number oxygen ligands within 3.0 Å away from K+ and within 2.8 Å away from Na+). The results for the 1077 reduced models are shown. The color code corresponding to different range of the Na+/K+ relative free energies is the same as in Fig. 4: highly selective for K+ over Na+ (ΔΔGNa,K > +3 kcal/mol, blue circles), weakly selective for K+ over Na+ (0 kcal/mol < ΔΔGNa,K < 3 kcal/mol, magenta circles), weakly selective for Na+ over K+ (−3 kcal/mol < ΔΔGNa,K < 0 kcal/mol, green circles), and highly selective for Na+ over K+ (ΔΔGNa,K < −3 kcal/mol, red circles).
The emergence of robust trends about ion solvation free energies in these confined microdroplet systems is fascinating. For example, size selectivity observed for divalent ions (Ca2+ versus Ba2+) seems to be correlated with the selectivity observed for monovalent ions (Na+ versus K+). As shown in Fig. 7, many of the reduced binding site that favoring K+ over Na+ also favor Ba2+ over Ca+2, and vice versa. The correlation is not perfect, but unambiguous (the calculated correlation coefficient is 0.80). Thus, one hallmark of size selectivity traditionally associated with the existence of a rigid cavity of a given radius can actually be reproduced in binding site models without the need to invoke a cavity size at sub-ångströms precision. Despite the neglect of induced polarization, which would required for a quantitatively accurate treatment of divalent cations, these results are suggestive. For example, observing that Ba2+ (about the size of K+) binds to the selectivity filter of the KcsA channel, but not Ca2+ (about the size of Na+), does not imply that ion selectivity is governed by a fixed cavity size as previously suggested (40). Similar trends can also appear in a reduced model without imposed precise geometry (Fig. 7).
Figure 7.
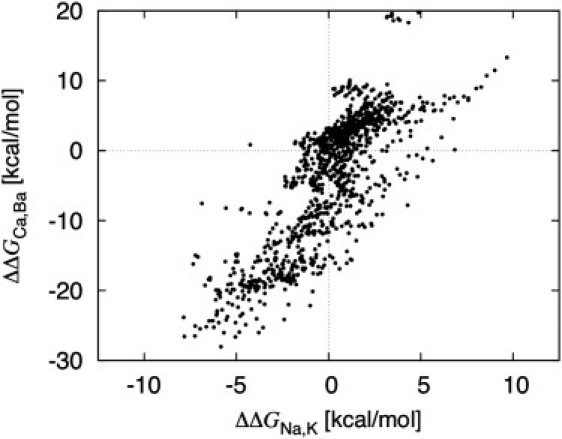
Comparison of the free energy for divalent cations (Ba2+ relative to Ca2+) with the free energy for monovalent cations (Na+ relative to K+) for the 1077 reduced models (the calculated correlation coefficient is 0.80).
As a last example, one can construct a simple toy model to represent the familiar DEKA motif of Na+ channels (41) using one N-methylacetamide, one methyl-ammonium, two acetate solvated by eight water molecules (not included in Fig. 2). Those 12 ligands were confined within a spherical region of 5 Å. Free energy simulations shows that this reduced model of DEKA with hydration displays selectivity for Na+ over K+ by 3.3 kcal/mol. Similar models of the familiar EEEE motif of Ca+2-channels, in which the negatively charged carboxylate oxygens are freely fluctuating within a confining region and the aqueous phase is represented by a continuum dielectric, have been introduced and investigated by Boda et al. (42,43). Those models, called charge-space competition, have been very successful to explain the observed behavior in L-type Ca+2 channels for a wide range of conditions without the need to enforce a precise geometry of the coordinating ligands. A model of the ryanodine receptor calcium channel build on these principles has been show to reproduce all the available permeation and selectivity data (44,45). The physical principles giving rise to ion selectivity in the charge-space competition are very similar to those at play in the confined microdroplet, with one notable difference. An assessment of Ca2+/Na+ selectivity involves two ions with different charges. For this reason, long-range electrostatic contributions, which can be safely neglected in the case of ions of the same valence, must be accounted for in idealized models of Ca2+-channels.
