

Abstract
Sleep deprivation can impair human health and performance. Habitual total sleep time and homeostatic sleep response to sleep deprivation are quantitative traits in humans. Genetic loci for these traits have been identified in model organisms, but none of these potential animal models have a corresponding human genotype and phenotype. We have identified a mutation in a transcriptional repressor (hDEC2-P385R) that is associated with a human short sleep phenotype. Activity profiles and sleep recordings of transgenic mice carrying this mutation showed increased vigilance time and less sleep time than control mice in a zeitgeber time– and sleep deprivation–dependent manner. These mice represent a model of human sleep homeostasis that provides an opportunity to probe the effect of sleep on human physical and mental health.
Although sleep is an essential process for life, the brain circuits regulating sleep and the cellular and/or molecular mechanisms involved in this complex process are still enigmatic (1–3). Sleep or a “sleeplike” behavior is present in virtually every animal species where it has been studied. Total sleep deprivation can be fatal, and partial deprivation of sleep has serious consequences on cognition, mood, and health (4–6). It is obvious that situational increases in behavioral drive can transiently delay sleep, but very little is known about chronic partial sleep curtailment as a possible consequence of a persistent elevation in waking behavioral drive. The latter trait, sometimes referred to as a “hyperthymic” temperament (7), is a theoretical third influence on sleep habits.
Murine Dec2 (mDec2) is a negative component of the circadian clock (8–10). It belongs to a basic helix-loop-helix (bHLH) protein family in which members can dimerize with each other and can affect gene transcription by binding to specific DNA sequences (11). While performing candidate gene resequencing in DNAs from human families, segregating alleles for extremely early wake up times, we identified an hDEC2 point mutation in a small family with two affected individuals (Fig. 1A) (12). Subjects carrying this mutation had lifelong shorter daily sleep times than normal individuals (Table 1). The self-reported nonworkday habitual sleep-offset times of the mutation carriers were much earlier than those of the noncarriers (including noncarrier family members and general controls). However, these two individuals have sleep-onset times that are similar to that of conventional sleepers. The habitual self-reported total sleep time per 24-hour day was much shorter in mutation carriers (average 6.25 hours) compared with the noncarriers (average 8.06 hours) in this family. Thus, they represent “natural short sleepers” who routinely sleep less than individuals with familial advanced sleep-phase syndrome (FASPS) or general controls (Table 1). The average total sleep time for American adults on nonworkdays is ∼7.4 hours (www.sleepfoundation.org). The mutation changes a C to G in the DNA sequence of DEC2, which is predicted to cause a proline-to-arginine alteration at amino acid position 385 of DEC2 (Fig. 1B). This change was not found in over 250 control DNA samples. The proline at position 385 of DEC2 (P385) is conserved in mammals but not invertebrates. P385 is located in a highly conserved region within a proline-rich domain of unknown function and is close to the C-terminal histone deacetylase (HDAC)–interacting region of DEC2 (Fig. 1B). Activity-rest recording in one mutation carrier using 10-day sleep logs with coincident wrist actigraphy demonstrated an extended active period each day (Fig. 1C).
Fig. 1.
