

Abstract
The triple helix of collagen shows a steep unfolding transition upon heating, whereas less steep and more gradual refolding is observed upon cooling. The shape of the hysteresis loop depends on the rate of temperature change as well as the peptide concentration. Experimental heating and cooling rates are usually much faster than rates of unfolding and refolding. In this work, collagen model peptides were used to study hysteresis quantitatively. Their unfolding and refolding profiles were recorded at different heating and cooling rates, and at different peptide concentrations. Data were fitted assuming kinetic mechanisms in which three chains combine to a helix with or without an intermediate that acts as a nucleus. A quantitative fit was achieved with the same kinetic model for the forward and backward reactions. Transitions of exogenously trimerized collagen models were also analyzed with a simplified kinetic mechanism. It follows that true equilibrium transitions can only be measured at high concentrations of polypeptide chains with slow scanning rates, for example, 0.1°C/h at 0.25 mM peptide concentration of (Gly-Pro-Pro)10. (Gly-Pro-4(R)Hyp)10 folds ∼2000 times faster than (Gly-Pro-Pro)10. This was explained by a more stable nucleus, whereas the rate of propagation was almost equal. The analysis presented here can be used to derive kinetic and thermodynamic data for collagenous and other systems with kinetically controlled hysteresis.
Introduction
The phenomenon of hysteresis is observed in ferromagnetism, mechanical elasticity, electronics, and biological systems. The term “hysteresis” comes from the Greek word “hysteros” (for later, lagging behind) and means that an effect persists even after its cause is abolished. Many conformational transitions and assembly processes of proteins and DNA exhibit hysteresis loops in which refolding occurs at a lower temperature or lower denaturant concentration than unfolding. Mergny and Lacroix (1) found hysteresis for the formation and dissociation of the tetrameric i-motif of DNA when rates of heating and cooling were of the same magnitude as the rate of the association-dissociation reaction. Related effects were found for protein transthyretin (2) and HBV-virus capsid disassembly (3). Hysteresis of the assembly and disassembly of a coiled-coil structure is believed to be important in the bilayer fusion of SNARE (4). An interesting time dependence of the hysteresis of thermal unfolding was observed for the complex of apolipoprotein C-1 with phospholipids (5).
Unfolding and refolding of collagen molecules is another well-known example of hysteresis (6,7). Here the sharpness and position of the unfolding and the refolding profile depend on the rate of temperature change as long as this rate is of comparable magnitude to the rate of conformational changes. Triple-helix formation is slower than many other conformational transitions because of rate-determining cis-trans isomerization steps in helix propagation (8–10). A clear hysteresis is observable at 2°C/h and higher rates of heating and cooling for pN type III collagen and collagen III. Collagen III is used as a convenient experimental system for collagen folding because of its interchain cystine knot at the C-terminus, where refolding starts (8). For processed collagens lacking a disulfide knot or other trimerization domains, refolding of the triple helix is virtually impossible. The three α-chains are not aligned, and slippage may occur because of the repeating sequence (9). Repeated sequences with Gly-Xaa-Yaa (where Xaa and Yaa stand for any amino acid) may then combine in many different combinations. Furthermore, a pronounced concentration dependence of folding and hysteresis has been observed (10). For short synthetic collagen model peptides or short natural collagen triple helices, the number of misaligned structures is reduced, but the concentration dependence remains as long as the chains are not linked.
For collagens, experimental observations of hysteresis are often of a rather qualitative nature. In many studies, investigators interpreted the measured transition profiles as equilibrium curves, ignoring the possible effects of heating rate or protein concentration. This led to severe errors in measured values, as shown by a literature comparison of the transition properties of collagen model peptides (10). Information on the heating rate and protein concentration is even lacking in many publications. The importance of establishing the real equilibrium transitions of collagen triple helices was recently noted by Persikov et al. (11).
In this work, the hysteresis of two collagen model peptides, (Gly-Pro-Pro)10 and (Gly-Pro-4(R)Hyp)10, were studied quantitatively to elucidate the effects of heating rate and peptide concentration. Furthermore, the model peptides (Gly-Pro-Pro)10-foldon, (Gly-Pro-Pro)10-NC2(XIX), and NC2(XIX)-(Gly-Pro-Pro)10 were studied. The three (Gly-Pro-Pro)10 chains are linked by the trimeric phage protein foldon (12–14) or by the NC2 trimerization domain of type XIX collagen at the N- or C-terminus (15).
Hysteresis experiments provide simultaneous information regarding the equilibrium and kinetic parameters of a system. Three simple kinetic model mechanisms were applied in this study. In the first model, which is applied to exogenously trimerized model peptides, the triple helix is assumed to be formed by propagation with a rate constant kp and unfolded with a rate constant kd in first-order reactions. In the second model, monomeric chains are assumed to fold with an apparent third-order rate constant ka,app and the triple helix is unfolded with an apparent rate constant kd. In the third model, monomeric chains are assumed to form an intermediate unstable trimer in fast preequilibrium, which then forms the triple helix by slow propagation steps with rate constant kp.
The above mechanisms were successfully applied to fit experimental curves within small error limits, even when the assumption of identity of the mechanism for the forward and backward scans was used. Only the initial conditions of integration of the differential equations were changed from folded helix to unfolded coiled state at time zero. This observation suggests that the apparent hysteresis of collagen-like peptide transitions is of a kinetic nature. The enthalpy and entropy values fitted those determined more directly by calorimetry and confirmed that (Gly-Pro-4(R)Hyp)10 is more stable than (Gly-Pro-Pro)10. It was also confirmed that the rate of folding is increased ∼2000-fold by the replacement of Pro by 4(R)Hyp in the Yaa position (12). Of importance, our data suggest that this acceleration originates not from the propagation rate, but from a higher stability of the nucleus. As outlined above, a number of other systems exhibit biologically relevant hysteresis behavior, but quantitative fits are lacking in most cases. We therefore regard this work as a stimulus to adopt the proposed formalism for other systems, perhaps in the context of different kinetic models.
Materials and Methods
Model peptides
Acetyl(Gly-Pro-Pro)10NH2 (designated (Gly-Pro-Pro)10) and acetyl(Gly-Pro-4(R)Hyp)10NH2 (designated (Gly-Pro-4(R)-Hyp)10) were synthesized by means of the Fmoc solid-phase peptide synthesis method as described previously (16,17). The chimeric peptides (Gly-Pro-Pro)10-foldon, (Gly-Pro-Pro)10-NC2(XIX), and NC2(XIX)-(Gly-Pro-Pro)10 with exogenously trimerized collagen triple helices were expressed recombinantly and purified as described previously (12,15). The amino acid sequences of these peptides are GS(GPP)10GSGYIPEAPRDGQAYVRKDGEWVLLSTFL, GS(GPP)10GIPADAVSFEEIKKYINQEVLRIFEERMAVFLSQLKLPAAMLAAQAYG, and GSPADAVSFEEIKKYINQEVLRIFEERMAVFLSQLKLPAAMLAAQAYGRP(GPP)10 for (Gly-Pro-Pro)10-foldon, (Gly-Pro-Pro)10-NC2(XIX), and NC2(XIX)-(Gly-Pro-Pro)10, respectively.
