

Abstract
TREM like transcript-1 (TLT-1) is a membrane protein receptor found in α-granules of platelets and megakaryocytes. Upon platelet activation TLT-1 is rapidly brought to the surface of platelets. Recently, we demonstrated that activated platelets release a soluble form of TLT-1 (sTLT-1) that is found in serum but not in the plasma of healthy individuals and can enhance platelet aggregation in vitro. Furthermore, evaluation of patients diagnosed with inflammatory diseases, such as sepsis, show that these patients have significantly elevated levels of sTLT-1 in their blood. Accordingly, mice deficient in TLT-1 are predisposed to bleeding in response to an inflammatory challenge; however the mechanism of TLT-1 function remains unknown. In this investigation we demonstrate an increase in the amount of platelets that adhere to endothelial cell monolayers in the presence of recombinant sTLT-1 (rsTLT-1). Additionally we present evidence that rsTLT-1 increases platelet adherence to glass slides by stimulating actin polymerization in platelets as determined by increased staining of rodamine phalloidin. These results suggest that during inflammation, sTLT-1 may mediate hemostasis by enhancing actin polymerization, resulting in increased platelet aggregation and adherence to the endothelium.
Introduction
Inflammation during the progress of sepsis and viral infections such as dengue fever often invoke the activation of our hemostatic system[1-3]. The clinical manifestations are associated with a hypovolemic shock that promotes hemoconcentration and low blood pressure, resulting from an acute increase in capillary permeability, and possibly death[2,4,5]. Release of cytokines such as tissue necrosis factor (TNF-a) and interleukin 1b (IL-1b) enhance neutrophil evagination into the tissues, subsequently leading to vascular leakage[6]. Both platelets and endothelial cells are called upon to control the loss of blood and plasma from the vessels through receptor engagement, release of granule contents, and remodeling of their actin cytoskeletons[7-9]. Storage granules in endothelial cells (weibel palade bodies), and in platelets (alpha and dense granules) play a major role in maintaining vascular integrity. Upon activation, platelets and endothelial cells release proteins from the storage granules into the plasma that enhance their ability to stop blood leakage. The release reaction from platelet granules is accompanied by an increase in the presence of several receptor proteins on the surface of the cells[10]. It is been shown that receptors such P-selectin[11,12] and TREM like transcript-1 (TLT-1) can be found in high concentrations on the surface of the platelet membrane upon platelet activation [13] as the granules of the resting platelet merge with the cell membrane, releasing their soluble contents and displaying membrane bound receptors previously undisplayed at the cell surface. Although much work has gone into the understanding the role of P-selectin in inflammation and hemostasis, the role of TLT-1 in platelet function remains somewhat an enigma[14,15].
Studies in TLT-1 null mice (TLT-/-) have demonstrated an inability of their platelets to aggregate efficiently[16]. Additionally, TLT-/- mice were shown to have extended tail-bleeding times. Lipopolysaccharide (LPS) treated TLT-1-/- mice showed higher plasma levels of D dimers compared to wild-type mice and they were more susceptible to death from the endotoxin challenge[16]. Accordingly, TLT-1-/- mice were predisposed to hemorrhage associated with the Shwartzman reaction, a specific localized inflammatory lesion. Collectively these results suggest that TLT-1 plays an important role in maintaining hemostasis during inflammation.
Recently, our lab demonstrated that activated platelets release a soluble form of TLT-1 (sTLT-1) that is found in serum but not in the plasma of healthy individuals. However, upon investigation of plasma from patients diagnosed with sepsis[16] or acute viral infections (unpublished observations), we demonstrated that these patients had significantly increased levels of sTLT-1 in their plasma in contrast to healthy individuals. In septic individuals, the presence of sTLT-1 correlated the presence of disseminated intravascular coagulation [16]. Previous studies have shown that thrombin-induced platelet aggregation can be specifically inhibited using human single-chain Fv antibodies against TLT-1[17]. Our studies further demonstrated that a recombinant, soluble (rs)TLT-1 increases platelet aggregation when using washed platelets and various agonists[16]. The ability of sTLT-1 to increase platelet reactivity to agonists suggests that TLT-1 may modulate hemostatic function in part by release of a soluble fragment. To gain insight into the mechanism of sTLT-1's affect on hemostasis we examined the effect of rsTLT-1 on platelet adhesion to endothelial cells and evaluated the effects of rsTLT-1 on actin polymerization to identify clues the mechanisms of TLT-1 function.