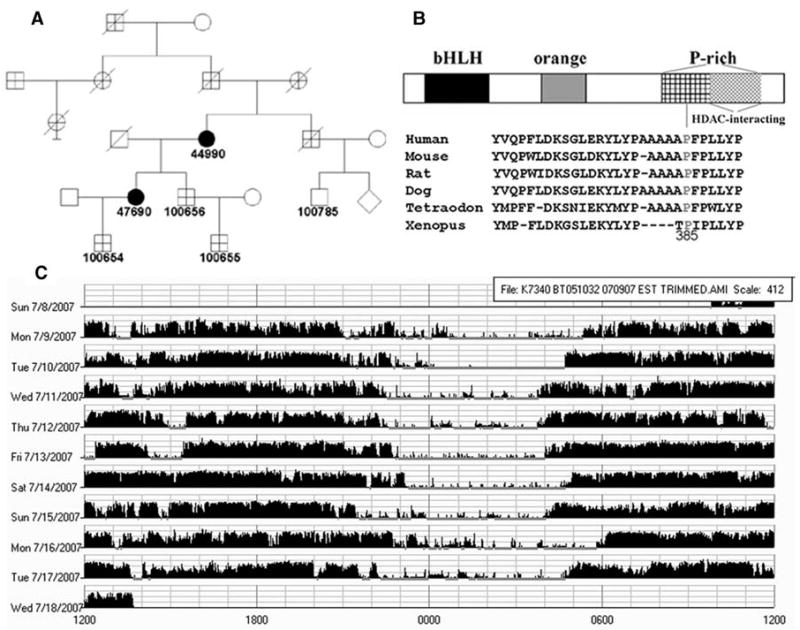
A DEC2 point mutation was identified in a short sleep family. (A) Pedigree of K7430 family carrying DEC2 mutation (P385R). (B) P385 is localized in the C-terminal proline-rich domain and its flanking sequences are highly conserved among mammalian DEC2 orthologs. (C) Activity recording by wrist actigraphy for one mutation carrier demonstrates the extended active period each day.
Table 1.
Sleep schedule comparison for human subjects. Age refers to when data were collected. Status: C, mutation carrier; NC, nonmutation carrier. Sleep offset is local standard clock time of “average” final morning awakening, and sleep onset is evening time of first falling asleep as stated by individuals recalling extended vacations based on structured interviews. Values are ± SD.
| Subjects | Age | Status | Sleep offset | Sleep onset | Sleep length (hour) |
|---|---|---|---|---|---|
| 44990 | 69 | C | 4:00 | 22:00 | 6.0 |
| 47690 | 44 | C | 4:30 | 22:00 | 6.5 |
| 101174 | 51 | NC | 6:00 | 22:35 | 7.4 |
| 100785 | 51 | NC | 7:00 | 22:45 | 8.3 |
| 100656 | 44 | NC | 5:00 | 21:30 | 7.5 |
| 100654 | 16 | NC | 7:45 | 24:00 | 7.7 |
| 100655 | 10 | NC | 7:00 | 21:35 | 9.4 |
| FASPS | 4:30 ± 1.33 | 19:45 ± 1.33 | 8.66 ± 0.80 (n = 16) |
||
| Control | 6:12 ± 2.45 | 21:50 ± 1.76 | 8.37 ± 1.67 (n = 15) |
To examine the effect of the P385R mutation on Dec2 repressor activity, a wild-type (WT) or a P385R mDec2 construct was used in a luciferase assay, and the results showed that P385R attenuated Dec2 repressive activity of Clk/Bmal1-mediated transactivation (fig. S1A). The reduction in Dec2 repressive activity was moderate compared with that of the R57A/K mutations (in which arginine 57 was replaced by alanine or lysine) reported before (13). Dec2 was previously shown to preferentially bind to class B E-box elements (CACGTG) as a homodimer and to repress the transcription of target genes in an HDAC-dependent manner (13). The effect of HDAC on the mutant Dec2 repression was then analyzed by monitoring mPer2 promoter-driven luciferase activity with or without a general HDAC inhibitor trichostatin A (TSA) (fig. S1B). HDAC inhibition resulted in similar increases in luciferase activity for both WT and mutant Dec2. Coimmunoprecipitation was then performed for mDec2 (WT or P385R) and human sirtuin-1 (hSIRT1). HEK293 cells were transiently cotransfected with green fluorescent protein (GFP)–tagged (WT or mutated) mDec2 and FLAG-tagged hSIRT1, followed by FLAG-peptide pull-down and detection of GFP with antibodies on Western blots. The results showed similar physical interactions between WT or P385R mDec2 and hSIRT1 (fig. S1C). Taken together, these results suggest that the P385R mutation affects Dec2 transcriptional repression activity independently from its interaction with HDAC/SIRT.