Recording of transition curves
The thermal transitions of (Gly-Pro-Pro)10, (Gly-Pro-4(R)Hyp)10, and the trimerized model peptides were measured in a circular dichroism (CD) spectrometer (model 202; Aviv, Lakewood, NJ) equipped with an electronic temperature control using a 1-mm path length quartz cell (Starna Cells, Atascadero, CA). Linearity of the temperature increase with time was achieved using appropriate settings for the temperature dead band, temperature equilibration time, and signal averaging time for a given desired rate in the Aviv software for data acquisition. Ellipticity at 225, 230, or 235 nm was measured at different peptide concentrations as a function of temperature. The peptides (Gly-Pro-Pro)10 and (Gly-Pro-4(R)Hyp)10 were measured in water. (Gly-Pro-Pro)10-foldon was measured in 10 mM phosphate buffer (pH 7.0) with 150 mM NaCl. (Gly-Pro-Pro)10-NC2(XIX) and NC2(XIX)-(Gly-Pro-Pro)10 were measured in 40 mM phosphate buffer (pH 7.0) with 135 mM NaCl. The protease inhibitor phenylmethanesulphonylfluoride (0.5 mM) was added to the recombinantly prepared samples. Sample stock solutions were stored at 4°C for 3 days or more. The diluted solutions were equilibrated at the starting temperature (TSTART) for 1–2 h. The temperature was increased at a defined rate. The unfolding profile was monitored to the state of complete unfolding of the triple helix. The cooling scan was started at a temperature (TSTARTR, where R stands for reverse; see the Supporting Material) with the same rate as the heating. For (Gly-Pro-Pro)10-foldon, the ellipticity was measured at 210 nm to avoid contributions of the conformational change of the foldon domain (12).
Fit of the hysteresis loop by model mechanisms
Sets of differential equations for all three mechanisms were iteratively solved by the MicroMath Scientist algorithm for Windows (version 2.01; MicroMath, St. Louis, MO) with the starting conditions F = 1 at t = 0 for heating, and F = 0 at t = 0 for cooling, where F is the fraction of triple helix and t is time. Ellipticities were then calculated from F and reference values for the linear temperature dependencies of ellipticities for folded and unfolded states. To allow accurate determination of their amplitudes and slopes, the measured transition profiles included sufficiently large ranges of these linear regions. For the peptides (Gly-Pro-Pro)10 and (Gly-Pro-4(R)Hyp)10, fitting for models 2 and 3 was performed by global analysis of three data sets, i.e., three concentrations at a constant temperature scanning rate or three rates at a constant peptide concentration.
Kinetic model 1 applied to (Gly-Pro-Pro)10-foldon, (Gly-Pro-Pro)10-NC2(XIX), and NC2(XIX)-(Gly-Pro-Pro)10
The foldon domain or the NC2 domain of type XIX collagen keeps the termini of the three collagenous chains together even upon unfolding. For this model (model 1, Eq. 1), we assume just two states (an all-or-none reaction): a trimeric molecule with an unfolded, C, or folded, H, triple helix. The model assumes an all-or-none reaction with two rate constants, kp for folding and kd for unfolding:
| (1) |
The differential of the concentration of the folded trimeric molecule, [H], is:
| (2) |
The fraction of triple helix is , where the total concentration is .
From Eq. 2, it follows that:
| (3) |
The temperature was calculated from the starting temperature, , the time, t, and the rate of scanning, ρ (K/s). For the reverse scan, the temperature was calculated according to , where Trev is the starting temperature of the cooling scan.
The rate constant of folding at different temperatures, kp, was calculated from the rate constant kp,7°C at a reference temperature of 7°C (280.15 K). The activation energy Ea was determined from the Arrhenius equation in which R is the gas constant:
| (4) |
Furthermore, the equilibrium constant K was calculated from the standard values of enthalpy ΔH° and entropy ΔS°:
| (5) |
and the rate constant of unfolding kd was:
| (6) |
Ellipticities at a given temperature, θ(T), were calculated from:
| (7) |
The ellipticities of the helical state (T) and the unfolded state (T) are assumed to be linearly temperature-dependent before and after the triple-helix-to-coil transition (10). Consequently,
| (8) |
| (9) |
where θn,0 and θu,0 are the amplitudes at 0°C; Sn and Su are the slopes of these dependencies; ΔH°, ΔS°, Ea, and kp at 7°C were used as fitting parameters; and Sn, Su, θn,0 and θu,0 were obtained from the linear parts of the experimental curves.
Kinetic model 2 applied to (Gly-Pro-Pro)10
In model 2, three polypeptide chains in the unfolded state, C, combine to a triple helix, H:
| (10) |
It is assumed that the kinetics is third-order for the folding direction and first-order for unfolding. Since elementary third-order reactions rarely occur, the rate constant ka,app is an apparent folding rate constant (12). The differential of [H] with respect to time t is:
| (11) |
The fraction of triple helix F is , where c0 is the total concentration of the peptide: . It follows that:
| (12) |
The apparent rate constant at temperature T was calculated with the Arrhenius equation from the apparent activation energy of the folding reaction Ea,app using a reference rate constant (7°C) at 280.15 K. The equilibrium constant K = [H]/[C]3 and the rate constant of unfolding kd were calculated in analogy to model 1. The ellipticities from the fraction of triple helix were calculated as defined in model 1. Standard values of enthalpy ΔH° and entropy ΔS°, the activation energy Ea,app, and ka,app at 7°C were used as fitting parameters.
Kinetic model 3 (preequilibrium model) applied to (Gly-Pro-Pro)10 and (Gly-Pro-4(R)Hyp)10
In model 3, three chains in the unfolded state, C, first form a trimeric nucleus, H∗, from which the triple helix is formed by propagation steps with rate constants kp and kd:
| (13) |
The fraction of triple helix is F = 3[H]/c0 and the fraction of the nucleus is F∗ = 3[H∗]/c0 with . It is assumed that H∗ is in fast equilibrium with the unfolded chains and the nucleus does not contribute to the ellipticity signal. The preequilibrium constant Q is defined as:
| (14) |
where ΔHQ° and ΔSQ° are the standard enthalpy and entropy, respectively.
