

Abstract
Genes that are differentially expressed in pancreatic cancers and under epigenetic regulation are of considerable biological and therapeutic interest. We employed global gene expression profiling and epigenetic treatment of pancreatic cell lines including pancreatic cancer cell lines, pancreatic cancer associated fibroblasts, and cell lines derived from non-neoplastic pancreata. We examined expression and epigenetic alterations of Cox-1 and Cox-2 in pancreatic cancers and normal pancreas and performed proliferation, knockdown and co-culture experiments to understand the role of stromal sources of prostaglandins for pancreatic cancers. We identify COX-1 as a gene under epigenetic regulation in pancreatic cancers. We find that COX-1 expression is absent in many pancreatic cancer cells and some of these cancers also lack COX-2 expression. Suspecting that such cancers must rely on exogenous sources of prostaglandins, we show that pancreatic cancer stromal cells, such as fibroblasts express COX-1 and COX-2, are a likely source of prostaglandins for pancreatic cancer cells deficient in cyclooxygenases. Knocking down the prostaglandin transporter, Mrp4, in fibroblasts suppresses the proliferation of co-cultured pancreatic cancer cells lacking cyclooxygenases. Pancreatic cancers that lack cyclooxygenases can utilize exogenous sources of prostaglandins. Blocking Mrp4 may be a useful therapeutic strategy to deplete cyclooxygenase-deficient pancreatic cancers of prostaglandins.
INTRODUCTION
Pancreatic cancer remains the fourth leading cause of cancer-related death in the USA. There are only a few agents that have chemotherapeutic activity against pancreatic cancer cells. One therapeutic target investigated for treatment and prevention of pancreatic and other cancers is the cyclooxygenases. Cyclooxygenases are the rate limiting step in prostaglandin synthesis and are encoded by the Cox genes (COX-1 and COX-2, also known as prostaglandin H synthases (PTGS) or prostaglandin endoperoxide synthases) (figure 1A). COX-1 has been generally considered the ‘constitutive’-gene, expressed in most tissues under basal conditions, while COX-2 is considered the ‘inducible’ gene- 1, undetectable in most normal tissues 2, 3, but highly expressed in a number of human cancers, including pancreaticcancer and its precursors 4–6. Overexpression of COX-2 results in excess prostaglandin E2 (PGE2) production, which promotes cell survival and proliferation, and angiogenesis 7. The importance of cyclooxygenases in cancer development and progression led to many studies examining the role of nonsteroidal anti-inflammatory drugs (NSAIDS) in the prevention and treatment of cancers and precursor neoplasms. Thus, many observational studies have confirmed a 40–50% relative risk reduction of developing colorectal adenomas and cancer, when comparing regular users of aspirin or NSAIDS to non-users 8, 9. Furthermore, randomized, double-blinded, placebo-controlled trials of Cox inhibition in patients with familial adenomatous polyposis (FAP) have shown that these agents induce polyp regression 10–13.
Figure 1.

(A) The arachidonic acid cascade. (B) PCA plots of Affymetrix exon array gene expression of pancreatic cancers and non-neoplastic pancreatic samples. Each data point represents one sample and the ellipse is drawn at two standard deviations around the centroid of the samples; 6 pancreatic cancer lines (blue), 7 pancreatic cancer associated fibroblasts (CAFs) (red), 1 non-neoplastic pancreatic duct line (green), and two non-neoplastic pancreatic fibroblasts (purple). The x-axis represents the first principle component (PC), the y-axis represents the second PC, and the z-axis represents the third PC (C)PTGS1 (Cox-1) expression by Affymetrix exon array. Each cell line was treated with 5Aza-dC (red), TSA (blue), the combination (green) or mock-treated (purple). Each dot represents one sample and each cell line is connected by a solid line. The box plot shows the variance among each type of cell lines. Signal intensity is plotted in log2 scale.