Because there are only two human mutation carriers in this study, the question remained whether the natural short sleep phenotype was caused by the DEC2 mutation. Thus, we generated WT and P385R DEC2 transgenic (Tg) mice using a human bacterial artificial chromosome (BAC) clone (RP11-288E19) carrying the entire hDEC2 gene to test this hypothesis. As DEC2 has been established as a component of circadian clock (9, 14), we first set out to determine the circadian period (τ) of DEC2-P385R mice. Mice with Dec2 deleted [knockout (KO) mice] (10) and WT littermates were tested in parallel as controls. No significant differences in τ were detected among mice of different genotypes (table S1).
Because the mutation was identified in human short sleepers who, presumably, have correspondingly longer total daily activity periods, we next determined the duration of the activity period (α) for these mice. DEC2-P385R mice retained the WT pattern of rest and activity (running primarily during the dark phase). However, α was ∼1.2 hours longer for DEC2-mutant transgenic mice (Fig. 2A) than for wild-type mice, DEC2-WT Tg mice, and Dec2 KO mice, which suggests that the expression of the DEC2-P385R allele leads to a dominant increase in the quantity of wakefulness in mice. In agreement with this notion, the α was lengthened further (∼2.5 hours) when the endogenous Dec2 alleles were removed by crossing DEC2-P385R mice onto the Dec2 KO background.
Fig. 2.
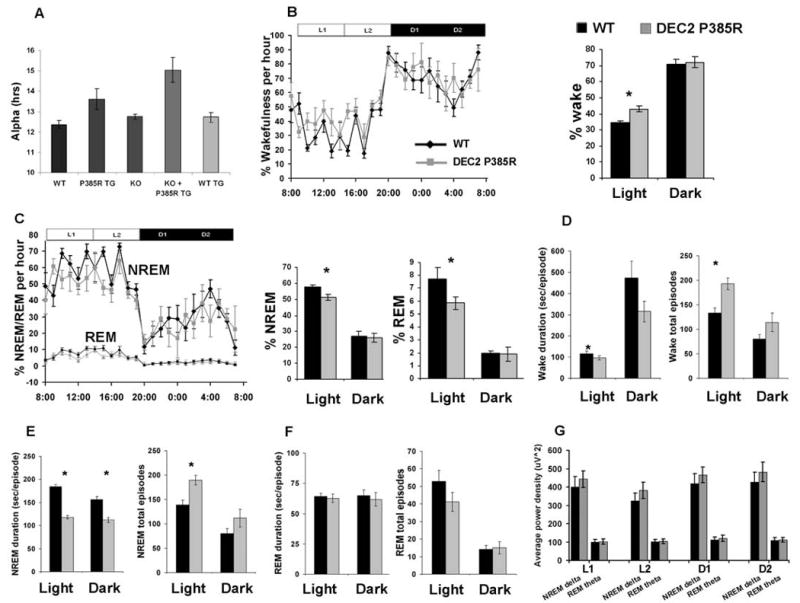
Baseline sleep-wake characterizations of DEC2-P385R and WT littermate mice. (A) Duration of the active phase (α) for mice of the indicated genotype. (B) Percentage of time in a wakeful state was significantly increased in DEC2-P385R (n = 5) compared with their WT littermates (n = 8), especially in light phase (ZT 0–12, ZT 0 = 8 A.M.). Horizontal bar indicates light and dark phases. L1, ZT 0–6; L2, ZT 6–12; D1, ZT 12–18; D2, ZT 18–24. (C) Percentage of time spent in NREM and REM during the light phase was shortened in DEC2-P385R mice. Comparisons of mean duration time and episode number of wakefulness (D), NREM (E), and REM (F) for DEC2-P385R (gray bar) and WT littermates (dark bar). (G) Average spectral power was calculated for each of the 6-hour periods (from left to right: L1, L2, D1, and D2) as indicated by the horizontal bar in (B). NREM delta and REM theta power were compared for the two genotypes. Significant differences were marked with asterisks. P < 0.05, by one-tailed and two-tailed Student's t test. All data are expressed as means ± SEM. Error bars represent SEM.
