

Abstract
Although the endogenous cannabinoid system modulates a variety of physiological and pharmacological processes, the specific role of cannabinoid CB1 receptors in the modulation of glutamatergic neurotransmission and neural plasticity is not well understood. Using whole-cell patch clamp recording techniques, evoked or spontaneous excitatory postsynaptic currents (eEPSCs or sEPSCs) were recorded from visualized, layer II/III pyramidal cells in frontal cortical slices from rat brain. Bath application of the CB1 receptor agonist, WIN 55212-2 (WIN), reduced the amplitude of NMDA receptor-mediated EPSCs in a concentration-dependent manner. When co-applied with the specific CB1 antagonists, AM251 or AM281, WIN did not suppress NMDA receptor mediated EPSCs. WIN also reduced the amplitude of evoked AMPA receptor-mediated EPSCs, an effect that was also reversed by AM251. Both the frequency and amplitude of spontaneous AMPA receptor-mediated EPSCs were significantly reduced by WIN. In contrast, WIN reduced the frequency, but not the amplitude of miniature EPSCs, suggesting that the suppression of glutmatergic activity by CB1 receptors in the frontal neocortex is mediated by a pre-synaptic mechanism. Taken together, these data indicate a critical role for endocannabinoid signaling in the regulation of excitatory synaptic transmission in frontal neocortex, and suggest a possible neuronal mechanism whereby THC regulates cortical function.
Keywords: Endocannabinoid, Glutamate, EPSC, Temporal Summation, Pyramidal cells, Neocortex
Introduction
Delta9-tetrahydrocannabinol (THC) exerts its principal biological action via endogenous cannabinoid receptors, and modulates synaptic transmission (Freund et al., 2003; Alger, 2002). The specific receptors involved in mediation of synaptic activity by THC are CB1 receptors, which are mainly expressed in the nervous system (Herkenham et al., 1990; Egertova and Elphick 2000), while CB2 receptors are mainly located on cells in the immune system (Howlett et al., 2002). In the nervous system, endogenous cannabinoids are released from postsynaptic neurons upon depolarization, and act in a retrograde fashion to activate CB1 receptors on the presynaptic terminals, thus inhibiting neurotransmitter release (Alger, 2002). This has recently been demonstrated in the frontal cortex of mice (Lafourcade et al, 2007).
Cannabinoid effects on neuronal function in the frontal neocortex and hippocampus are hypothesized to underlie the disruptive action of marijuana on higher cognitive processes, including memory (Ranganathan and D’Souza, 2006). Indeed hippocampal-neocortical interactions, including frontal lobe interactions, represent a key component of the process of memory consolidation (Eichenbaum et al, 1998; Fletcher and Henson, 2001; Fletcher et al, 1998a,b). Although the mediation of excitatory synaptic activity by CB1 receptors has been well studied in the hippocampal formation (Hoffman et al, 2003; Hofman et al, 2006), the role of CB1 receptors in the inhibition of glutamatergic transmission in the frontal neocortex has not been extensively studied. The available evidence indicates that CB1 activation suppresses excitatory neurotransmission by reducing glutamate release or uptake in the cortico-striatal pathway (Brown et al 2003), and functional CB1 receptors are found on neocortical glutamatergic neurons (Hill et al, 2007). Furthermore, in mice lacking CB1 receptors on principal neurons, the CB1 agonist WIN 55212-2 did not reduce glutamatergic synaptic transmission in the hippocampus, the basolateral amygdala, or the primary somatosensory cortex (Domenici et al, 2006), providing strong evidence of the existence of CB1 receptors located on glutamatergic synapses in those regions. Despite the fact that CB1 receptors are expressed in the frontal cortex, and that this region is involved in memory processes, cannabinoid modulation of excitatory synaptic input to these cortical layers, especially that mediated by NMDA receptors, has not been extensively studied. Lafourcade, et al (2007) studied CB1 receptor modulation of excitatory postsynaptic currents (EPSCs) in layer V and VI of the mouse prefrontal cortex, showing that CB1 activation reduced evoked EPSCs and that depressing CB1 function inhibited long term depression (LTD). Other electrophysiological studies indicate that excitatory synaptic activity can be reduced by activation of CB1 receptors in some neocortical regions such as layer V pyramidal neurons from the prelimbic area of the neocortex (Auclair et al, 2000), the visual field of rats (Sjostrom et al 2003), and layer II/III pyramidal cells in the auditory field of mice (Trettel and Levine, 2002). Thus, the present experiments were designed to more fully assess the role of CB1 receptors in mediating excitatory neuronal function within the frontal neocortex of the rat.
Results
Images of a layer III pyramidal cell filled with florescent dye before, and immediately after the establishment of the whole-cell configuration are shown in Figure-1A. In addition, the morphology of the cortical pyramidal cells was visualized by confocal laser scanning microscopy. Only one experiment was performed with each slice treated with WIN55212-2. Pharmacologically isolated NMDA or AMPA receptor-mediated EPSCs were completely blocked by APV (50 μM) or DNQX (20 μM), respectively, confirming that they were mediated exclusively by activation of postsynaptic NMDA or AMPA receptors (data not shown).
Figure 1.

Activation of CB1 receptors inhibits NMDA EPSCs in cortical pyramidal cells in a dose-dependent manner.
(A) Photomicrographs showing a layer III pyramidal cell in the bright field (A1), and under DIC (A2). A3 shows the same cell revealed with florescent dye two minutes after whole-cell configuration was established. A4 shows detailed morphology of the same cell using laser scanning confocal microscopy. Scale bar: 10μm.