It follows that:
| (15a) |
with
or
| (15b) |
where F∗ is the fraction of the trimeric nucleus H∗. F∗ is calculated by solving this cubic equation according to the method of Cardano in Bartsch (18):
| (16) |
with
| (17) |
| (18) |
The overall equilibrium constant K is defined as in model 2. The rate constant kp is calculated by the Arrhenius equation from reference values kp at 7°C and the activation energy Ea as in model 1. The rate constant of unfolding is:
| (19) |
Finally,
| (20) |
F is calculated from ellipticities as described in models 1 and 2, assuming that H∗ does not contribute to the ellipticity signal. This assumption is valid because of the very low fraction in which the intermediate occurs (see Discussion). ΔH°, ΔS°, ΔHQ°, ΔSQ°, Ea, and kp at 7°C are used as fitting parameters. The SCIENTIST software algorithms of all three model mechanisms are shown in the Supporting Material.
Relationship between models 2 and 3
Model 2 is a limiting case of model 3 for large P (small Qc02). For P >> 1, the first term in Eq. 15a can be neglected, and it follows that F∗ ≈ (1-F)3/P. Substitution of this equation into Eq. 20 leads to Eq. 12 with:
| (21) |
The temperature dependence of ka,app is given by:
It follows that:
| (22) |
Results
Hysteresis of (Gly-Pro-Pro)10 in the trimerized state
The collagenous part of (Gly-Pro-Pro)10-foldon, (Gly-Pro-Pro)10-NC2(XIX), and NC2(XIX)-(Gly-Pro-Pro)10 show detectable hysteresis only at high heating and cooling rates of ≥30°C/h. The thermal transition curves and best fits with model 1 are shown in Fig. 1. The fitting parameters are summarized in Table 1. The activation energy Ea was set to 53.5 kJ/mol, the value determined for (Gly-Pro-Pro)n folding by direct kinetic experiments (12). Inclusion of Ea as a free fitting parameter introduced problems originating from a too-large number of parameters, but resulted in a similar value. The transition enthalpies of the (Gly-Pro-Pro)10 regions in the three peptides are similar within 10%. The average value of −300 kJ · mol−1 (per mol trimer) may be compared with calorimetric values of (Gly-Pro-Pro)10 of −216 kJ · mol−1 (23) and −270 kJ · mol−1 (K. Mizuno, and H. P. Bächinger. unpublished data). The average rate constant for the three peptides is 4.2 × 10−5s−1 at 7°C. This value compares with the rate constant of 7.3 × 10−4 s−1 previously determined for (Gly-Pro-Pro)10-foldon by direct kinetic measurements that monitored ellipticity change with time after a temperature jump from 70°C to 7°C (12). The agreement between directly measured and hysteresis-derived equilibrium and kinetic values is satisfactory in view of the experimental errors in both methods.
Figure 1.
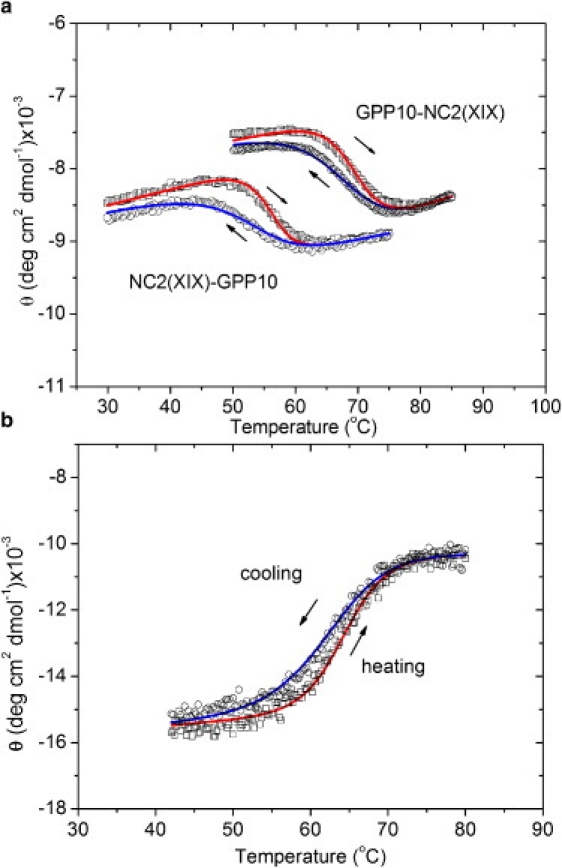
Hysteresis loops of (Gly-Pro-Pro)10-NC2(XIX), NC2(XIX)-(Gly-Pro-Pro)10, and (Gly-Pro-Pro)10-foldon. The mean molar ellipticities of heating curves are red (right arrow) and those of cooling curves are blue (left arrow), experimental values are indicated by squares (heating) and circles (cooling), and curves obtained by a best fit with model 1 are indicated as lines. Heating and cooling rates were 30°C/h. Results for (Gly-Pro-Pro)10-NC2(XIX) ((GPP)10-NC2(XIX)) and NC2(XIX)-(Gly-Pro-Pro)10 (NC2(XIX)-GPP10) are shown in panel a, and (Gly-Pro-Pro)10-foldon curves are shown in panel b.
Table 1.
Parameters of model 1 describing the hysteresis loops of trimerized peptides
| Peptide | ΔH° (kJ · mol−1) | ΔS° (J · K−1mol−1) | kp (7°C) (s−1) | Coefficient of determination (R2) |
|---|---|---|---|---|
| (Gly-Pro-Pro)10-NC2(XIX) | −310 | −915 | 3.5×10−5 | 0.9897 |
| NC2(XIX)-(Gly-Pro-Pro)10 | −304 | −939 | 3.3×10−5 | 0.9806 |
| (Gly-Pro-Pro)10-foldon | −288 | −865 | 5.8×10−5 | 0.9888 |
The activation energy Ea was fixed at 53.5 kJ · mol−1 in fits of individual hysteresis loops to avoid having too many fitting parameters. This value was confirmed by variations of Ea with the other parameters fixed. The four additional parameters Sn, θn,0, Su, and θu,0 were determined from the linear dependencies of ellipticities of the peptides in either the helical or unfolded state.
Hysteresis of (Gly-Pro-Pro)10: dependence on heating rate and chain concentration
The hysteresis loops of thermal transitions of (Gly-Pro-Pro)10 at different scanning rates and concentrations were fitted by model 2. All hysteresis loops at the same rate and three different concentrations were fitted simultaneously with the same parameter set (Fig. 2). The same type of global fit was applied to the data at constant concentration and different scanning rates (Fig. 3). Global fits were found to be more reproducible than fits of the individual loops. The fitting parameters are listed in Table 2. The fits of calculated curves to experimental data were very good in both sets of experiments, as judged by coefficients of determination (R2) > 0.9954. The good fits suggest that (Gly-Pro-Pro)10 folding can be interpreted as apparent third-order kinetics under the limited peptide concentration range used in our experiments. In a model using multiple fitting parameters, a good value of the coefficient of determination (R2) does not prove that the model is correct. In our case, the model is supported by the good correspondence of fitted parameters with values obtained by direct kinetic determination.
Figure 2.