Conclusions
This study shows that simplified binding site models comprising a small number of ligands confined to a small subvolume (crowded microdroplet of ligands) are able to display nontrivial ion selectivity. Within the context of those spatially confined binding sites, ion selectivity is controlled by the number of coordinating ligands and their physical properties (12,17,28). The FEP/MD calculations were carried out using an additive force field that does not account for induced polarization effects explicitly. Despite this approximation, there is confidence that the basic physical trends regarding the mechanism of ion selectivity in flexible reduced binding site models that have been identified are robust and independent of the force field used. Although the unique identification of ion-ligand and ligand-ligand interactions becomes nontrivial in the case of nonadditive potential functions, a similar analysis can be carried out and it is observed that induced polarization of the coordinating ligands by an ion actually increases the ligand-ligand repulsion and reinforce the trends described in this study (this was shown with quantum mechanical calculations in the Supporting Material of Noskov et al. (12)). Nevertheless, it is likely that induced polarization could alter the detailed behavior of specific reduced binding sites, particularly in the case of divalent ions. Therefore, it is better to interpret the results for any specific system with caution.
It is clear from existing studies that there are multiple ways to achieve ion selectivity at the atomic level in protein binding sites (30). One important goal of theoretical efforts ought to be the identification of the microscopic conditions under which a given mechanism is predominant. In these efforts, computational studies based on simplified reduced toy models can serve to illuminate the mechanism of ion selectivity that exists in these complex macromolecular systems. Studies of reduced toy models can also raise interesting questions of their own, particularly concerning the structure and thermodynamics of ion solvation in flexible binding sites surrounded by restricted ligands (the confined microdroplet illustrated in Fig. 1). For meaningful studies, however, it is important that the information extracted from MD simulations carried out on full all-atom systems (10–12,24,28,30,46–52) be used to guide the choices made when constructing reduced models. The example of LeuT highlights the fact that for any protein, architectural rigidity or dynamical flexibility cannot be postulated a priori, and that all-atom MD simulations are ultimately necessary to ascertain the microscopic character of a given binding site.
Acknowledgments
Discussions with Olaf S. Andersen are gratefully acknowledged.
This work was supported by the National Institutes of Health (GM-62342).
References
- 1.Doyle D.A., Morais Cabral J., MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 2.Zhou Y., Morais-Cabral J.H., MacKinnon R. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 A resolution. Nature. 2001;414:43–48. doi: 10.1038/35102009. [DOI] [PubMed] [Google Scholar]
- 3.Bezanilla F., Armstrong C.M. Negative conductance caused by entry of sodium and Cesium ions into the K channels of squid axon. J. Gen. Physiol. 1972;53:342–347. doi: 10.1085/jgp.60.5.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hille B., Armstrong C.M., MacKinnon R. Ion channels: from idea to reality. Nat. Med. 1999;5:1105–1109. doi: 10.1038/13415. [DOI] [PubMed] [Google Scholar]
- 5.Pedersen C.J., Frensdor H.K. Macrocyclic polyethers and their complexes. Angew. Chem. Int. Ed. 1972;11:16. doi: 10.1002/anie.197200161. [DOI] [PubMed] [Google Scholar]
- 6.Guidoni L., Torre V., Carloni P. Potassium and sodium binding to the outer mouth of the K+ channel. Biochemistry. 1999;38:8599–8604. doi: 10.1021/bi990540c. [DOI] [PubMed] [Google Scholar]
- 7.Bernèche S., Roux B. Molecular dynamics of the KcsA K(+) channel in a bilayer membrane. Biophys. J. 2000;78:2900–2917. doi: 10.1016/S0006-3495(00)76831-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Biggin P.C., Smith G.R., Sansom M.S. Potassium and sodium ions in a potassium channel studied by molecular dynamics simulations. Biochim. Biophys. Acta. 2001;1510:1–9. doi: 10.1016/s0005-2736(00)00345-x. [DOI] [PubMed] [Google Scholar]
- 9.Allen T.W., Bliznyuk A., Chung S.H. The potassium channel: structure, selectivity and diffusion. J. Chem. Phys. 2000;112:8191–8204. [Google Scholar]