(B) Upper panel, average traces of NMDA EPSCs were isolated from a cell held at −40mV. 1μM WIN 55212-2 reduced the peaks of NMDA-EPSCs. Scale bar: 100ms/50pA. Lower panel: Time course of WIN55212-2 induced inhibition of the amplitude of NMDA EPSCs recorded from the same cell.
(C) WIN 55212-2 induced a dose-dependent inhibition of the mean amplitude of NMDA EPSCs (0.5 μM, n=6) and 1μM, n=8) (p<0.05).
Activation of CB1 receptors by WIN 55212-2 reduced NMDA receptor-mediated EPSCs
We first tested whether the CB1 receptor agonist, WIN 55122-2, could affect NMDA-mediated transmission during an evoked synaptic event. An example of the suppression of NMDA EPSCs by WIN55212-2 is shown in Figure-1B. At a holding potential of −40mV, the recorded EPSCs were inward currents. Bath application of 1μM WIN55212-2 caused a significant decrease in amplitude of NMDA EPSCs, which was reduced to 54% of control. This suppression generally occurred within 4–7 minutes after bath application of the agonist. Moreover, WIN55212-2 suppressed NMDA eEPSCs in a concentration-dependent manner as demonstrated in Figure-1C.
To confirm that CB1 receptors were mediating the inhibition of NMDA eEPSCs observed with WIN 55212-2, we used two highly specific CB1 antagonists AM251 (n=7) and AM281 (n=6) in the following experiments. After stable NMDA eEPSCs were isolated, the slices were superfused with 2μM AM281 in combination with 1μM WIN55212-2. The suppressive effect of WIN 55212-2 on NMDA EPSCs was blocked by the antagonist and the result is shown in Figure-2A. Similarly, in the presence of 2μM AM251, WIN 55212-2 failed to attenuate the peak of NMDA eEPSCs (Figure-2B). The results of these experiments are summarized in Figure 2C.
Figure 2.
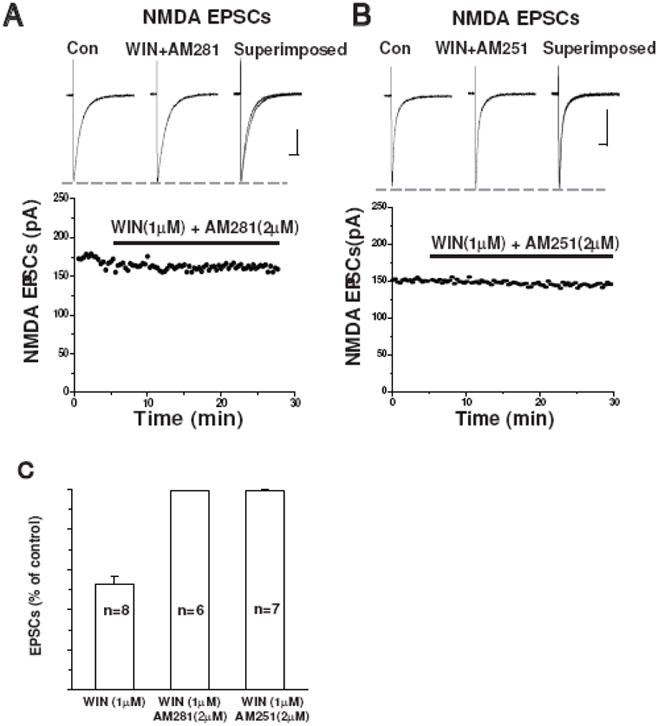
Selective CB1 receptor antagonists prevent WIN-induced inhibition of NMDA EPSCs
(A) The CB1 receptor antagonist AM281 (2μM) blocked the inhibitory effect of WIN on NMDA EPSCs. Time course of the amplitude of NMDA EPSCs recorded from the same cell shown in lower panel. Scale bar 100ms/50pA.
(B) The CB1 receptor antagonist AM215 (2μM) also abolished WIN-induced inhibition of NMDA EPSCs. Time course of the amplitude of NMDA EPSCs recorded from the same cell shown in lower panel. Scale bar: 100ms/50pA.
(C) Bar graph showing the average percent inhibition of NMDA EPSCs by 1μM WIN alone (n=8; unpaired t-test, p<0.05), or in the presence of AM281 (2μM, n=6) or AM251 (2μM, n=7).
WIN55212-2 reduction of evoked AMPA EPSCs
We next assessed the effect of CB1 receptor activation on the fast synaptic component mediated by AMPA receptors. As illustrated in Figure 3A, 1μM WIN55212-2 reduced the peak of evoked AMPA receptor-mediated EPSCs by an average of 44.3 ± 3.0% (n=9, p<0.05, Figure 3C). The effects of WIN55212-2 on AMPA EPSCs were blocked by 2 μM AM251 (Figure 3B). This is illustrated in Figure 3C. In a separate experiment, we found that bath application of 2μM AM-251 reversed the WIN-induced suppression of AMPA EPSC amplitude in all four cells tested (Figure 3D). However, there was no significant difference in the magnitude of the reduction of NMDA, compared to AMPA receptor-mediated EPSCs by WIN 55212-2 (NMDA 52.9 ± 3.5%, n=8., AMPA 44.3 ± 3.0%, n=9, p> 0.05). These findings indicate that activation of CB1 receptors by WIN produces a similar inhibitory action on two major components (slow vs. fast) of excitatory synaptic transmission in the frontal cortex.
Figure 3.

Activation of CB1 receptors inhibits AMPA EPSCs in cortical pyramidal cells.