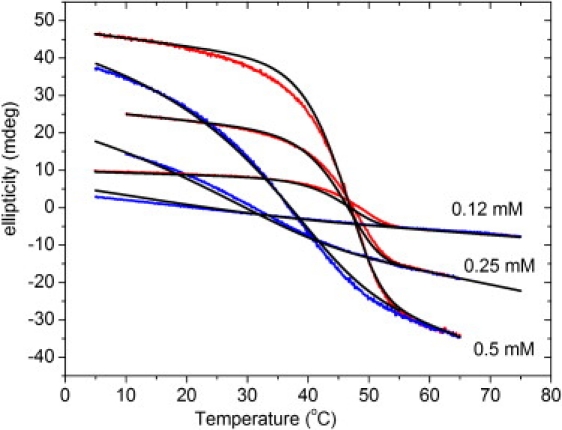
Fitting of the hysteresis loops of peptide (Gly-Pro-Pro)10 at three different concentrations. Data for the three concentrations were fitted simultaneously (see text). The ellipticity was recorded at a heating or cooling rate of 10°C/h with different concentrations (0.12 mM, 0.25 mM, and 0.55 mM). The experimental heating and cooling curves are plotted in red (light gray) and blue (dark gray), respectively. The curves fitted by model 2 are indicated as black lines.
Figure 3.

Fitting of the hysteresis of peptide (Gly-Pro-Pro)10 at different scanning rates. Data for the three rates were fitted simultaneously (see text). Mean molar ellipticities θ were recorded as a function of temperature at c0 = 0.25 mM and scanning rates of 2, 5, and 10°C/h. The experimental heating and cooling curves are plotted in red (light gray) and blue (dark gray), respectively. The curves fitted by model 2 are indicated as black lines.
Table 2.
(A) Parameters of model 2 for the hysteresis of (Gly-Pro-Pro)10 at different total chain concentrations and constant scanning rate at 10°C/h
| c0 (mM) | ΔH° (kJ · mol−1) | ΔS° (J · K−1mol−1) | ka,app (7°C) (M−2s−1) | Ea,app (kJ · mol−1) | Coefficient of determination (R2) |
|---|---|---|---|---|---|
| 0.12 | 0.9954 | ||||
| 0.25 | 0.9996 | ||||
| 0.5 | 0.9997 | ||||
| Global fit | −229 | −654 | 4.7 × 103 | −60.1 | 0.9977 |
|
(B) Parameters of model 2 for the hysteresis of (Gly-Pro-Pro)10 at different scanning rates and constant chain concentration | |||||
|---|---|---|---|---|---|
| Scanning rate (°C/h) | ΔH° (kJ · mol−1) | ΔS° (J · K−1mol−1) | ka,app (7°C) (M−2s−1) | Ea,app (kJ · mol−1) | Coefficient of determination (R2) |
| 10 | 0.9994 | ||||
| 5 | 0.9995 | ||||
| 2.5 | 0.9995 | ||||
| Global fit | −242 | −644 | 6.7 × 103 | −60.1 | 0.9995 |
An independent variation of all four fitting parameters ΔH°, ΔS°, ka, and Ea was possible for this model. The four additional fitting parameters Sn, θn,0, Su, and θu,0 were fitted from the linear dependencies of ellipticities of the peptides in either the helical or unfolded state.
The average value of ka,app from experiments at constant rate and constant concentration is 5.7 103 M−2s−1. This is six times larger than the value of 0.9 103 M−2s−1 obtained by direct kinetic measurements. Hysteresis loops of the (Gly-Pro-4(R)Hyp)10 could not be fitted with model 2 (data not shown), implying that the apparent reaction order of folding is significantly different from third-order, or that the mechanism is much more complicated. As shown further below, fits with reasonable parameter values are obtained with model 3, in which a fast preequilibration is followed by a first-order propagation.
The dependency of the midpoint temperature of the transition (Tm) at various scanning rates and a chain concentration of 0.25 mM is shown in Fig. 4. The Tm was calculated according to model 2 using the average values of the parameters listed in Table 2. The approach to equilibrium with decreasing rates below 1°C/h can be clearly seen.
Figure 4.
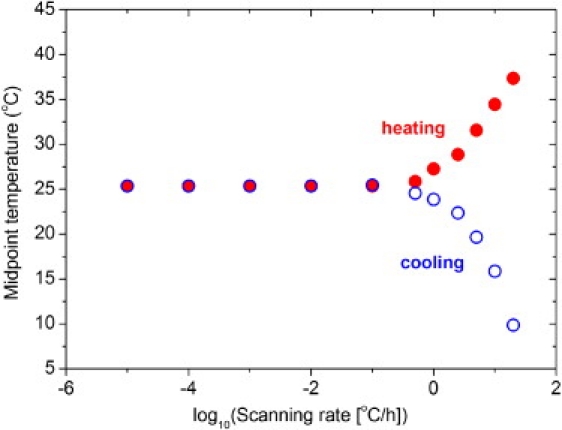
Calculated midpoint temperatures of the transition curves for (Gly-Pro-Pro)10 at constant total peptide concentration c0 = 0.25 mM in a forward (heating, solid circle) or backward (cooling, open circle) direction at different heating and cooling rates. Calculations were performed with model 2 and the average parameters of Table 2, that is, ΔH° = −235.5 kJ/mol, ΔS° = −649 Jmol−1K−1, Ea,app = −60.1 kJ/mol, and ka,app = 5.7 × 102M−2s−1.
Fits with a fast preequilibrium mechanism: model 3
In model 2, the apparent rate constant ka,app and apparent activation energy Ea,app are overall parameters that do not reflect the rate constants of elementary reactions. Furthermore, model 2 was not able to fit the scanning curves of the 4(R) Hyp-containing peptide. For these reasons, the preequilibrium model (model 3) was applied. This model is based on folding kinetic measurements on (Gly-Pro-Pro)10 and (Gly-Pro-4(R)Hyp)10 (10,12,19). A model mechanism was proposed assuming that propagation is preceded by the formation of an intermediate, in which the three chains combine first to an intermediate trimer. The enthalpy and entropy of the precomplex formation were termed ΔH°Q and ΔS°Q. These thermodynamic parameters determine the magnitude of the preequilibrium constant Q. If the equilibrium is on the side of free chains (fraction of the precomplex F∗ close to 0), the apparent order of the total folding reaction can be approximated by third order. If H∗ is dominating (F∗ near to 1), it will approximate first order. Model 3 can therefore describe different apparent reaction orders, which were previously observed by direct kinetic measurements (12). A problem with model 3 is the larger number of parameters compared to model 2. For (Gly-Pro-Pro)10, both models yield reasonable results. A comparison of the global fits is shown in Fig. 5.
Figure 5.