Materials and Methods
Platelet Isolation
The Universidad Central del Caribe, Escuela del Medicina (Institutional Review Board (Federalwide Assurance no. FWA00001103) approved the study, and volunteers provided written informed consent before enrollment (protocol no. 200619; primary investigator, A. Valance Washington). Platelets were isolated as described previously[16]. Briefly, blood was obtained by venipuncture from healthy volunteers who had not have taken any anti-inflammatory drugs for the previous 7 days. Blood was drawn into polypropylene syringes containing one-sixth volume of 3.8% trisodium citrate and centrifuged at 800 g for 20 min to obtain platelet-rich plasma (PRP). Platelets were sedimented by centrifugation at 2,200 g for 10 min and washed twice with 10 ml of Hepes Tyrode's buffer (10 mM Hepes, 0.5 mM MgCl2, 130 mM NaCl, 4 mM KCl, 1 mM CaCl2, and 5 mM glucose, pH 7.4). Platelets stained with calcein were incubated with 1 μM calcein-acetoxymethyl ester (Molecular Probes Inc., Eugene, OR) in the dark for 30 min at 37°C. All centrifugations were done at room temperature in the presence of 1 μM prostaglandin E1 and 0.02 U/ml of asparase. To remove excess of calcein, labeled platelets were washed once with Hepes-Tyrode's buffer containing 0.3% BSA. Then the density of platelets was adjusted to 1.3× 108 per ml in Hepes–tyrode's buffer containing 10% newborn calf serum. Platelets were activated with 0.5 U/ml human thrombin for 10 min. Hirudin (1U/ml) was added to stop the reaction.
Bovine Aortic Endothelial cells (BAEC)
Bovine aortic endothelial cells (BAEC) were prepared as described previously[18], briefly the cells were maintained under a humidified atmosphere of 95% air and 5% CO2 at 37°C. The cells were seeded on 12 well plates 4 days before each assay. Cells were cultured in RPMI 1640 medium (20% bovine calf serum, 90 ug/ml Heparin, 50 ug/ml endothelial growth factor) and allowed the cells to grow to confluence in 12 well plates.
Adherence assay
BAEC were activated with of 150 nM of Thromboxane A2 (TxA2) for 15 minutes at 37°C in RPMI media. After 15 minutes BAEC were washed twice with RPMI media and incubated with 600μl of Hepes-Tyrode's buffer with 2mM CaCL2 and 40μl of the solution of calcein treated human platelets. The platelets were either resting or thrombin activated and incubation was carried out either in the presence or absence of rsTLT-1 for 30 min at 37°C. Unbound platelets were removed by two washes with PBS. BAEC were harvested mechanically, washed once and then fixed with 80% ethanol on ice for 30 minutes. The cells were resuspended in 500 μl of PBS containing 0.1% Triton X-100, 5μg/ml propidium iodine and 50 μg/ml ribonuclease A. We analyzed adherence by flow cytometery. Bound platelets were identified by the increase in events in the endothelial cell platelet (PEC) gate (shown in figure 2c). Results are expressed as the average number of events in the PEC gate from at least three experiments. At least 20,000 events were counted per sample.
Figure 2. rsTLT-1 effect on platelet adhesion to resting and activated endothelial cells.