(A) Upper panel, average traces of AMPA EPSCs isolated from a cell held at −40mV are shown. 1μM WIN 55212-2 reduced the amplitude of NMDA-EPSCs. Scale bar: 100ms/50pA. Inset: Scale bar: 20ms/50pA. Lower panel: Time course of WIN 55212-2 induced inhibition of the amplitude of AMPA EPSCs recorded from the same cell.
(B) Upper panel, average traces of AMPA EPSCs isolated from a cell held at −70mV. 1μM WIN 55212-2 reduced the amplitude of NMDA-EPSCs, and this effect was abolished by AM251 (2μM) Scale bar: 100ms/50pA. Lower panel: Time course of the amplitude of AMPA EPSCs recorded from the same cell.
(C) Bar graph showing the effects of AM251 (2μM) on WIN 55212-2 induced inhibition of AMPA EPSCs (n=4; paired t-test, p<0.05).
(D) Bar graph showing WIN 55212-2 induced inhibition of the mean amplitude of AMPA EPSCs (1μM, n=9; paired t-test, p<0.05). Activation of CB1 receptors by 1μM WIN55212-2 inhibits NMDA and AMPA EPSCs to comparable degrees (unpaired t-test, p>0.05).
WIN55212-2 reduced spontaneous AMPA receptor-mediated EPSCs
We also assessed the effects of WIN55212-2 on the frequency and amplitude of action potential-dependent, AMPA receptor-mediated EPSCs. A representative example of the suppression of AMPA receptor-mediated sEPSCs by WIN55212-2 is shown in Figure 4A. In this example, 1μM WIN55212-2 reduced the mean frequency of sEPSCs to 2.3 ± 0.4Hz from 4.7 ± 0.7Hz. A cumulative probability analysis indicated that there was a significant increase of the inter-event interval during WIN55212-2 exposure, and the inter-event interval distribution was shifted to the right (K-S test, p<0.0001, Figure 4B). Furthermore, the mean amplitude of sEPSCs was decreased from 64.6 ± 5.7pA to 40.4 ± 4.4pA. The cumulative probability of sEPSC amplitude decreased significantly (K-S test, p<0.001, Figure 4C). Figure 4D illustrates the effects of 1μM WIN55212-2 on AMPA sEPSC frequency and amplitude in the seven pyramidal cells tested. WIN reduced sEPSC frequency by 52.8 ± 4.8% (n=7, p<0.05) and sEPSC amplitude by 37.9 ± 2.3% (n=7, p<0.05). These findings suggested that CB1 receptor-mediated signaling could modulate AMPA receptor-mediated fast excitatory synaptic activity in the frontal cortex.
Figure 4.

Activation of CB1 receptors inhibits spontaneous AMPA-EPSCs in cortical pyramidal cells.
(A) Representative traces of spontaneous AMPA EPSCs were isolated from a cortical cell held at −70mV in the presence of picrotoxin (75μM) and APV (50μM) before, and during bath application of 1μM WIN 55212-2. Scale Bar: 500ms/80pA.
(B) Cumulative probability analysis of the distribution of inter-event intervals between sEPSCs before, and after bath application of 1.0μM WIN 55212-2 (K-S test, p<0.0001).
(C) Cumulative probability analysis of sEPSC amplitude before, and after bath application of 1.0μM WIN 55212-2 (K-S test, p<0.001).
(D) Bar graph showing the average percent inhibition of sIPSCs by 1.0μM WIN 55212-2 (n=7; paired t-test, p<0.05).
WIN55212-2 reduced the frequency of AMPA receptor-mediated mEPSCs
We next tested the effects of WIN on action potential-independent, spontaneous miniature AMPA EPSCs (mEPSCs). In the presence of 1μM TTX, mEPSCs were isolated from frontal cortical cells held at a potential of −70mV. Application of 1μM WIN 55212-2 significantly inhibited mEPSC frequency, as illustrated in the data from a neuron in Figure 5A. Cumulative analysis of the inter-event interval in this cell showed that WIN shifted the distribution curve significantly to the right (K-S test, p<0.001, Figure 5B), but had no effect on mEPSCs amplitude (K-S test p>0.05, Figure 5C). Figure 5D shows the effects of WIN on mEPSC frequency and amplitude across all neurons tested (n=6). These data strongly suggest that WIN inhibits glutamatergic synaptic transmission in frontal pyramidal cells through a presynaptic mechanism.
Figure 5.

WIN reduced the frequency of AMPA receptor mediated mEPSCs.
(A) Spontaneous mEPSCs were recorded from a cortical pyramidal neuron held at −70mV in the presence of 1.0μM TTX. Bath application of 1.0μM WIN55212-2 reduced the frequency of AMPA receptor mediated mEPSCs. Scale bar: 200ms/30pA.
(B) Cumulative probability analysis of the inter-event interval distribution of mEPSCs before, and after bath application of 1.0μM WIN 55212-2 (K-S test, p<0.0001).
(C) Cumulative probability analysis of sEPSC amplitude before, and after bath application of 1.0μM WIN 55212-2 (K-S test, p>0.05).
(D) Bar graph showing the average percent inhibition by 1.0μM WIN 55212-2 of the frequency (n=6; paired t-test, p<0.05) and amplitude of sIPSCs (n=6; paired t-test, p>0.05).
Discussion
These experiments were designed to assess the role of CB1 receptor-mediated function in the modulation of excitatory neurotransmission in the frontal cortex. Our principal findings are (1) activation of CB1 receptors by WIN55212-2 inhibited NMDA and AMPA-mediated excitatory synaptic transmission in the frontal cortex, (2) analysis of spontaneous miniature EPSCs indicates that this inhibition was mediated through a presynaptic mechanism, and (3) temporal summation mediated by glutamatergic signals in frontal cortex was also altered by activation of CB1 receptors. Taken together, these findings indicate that cannabinoids act through a presynaptic mechanism to suppress excitatory synaptic activity in the frontal cortex.