- 10.Bernèche S., Roux B. Energetics of ion conduction through the K+ channel. Nature. 2001;414:73–77. doi: 10.1038/35102067. [DOI] [PubMed] [Google Scholar]
- 11.Luzhkov V.B., Aqvist J. K(+)/Na(+) selectivity of the KcsA potassium channel from microscopic free energy perturbation calculations. Biochim. Biophys. Acta. 2001;1548:194–202. doi: 10.1016/s0167-4838(01)00213-8. [DOI] [PubMed] [Google Scholar]
- 12.Noskov S., Bernèche S., Roux B. Control of ion selectivity in K(+) channels by dynamic and electrostatic properties of carbonyl ligands. Nature. 2004;431:830–834. doi: 10.1038/nature02943. [DOI] [PubMed] [Google Scholar]
- 13.Asthagiri D., Pratt L.R., Paulaitis M.E. Role of fluctuations in a snug-fit mechanism of KcsA channel selectivity. J. Chem. Phys. 2006;125:24701. doi: 10.1063/1.2205853. [DOI] [PubMed] [Google Scholar]
- 14.Varma S., Rempe S.B. Tuning ion coordination architectures to enable selective partitioning. Biophys. J. 2007;93:1093–1099. doi: 10.1529/biophysj.107.107482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thomas M., Jayatilaka D., Corry B. The predominant role of coordination number in potassium channel selectivity. Biophys. J. 2007;93:2635–2643. doi: 10.1529/biophysj.107.108167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bostick D.L., Brooks C.L. Selectivity in K+ channels is due to topological control of the permeant ion's coordinated state. Proc. Natl. Acad. Sci. USA. 2007;104:9260–9265. doi: 10.1073/pnas.0700554104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dixit P.D., Merchant S., Asthagiri D. Ion selectivity in the KcsA potassium channel from the perspective of the ion binding site. Biophys. J. 2009;96:2138–2145. doi: 10.1016/j.bpj.2008.12.3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bostick D.L., Arora K., Brooks C.L. K+/Na+ selectivity in toy cation binding site models is determined by the ‘host’. Biophys. J. 2009;96:3887–3896. doi: 10.1016/j.bpj.2008.12.3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bostick D.L., Brooks C.L. Statistical determinants of selective ionic complexation: ions in solvent, transport proteins, and other “hosts”. Biophys. J. 2009;96:4470–4492. doi: 10.1016/j.bpj.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roux B., Karplus M. Potential-energy function for cation-peptide interactions—an ab-initio study. J. Comput. Chem. 1995;16:690–704. [Google Scholar]
- 21.Brooks B.R., Brooks C.L., Karplus M. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.MacKerell A.D.J., Bashford D., Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 23.Jorgensen W.L., Chandrasekhar J., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 24.Noskov S.Y., Roux B. Importance of hydration and dynamics on the selectivity of the KcsA and NaK channels. J. Gen. Physiol. 2007;129:135–143. doi: 10.1085/jgp.200609633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamashita M.M., Wesson L., Eisenberg D. Where metal ions bind in proteins. Proc. Natl. Acad. Sci. USA. 1990;87:5648–5652. doi: 10.1073/pnas.87.15.5648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aqvist J., Alvarez O., Eisenman G. Ion-selective properties of a small ionophore in methanol studied by free energy perturbation simulations. J. Phys. Chem. 1992;96:10019–10025. [Google Scholar]
- 27.Marrone T.J., Merz K.M. Molecular recognition of K+ and Na+ by valinomycin in methanol. J. Am. Chem. Soc. 1995;117:779–791. [Google Scholar]
- 28.Noskov S.Y., Roux B. Ion selectivity in potassium channels. Biophys. Chem. 2006;124:279–291. doi: 10.1016/j.bpc.2006.05.033. [DOI] [PubMed] [Google Scholar]
- 29.Varma S., Sabo D., Rempe S.B. K+/Na+ selectivity in K channels and valinomycin: over-coordination versus cavity-size constraints. J. Mol. Biol. 2008;376:13–22. doi: 10.1016/j.jmb.2007.11.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Noskov S.Y., Roux B. Control of ion selectivity in LeuT: two Na+ binding sites with two different mechanisms. J. Mol. Biol. 2008;377:804–818. doi: 10.1016/j.jmb.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu H., Roux B. On the utilization of energy minimization to the study of ion selectivity. Biophys. J. 2009;97:L15–L17. doi: 10.1016/j.bpj.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gouaux E., Mackinnon R. Principles of selective ion transport in channels and pumps. Science. 2005;310:1461–1465. doi: 10.1126/science.1113666. [DOI] [PubMed] [Google Scholar]
- 33.Eisenman, G., and S. Krasne. 1973. Further considerations on the ion selectivity of carrier molecules and membranes. IV International Biophysics Congress Symposium on Membrane Structure and Function., Moscow.