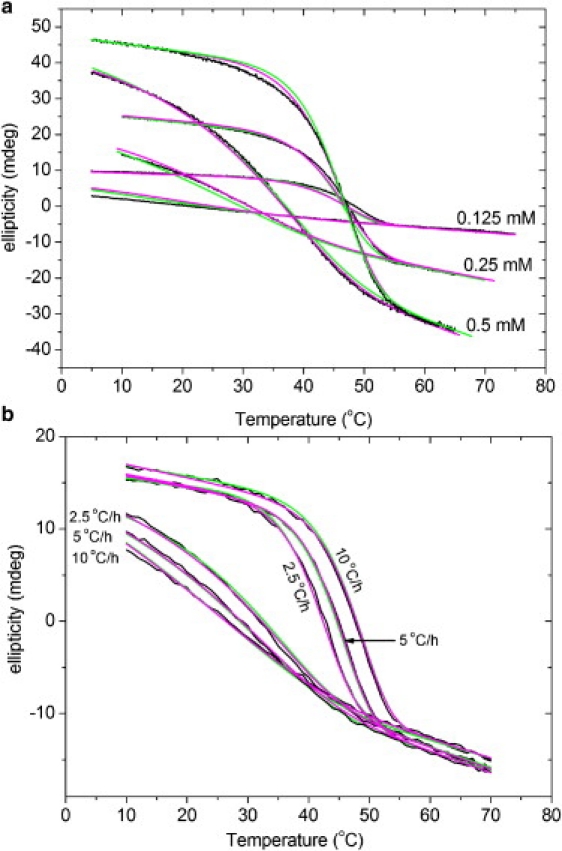
Fitting of (Gly-Pro-Pro)10 hysteresis loops with models 2 and 3 at different peptide concentrations (a) and scanning rates (b). The experimental data and fitting curves with models 2 and 3 are indicated in black, green (light gray), and magenta (dark gray), respectively. The scanning rate was constant at 10°C/h in panel a, and the peptide concentration was constant at 0.2 mM in panel b.
The fits with model 3 yield reasonable results for the different peptides. Representative global fits are shown in Fig. 6. We analyzed the peptides (Gly-Pro-Pro)10 and (Gly-Pro-4(R)Hyp-10 with scanning rates varied from 2.5 to 10°C/h, and concentrations varied from 0.1 to 0.5 mM. The fitting parameters are summarized in Table 3. We used the previously reported (12) value of 53.5 kJ · mol−1 for Ea. This value was determined by direct kinetic measurements and is more reliable than values obtained by fitting of the hysteresis loops, where the temperature dependence is determined not only by Ea but also by the change of equilibrium. Nevertheless, Ea values between 50 and 60 kJ/mol were also obtained by fits, keeping all other parameters fixed.
Figure 6.
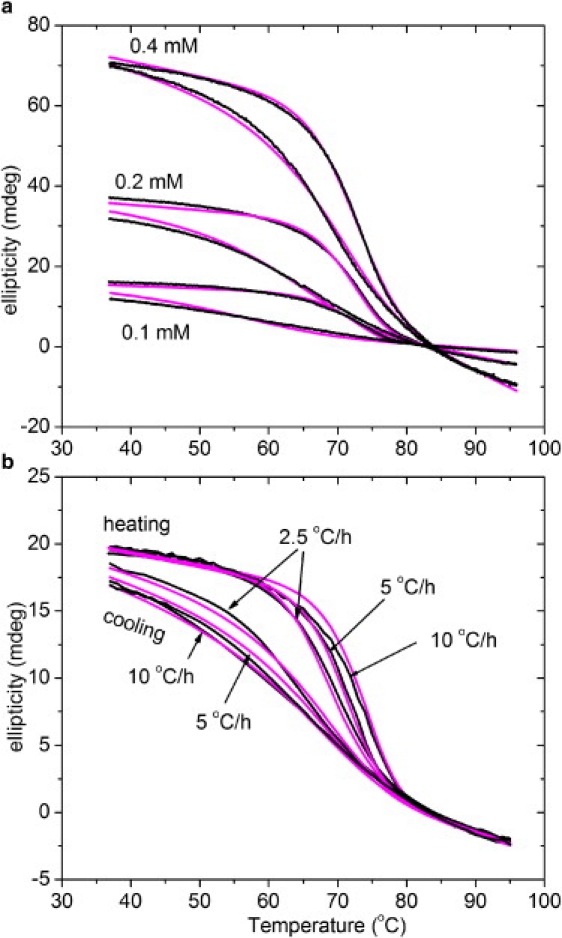
Fitting of hysteresis loops of (Gly-Pro-4(R)Hyp)10 with model 3. (a) Effect of concentration with constant scanning rate of 10°C/h. (b) Effect of scanning rate with constant peptide concentration of 0.1 mM. The experimental data and fitted results are indicated by black and magenta (light gray) lines, respectively.
Table 3.
Parameters of model 3 describing the hysteresis of (Gly-Pro-4(R)Hyp)10, and (Gly-Pro-Pro)10
| Peptide | c0 (mM) | rate (°C/h) | ΔH° (kJ · mol−1) | ΔS° (J · mol−1K−1) | ΔG° (kJ · mol−1) | ΔHQ° (kJ · mol−1) | ΔSQ° (J · mol−1K−1) | ΔGQ° (kJ · mol−1) | kp (7°C) (s−1) | Coefficient of determination (R2) |
|---|---|---|---|---|---|---|---|---|---|---|
| (Gly-Pro-Pro)10 | 0.125 | 10 | 0.9956 | |||||||
| (Gly-Pro-Pro)10 | 0.25 | 10 | 0.9982 | |||||||
| (Gly-Pro-Pro)10 | 0.5 | 10 | 0.9983 | |||||||
| (Gly-Pro-Pro)10 | 10 | −262 | −711 | −51 | −128 | −309 | −36 | 2.9×10−4 | 0.9978 | |
| (Gly-Pro-Pro)10 | 0.2 | 10 | 0.9995 | |||||||
| (Gly-Pro-Pro)10 | 0.2 | 5 | 0.9994 | |||||||
| (Gly-Pro-Pro)10 | 0.2 | 2.5 | 0.9995 | |||||||
| (Gly-Pro-Pro)10 | 0.2 | −258 | −695 | −51 | −116 | −280 | −32 | 6.7×10−4 | 0.9995 | |
| (Gly-Pro-4(R)Hyp)10 | 0.1 | 10 | 0.9898 | |||||||
| (Gly-Pro-4(R)Hyp)10 | 0.2 | 10 | 0.9969 | |||||||
| (Gly-Pro-4(R)Hyp)10 | 0.4 | 10 | 0.9992 | |||||||
| (Gly-Pro-4(R)Hyp)10 | 10 | −360 | −923 | −86 | −172 | −417 | −47 | 1.2×10−3 | 0.9989 | |
| (Gly-Pro-4(R)Hyp)10 | 0.1 | 2.5 | 0.9957 | |||||||
| (Gly-Pro-4(R)Hyp)10 | 0.1 | 5 | 0.9960 | |||||||
| (Gly-Pro-4(R)Hyp)10 | 0.1 | 10 | 0.9991 | |||||||
| (Gly-Pro-4(R)Hyp)10 | 0.1 | −344 | −861 | −88 | −159 | −361 | −52 | 3.0×10−4 | 0.9968 | |
The fitting parameters are ΔH°, ΔS°, ΔHQ°, ΔSQ°, Ea, and kp (7°C). Ea was fixed to 53.5 kJ · mol−1. The values of ΔG° and ΔGQ° were calculated from and , with T = 298.15 K. The four additional fitting parameters Sn, θn,0, Su, and θu,0 were fitted from the linear dependencies of ellipticities of the peptides in either the helical or unfolded state.