CB1 receptors are expressed in axonal terminals of GABAergic interneurons (Katona et al, 1999; Tsou et al., 1998), and endocannabinoids bind to these presynaptic CB1 receptors and thereby modulate neurotransmitter release (Wilson and Nicoll, 2002; for reviews see Alger 2002; Howlett, 2005). However, controversy remains about the possible involvement of CB3 receptors (Hajos et al, 2001) in these processes despite the high concentration of CB1 receptors in multiple neocortical regions (Oropeza et al, 2007; Tsou et al, 1998; Egertová and Elphick, 2000). Further support for the primary involvement of CB1 receptors in the mediation of the above pharmacological effects is derived from a recent study indicating the existence of CB1 receptors on the terminals of glutamatergic neurons in sensorimotor cortex (Hill et al, 2007). Monosynaptic EPSCs evoked in layer V pyramidal cells in the frontal cortex of rats are also potently inhibited by CB1 receptor activation, an effect that is reversed by the CB1 antagonist, SR141617 (Auclair et al, 2000; Barbara et al, 2003). And, interestingly, WIN 55212-2 does not reduce evoked, glutamatergic synaptic responses in the cortical layer II principal neurons, which lack CB1 receptors (Domenici et al, 2006). Thus, there is considerable evidence that cannabinoids, released from depolarized postsynaptic neocortical neurons, decrease glutamate release through presynaptic CB1 receptors. The present data are not only consistent with those findings, but also, to our knowledge, provide the first whole cell assessment of interactions between the glutamatergic and endocannabinoid systems in layer II/III of the frontal cortex of rats, pivotal brain regions involved in cognition, associative learning, and drug addiction.
The fact that WIN 55212-2 reduced the frequency, but not the amplitude of TTX-insensitive mEPSCs in the frontal cortex, strongly suggests that cannabinoid mediated presynaptic mechanism modulating glutamate release in the frontal cortex and did not alter the postsynaptic efficacy of released glutamate and did not affect quantal size or probability of spontaneous release Miniature EPSCs are mediated by action potential-independent, quantal release of neurotransmitters from presynaptic terminals. Therefore, the observed decrease in the frequency of mEPSCs is due to the effects of cannabinoid receptor activation on the probability of quantal release from presynaptic glutamatergic terminals originating from intra- and inter-cortical sources. Moreover, in these experiments the postsynaptic, voltage-sensitive potassium A conductance which mediates the postsynaptic effects of cannabinoids (Deadwyler et al., 1995), was blocked by QX-314 and cesium contained in our internal recording solution, further indicating a presynaptic mechanism.
The endogenous cannabinoid system is known to suppress both GABAergic and glutamatergic neurotransmission in multiple CNS regions (Hoffman and Lupica, 2000; Hajos et al, 2000; Diana et al, 2002; Wilson and Nicoll, 2002), including the hippocampus (Kawamura et al., 2006), ventral tegmental area (Szabo et al, 2004; Melis et al., 2004), amygdala (Katona et al, 2001; Azad et al., 2003), and cerebellum (Kawamura et al, 2006). The present results are consistent with, and extend these findings. In the present study, activation of CB1 receptors by the exogenous agonist WIN 55212-2 markedly suppressed both NMDA and AMPA receptor-mediated EPSCs in the frontal neocortex, effects that were blocked by the selective CB1 receptor antagonists AM251 and AM281, confirming the involvement of CB1 receptors in modulating excitatory synaptic transmission in the frontal cortex. These results are consistent with others showing regulation of excitability and synaptic plasticity by endocannabinoids in other neocortical regions (Fortin et al, 2004). In addition, they support the notion that activation of CB1 receptors results in presynaptic inhibition of glutamate release in the frontal cortex as has been observed in several other brain regions (Szabo et al, 2000; Robbe et al, 2001; Azad et al, 2003; Riegel and Lupica, 2004; Dominici et al, 2006). Furthermore, the magnitude of the WIN-induced depression of NMDA and AMPA receptor mediated synaptic currents in frontal cortex appears to be similar at these excitatory synapses. Taken together, these data demonstrate a broad role for endocannabinoids in the regulation of neuronal activity in the neocortex. Given the role of pyramidal cells in originating cortico-cortical connections, cannabinoid induced inhibition of glutamatergic synaptic transmission may provide, in conjunction with other neurotransmitters and neuromodulators, precise control of functions of the frontal cortex.
Neocortical cannabinoid-NMDA interactions may have implications for both physiological and pathological processes. Although NMDA receptors are found in all layers of the neocortex, they are concentrated in layers II/III. Moreover, precise co-localization of NMDA and CB1 receptors in superficial layer II/III pyramidal cells has been described (Oropeza et al, 2007; Tsou et al, 1998), suggesting a close interaction between endocannabinoid signaling and NMDA receptor function in the neocortex. Such an interaction would likely be of great physiological significance as the excitatory synaptic transmission is critical for maintaining normal function in local networks. For example, it has been proposed that some neuropathological processes related to psychiatric disorders, such as schizophrenia, may be associated with NMDA hypofunction (Olney et al, 1999), and some schizophrenic patients display a significant increase in CB1 binding in the superficial layers (I and II) of the posterior cingulate cortex relative to controls (Newell et al, 2006).