Interestingly, epidemiological studies do not support a role for NSAIDs in the prevention of pancreatic cancer 14, 15. In addition, although in vitro studies suggest that Cox-2 inhibitors are effective against pancreatic cancer cells that express Cox-2 16, clinical trials have not found these agents to improve the treatment of patients with pancreatic cancer 17. The reasons for this apparent lack of benefit are not certain. NSAIDs also have COX independent mechanisms, but these effects are not thought to explain the benefit of these agents in chemoprevention 11, 18–20, 21, 22. NSAID inhibition can shunt arachidonic acid metabolites down the 5-lipoxygenase pathway and 5-lipoxygenase is overexpressed in pancreatic cancers. Indeed, 5-lipoxygenase inhibitors have been evaluated as therapies for pancreatic cancer 23, 24. Pancreatic cancer precursors such as PanINs and IPMNs overexpress Cox-2 25, and Cox-2 inhibitors reduce the development of PanINs in a mutant Kras driven mouse model of pancreatic neoplasia 26. Polymorphisms in COX-2 influence the level of COX-2 transcripts 27 and may contribute to sensitivity to Cox-2 inhibitors, and the development of intestinal polyps, although do not appear to contribute to pancreatic cancer risk 28 (figure 1A).
The putative housekeeping function of Cox-1 suggests an essential role of Cox enzymes and prostaglandins for normal cell functions. Mice with knockout of either Cox-1 or Cox-2 display a variety of phenotypes 29. Cyclooxygenases are also thought to be important in the development of pancreatitis. Cox-2 is overexpressed in chronic pancreatitis tissues and mice lacking Cox-2 develop minimal pancreatitis while mice lacking Cox-1 develop severe pancreatitis 30, 31. Yet interestingly, recent studies indicate that Cox-1 expression is more restricted in normal tissues than previously appreciated 2, 3. Although many cancers overexpress Cox-2, epigenetic silencing of COX-2 occurs in some pancreatic and other cancers 16. During an investigation of genes silenced in pancreatic cancers, we identified cancers lacking Cox-1 expression as well as cancers lacking expression of both Cox-1 and Cox-2. We find that Cox-1 is epigenetically silenced in many pancreatic cancers. We also find evidence that pancreatic cancers lacking Cox enzymes can use stromal fibroblasts as a source of prostaglandins and demonstrate that targeting the prostaglandin transporter, Mrp4 in fibroblasts can diminish the proliferation of pancreatic cancer cells.
MATERIALS AND METHODS
Cell lines and tissue samples
Fourteen human pancreatic cancer cell lines (AsPC1, BxPC3, Capan1, Capan2, CFPAC1, MiaPaCa2, Panc1, su86.86, panc215, A32-1, A38-5, panc2.5, panc2.8 and panc3.014). AsPC1, BxPC3, Capan1, Capan2, CFPAC1, MiaPaCa2, Panc1, su86.86 were obtained from ATCC. Panc215, A32-1, A38-5, panc2.5, panc2.8 and panc3.014 were obtained from the investigator who created them (Dr. James Eshleman, JHU for Panc215, A32-1, A38-5 and Dr. Elizabeth Jaffee for Panc215, A32-1, A38-5). Cancer associated fibroblasts (CAFs) and immortalized normal fibroblasts (SC-2) were established previously in our laboratory 32, 33. Immortalized cell lines, non-neoplastic human pancreatic ductal epithelium (HPDE) and human pancreatic Nestin-expressing cells (HPNE) were generously provided by Dr Ming-Sound Tsao (University of Toronto, Ontario, Canada) and Dr Michel Ouellette (University of Nebraska Medical Center, Omaha, NE), respectively.
Discarded frozen normal and neoplastic tissues were obtained from patients who had undergone pancreatic resection for pancreatic adenocarcinoma or pancreatic neuroendocrine neoplasm at Johns Hopkins Hospital. We included sixteen previously established pancreatic cancer xenografts as described 34. In addition, tissue microarrays (TMAs) of formalin-fixed paraffin-embedded tissues were retrieved from 144 patients who underwent surgical resection at our institution. Specimens were collected and analyzed with the approval of the Johns Hopkins Committee for Clinical Investigation.