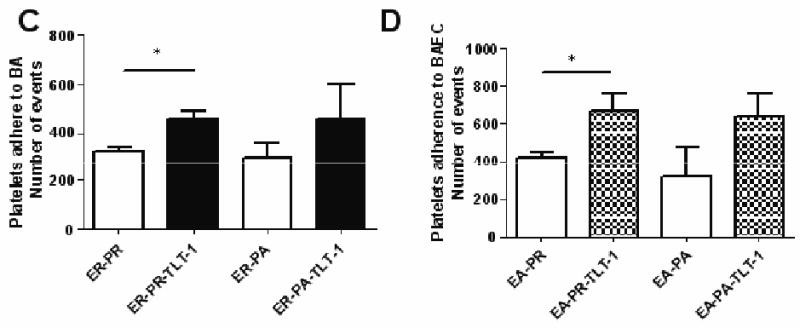
(A, B) Flow cytometric analysis of platelets within the PEC gate shown in figure 1A after incubation with activated endothelial cells either without rsTLT-1 (A), or with rsTLT-1 (B). Quantification of platelet adhesion to (C) resting BAEC (D) TxA2 activated BAEC incubated with either resting or thrombin activated human platelets. Binding was expressed as the number of platelets stained with calcein bound to the endothelial cell monolayer. The data presented here is an average of three independent experiments. PEC (platelet-endothelial cell) ER (BAEC resting), PR (resting platelets), and PA (activated platelets) EA (BAEC activated), PR (resting platelets), and PA (activated platelets). (*) stars represent a P≤0.05
Confocal Microscopy
Platelets were seeded on glass slides with fibrinogen matrixes (100μg/ml) in the presence of different concentrations of rsTLT-1 (0, 25, 50, and 100μg/ml). Platelets were allowed to adhere for 5 minutes then the slides were washed with Tyrodes and fixed with Cytofix/Cytoperm (BD Science) for 20 min at 4 °C. Platelets were stained with rhodamine phalloidin to determine the changes in actin polymerization (red) and counter stained with anti-CD41 (the integrin αIIb also known as platelet GPIIb: US Biologicals: Swampscott, MA cat#C2394-10 clone 7H138). Because phalloidin only binds to polymerized actin we correlated platelet platelet spreading with increased rodamine intensity. Ten fields of each slide were counted in each of three experiments. The quantification of platelet binding and spreading was completed by using the Metamorph program (Molecular Devices, Downingtown, PA).
Statistical Analysis
Analysis was completed with Prism software (version 5.01, Graph-Pad Software). Platelet binding to endothelial cells, platelet spreading, and platelet binding to the fibrinogen matrixes were expressed as means ± SD. We used a 2 tailed Student's t test and a P < 0.05 was considered statistically significant.
Results
Soluble TLT-1 enhances platelet-endothelial cells binding interactions
We have recently demonstrated that the addition of rsTLT-1 to platelet aggregation assays significantly increases the ability of platelets to aggregate [16]. We hypothesized that the rsTLT-1 mediated enhanced aggregation would translate to increased platelet adhesion to endothelial cells. To address this possibility, we modified the protocol of Kakutani et. Al [18] which used bovine aortic endothelial cells (BAEC) to measure platelet adhesion to endothelium in a static assay. Recent reports demonstrate that TxA2 plays a formidable role in mediating neutrophil and platelet adhesion to endothelial cells[19]. Because we have consistently seen the greatest TLT-1 mediated platelet inhibition when we used TxA2 to activate platelets and recent reports demonstrate that TxA2 plays a formidable role in mediating leukocyte and platelet adhesion to and vascular leakage from the endothelium, we used TxA2 to activate the endothelial cells. TxA2 causes cellular withdrawal and shrinkage and as expected TxA2 activation of endothelial cells caused greater endothelial cell loss during harvesting. The greater cell loss is reflected in the flow cytometry shown in figure 1 (A and B) where we consistently counted less endothelial cells from wells where activated endothelial cells were harvested.
Figure 1. Quantification of platelet adherence to endothelial cells.

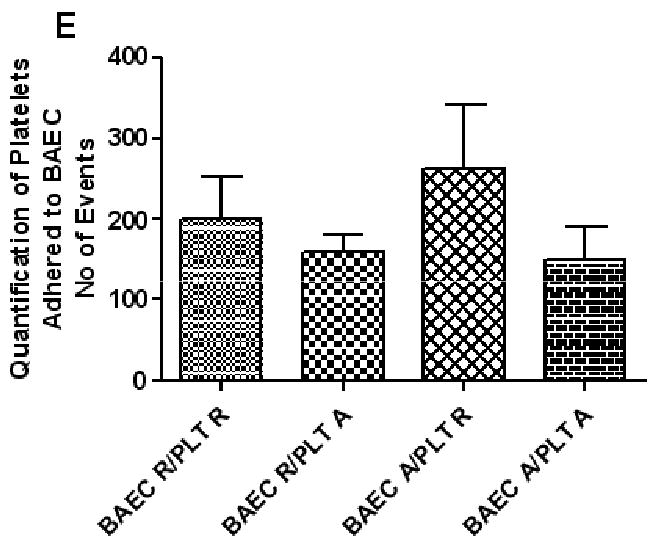
(A-F) Flow cytometric analysis of platelet adherence. FL-1 is shown on the Y-axis (calcein) and FL-2 (PE) is shown on the X-axis. Scatter profile of resting endothelial cells showing gate (PEC) used to identify adherent platelets (A), activated endothelial cells (B), resting platelets (C), and activated platelets (D). (E) Quantification of adherent platelets by flow cytometry. Results are from four independent experiments. PEC (platelet-endothelial cell) ER (BAEC resting), PR (resting platelets), and PA (activated platelets) EA (BAEC activated), and PR (resting platelets).