Furthermore, involvement of NMDA receptors in the generation of neocortical synaptic plasticity is likely one neural mechanism underlying the development of addiction. We have reported that NMDA receptor-mediated EPSCs in the posterior cingulate cortex are potently suppressed by ethanol (Li et al, 2002b), and other studies have indicated that endocannabinoid signaling may play critical role in mediating alcohol-seeking behavior in animals (Hungund et al, 2002). Thus, the physiological significance of neocortical CB1-NMDA interactions may be multifaceted and include pathology related to both memory and addiction.
In summary, the present study demonstrates that frontal neocortical excitatory synaptic activity is modulated by activation of CB1 receptors, and provides strong evidence that endocannabinoid signaling is involved in regulation of synaptic transmission in the frontal neocortex. By modulating glutamate release from excitatory presynaptic terminals, endocannabinoids may play a role in shaping the efficiency of synaptic transmission and plasticity, including plasticity related to memory formation and addiction. These findings also suggest a potential neuronal mechanism whereby marijuana, and its principal psychoactive component, regulate cortical and cognitive function.
Experimental Procedure
Cortical slice preparation
Frontal cortical slices were prepared from male, Sprague-Dawley rats (PD15–25). The animals were handled and housed according to the guidelines of the National Institutes of Health Committee on Laboratory Animal Resources. All experimental procedures and protocols were approved by the Animal Care and Use Committees of Duke University and the Durham VA Medical Center. The rats were anesthetized with isoflurane and decapitated. The brains were quickly removed from the skulls and placed in a cold (4°C) standard artificial cerebrospinal fluid (aCSF) containing (in mM) 120 NaCl, 3.3 KCl, 1.23 NaH2PO4, 26 NaHCO3, 1.2 MgSO4, 1.8 CaCl2 and 10 D-Glucose at pH 7.3, previously saturated with 95%O2/5%CO2. Frontal cortical slices (300μm thickness) were cut on a vibratome (100PLUS, Sectioning System, Ted Pella, Inc., Redding CA) and incubated in a holding chamber containing aCSF continuously bubbled with 95% O2/5% CO2 at room temperature (20–24°C).
Patch pipettes were pulled from borosilicate glass capillary tubing (1.5mm O.D., 1.05 mm I.D., World Precision Instruments, Sarasota, FL) on a Flaming-Brown horizontal microelectrode puller (Model P-97, Sutter Instrument Co, Novato, CA). The pipettes were filled with an intracellular solution containing (in mM) 130 Cs-gluconate, 7 CsCl, 10 HEPES, 4 Mg-ATP, 0.5 Tris-GTP (pH=7.25). The quaternary lidocaine derivative QX-314 (4mM) (Sigma Chemical Co., St Louis, MO) was also included to suppress fast sodium currents and GABAB receptor-mediated currents. In some experiments, fluorescence dye Alexa Fluor-568 (50–80 μM, Molecular Probes, Carlsbad, CA) was also included in the internal solution. Osmolarity was adjusted to 285 mOsm. The pipette resistances were in the range of 4–7Mohm.
After one hour of incubation in the holding chamber, slices were transferred into a submersion recording chamber and secured in place with a bent piece of platinum wire resting on the top of the slice. The chamber temperature was maintained at 34°C during recording. Whole cell voltage patch clamp recordings were made from the visualized pyramidal cells in frontal cortex layer II/III. Individual cells were visualized on an upright Zeiss microscope equipped with an infrared differential interference contrast optics (IR-DIC) and a 40X water immersion objective (Zeiss, Oberkochen, Germany), and were confirmed real-time by examining the images of the recorded pyramidal cell filled with fluorescent dye. After the establishment of the whole-cell recording configuration, stable long lasting tight-seal recordings were achieved in most cases. Evoked, NMDA receptor-mediated EPSCs (eEPSCs) were recorded using an Axopatch 200B amplifier (Molecular Devices, Union City, CA). Output current signals were DC-coupled to a digital storage oscilloscope (TDS 2014, Tektronix, Inc., Beaverton, OR). Series resistance was monitored throughout the recording period and cells were not used if the series resistance changed by more than 20%. The digitized data were also acquired and stored using Strathclyde Electrophysiology Software, Whole Cell Program (WINWCP) (Courtesy of Dr. John Dempster) with an interface (BNC-2090, National Instruments, Austin, TX) to a PC-computer. Additionally, real–time measurements of the amplitude of NMDA-EPSCs were performed and displayed simultaneously on the second PC computer using a custom-written program developed with Labview (Version 6i, National Instrument) by Dr. Maeng-Hee Kang-Park in our laboratory.
Electrical stimulation and isolation of NMDA and AMPA receptor-mediated EPSCs
In the presence of the GABAA receptor antagonist, picrotoxin (75μM), and the a-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptor antagonist, 6,7-dinitroquinoxaline-2,3-dione (DNQX)(20μM), NMDA receptor-mediated EPSCs were evoked at a holding potential of −40mV by electrical stimulation through a monopolar tungsten electrode (A-M systems, Inc., Carlsborg, WA) placed 50~70μm from the soma of the recorded pyramidal cells located in layer II/III of the frontal neocortical slices. AMPA receptor-mediated eEPSCs were isolated at a holding potential of −70mV in the presence of picrotoxin (75μM) and the NMDA receptor antagonist, D-(−)-2-amino-5-phosphonovaleric acid (APV)(50μM). The stimulus threshold was first determined by increasing the intensity of constant current rectangular wave pulses generated by an isolated stimulator (Grass S88, Grass Instrument CO, Quincy, MA) until detectable responses occurred. Next, constant current rectangular stimulus pulses, 50% higher than threshold intensity, with duration of 0.1ms and inter-stimulus interval of 0.033Hz, were applied and eEPSCs were recorded for 50 to 70 minutes to allow for completion of the experimental protocols.