Treatment with 5-aza-2′-deoxycytidine (5-aza-dC) and Trichostatin A (TSA)
Cells were treated with 5-aza-dC (Sigma Chemical Co) at 1μmol/L for 4 days and/or 1μmol/L of TSA for 24 hours as previously described35.
RNA isolation
Total RNA from frozen tissues or cell lines was extracted using mirVana miRNA Isolation Kit (Ambion, Austin, TX) following the manufacturer’s protocol. Isolated total RNA were treated with DNA-free kit (Ambion) to eliminate possible DNA contamination.
Affymetrix Exon Arrays
The Affymetrix Exon Array ST 1.0 (Affymetrix, Santa Clara, CA) was used to define gene expression profiles. Using the GeneChip Whole Transcript Sense TargetLabeling Assay, labeling and hybridization was performed following manufacturer’s recommendations. Data analysis was performed using Partek® Genomics Suite v6.3 beta (Partek Inc., St. Louis MO). We are in compliance with the Minimum Information about a Microarray Experiment (MIAME) guidelines and have submitted our microarray data set to the Gene Expression Omnibus (GEO) repository (ref # GSE21163).
Quantitative reverse-transcriptase PCR (qRT-PCR)
2μg of total RNA were reverse transcribed using Superscript® III Reverse Transcriptase and random hexamers (Invitrogen Life Technologies; Carlsbad, CA) for qRT-PCR. COX-1 and COX-2 cDNAs were quantified using SYBR Green PCR Master Mix for SYBR green I or Taqman® Universal PCR Master Mix (Applied Biosystems, Foster City, CA). PCR was performed on an ABI 7300 real-time thermocycler. Primers and probes for COX-1 and COX-2 were as previously described 36. The housekeeping genes GAPDH or PGK1 were used as a reference for SYBR green and 18s-rRNA (Applied Biosystems) was used for Taqman® respectively (supplemental table 1).
Bisulfite modified sequencing and Methylation specific PCR
The methylation status of the 5′ CpG sites of COX-1 was determined by bisulfite modified sequencing (BMS) and methylation-specific PCR (MSP) as previously described 37 (primer sequences are provided in supplemental Table 1). For MSP, by SssI methylase (SSSI)-treated DNA (New England Biochemicals) and whole-genome amplified DNA (WGA) (Qiagen Inc.) were used as controls for methylated and unmethylated DNA, respectively.
Chromatin Immunoprecipitation (Chip)
Chip was performed as previously described 38. Briefly, cells were treated with 1% (v/v) formaldehyde and cross-links were quenched with glycine. Cells were rinsed with ice-cold PBS with protease inhibitors (Roche Applied Science), scraped and collected by centrifugation, before being resuspended in lysis buffer plus protease inhibitors. Chromatin was sheared with the Bioruptor system (Diagenode). Antibodies to the repressive mark H3K27m3 and the active mark, acetylated H3, or normal IgG as a control were used. DNA was PCR amplified quantified using SYBR Green with Cox-1 primers (Fwd CACCAGGCATCAGAAACGTA, Rev CTCCCTCCAGCTGTCACC). The percentage enrichment of immunoprecipitated DNA was calculated input DNA (20ng) for immunoprecipitation is (i), the histone mark (H), and IgG (IgG)), then dCt(H) = Ct(H)–Ct(i), dCt(IgG) = Ct(IgG)–Ct(IgG), and ddCt = dCt(H)–ddCt(IgG).
Immunohistochemistry
Immunohistochemical labeling was performed using HRP EnVision+ System (DAKO Corp.) on TMA slides as previously described39. Deparaffinized slides were subjected to heat-induced epitope retrieval using a steamer and DAKO Target retrieval solution (pH 6.0–6.2; DAKO Corp.). Slides were incubated with rabbit polyclonal anti-ovine Cox-1 (Cat# 160112, Cayman Chemical, Ann Arbor, MI) diluted to 1:1000 at 4°C overnight. Collecting ducts of kidney and in glial cells express Cox-1 and were used as positive controls for inter-slide normalization3. The immunostaining area was categorized into 4 scores as follows: 0, 0–5% of labeled tumor cells; 1, 6%–25%; 2, from 26%–50%; and 3, from 51–75%; 4, above 76%. A score of 0 to 2 was attributed to the immunostaining intensity as follows: 0, no appreciable labeling; 1, mild; 2, strong intensity. The scoring index was determined by multiplying the area score and the intensity score.