Platelets were treated with the fluorescent intercellular cell dye calcein before addition to endothelial cells. Resting and activated calcein treated platelets demonstrated a typical platelet scatter with a classical subtle shift in fluorescence once activated as shown in figures 1 C and D. Resting or activated platelets were incubated with either activated or resting endothelial cells for 30 min at 37°C. Adherent platelets were monitored by flow cytometry as described in materials and methods. Endothelial cell activation caused a 30% increase in the total numbers of resting platelets that adhered compared to resting platelets that adhered to resting endothelial cells (figure 1g). The addition of previously activated platelets to resting or activated endothelial cells show a 25 and 43% reduction respectively in average total numbers of adherent platelets.
Once we established the parameters of our experimental system, we repeated these experiments in either the presence or absence of rsTLT-1. Figures 2A and B give an example of the changes seen within the PEC gate with the addition of rsTLT-1 (figure 2B) to activated BAEC and resting platelets. Examination of platelet-endothelial cell aggregates demonstrated that rsTLT-1 has an augmentative effect on platelet adherence to BAEC in all the conditions tested when compared to controls (Figure 2C and D). Using resting platelets and resting endothelial cells, addition of rsTLT-1 increased the average observed binding events from 320±46 to 460± 51. Using activated platelets with resting endothelial cells the observed binding events increased from 297±109 to 456±248. The effect of added rsTLT-1 was much more pronounced on platelet binding to activated BAEC. Using resting platelets, the addition of rsTLT-1 increased platelet numbers from 423±43 to 676±148 and with activated platelets the number of binding events without rsTLT-1 was 314±286, and 636±219 in the presence of 50ug/ml rsTLT-1. Although TxA2 stimulated endothelial cells incubated with thrombin activated platelets show an increase in platelet adherence, the increase in binding seen with resting platelets to either resting or activated endothelial cells demonstrated a greater and significant (P=0.05) increase in the number of platelets that remained bound to the monolayer.
STLT-1 enhances actin polymerization and platelet binding to fibrinogen matrixes
Previous work with TLT-1 using scFvs[17] to inhibit platelet aggregation showed that the scFv mediated inhibition fails to stop P-selectin expression suggesting that the TLT-1 mediated inhibition is downstream of the calcium signal required for α-granule/membrane fusion. These results open the possibility that actin polymerization, which plays an important role downstream of the calcium signal in both platelet activation and adhesion could provide key mechanistic insights to TLT-1 function. To evaluate the potential of actin polymerization playing a role in TLT-1 mediated platelet function, we seeded platelets at 1× 108 cells on glass slides coated with bovine serum albumin (BSA), fibrinogen, TLT-1, and/or a fibrinogen/TLT-1 mixture and slides were washed after five minutes of incubation at 37°C. Slides were subsequently stained with rodamine phalloidin, which binds only polymerized actin. Slides were examined by confocal microscopy and evaluated for platelet spreading and adhesion. Confocal microscopy revealed that actin polymerization in platelets increased with increasing TLT-1 concentration (25, 50, or 100 μg/ml) when compared to those allowed to adhere to fibrinogen alone (figure 3). There was no difference in actin polymerization between the slides that contained fibrinogen-only (100μg/ml), fibrinogen (100μg/ml)/BSA (50μg/ml) or BSA-only (100μg/ml) (data not shown). Recombinant sTLT-1 concentrations of 25, 50, 100 μg/ml yielded a significant difference in the increase of spreading compared to fibrinogen only controls. There was an increase in average platelet radius (figure 3f) and area (figure 3e). Average platelet areas observed in photomicrographs increased from on average from 33 μm2 ±0.3 with fibrinogen-only to 53.38 μm2 ±5.2, 45.7±3.1, or 57.8 μm2±12.1 with addition of 25, 50, or 100 μg/ml of rsTLT-1 respectively (figure 3e). Platelets allowed to adhere for 15 minutes did not show the dose dependant difference in actin polymerization (data not shown) suggesting that TLT-1 plays a role early during the process of platelet aggregation and adhesion.