Spontaneous AMPA receptor-mediated EPSCs (sEPSCs) were recorded at a holding potential of −70mV in the presence of picrotoxin (70μM) and the NMDA receptor antagonist, APV (50μM). Action potential-independent miniature EPSCs (mEPSCs) were isolated at a holding potential of −70mV in the presence of picrotoxin (70μM), APV (50μM), and tetrodotoxin (TTX)(1.0μM).
Histological identification of pyramidal neurons
After recording, the pyramidal cells were filled with Alexa Fluor 568 hydrazide (50–80 μM, Invitrogen, Carlsbad, CA) to reveal their morphological characteristics. The florescence filled slices were fixed with 4% paraformaldehyde for 20–30 minutes, and rinsed three times using a 0.2 M phosphate buffer. The slices were then mounted on gelatin-coated slides with Prolong Gold Antifade (Invitrogen). Fluorescence images were then examined with a Leica confocal laser scanning microscope (TCS SP5, Leica Microsystems, Inc., Exton, PA).
Drug application
All drugs were bath applied. CB1 agonists and antagonists were first dissolved in dimethyl sulfoxide (DSMO) and then diluted in the bath solution to the designed concentration. TTX, DNQX and Picrotoxin were purchased from Sigma (Sigma Chemical Co., St Louis, MO). APV was from ACROS (Geel, Belgium). WIN 55122-2 ((R)-(+)-[2,3-Dihydro-5-methyl-3-(4-morpholinylmethyl) pyrrolo[1,2,3-de)-1,4-benzoxazin-6-yl]-1-napthalenylmethanone, and AM 251 (N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide), AM 281 (1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-4-morpholinyl-1H-pyrazole-3-carboxamide) were purchased from Tocris (Tocris, Cookson, UK). SR 141716 (N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide) was obtained from National Institute of Mental Health Chemical Synthesis and Drug Supply Program.
Statistical Analyses
Evoked EPSCs were analyzed off-line using Mini Analysis software 6.0.3 (Synaptosoft, Decatur GA) or Clampfit 9.2 (Molecular Devices, Union City, CA). The frequency and amplitude of AMPA receptor-mediated sEPSCs were calculated and analyzed using Mini Analysis programs, followed by visual inspection of recorded traces to corroborate the accuracy of measurements. Cumulative probability distributions were compared statistically using the Kolmogolov-Smirnov test. We used Student’s t-tests or ANOVA with repeated measures, as appropriate, to generate statistical inferences for grouped data sets.
Acknowledgments
Grants: This work was supported by the National Institute of Drug Abuse (grant # DA019346 to HSS), the Institute for Medical Research (IMR) of the Durham VA Medical Center (to QL), and VA research career scientist awards to HSS and WAW.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alger B. Retrograde signaling in the regulation of synaptic transmission: focus on endocannabinoids. Prog Neurobiol. 2002;68:247–286. doi: 10.1016/s0301-0082(02)00080-1. [DOI] [PubMed] [Google Scholar]
- Auclair N, Otani S, Soubrie P, Crepel F. Cannabinoids modulate synaptic strength and plasticity at glutamatergic synapses of rat prefrontal cortex pyramidal neurons. J Neurophysiol. 2000;83:3287–3293. doi: 10.1152/jn.2000.83.6.3287. [DOI] [PubMed] [Google Scholar]
- Azad S, Eder M, Marsicano G, Lutz B, Zieglgansberger W, Rammes G. Activation of the cannabinoid receptor type 1 decreases glutamatergic and GABAergic synaptic transmission in the lateral amygdala of the mouse. Learn Mem. 2003;10:116–128. doi: 10.1101/lm.53303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbara JG, Auclair N, Roisin MP, Otani S, Valjent E, Caboche J, Soubrie P, Crepel F. Direct and indirect interactions between cannabinoid CB1 receptor and group II metabotropic glutamate receptor signalling in layer V pyramidal neurons from the rat prefrontal cortex. Euro J Neurosci. 2003;17:981–990. doi: 10.1046/j.1460-9568.2003.02533.x. [DOI] [PubMed] [Google Scholar]
- Brown TM, Brotchie JM, Fitzjohn SM. Cannabinoids decrease corticostrial synaptic transmission via an effect on glutamate uptake. J Neurosci. 2003;23:11073–11077. doi: 10.1523/JNEUROSCI.23-35-11073.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deadwyler SA, Hampson RE, Mu J, Whyte A, Childers S. Cannabinoids modulate voltage sensitive potassium A-current in hippocampal neurons via a cAMP-dependent process. J Pharm Exp Ther. 1995;273(2):734–743. [PubMed] [Google Scholar]