Western blotting
Western blotting was performed as previously described40 using primary antibodies; rabbit polyclonal anti-ovine COX-1 diluted at 1:2000, mouse monoclonal anti-human COX-2 diluted at 1:1000 (Cat# 160112, Cayman Chemical), or rabbit polyclonal anti-GAPDH (Sigma).
MTS Assay
Aspirin was used as a COX inhibitor, (0.4–10 μM) (Sigma) and nordihydroguaiaretic acid as a LOX inhibitor, (CalBiochem, La Jolla, CA) were used. 4,000 cells/well) were seeded onto 96-well plates, incubated overnight, washed twice with PBS and serum-starved for 24h before replacing with 1% FCS media containing aspirin (0.4–10μM), NDGA (1.01–100 μM), or DMSO for another 72 h. Cell proliferation was quantified by adding 20 μl of CellTiter AQueous One Solution (Promega) into each well containing 100 μl culture medium, and incubated for 2h at 37 °C. Absorbance at 490 nm was measured using a microplate reader (Perkin Elmer). Dose–response graphs were performed in triplicate and linear regression lines were used to calculate I.C.50 values, the dose required to kill 50% of the cells.
Measurement of prostaglandin E2 (PGE2) levels
1×104 cells/500 μl 1%FCS-DMEM) were seeded into 24-well microplates grown for 48hr and PGE2 in the culture medium determined (Prostaglandin E2 Express EIA kit, Cayman Chemical) according to manufacturer’s instructions.
PGE2 Treatment
10,000 cells/well) were seeded onto 96-well plates and incubated overnight, washed twice with PBS and serum-starved for 48 hr before replacing with medium (1% FCS) containing 1μM or 10μM of PGE2 (P0409, Sigma). Cell proliferation was quantified by MTS assay.
Small Interfering RNA Transfection and Pancreatic Cancer/Fibroblast co-culture
A siRNA targeting MRP4 (SMARTpool, L-007313-00-0005) and a non-targeting control siRNA were obtained from Dharmacon (Lafayette, CO). 1×105 cells/well of CAF19 fibroblasts, were seeded in the lower wells of 24 well plates (BD Biosciences) and incubated for 24 hrs. Cells were transfected with MRP4 siRNA or control siRNA (100 nmol/L) using DharmaFECT4 transfection reagent, incubated for 24hr and subjected to co-culture. RT-PCR primers used to amplify MRP4: (F:CGAGTAGCCATGTGCCATATGA. R: TGACTATCTGGCCTGTGGTTGTCT)
Separately pancreatic cancer cells (1×104), AsPC-1, MiaPaCa2, or BxPC3 were seeded on polycarbonate membrane with 0.45-μm pores in the Transwell® permeable support inserts (Corning Inc.) and serum-starved for 48 hr. Cells were cocultured with fibroblasts for 48 hr in 1% FCS-DMEM with or without PGE2. Culture media PGE2 was quantified as above and cell number quantified using the MTS assay.
Statistical Analysis
Values reported are means ± SD. All data were normally distributed and underwent equal variance testing. Statistical analysis of gene expression array data was determined by Partek® Genomics Suite v6.3B. Raw Affymetrix intensity measurements of all probe sets were background-corrected and normalized by the Robust Multichip Average (RMA) method. Sample relationships were examined using principal components analysis to reveal any technical effects that would encumber the subsequent analysis. Gene expression intensities were summarized by the one-step Tukey’s biweight method. 2-way ANOVA analysis was performed to identify significant expression changes between cancer cells vs. the non-cancer cell lines and 5-aza-dC and/or TSA treated cancer cells vs. untreated cancer cells, based on a fold-change criteria of ±5 fold and a P-value <0.001.