To study sleep directly (versus activity rhythms) and to investigate a possible role for DEC2 in sleep-quantity regulation, electroencephalography (EEG) and electromyography (EMG) were performed. Because we did not observe a change in α for DEC2-WT Tg mice (Fig. 2A) and because human mutation carriers have one normal allele with one mutant allele, we chose to perform EEG and EMG on DEC2-P385R mice and their WT littermates. Mice of both genders (female:male/1:1) were included in all EEG studies to exclude the possibility of sex differences noticed in other reports (15). DEC2-P385R mice were awake (as defined by EEG) ∼8% longer than WT mice in the light phase (Fig. 2B, table S2). The short-sleep phenotype of these mice was reflected in the significant shortening of both non–rapid eye movement (NREM) and rapid eye movement (REM) during sleep in the light phase for DEC2-P385R when compared with control mice (Fig. 2C and table S2). NREM sleep was ∼6% less and REM sleep was ∼2% less in DEC2-P385R versus WT mice during the light phase. Sleep architecture was further characterized by counting sleep and wakefulness episodes. Over a 12-hour period, DEC2-P385R mice showed more episodes of wakefulness than WT mice (193 ± 12 versus 133 ± 10, P < 0.05), but the mean duration of each episode was slightly shorter during the light phase (97 ± 10 s versus 116 ± 12, P < 0.05) (Fig. 2D and table S3). Consistent with this, DEC2-P385R mice also showed more NREM episodes during light periods (190 ± 10 versus 139 ± 9, P < 0.05) but each episode was shorter (118 ± 3 s versus 184 ± 5, P < 0.05) (Fig. 2E and table S3). REM episodes were similar in abundance (41 ± 5 versus 53 ± 6) and duration (63 ± 3 s versus 64 ± 3) for DEC2-P385R and WT mice (Fig. 2F and table S3). These results suggest that the sleep structure of DEC2-P385R mice is more fragmented than that of WT mice (particularly NREM sleep). Although a slightly increased NREM delta power in the EEG is visible in DEC2-P385R mice, we did not see a statistically significant difference in NREM delta or REM theta power during daytime or nighttime sleep, which suggested that, despite decreased sleep duration and continuity, sleep depth under baseline conditions is not significantly affected in mutant transgenic mice (Fig. 2G). These results indicate that the amount and consolidation of wakefulness, NREM, and REM in DEC2-P385R mice are affected mainly during the light phase. The only significant difference observed in the dark phase was the mean duration of NREM. Together, these results strongly suggest that the changes in sleep measurements for DEC2-P385R mice are caused by the P385R point mutation.
We next examined the response of DEC2-P385R mice to sleep deprivation to further probe the role of DEC2 in sleep regulation. DEC2-P385R and WT littermate mice were subjected to 6 hours of continuous sleep deprivation. Both genotypes showed a rebound in NREM and REM sleep in the remaining 6 hours of the light period and the subsequent 12 hours of darkness, although the degree of NREM and REM elevation was much smaller in the DEC2-P385R mice during the 12 hours of darkness (Fig. 3, A and B). During the dark phase (D1 and D2), NREM sleep was increased less in DEC2-P385R (ΔD1, 17.1% and ΔD2, 2.0%) versus WT mice (ΔD1, 71.8% and ΔD2, 27.2%) (Fig. 3B and table S2). Similarly, REM sleep was increased less for DEC2-P385R (ΔD1, 74.4%, ΔD2, 82.3%) than for WT mice (ΔD1, 175.2%, ΔD2, 122.4%). As seen in Fig. 3C, cumulative sleep loss and recovery data relative to the baseline hours of sleep across the deprivation and recovery periods show that WT mice lost more sleep during the L1 sleep deprivation period, probably because they had a baseline of more sleep during L1 periods. The slower recovery of acutely lost sleep in the Dec2-P385R mice is therefore even more remarkable because they started the experimental sleep deprivation with chronically less sleep. Immediately after sleep deprivation, mutant DEC2-P385R mice also had less compensatory gain in NREM compared with WT mice (Fig. 3C), consistent with a role for Dec2 in sleep homeostasis. A significantly lower NREM delta power density change after sleep deprivation was found in DEC2-P385R compared with WT mice, which further confirmed the altered NREM homeostasis in mutant mice (Fig. 3D). The differences of REM qualities between WT and DEC2-P385R mice were much less than those of the NREM and did not reach statistical significance. These results demonstrate that the P385R mutation leads to alterations in sleep rebound and NREM intensity after sleep deprivation in mice. These changes in sleep homeostasis in the mutant mice provide a testable hypothesis for future work examining why human subjects with the mutation lead such active lives despite their persistently shorter sleep.