- Diana MA, Levenes C, Mackie K, Marty A. Short-term retrograde inhibition of GABAergic synaptic currents in rat Purkinje cells is mediated by endogenous cannabinoids. J Neurosci. 2002;22:200–208. doi: 10.1523/JNEUROSCI.22-01-00200.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domenici M, Azad S, Marsicano G, Schierloh A, Wotjak, Dodt H-U, Zieglgansberger W, Lutz B, Rammes G. Cannabinoid Receptor Type 1 Located on Presynaptic Terminals of Principal Neurons in the Forebrain Controls Glutamatergic Synaptic Transmission. J Neurosci. 2006;26:5794–5799. doi: 10.1523/JNEUROSCI.0372-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egertová M, Elphick M. Localisation of cannabinoid receptors in the rat brain using antibodies to the intracellular C-terminal tail of CB1. J Comp Neurol. 2000;422:159–171. doi: 10.1002/(sici)1096-9861(20000626)422:2<159::aid-cne1>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- Eichenbaum H, Schoenbaum G, Young B, Bunsey M. Functional organization of the hippocampal memory system. Proceedings of the National Academy of Sciences, USA. 1998;93:13500–13507. doi: 10.1073/pnas.93.24.13500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher PC, Henson RN. Frontal lobes and human memory: Insights from functional neuroimaging. Brain. 2001;124:849–881. doi: 10.1093/brain/124.5.849. [DOI] [PubMed] [Google Scholar]
- Fletcher PC, Shallice T, Dolan RJ. The functional roles of prefrontal cortex in episodic memory: I. Encoding. Brain. 1998a;121:1239–1248. doi: 10.1093/brain/121.7.1239. [DOI] [PubMed] [Google Scholar]
- Fletcher PC, Shallice T, Frith CD, Frackowiak RS, Dolan RJ. The functional roles of prefrontal cortex in episodic memory: II. Retrieval. Brain. 1998b;121:1249–1256. doi: 10.1093/brain/121.7.1249. [DOI] [PubMed] [Google Scholar]
- Fortin D, Levine E. Differential Effects of Endocannabinoids on Glutamatergic and GABAergic Inputs to Layer 5 Pyramidal Neurons. Cereb Cortex. 2007;17:163–174. doi: 10.1093/cercor/bhj133. [DOI] [PubMed] [Google Scholar]
- Fortin D, Trettel J, Levine E. Brief Trains of Action Potentials Enhance Pyramidal Neuron Excitability Via Endocannabinoid-Mediated Suppression of Inhibition. J Neurophysiol. 2004;92:2105–2112. doi: 10.1152/jn.00351.2004. [DOI] [PubMed] [Google Scholar]
- Freund TF, Katona I, Piomelli D. Role of endogenous cannabinoids in synaptic signaling. Physiol Rev. 2003;83:1017–1066. doi: 10.1152/physrev.00004.2003. [DOI] [PubMed] [Google Scholar]
- Hájos N, Katona I, Naiem SS, MacKie K, Ledent C, Mody I, Freund TF. Cannabinoids inhibit hippocampal GABAergic transmission and network oscillations. Euro J Neurosci. 2000;12:3239–3249. doi: 10.1046/j.1460-9568.2000.00217.x. [DOI] [PubMed] [Google Scholar]
- Hájos N, Ledent C, Freund T. Novel cannabinoid-sensitive receptor mediates inhibition of glutamatergic synaptic transmission in the hippocampus. Neuroscience. 2001;106:1–4. doi: 10.1016/s0306-4522(01)00287-1. [DOI] [PubMed] [Google Scholar]
- Herkenham M, Lynn AB, Little MD, Johnson MR, Melvin LS, de Costa BR, Rice KC. Cannabinoid receptor localization in brain. Proc Natl Acad Sci USA. 1990;87:1932–1936. doi: 10.1073/pnas.87.5.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill EL, Gallopin T, Ferezou I, Cauli B, Rossier J, Schweitzer P, Lambolez B. Functional CB1 receptors are broadly expressed in neocortical GABAergic and glutamatergic neurons. J Neurophysiol. 2007;97:2580–2589. doi: 10.1152/jn.00603.2006. [DOI] [PubMed] [Google Scholar]
- Hoffman A, Lupica C. Mechanisms of cannabinoid inhibition of GABA(A) synaptic transmission in the hippocampus. J Neurosci. 2000;20:2470–2479. doi: 10.1523/JNEUROSCI.20-07-02470.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman A, Riegel A, Lupica C. Functional localization of cannabinoid receptors and endogenous cannabinoid production in distinct neuron populations of the hippocampus. European Journal of Neuroscience. 2003;18:524–534. doi: 10.1046/j.1460-9568.2003.02773.x. [DOI] [PubMed] [Google Scholar]
- Hofmann M, Nahir B, Frazier C. Endocannabinoid mediated depolarization-induced suppression of inhibition in hilar mossy cells of the rat dentate gyrus. J Neurophysiol. 2006;96:2501–2512. doi: 10.1152/jn.00310.2006. [DOI] [PubMed] [Google Scholar]
- Howlett AC. Cannabinoid receptor signaling. Handbook of Experimental Pharmacology. 2005;168:53–79. doi: 10.1007/3-540-26573-2_2. [DOI] [PubMed] [Google Scholar]
- Howlett AC. The cannabinoid receptors Prostaglandins & Other Lipid Mediators. 2002;68–69:619–631. doi: 10.1016/s0090-6980(02)00060-6. [DOI] [PubMed] [Google Scholar]
- Hungund BL, Basavarajappa BS, Vadasz C, Kunos G, Rodriguez de Fonseca F, Colombo G, Serra S, Parsons L, Koob GF. Ethanol, endocannabinoids, and the cannabinoidergic signaling system. Alcoholism: Clinical & Experimental Research. 2002;26(4):565–74. [PubMed] [Google Scholar]