RESULTS
Identification of epigenetically regulated genes
To identify genes that are silenced in pancreatic cancers and regulated by epigenetic mechanisms, we compared the gene expression profiles of 6 pancreatic cancer cell lines (panc215, A32-1, A38-5, panc2.5, panc2.8, and panc3.014), to the non-neoplastic pancreas cell line, HPDE and to 9 pancreatic fibroblast lines including 7 pancreatic cancer associated fibroblast lines and fibroblast lines derived from non-neoplastic pancreas (HPNE, and SC2). We also compared the baseline gene expression of the pancreatic cancer cell lines to expression patterns after treatment with 5-aza-dC alone, TSA alone, and to a combination of 5-aza-dC/TSA. Gene expression profiles were obtained using the Affymetrix Exon Array ST 1.0, which contains1.4 million probe sets representing known full length and alternate spliced mRNAs.
The data quality was visualized using principal components analysis (PCA) using the distribution of probe intensities for all samples (see Materials and methods). For example, the cancer cell lines robustly separated from CAFs, HPNE, and SC-2, but were relatively similar to the non-neoplastic epithelial cell line, HPDE (Figure 1B).
To search for epigenetically silenced genes in pancreatic cancer we identified genes with reduced expression in the pancreatic cancer cell lines relative to the non-neoplastic cell lines, and then merged this list to the list of genes significantly induced by epigenetic drug treatment using Tukey’s biweight method. As shown in the 3-dimensional (3-D) volcano plot (Supplemental Figure 1), subsequent fold-change and ANOVA analyses showed that 702 (3.19%) of 21,980 genes were expressed at significantly lower levels (less than 5-fold lower and P < .001 by ANOVA test) in the 6 pancreatic cancers compared to the non-neoplastic pancreatic samples. Among the 702 genes under-expressed in the pancreatic cancer cell lines, 10 genes were up-regulated (more than 5-fold) in response to the combination epigenetic treatment with (5-aza-dC), a histone deacetylase inhibitor (TSA) in these pancreatic cancer cell lines. This criterion identified TFPI-2, a gene previously identified as epigenetically silenced in pancreatic and other cancers41 (Supplemental Figure 1).
The most interesting of the 10 genes identified was Cox-1 (prostaglandin H synthase-1, PTGS-1). COX-1 expression was absent in most pancreatic cancers by microarray compared to modest expression in HPDE and higher levels in the fibroblast lines (Figure 1C). By array, several pancreatic cancer cell lines demonstrated re-expression by treatment either with the DNA methyltransferase inhibitor, 5-azadeoxycytidine (5aza-dC) or with the histone deacetylase inhibitor, TSA, (Figure 1C, red or blue dots, respectively, compared to purple dots). Treatment of cancer cell lines with a combination of 5-aza-dC and TSA further increased the re-expression of COX-1 in five out of the six pancreatic cancer cell lines studied compared to 5-aza-dC and/or TSA treatment alone (Figure 1C, green dots).
We also found that either 5-azaC or the combination of 5-aza and TSA treatments increased COX-2 mRNA expression in five out of six pancreatic cancer cell lines (3.69-fold change, p=0.0014, Supplemental Figure 2). Since previous studies have examined the epigenetic regulation of Cox-2, we focused our attention on Cox-1 expression.
We confirmed the differential expression of Cox-1 treatment in response to 5-aza-dC or TSA treatment by qRT-PCR. Pancreatic cancer cell lines treated with these agents either alone or in combination led to robust induction of COX-1 mRNA in several cancer lines after drug treatment, but not in the non-neoplastic pancreatic samples, HPDE and HPNE (Figure 2A). The effect of epigenetic treatment on COX-2 expression was also examined (see Supplemental Figure 2).
Figure 2.