Figure 3. Actin polymerization and platelet spreading on sTLT-1 and/or fibrinogen matrixes.
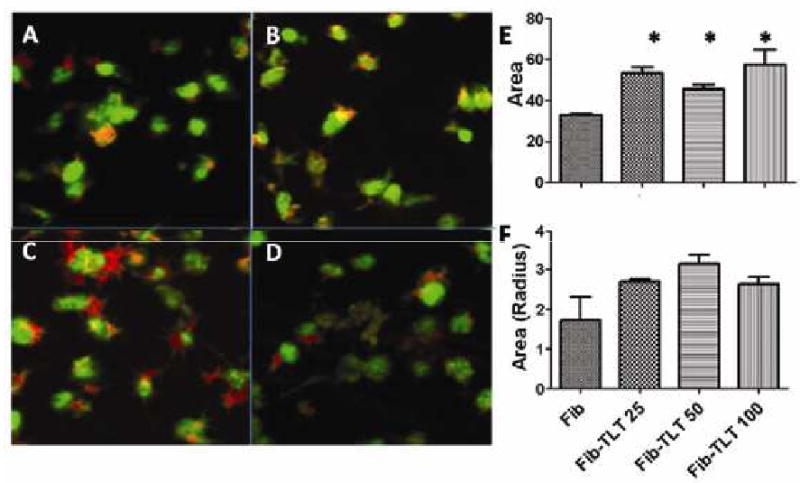
(A-D) Representative photomicrographs demonstrating changes in platelet spreading on fibrinogen matrixes (100μg/ml) in the absence of rsTLT-1 (A) or with 25μg/ml rsTLT-1 (B), 50μg/ml rsTLT-1 (C), or 100μg/ml rsTLT-1 (D). (E) Quantification of platelet spreading and (F) average platelet radius - TLT-1 as evaluated and determined using quantification of rhodamine phalloidin. Platelets are counter stained with anti gpIIβ IIIα (green). These are representative of at least 3 independent experiments. (*) stars represent a P≤0.05
We subsequently measured the amount of platelets that adhered to the fibrinogen and fibrinogen/TLT-1 matrixes (figure 4 a-d). Platelets were seeded at 1×108 platelets/ml and allowed to adhere for 5 minutes. Platelets were then stained with rodamine phalloidin and subjected to counting using Metamorph. Consistent with the results from the platelet spreading we saw an increase in the amount of platelets that adhered in the presence of rsTLT-1 compared to fibrinogen alone. Accordingly, the increase was greatest at the rsTLT-1 concentration 50 μg/ml showing an average increase of 217±12 adherent platelets per 10 fields counted (p=0.02)(figure 4 e. The results are consistent with the results from the endothelial cell experiments.
Figure 4. Quantification of the binding of human platelets to fibrinogen rsTLT-1 matrixes.

(A-D) Representative photomicrographs demonstrating changes in platelet binding on fibrinogen matrixes (100μg/ml) in the absence of rsTLT-1 (A) or with 25μg/ml rsTLT-1 (B), 50μg/ml rsTLT-1 (C), or 100μg/ml rsTLT-1 (D). (E) Quantification of platelet:rsTLT-1:fibrinogen matrices as evaluated by quantification of rhodamine phalloidin. Platelets are counter stained with anti gpIIβ IIIα (green). These are representative of at least 3 independent experiments.
Discussion
The TLT-1 receptor, to date, has only been identified on platelets. The lineage restriction of TLT-1 suggests that TLT-1 plays a specific role in platelet biology. Current work on the TLT-1 receptor shows that antibodies specific to TLT-1 can inhibit platelet aggregation implying that TLT-1 enhances platelet aggregation. Accordingly, the addition of rsTLT-1 significantly enhances platelet aggregation further supporting an important role for TLT-1 during platelet activation and maintenance of vascular integrity. Consistent with this data, studies with platelets from TLT-1 null mice show that these mice are susceptible to bleeding when challenged with lipopolysaccaride. In our recent publication we demonstrated that rsTLT-1 binds to fibrinogen suggesting that interaction with fibrinogen may play a key role to understanding how TLT-1 regulates platelet function. Furthermore, our laboratory has found prominent concentrations of sTLT-1 in patient populations suffering from either sepsis or viral syndrome compared to healthy individuals suggesting a potential role for sTLT-1 in maintenance of vascular hemostasis during inflammatory diseases [16] (Washington AV, Gibot S, et al; unpublished observations). Here we provide additional evidence for a role for sTLT-1 in platelet-endothelial cell binding interactions and gain insights to the mechanisms of TLT-1 regulation of platelet function.