Fig. 3.
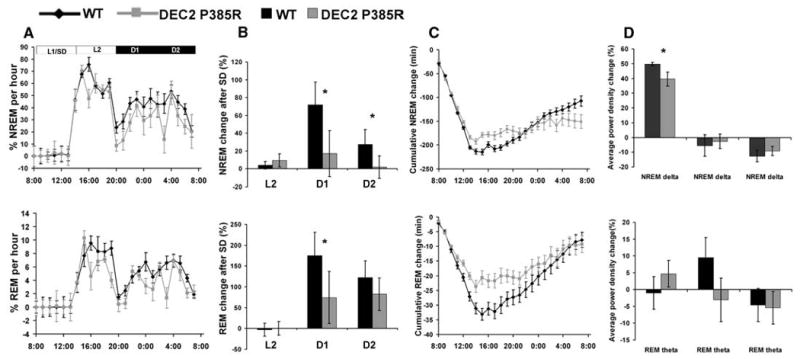
Altered sleep regulation in DEC2-P385R mice. Data for NREM sleep are shown in top panels and for REM sleep are shown in bottom panels. (A) Time course of NREM and REM sleep as percentage of time spent every hour during and after sleep deprivation for one day. (B) Percentage changes of time after sleep deprivation in comparison with the baseline condition for NREM and REM in L2, D1, and D2 for DEC2-P385R and WT mice. (C) Cumulative NREM and REM sleep loss and gain compared with baseline conditions for the sleep deprivation experiment. (D) Analysis of spectral sleep power changes compared with baseline conditions for L2, D1, and D2. Significant differences are marked with asterisks. P < 0.05, by one-tailed and two-tailed Student's t test. Error bars represent SEM.
We performed EEG and EMG on Dec2 KO mice and their WT littermates. Baseline wakefulness, NREM, and REM percentages showed that Dec2 KO mice sleep slightly more than the WT mice and that the difference is mostly in the dark period (table S4 and fig. S2). During the light period, only the NREM sleep of Dec2 KO mice was more abundant than that of WT mice and only slightly so. NREM rebound after sleep deprivation for Dec2 KO mice was much slower, which implied that Dec2 is an important factor regulating sleep recovery.
We next set out to test whether mDec2P385R can cause a similar rest-sleep phenotype in Drosophila. The closest homologous protein to DEC2 in Drosophila [CG17100, clockwork orange (16, 17)] shares <18% amino acid sequence similarity, <11% identity, and P385 is not conserved. We therefore generated transgenic flies with expression-inducible UAS-mDec2WT and UAS-mDec2P385R on the w1118 background. When these flies were crossed with elav-GAL4, driving pan-neuronal overexpression (18), mDec2P385R flies showed significantly lower daytime sleep-like behavior with reduced rest bout number and lengthened rest bout durations compared with WT flies (Fig. 4A). Because mushroom bodies were shown to be the likely sleep-rest behavior center in Drosophila (19), we also overexpressed P385R and WT mDec2 under the control of a mushroom body 30Y-GAL4 driver (20). It was noteworthy that mDec2P385R transgenic flies driven by the 30Y-GAL4 showed significantly less sleeplike behavior with significantly shorter sleep bout duration in both light and dark phases than mDec2WT flies (Fig. 4B). However, rest or sleep bout number was significantly higher only in the dark phase for mDec2P385R transgenic flies. These results indicate that the behavior of flies with mushroom body driver expression of mDec2P385R echoed those of the DEC2-P385R transgenic mice (Fig. 2, B, C, and E, and Fig. 4B).