The average rate constants derived from measurements at constant rate and constant concentration are 4.8 10−4 s−1 for (Gly-Pro-Pro)10 and 7.5 10−4 s−1 for (Gly-Pro-4(R)Hyp)10 (Table 3), which implies that the rate of propagation is only 1.6-fold accelerated by hydroxyproline. The rate constants of propagation kp in model 3 correspond reasonably well with previously measured rate constants of propagation. The average value for (Gly-Pro-Pro)10 of kp = 4.8 10−4 s−1 is 11 times larger than the value obtained from the hysteresis of (Gly-Pro-Pro)10 peptides trimerized by linker domains (see Table 1) and 1.5 times smaller than directly measured values for trimerized peptides, kp = 7 10−4 s−1 (10,15). This agreement indicates that it is reasonable to analyze the hysteresis of the collagen-like peptides by applying a preequilibrium mechanism with an intermediate.
The preequilibrium constant Q follows from Eq. 14 for ΔHQ° and ΔSQ° in Table 3. Values of Q at 7°C are 3 107 M−2 for (Gly-Pro-Pro)10 and 4.5 1010 M−2 for (Gly-Pro-4(R)Hyp)10. It follows from Eq. 21 that the apparent rate constants are approximately ka,app = Q kp. Values calculated by this equation are ka,app = 1.4 104 M−2s−1 and 3.4 107 M−2s−1 for the two peptides. The calculated value for (Gly-Pro-Pro)10 is two times larger than the value obtained by model 2. For (Gly-Pro-4(R)Hyp)10, model 2 was not applicable. However, a value of ka,app > 106 M−2s−1 was estimated from direct kinetic experiments (10).
Of interest, the ∼2000-fold faster overall folding rate at 7°C of (Gly-Pro-4(R)Hyp)10 as compared to (Gly-Pro-Pro)10 originates from a more stable preequilibrium complex, whereas the rates of propagation, kp, are about the same. It is also expected according to Eq. 22 that the apparent activation energy Ea,app is the sum of ΔHQ and Ea, the activation energy of propagation. A comparison of values in Table 2 with those in Table 3 shows that this approximation is fulfilled for the fitting parameters. For example, for (Gly-Pro-Pro)10, ΔHQ + Ea = −68.5 kJ · mol−1 and Ea,app = −60.1 kJ · mol−1.
The temperature dependence of the fraction of H∗ (F∗) is plotted for (Gly-Pro-4(R)Hyp)10 and (Gly-Pro-Pro)10 in Fig. 7. The fractions of H∗ are small in all cases, justifying the assumption that H∗ does not contribute to the CD signal.
Figure 7.
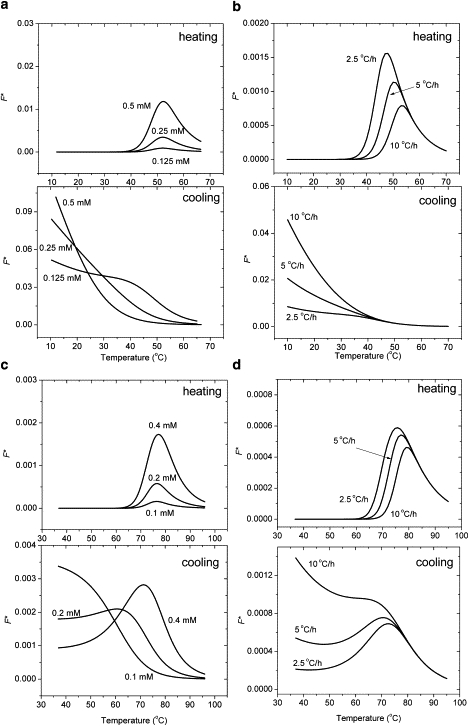
Calculated fractions F∗ of the intermediate H∗ during heating and cooling of (Gly-Pro-Pro)10 (a and b) and (Gly-Pro-4(R)Hyp)10 (c and d) with model 3. The upper and lower panels show the F∗ of heating and cooling scans, respectively, at a scanning rate of 10°C/h as predicted by model 3. The peptide concentration is c0 = 0.2 mM in panel a, and c0 = 0.1 mM in panel c. The scanning rate is 10°C/h in panels b and d.
Comparison with calorimetric data
Published data for the van't Hoff enthalpy obtained from thermal transition curves range from −224 to −526 kJ · mol−1 for (Pro-Pro-Gly)10, and from −375 to −652 kJ · mol−1 for (Gly-Pro-4(R)Hyp)10 (10). The large variations are probably explained by concentration errors, baseline errors, and nonequilibrium conditions originating from too-fast scanning. The data derived here from the hysteresis loops can be considered to be more accurate. Within 15% deviations, these data agree with previously published values derived from scanning differential calorimetry (Table 4).
Table 4.
Comparison of enthalpy values from hysteresis loops and published enthalpy values derived from differential scanning calorimetry
| Peptide | ΔH° by model 2 (kJ · mol−1) | ΔH° by model 3 (kJ · mol−1) | ΔHcal° (kJ · mol−1) |
|---|---|---|---|
| (Gly-Pro-Pro)10 | −236 | −260 | −215 (24)∗−270 (K. Mizuno, and H. P. Bächinger, unpublished data) |
| (Gly-Pro-4(R)Hyp)10 | not determined | −352 | −337 (17)−367 (24)† |
Measured with H-(Pro-Pro-Gly)10-OH.
Measured with H-(Pro-4(R)Hyp-Gly)10-OH.
Discussion
The aim of this work was to provide a quantitative description of the hysteresis of the collagen triple helix, which up to now have only been explored qualitatively. This was achieved for trimerized collagen-like peptides and for peptides in which three separated chains assemble to a triple helix. Good fits of the hysteresis loops were obtained by integrating differential equations for a given mechanism and the same set of parameters for both heating and cooling. There was no need to assume different mechanisms for heating and cooling transitions. This confirms an earlier suggestion that the hysteresis is exclusively controlled kinetically (10,11). The extent of hysteresis as measured by the difference in apparent midpoint temperatures in the forward and backward reaction depends on the difference between the rate of temperature change and the rate at which the system follows the external change. For example, to obtain equilibrium transition curves for (Gly-Pro-Pro)10 at a total chain concentration of 0.25 mM, unfolding profiles should be measured at a rate of <0.1°C/h. The 4(R)Hyp-containing peptides equilibrate faster at the same concentration. Persikov et al. (11) obtained related information by following the kinetics at a constant temperature from the unfolded as well as the folded state.