- 34.Yu H.A., Karplus M. A thermodynamic analysis of solvation. J. Chem. Phys. 1988;89:2366–2379. [Google Scholar]
- 35.Born M. Volumen Und Hydratationswarme Der Ionen. Z. Phys. 1920;1:45–48. [Google Scholar]
- 36.Roux B., Yu H.A., Karplus M. Molecular basis for the Born model of ion solvation. J. Phys. Chem. 1990;94:4683–4688. [Google Scholar]
- 37.Eisenman G., Horn R. Ionic selectivity revisited: the role of kinetic and equilibrium processes in ion permeation through channels. J. Membr. Biol. 1983;76:197–225. doi: 10.1007/BF01870364. [DOI] [PubMed] [Google Scholar]
- 38.Rempe S.B., Asthagiri D., Pratt L.D. Inner shell definition and absolute hydration free energy of K+(Aq) on the basis of quasi-chemical theory and ab initio molecular dynamics. Phys. Chem. Chem. Phys. 2004;6:1966–1969. [Google Scholar]
- 39.Yu H., Noskov S.Y., Roux B. Hydration number, topological control, and ion selectivity. J. Phys. Chem. B. 2009;113:8725–8730. doi: 10.1021/jp901233v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lockless S.W., Zhou M., MacKinnon R. Structural and thermodynamic properties of selective ion binding in a K+ channel. PLoS Biol. 2007;5:e121. doi: 10.1371/journal.pbio.0050121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lipkind G.M., Fozzard H.A. Voltage-gated Na channel selectivity: the role of the conserved domain III lysine residue. J. Gen. Physiol. 2008;131:523–529. doi: 10.1085/jgp.200809991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boda D., Nonner W., Gillespie D. Volume exclusion in calcium selective channels. Biophys. J. 2008;94:3486–3496. doi: 10.1529/biophysj.107.122796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boda D., Valiskó M., Nonner W. Ionic selectivity in L-type calcium channels by electrostatics and hard-core repulsion. J. Gen. Physiol. 2009;133:497–509. doi: 10.1085/jgp.200910211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gillespie D., Xu L., Meissner G. (De)constructing the ryanodine receptor: modeling ion permeation and selectivity of the calcium release channel. J. Phys. Chem. B. 2005;109:15598–15610. doi: 10.1021/jp052471j. [DOI] [PubMed] [Google Scholar]
- 45.Gillespie D. Energetics of divalent selectivity in a calcium channel: the ryanodine receptor case study. Biophys. J. 2008;94:1169–1184. doi: 10.1529/biophysj.107.116798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Caplan D.A., Subbotina J.O., Noskov S.Y. Molecular mechanism of ion-ion and ion-substrate coupling in the Na+-dependent leucine transporter LeuT. Biophys. J. 2008;95:4613–4621. doi: 10.1529/biophysj.108.139741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vijayan R., Plested A.J., Biggin P.C. Selectivity and cooperativity of modulatory ions in a neurotransmitter receptor. Biophys. J. 2009;96:1751–1760. doi: 10.1016/j.bpj.2008.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Plested A.J., Vijayan R., Mayer M.L. Molecular basis of kainate receptor modulation by sodium. Neuron. 2008;58:720–735. doi: 10.1016/j.neuron.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Luzhkov V.B., Almlöf M., Aqvist J. Computational study of the binding affinity and selectivity of the bacterial ammonium transporter AmtB. Biochemistry. 2006;45:10807–10814. doi: 10.1021/bi0610799. [DOI] [PubMed] [Google Scholar]
- 50.Shi W., Inamdar M.V., Lastoskie C.M. Divalent cation adsorption on the actin monomer. J. Phys. Chem. C. 2007;111:15642–15652. [Google Scholar]
- 51.Khalili-Araghi F., Gumbart J., Schulten K. Molecular dynamics simulations of membrane channels and transporters. Curr. Opin. Struct. Biol. 2009;19:128–137. doi: 10.1016/j.sbi.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lindahl E., Sansom M.S. Membrane proteins: molecular dynamics simulations. Curr. Opin. Struct. Biol. 2008;18:425–431. doi: 10.1016/j.sbi.2008.02.003. [DOI] [PubMed] [Google Scholar]