Fig. 4.
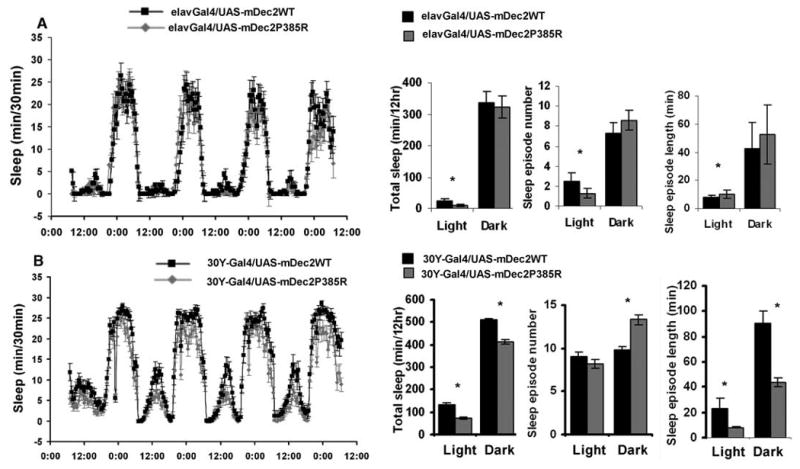
Characterization of sleeplike behavior in transgenic flies overexpressing WT (black) or P385 (gray) mDec2 with elav-GAL4 (A) or 30Y-GAL4 (B) drivers. Profile of daily sleep-like behavior (amount of sleep during each 30-min period), total sleep time, sleep episode number, and mean sleep episode length in the light and dark periods of the day were plotted. Significant differences by one-tailed Student's t test are marked with asterisks (P < 0.05). Error bars represent SEM.
The power of human genetics in studying human behavioral traits was demonstrated in the identification of mutations and the subsequent molecular characterization of FASPS (21–23). As currently understood, FASPS is primarily a circadian rhythm variant leading to altered phase; total daily sleep time is normal (21, 23, 24). We have applied a similar approach and identified a gene involved in regulation of sleep quantity. This provides a unique opportunity for exploring human sleep quantity regulation. DEC2-P385R mutation gave a short sleep phenotype, which was recapitulated in transgenic mouse and fly models but was not found in Dec2 KO mice. In addition, this phenotype was enhanced by the absence of endogenous Dec2 alleles, which suggested that P385R leads to a dominant-negative mutation. Our results demonstrate that DEC2 plays an important role in regulating daily total sleep time in mammals and that the control of sleep-like behavior may be conserved and regulated in a similar manner as far back in evolution as invertebrates.
We did not see statistically significant differences in the NREM delta or REM theta power in the DEC2-P385R mice during day- or night-time sleep, although there is a trend toward increased NREM delta in these mice. It is possible that a small deficit of sleep in the short term does not significantly affect sleep power, whereas a long-term accumulation will. Alternatively, the attenuated NREM delta power enhancement, together with slow and incomplete NREM sleep recovery after sleep deprivation in the DEC2-P385R mice, suggests that these mice are able to cope with shorter sleep because of altered sleep homeostasis. It is noteworthy that recent data on human sleep emphasizes the importance of cumulative sleep debt even if it is only due to partial sleep deprivation. Understanding the regulatory mechanisms of sleep quality and quantity will facilitate the development of interventions to alleviate pathologies associated with sleep disturbance.