The risk of determining nonequilibrium transitions is high at very low concentrations. At chain concentrations < 0.1 mM, unlinked (Gly-Pro-Pro)10 chains may stay in the unfolded state for a long time even under conditions in which the triple helix is formed readily under high peptide concentrations, because of the strongly concentration-dependent hysteresis. In many publications, however, information on the rate of heating and concentration is lacking, and the state in which the peptides were studied remains unclear.
It is important to note that all simple models on collagen folding include simplifications. Simplifying assumptions include the all-or-none nature of the transitions, the assumption of only a single nucleation event of folding, and the exclusion of wrong products, possibly with mismatched chains. More sophisticated models that include these effects suffer from large parameter sets, which make fitting unstable or impossible. Therefore, the goal is to find a simple mechanism that describes the data reasonably well.
In this work, we applied an all-or-none mechanism for the transition of trimerized model peptides. Of more critical importance are the transitions from separated chains, which first have to meet and form a trimerized complex. Here, we applied the most simple model—model 2—because this model is frequently employed for collagenous peptides. Association rate constants defined by model 2 have been published for (Gly-Pro-Pro)10, (Gly-Pro-4(R)Hyp)10 (12), and other collagenous model peptides (10). They are apparent rate constants because third-order reactions are highly unlikely to occur in solution.
Model 3 describes the complex formation and folding of the triple helix in separate steps. It is assumed that the formation of the precomplex is a prerequisite for the following propagation. This assumption is in agreement with the propagation rates measured for the triple-helix propagation of collagen model peptides with crosslinks (12,21). It should be emphasized that models 2 and 3 do not describe different mechanisms; model 3 just adds a special assumption for the complex formation. With this assumption, the apparent rate constant of association in model 2 becomes the product of the equilibrium constant of complex formation and the propagation rate constant of folding.
Of interest, we found that the equilibrium constant of complex formation Q at 7°C is ∼1500-fold higher for (Gly-Pro-4(R)Hyp)10 than for (Gly-Pro-Pro)10, whereas the rate constant of helix propagation was about the same (∼1.6 times higher for (Gly-Pro-4(R)Hyp)10). Similar conclusions and a similar ratio between equilibrium constants of precomplex formation were obtained by direct kinetic measurements of the two peptides (12). The intermediate trimer acts as a nucleus from which the triple helix is formed by propagation steps with rate constants and activation energies similar to those observed for propagation of exogenously trimerized model peptides. In the case of folding of three chains without a trimerization domain, earlier kinetic results (10,12) and our analysis strongly suggest that the formation of the nucleus is the rate-limiting step under typical temperature-scanning experiments with peptide concentrations < 1 mM. The fraction of the nucleus during the folding and unfolding reactions is not larger than 0.1 (Fig. 7). The fits of hysteresis loops are therefore insensitive to assumptions about the CD signals provided by the nucleus. No signal from the intermediate was assumed to reduce the number of fitting parameters, but other values could also be used without affecting the results. In turn, no information on the size of the nucleus could be obtained from the results. Independent evidence indicates that the nucleus consists of 10 tripeptide units (three per chain) in a helical state (21), suggesting a maximal contribution of ∼3% to the CD signal at low temperatures and less in the transition region.
It has been known for a long time that hydroxyproline in the Yaa position increases the equilibrium stability of the collagen triple helix (10). This also follows from the thermodynamic values obtained from the hysteresis experiments (Table 3). The stabilizing effect can now be explained in part by an increased association potential in the precomplex. The formation of a trimeric complex is consistent with the observation that the single-chain peptide (Gly-Pro-4(R)Hyp)5 has a negative second virial coefficient at 15°C, indicating a tendency of the Gly-Pro-4(R)Hyp sequences to interact with each other, even though they are not long enough to form a triple helix (20).
The rate of triple-helix propagation is known to be determined by the slow cis-trans isomerization steps of peptide bond preceding proline or hydroxyproline (8–10). Its activation energy is high because of the high activation energy of individual cis-trans isomerizations of ∼80 kJ/mol (10). Lower values were observed for the two collagen model peptides, and the most reliable value of 53.5 kJ/mol determined for oxidized (Gly-Pro-Pro)11-Gly-Pro-Cys-Cys-Gly3 and (Gly-Pro-Pro)10-foldon (12) was used in this study.
The mechanism of triple-helix folding is still under investigation, and several somewhat controversial features have to be clarified. Recently, a kinetic mechanism featuring an all-trans peptide bond unfolded nucleus and a third-order rate constant was described for the T1 peptide (22). The nucleus in this peptide consisted of a (Gly-Pro-4(R)Hyp)4 sequence, and the rate-limiting step was the acquisition of an all-trans unfolded chain that then trimerized in a faster third-order reaction. Although we have no structural information about H∗, it seems unlikely that it consists of an all-trans peptide bond chain segment. If the acquisition of an all-trans chain or portion of a chain were rate-limiting, we would not observe a 2000-fold difference between the proline- and hydroxyproline-containing peptides, and the hydroxyproline-containing peptide could be fitted with model 2. The difference in cis content between the Gly-Pro-Pro and Gly-Pro-4(R)Hyp sequences is only ∼3% (23) (unpublished data). It would be interesting to study the T1 peptide with a Gly-Pro-Pro nucleus to test this model. A small decrease in the rate of nucleation should then be observed. Further studies are required to identify the structure of H∗.
The main lesson to be learned from this work can be summarized as follows: Kinetically controlled hysteresis can be described quantitatively by assuming different starting conditions for the rate equations. The formalism described here can be applied to any similar mechanism applicable to a system of interest. The results of our experiments with collagen model peptides show that one can derive useful new kinetic and equilibrium information from such a system. As described in the Introduction, many interesting biological systems show hysteresis, and a quantitative evaluation may yield valuable information. To help the reader perform such an analysis, the programs used in the MicroMath algorithm are described in the Supporting Material.
Supporting Material
An appendix is available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(10)00351-6.
Supporting Material
Acknowledgments
The authors thank Dr. Michael Kümin for a critical reading of the manuscript. The authors thank Jessica L. Hacker, Dr. B. Kerry Maddox, and Jesse M. Vance for expert technical assistance in peptide synthesis and amino acid analyses.
This work was supported by a grant from Shriners Hospital for Children (to H.P.B.).