We performed a platelet-endothelial adherence assay and demonstrated for the first time that TLT-1 plays a role in platelet adherence to endothelial cells in a static assay. Our results show that the addition of rsTLT-1 to either resting or TxA2 activated BAEC increased platelet adherence to the BAEC over controls. Using activated platelets appeared to cause a reduction of platelet adherence compared to incubations with resting platelets. Activated endothelial cells have been shown to shrink and lose cell to cell contact and detach. The addition of activated platelets may further increase the shrinkage and detachment of the endothelial cells, resulting in a greater variation in the numbers of platelets that adhere under those conditions. Although we consistently found lower numbers of adherent platelets when activated platelets were incubated with activated endothelial cells, the addition of rsTLT-1 increased the total amount of adherent platelets to similar levels seen when resting platelets were used. We have explored the possibility of sTLT-1 increasing adherence to endothelial cells, by investigating the ability of sTLT-1 to directly interact with the extracellular matrix proteins (ECM) vitronectin, collagen, and fibronectin by enzyme linked immunosorbant assay (ELISA). Using the ELISA assay we were, however, unable detect any interactions of sTLT-1 with any of these proteins (data not shown). Therefore we do not believe that the increased adhesion induced by sTLT-1 is mediated through any of these ECM proteins. Our study opens the possibility of a preserving effect of sTLT-1 on endothelial cells and is currently under investigation in our laboratory. This work also suggests that sTLT-1 may play a role in the maintenance of vascular integrity and that addition of rsTLT-1 to sites of vascular injury may accelerate the cession of bleeding.
Another interesting point is that the most significant difference was seen when rsTLT-1 was incubated with resting platelets. Although resting platelets bound activated endothelial cells more effectively than activated platelets, our results indicate that addition of rsTLT-1 resulted in a statistically significant effect on the interaction between resting platelets and endothelial cells that wasn't seen with activated platelets under the same conditions. Based on these results and recent demonstrations of the importance of α-granule components to platelet function[20] we hypothesize that rsTLT-1 amplifies activation signals from the endothelial cells and/or processing derived signals that may occur during the treatment. A possible mechanism for the increased adherence with resting platelets may lie in the generation of platelet microparticles. It is generally accepted that even with gentle manipulation of platelet samples, platelets will generate microparticles, which have been shown to lower the natural anti-coagulant properties of endothelial cells[21] as well as contain the procoagulant tissue factor[22]. The combination of lower endothelial resistance to platelet adherence and low levels of tissue factor in the presence of sTLT-1 may lead to increased platelet degranulation[23,24]. We hope to be able to discern some of these possibilities in the future. Our results are consistent with the finding that rsTLT-1 enhances platelet aggregation and may indicate that TLT-1 may enhance both platelet-platelet and platelet-endothelial interactions by a single mechanism.
Because actin polymerization plays a large role in platelet aggregation, adhesion, and functions downstream of the calcium signal needed for fusion of α-granules to the platelet membrane, we investigated the ability of TLT-1 to effect actin polymerization. Recombinant sTLT-1 increased platelet spreading and the amount of platelets that adhered to fibrinogen matrixes on glass slides. On the fibrinogen-only slides you can witness the beginnings of filopodia extension as evidence of increased actin polymerization at 5 min (figure3 a- d). However, on the slides with rsTLT-1 there was visually detectable increases in the amount of platelet structures such as filopodia and lamellipodia. These differences were most noticeable on the slides that contained 50 and 100 μg/ml. These results are consistent with the rsTLT-1 mediated adhesion of platelets to the endothelial cells. We attribute the enhanced binding and size of platelets to an increase in the amount of actin structures that were made in the presence of the rsTLT-1 compared to fibrinogen alone. Actin polymerization in platelets is a crucial step in this cascade of events; allowing for a rapid shape change in platelets that leads to the formation of filopodia and lamellipodia on activated platelets. Confocal microscopy revealed that rsTLT-1 promotes platelet cytoskeletal actin polymerization and allows for the formation of filopodia and lamellipodia at the mobile edges of the platelets. Collectively, our studies suggest that TLT-1 may play a visible role in the maintenance of vascular integrity and suggests that the addition of rsTLT-1 to sites of vascular injury may accelerate the cession of bleeding.