Supplementary Material
Acknowledgments
We thank A. Sehgal for elav-GAL4 Drosophila driver constructs; the Ishikawa laboratory in Kyoto University for pcDNA3-hSIRT1-FLAG constructs, H.-Y. Lee for technical support and L. J. Ptác(x0030C)ek for insightful discussions and careful editing of the manuscript. This work was supported by NIH grant HL059596, Conte Center grant (MH074924), and by the Sandler Neurogenetics fund.
Footnotes
References and Notes
- 1.Fuller PM, Gooley JJ, Saper CB. J Biol Rhythms. 2006;21:482. doi: 10.1177/0748730406294627. [DOI] [PubMed] [Google Scholar]
- 2.Saper CB, Chou TC, Scammell TE. Trends Neurosci. 2001;24:726. doi: 10.1016/s0166-2236(00)02002-6. [DOI] [PubMed] [Google Scholar]
- 3.Pace-Schott EF, Hobson JA. Nat Rev Neurosci. 2002;3:591. doi: 10.1038/nrn895. [DOI] [PubMed] [Google Scholar]
- 4.Durmer JS, Dinges DF. Semin Neurol. 2005;25:117. doi: 10.1055/s-2005-867080. [DOI] [PubMed] [Google Scholar]
- 5.Knutson KL, Van Cauter E. Ann N Y Acad Sci. 2008;1129:287. doi: 10.1196/annals.1417.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Dongen HP, Maislin G, Mullington JM, Dinges DF. Sleep. 2003;26:117. doi: 10.1093/sleep/26.2.117. [DOI] [PubMed] [Google Scholar]
- 7.Akiskal HS, Akiskal KK, Haykal RF, Manning JS, Connor PD. J Affect Disord. 2005;85:3. doi: 10.1016/j.jad.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 8.Hamaguchi H, et al. Biochem J. 2004;382:43. doi: 10.1042/BJ20031760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Honma S, et al. Nature. 2002;419:841. doi: 10.1038/nature01123. [DOI] [PubMed] [Google Scholar]
- 10.Rossner MJ, et al. PLoS One. 2008;3:e2762. doi: 10.1371/journal.pone.0002762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Massari ME, Murre C. Mol Cell Biol. 2000;20:429. doi: 10.1128/mcb.20.2.429-440.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Materials and methods are available as supporting material on Science Online.
- 13.Kondo J, et al. Int J Mol Med. 2006;17:1053. [PubMed] [Google Scholar]
- 14.Noshiro M, et al. J Biol Rhythms. 2005;20:404. doi: 10.1177/0748730405280195. [DOI] [PubMed] [Google Scholar]
- 15.Franken P, et al. Proc Natl Acad Sci USA. 2006;103:7118. doi: 10.1073/pnas.0602006103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kadener S, Stoleru D, McDonald M, Nawathean P, Rosbash M. Genes Dev. 2007;21:1675. doi: 10.1101/gad.1552607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lim C, et al. Curr Biol. 2007;17:1082. doi: 10.1016/j.cub.2007.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang YQ, Rodesch CK, Broadie K. Genesis. 2002;34:142. doi: 10.1002/gene.10144. [DOI] [PubMed] [Google Scholar]
- 19.Joiner WJ, Crocker A, White BH, Sehgal A. Nature. 2006;441:757. doi: 10.1038/nature04811. [DOI] [PubMed] [Google Scholar]
- 20.Yang MY, Armstrong JD, Vilinsky I, Strausfeld NJ, Kaiser K. Neuron. 1995;15:45. doi: 10.1016/0896-6273(95)90063-2. [DOI] [PubMed] [Google Scholar]
- 21.Jones CR, et al. Nat Med. 1999;5:1062. doi: 10.1038/12502. [DOI] [PubMed] [Google Scholar]
- 22.Toh KL, et al. Science. 2001;291:1040. doi: 10.1126/science.1057499. [DOI] [PubMed] [Google Scholar]
- 23.Xu Y, et al. Nature. 2005;434:640. doi: 10.1038/nature03453. [DOI] [PubMed] [Google Scholar]
- 24.Xu Y, et al. Cell. 2007;128:59. doi: 10.1016/j.cell.2006.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.