References
- 1.Mergny J.L., Lacroix L. Kinetics and thermodynamics of i-DNA formation: phosphodiester versus modified oligodeoxynucleotides. Nucleic Acids Res. 1998;26:4797–4803. doi: 10.1093/nar/26.21.4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lai Z., McCulloch J., Kelly J.W. Guanidine hydrochloride-induced denaturation and refolding of transthyretin exhibits a marked hysteresis: equilibria with high kinetic barriers. Biochemistry. 1997;36:10230–10239. doi: 10.1021/bi963195p. [DOI] [PubMed] [Google Scholar]
- 3.Singh S., Zlotnick A. Observed hysteresis of virus capsid disassembly is implicit in kinetic models of assembly. J. Biol. Chem. 2003;278:18249–18255. doi: 10.1074/jbc.M211408200. [DOI] [PubMed] [Google Scholar]
- 4.Fasshauer D., Antonin W., Jahn R. SNARE assembly and disassembly exhibit a pronounced hysteresis. Nat. Struct. Biol. 2002;9:144–151. doi: 10.1038/nsb750. [DOI] [PubMed] [Google Scholar]
- 5.Gursky O., Ranjana, Gantz D.L. Complex of human apolipoprotein C-1 with phospholipid: thermodynamic or kinetic stability? Biochemistry. 2002;41:7373–7384. doi: 10.1021/bi025588w. [DOI] [PubMed] [Google Scholar]
- 6.Davis J.M., Bächinger H.P. Hysteresis in the triple helix-coil transition of type III collagen. J. Biol. Chem. 1993;268:25965–25972. [PubMed] [Google Scholar]
- 7.Engel J., Bächinger H.P. Cooperative equilibrium transitions coupled with a slow annealing step explain the sharpness and hysteresis of collagen folding. Matrix Biol. 2000;19:235–244. doi: 10.1016/s0945-053x(00)00071-8. [DOI] [PubMed] [Google Scholar]
- 8.Bächinger H.P., Bruckner P., Engel J. Folding mechanism of the triple helix in type-III collagen and type-III pN-collagen. Role of disulfide bridges and peptide bond isomerization. Eur. J. Biochem. 1980;106:619–632. doi: 10.1111/j.1432-1033.1980.tb04610.x. [DOI] [PubMed] [Google Scholar]
- 9.Engel J., Prockop D.J. The zipper-like folding of collagen triple helices and the effects of mutations that disrupt the zipper. Annu. Rev. Biophys. Biophys. Chem. 1991;20:137–152. doi: 10.1146/annurev.bb.20.060191.001033. [DOI] [PubMed] [Google Scholar]
- 10.Bächinger H.P., Engel J. The thermodynamics and kinetics of collagen folding. In: Buchner J., Kiefhaber T., editors. Protein Folding Handbook. Wiley; New York: 2005. pp. 1059–1110. [Google Scholar]
- 11.Persikov A.V., Xu Y., Brodsky B. Equilibrium thermal transitions of collagen model peptides. Protein Sci. 2004;13:893–902. doi: 10.1110/ps.03501704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boudko S., Frank S., Engel J. Nucleation and propagation of the collagen triple helix in single-chain and trimerized peptides: transition from third to first order kinetics. J. Mol. Biol. 2002;317:459–470. doi: 10.1006/jmbi.2002.5439. [DOI] [PubMed] [Google Scholar]
- 13.Frank S., Boudko S., Bächinger H.P. Collagen triple helix formation can be nucleated at either end. J. Biol. Chem. 2003;278:7747–7750. doi: 10.1074/jbc.C200698200. [DOI] [PubMed] [Google Scholar]
- 14.Frank S., Kammerer R.A., Engel J. Stabilization of short collagen-like triple helices by protein engineering. J. Mol. Biol. 2001;308:1081–1089. doi: 10.1006/jmbi.2001.4644. [DOI] [PubMed] [Google Scholar]
- 15.Boudko S.P., Engel J., Bächinger H.P. Trimerization and triple helix stabilization of the collagen XIX NC2 domain. J. Biol. Chem. 2008;283:34345–34351. doi: 10.1074/jbc.M806352200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mizuno K., Hayashi T., Bächinger H.P. Hydroxylation-induced stabilization of the collagen triple helix. Acetyl-(glycyl-4(R)-hydroxyprolyl-4(R)-hydroxyprolyl)(10)-NH(2) forms a highly stable triple helix. J. Biol. Chem. 2004;279:38072–38078. doi: 10.1074/jbc.M402953200. [DOI] [PubMed] [Google Scholar]
- 17.Mizuno K., Peyton D.H., Bächinger H.P. Effect of the -Gly-3(S)-hydroxyprolyl-4(R)-hydroxyprolyl- tripeptide unit on the stability of collagen model peptides. FEBS J. 2008;275:5830–5840. doi: 10.1111/j.1742-4658.2008.06704.x. [DOI] [PubMed] [Google Scholar]
- 18.Bartsch H.-J. Taschenbuch mathematischer Formeln. Verlag Harri Deutsch; Thun, Germany: 1982. Potenzen und Wurzeln; p. 66. [Google Scholar]
- 19.Engel J., Bächinger H.P. Stability and folding of the collagen triple helix. In: Brinckmann J., Notbohm H., Müller P.K., editors. Topics in Current Chemistry. Springer; Berlin, Germany: 2005. pp. 7–34. [Google Scholar]
- 20.Terao K., Mizuno K., Bächinger H.P. Chain dimensions and hydration behavior of collagen model peptides in aqueous solution: [Glycyl-4(R)-hydroxyprolyl-4(R)-hydroxyproline]n, [Glycylprolyl-4(R)-hydroxyproline]n, and some related model peptides. Biomacromolecules. 2008;41:7203–7210. [Google Scholar]
- 21.Bachmann A., Kiefhaber T., Bächinger H.P. Collagen triple-helix formation in all-trans chains proceeds by a nucleation/growth mechanism with a purely entropic barrier. Proc. Natl. Acad. Sci. USA. 2005;102:13897–13902. doi: 10.1073/pnas.0505141102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu Y., Bhate M., Brodsky B. Characterization of the nucleation step and folding of a collagen triple-helix peptide. Biochemistry. 2002;41:8143–8151. doi: 10.1021/bi015952b. [DOI] [PubMed] [Google Scholar]
- 23.Holmgren S.K., Bretscher L.E., Raines R.T. A hyperstable collagen mimic. Chem. Biol. 1999;6:63–70. doi: 10.1016/S1074-5521(99)80003-9. [DOI] [PubMed] [Google Scholar]
- 24.Engel J., Chen H.T., Klump H. The triple helix in equilibrium with coil conversion of collagen-like polytripeptides in aqueous and nonaqueous solvents. Comparison of the thermodynamic parameters and the binding of water to (L-Pro-L-Pro-Gly)n and (L-Pro-L-Hyp-Gly)n. Biopolymers. 1977;16:601–622. doi: 10.1002/bip.1977.360160310. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.