Conclusions
Platelets have many receptors such as gpIIβ IIIα which binds fibrinogen[25], and P-selectin which is an adhesive molecule that promotes rolling and tethering of platelets and the recruitment of leukocytes to the site of vascular[26]. In addition to of all the known platelet receptors, TLT-1 arises as a new receptor which is capable of augmenting platelet aggregation and platelet-endothelial cell interactions. We conclude that rsTLT-1 functions to augment actin polymerization in platelets leading to increased aggregation and adhesion. Our data suggests that rsTLT-1 mediated actin polymerization may mediate enhanced platelet adhesion to the endothelium in vivo. Although more studies are needed to dissect TLT-1 function and its effects in relation with vascular integrity and its association with platelets behavior these studies give new insights to platelet-endothelial cell interactions. Since rsTLT-1 modulates actin formation it could affect not only platelets, but also other cells as well.
Acknowledgments
The authors would like to thank the staff of the Protein and Nucleic Acid and Optical Imaging Facilities at Universidad Central del Caribe. This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, and under contract N01-Co-12400. Additional funding was obtained through grants from the National Center for Research Resources (NCRR), a component of the NIH (2G12RR3035), the National Institute of General Medical Sciences (SC2GM081237), and the Aniara Corp. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the NIH or the US government.
Reference List
- 1.Cohen J. TREM-1 in sepsis. Lancet. 2001;358:776–778. doi: 10.1016/S0140-6736(01)06007-X. [DOI] [PubMed] [Google Scholar]
- 2.Cohen J. The immunopathogenesis of sepsis. Nature. 2002;420:885–891. doi: 10.1038/nature01326. [DOI] [PubMed] [Google Scholar]
- 3.Bente DA, Rico-Hesse R. Models of dengue virus infection. Drug Discov Today Dis Models. 2006;3:97–103. doi: 10.1016/j.ddmod.2006.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krishnamurti C, Peat RA, Cutting MA, Rothwell SW. Platelet adhesion to dengue-2 virus-infected endothelial cells. Am J Trop Med Hyg. 2002;66:435–441. doi: 10.4269/ajtmh.2002.66.435. [DOI] [PubMed] [Google Scholar]
- 5.Noisakran S, Perng GC. Alternate hypothesis on the pathogenesis of dengue hemorrhagic fever (DHF)/dengue shock syndrome (DSS) in dengue virus infection. Exp Biol Med (Maywood) 2008;233:401–408. doi: 10.3181/0707-MR-198. [DOI] [PubMed] [Google Scholar]
- 6.Colonna M, Facchetti F. TREM-1 (triggering receptor expressed on myeloid cells): a new player in acute inflammatory responses. J Infect Dis. 2003;187 2:S397–S401. doi: 10.1086/374754. [DOI] [PubMed] [Google Scholar]
- 7.Fox JE. The platelet cytoskeleton. Thromb Haemost. 1993;70:884–893. [PubMed] [Google Scholar]
- 8.Fox JE. Cytoskeletal proteins and platelet signaling. Thromb Haemost. 2001;86:198–213. [PubMed] [Google Scholar]
- 9.Hartwig JH, Italiano JE., Jr Cytoskeletal mechanisms for platelet production. Blood Cells Mol Dis. 2006;36:99–103. doi: 10.1016/j.bcmd.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 10.George JN. Platelets. Lancet. 2000;355:1531–1539. doi: 10.1016/S0140-6736(00)02175-9. [DOI] [PubMed] [Google Scholar]
- 11.Frenette PS, Denis CV, Weiss L, Jurk K, Subbarao S, Kehrel B, Hartwig JH, Vestweber D, Wagner DD. P-Selectin glycoprotein ligand 1 (PSGL-1) is expressed on platelets and can mediate platelet-endothelial interactions in vivo. J Exp Med. 2000;191:1413–1422. doi: 10.1084/jem.191.8.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geng JG, Chen M, Chou KC. P-selectin cell adhesion molecule in inflammation, thrombosis, cancer growth and metastasis. Current Medicinal Chemistry. 2004;11(16):2153–60. doi: 10.2174/0929867043364720. [DOI] [PubMed] [Google Scholar]