- Katona I, Rancz E, Acsady L, Ledent C, Mackie K, Hajos N, Freund T. Distribution of CB1 cannabinoid receptors in the amygdala and their role in the control of GABAergic transmission. J Neurosci. 2001;21:9506–9518. doi: 10.1523/JNEUROSCI.21-23-09506.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona I, Sperlagh B, Sik A, Kafalvi A, Vizi E, Mackie K, Freund T. Presynaptically located CB1 cannabinoid receptors regulate GABA release from axonal terminals of specific hippocampal interneurons. J Neurosci. 1999;19:4544–4558. doi: 10.1523/JNEUROSCI.19-11-04544.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona I, Urban G, Wallace M, Ledent C, Jung K, Piomelli D, Mackie K, Freund T. Molecular composition of the endocannabinoid system at glutamatergic synapses. J Neurosci. 1996;26:5628–5637. doi: 10.1523/JNEUROSCI.0309-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura Y, Fukaya M, Maejima T, Yoshida T, Miura E, Watanabe M, Ohno-Shosaku T, Kano M. The CB1 cannabinoid receptor is the major cannabinoid receptor at excitatory presynaptic sites in the hippocampus and cerebellum. J Neurosci. 2006;26:2991–3001. doi: 10.1523/JNEUROSCI.4872-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafourcade M, Elezgarai I, Mato S, Bakiri Y, Grandes P, Manzoni O. Molecular components and functions of the endocannabinoid system in mouse prefrontal cortex. PLoS ONE. 2007;2(8):e709. doi: 10.1371/journal.pone.0000709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Clark S, Lewis D, Wilson WA. NMDA receptor antagonists disinhibit rat posterior cingulate and retrosplenial cortices: a potential mechanism of neurotoxicity. J Neurosci. 2002a;22:3070–3080. doi: 10.1523/JNEUROSCI.22-08-03070.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Wilson WA, Swartzwelder HS. Differential Effect of Ethanol on NMDA EPSCs in Pyramidal Cells in the Posterior Cingulate Cortex of Juvenile and Adult Rats. J Neurophysiol. 2002b;87:705–711. doi: 10.1152/jn.00433.2001. [DOI] [PubMed] [Google Scholar]
- Melis M, Pistis M, Perra S, Muntoni A, Pillolla G, Gessa G. Endocannabinoids mediate presynaptic inhibition of glutamatergic transmission in rat ventral tegmental area dopamine neurons through activation of CB1 receptors. J Neurosci. 2004;24:53–62. doi: 10.1523/JNEUROSCI.4503-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newell KA, Deng C, Huang XF. Increased cannabinoid receptor density in the posterior cingulate cortex in schizophrenia. Experimental Brain Research. 2006;172(4):556–60. doi: 10.1007/s00221-006-0503-x. [DOI] [PubMed] [Google Scholar]
- Olney J, Newcomer J, Farber N. NMDA receptor hypofunction model of schizophrenia. J Psychiat Res. 1999;33:523–533. doi: 10.1016/s0022-3956(99)00029-1. [DOI] [PubMed] [Google Scholar]
- Oropeza VC, Mackie K, Van Bockstaele EJ. Cannabinoid receptors are localized to noradrenergic axon terminals in the rat frontal cortex. Brain Res. 2007;1127:36–44. doi: 10.1016/j.brainres.2006.09.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petwee RG. Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacol Ther. 1998;74:129–180. doi: 10.1016/s0163-7258(97)82001-3. [DOI] [PubMed] [Google Scholar]
- Ranganathan M, D’Souza DC. The acute effects of cannabinoids on memory in humans: a review. Psychopharmacology. 2006;188(4):425–444. doi: 10.1007/s00213-006-0508-y. [DOI] [PubMed] [Google Scholar]
- Riegel AC, Lupica CR. Independent presynaptic and postsynaptic mechanisms regulate endocannabinoid signaling at multiple synapses in the ventral tegmental area. J Neurosci. 2004;24:11070–11078. doi: 10.1523/JNEUROSCI.3695-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbe D, Alonso G, Duchamp F, Bockaert J, Manzoni OJ. Localization and Mechanisms of Action of Cannabinoid Receptors at the Glutamatergic Synapses of the Mouse Nucleus Accumbens. J Neurosci. 2001;21:109–116. doi: 10.1523/JNEUROSCI.21-01-00109.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez J, Mackie K, Pickel V. Ultrastructural Localization of the CB1 Cannabinoid Receptor in μ-Opioid Receptor Patches of the Rat Caudate Putamen Nucleus. J Neurosci. 2001;21:823–833. doi: 10.1523/JNEUROSCI.21-03-00823.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo B, Than M, Thorn D, Wallmichrath I. Analysis of the effects of cannabinoids on synaptic transmission between basket and Purkinje cells in the cerebellar cortex of the rat. J Pharmacol Exp Ther. 2004;310:915–925. doi: 10.1124/jpet.104.066670. [DOI] [PubMed] [Google Scholar]
- Sjostrom PJ, Turrigiano GG, Nelson SB. Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron. 2003;39:641–654. doi: 10.1016/s0896-6273(03)00476-8. [DOI] [PubMed] [Google Scholar]
- Trettel J, Levine ES. Cannabinoids depress inhibitory synaptic inputs received by layer 2/3 pyramidal neurons of the neocortex. J Neurophysiol. 2002;88:534–539. doi: 10.1152/jn.2002.88.1.534. [DOI] [PubMed] [Google Scholar]
- Tsou K, Brown S, Sanudopena M, Mackie K, Walker J. Immuohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience. 1998;83:393–411. doi: 10.1016/s0306-4522(97)00436-3. [DOI] [PubMed] [Google Scholar]
- Tsumoto T. Excitatory amino acid transmitters and their receptors in neural circuits of the cerebral neocortex. Neurosci Res. 1990;9:79–102. doi: 10.1016/0168-0102(90)90025-a. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA. Endocannabinoid signaling in the brain. Science. 2002;296:678–682. doi: 10.1126/science.1063545. [DOI] [PubMed] [Google Scholar]