- 13.Washington AV, Schubert RL, Quigley L, Disipio T, Feltz R, Cho EH, McVicar DW. A TREM family member, TLT-1, is found exclusively in the alpha-granules of megakaryocytes and platelets. Blood. 2004;104:1042–1047. doi: 10.1182/blood-2004-01-0315. [DOI] [PubMed] [Google Scholar]
- 14.Ford JW, McVicar DW. TREM and TREM-like receptors in inflammation and disease. Curr Opin Immunol. 2009 doi: 10.1016/j.coi.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Washington AV, Quigley L, McVicar DW. Initial characterization of TREM-like transcript (TLT)-1: a putative inhibitory receptor within the TREM cluster. Blood. 2002;100:3822–3824. doi: 10.1182/blood-2002-02-0523. [DOI] [PubMed] [Google Scholar]
- 16.Washington AV, Gibot S, Acevedo I, Gattis J, Quigley L, Feltz R, De La MA, Schubert RL, Gomez-Rodriguez J, Cheng J, Dutra A, Pak E, Chertov O, Rivera L, Morales J, Lubkowski J, Hunter R, Schwartzberg PL, McVicar DW. TREM-like transcript-1 protects against inflammation-associated hemorrhage by facilitating platelet aggregation in mice and humans. J Clin Invest. 2009;119:1489–1501. doi: 10.1172/JCI36175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giomarelli B, Washington VA, Chisholm MM, Quigley L, McMahon JB, Mori T, McVicar DW. Inhibition of thrombin-induced platelet aggregation using human single-chain Fv antibodies specific for TREM-like transcript-1. Thromb Haemost. 2007;97:955–963. [PubMed] [Google Scholar]
- 18.Kakutani M, Masaki T, Sawamura T. A platelet-endothelium interaction mediated by lectin-like oxidized low-density lipoprotein receptor-1. Proc Natl Acad Sci U S A. 2000;97:360–364. doi: 10.1073/pnas.97.1.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zarbock A, Ley K. The role of platelets in acute lung injury (ALI) Front Biosci. 2009;14:150–158. doi: 10.2741/3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ho-Tin-Noe B, Goerge T, Cifuni SM, Duerschmied D, Wagner DD. Platelet granule secretion continuously prevents intratumor hemorrhage. Cancer Res. 2008;68:6851–6858. doi: 10.1158/0008-5472.CAN-08-0718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morel O, Morel N, Freyssinet JM, Toti F. Platelet microparticles and vascular cells interactions: a checkpoint between the haemostatic and thrombotic responses. Platelets. 2008;19:9–23. doi: 10.1080/09537100701817232. [DOI] [PubMed] [Google Scholar]
- 22.Key NS. Platelet tissue factor: how did it get there and is it important? Semin Hematol. 2008;45:S16–S20. doi: 10.1053/j.seminhematol.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 23.Ardoin SP, Shanahan JC, Pisetsky DS. The role of microparticles in inflammation and thrombosis. Scand J Immunol. 2007;66:159–165. doi: 10.1111/j.1365-3083.2007.01984.x. [DOI] [PubMed] [Google Scholar]
- 24.Barry OP, Pratico D, Lawson JA, FitzGerald GA. Transcellular activation of platelets and endothelial cells by bioactive lipids in platelet microparticles. J Clin Invest. 1997;99:2118–2127. doi: 10.1172/JCI119385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Osdoit S, Rosa JP. Fibrin clot retraction by human platelets correlates with alpha(IIb)beta(3) integrin-dependent protein tyrosine dephosphorylation. J Biol Chem. 2001;276:6703–6710. doi: 10.1074/jbc.M008945200. [DOI] [PubMed] [Google Scholar]
- 26.Bergmeier W, Burger PC, Piffath CL, Hoffmeister KM, Hartwig JH, Nieswandt B, Wagner DD. Metalloproteinase inhibitors improve the recovery and hemostatic function of in vitro-aged or -injured mouse platelets. Blood. 2003;102:4229–4235. doi: 10.1182/blood-2003-04-1305. [DOI] [PubMed] [Google Scholar]