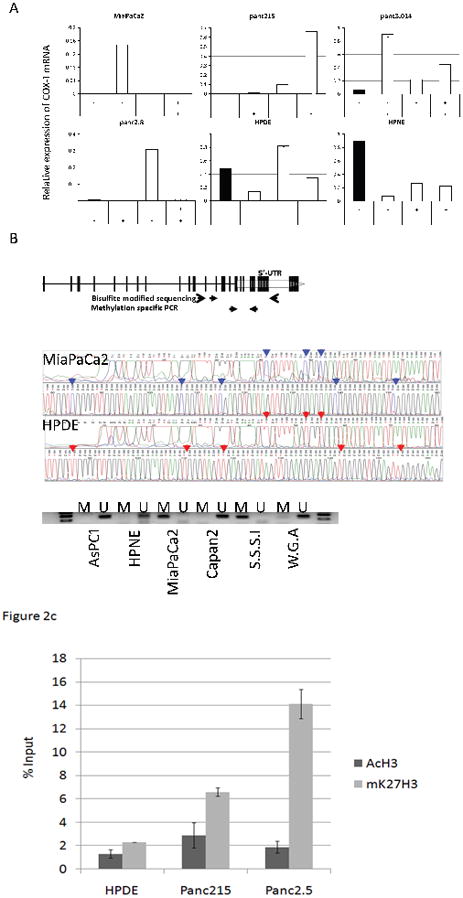
(A) Effect of Epigenetic treatment with 5-aza-dC and/or TSA on pancreatic Cox-1expression. Cox-1 mRNA was measured by qRT-PCR. Ct values were normalized by GAPDH and calibrated by the COX-1 level of BxPC-3. The data is represented as means ± SD (n=3) (B) Primers used in bisulfite sequencing and MSP. A broken arrow represents 5′ untranslated region (UTR) of Cox-1. Vertical lines represent each CpG dinucleotides and black solid arrows indicate primer locations. A representative bisulfite sequencing chromatograph is shown in a middle panel and MSP in the lower panel. After bisulfite treatment methylated cytosines remain unmodified (C) (represented by blue peaks and arrows), but unmethylated cytosines are converted to thymines (T) (red peak and arrow). By MSP, MiaPaCa2 and the S.S.S.I methylase treated DNA are methylated (M) but other samples are unmethylated (U). (C) Quantitative chromatin immunoprecipitation-PCR analysis of the Cox-1 promoter using antibodies to acetylated H3 and mK27H3. Y axis reveals the percentage enrichment of Cox-1 DNA with immunoprecipitation, relative to input DNA.
We then verified the DNA methylation of the COX-1 promoter by bisulfite sequencing. The COX-1 promoter has a CpG rich region (Length=343, %GC=62.8, Observed CpG/Expected CpG=0.652) around the 5’-UTR. Sequencing of 12 CpG sites within product 400 bp region (−188 to +212 of the transcription start site) (Figure 2B) revealed all CpG sites to be unmethylated (converted to T by bisulfite modification) in the non-neoplastic COX-1 expressing cell line HPDE, whereas the non-Cox-1 expressing cell line MiaPaCa2 was highly methylated in all of the CpGs sequenced. Several other non-COX-1 expressing pancreatic cancer cell lines (e.g. Panc215, panc2.8, and panc3.014) had unmethylated CpGs.
Further analysis of the COX-1 promoter for aberrant methylation using MSP revealed that MiaPaCa2 was the only completely methylated pancreatic cancer cell line of 11 lines examined, consistent with bisulfite sequencing data (representative data is shown in Figure 2B). MSP analysis of Cox-1 in pancreatic cancer xenografts and normal pancreas tissues found 3 of the 16 cancers (18%) but 0 of 7 normal pancreata were methylated (data not shown).
We next performed quantitative chromatin immunoprecipitation (Chip)-PCR of the Cox-1 promoter using antibodies to active and inactive chromatin marks. The repressive mark was more abundant by Chip PCR in the pancreatic cancer cell lines Panc215 and Panc2.5 that lacked Cox-1 expression compared to the Cox-1 expressing cell line, HPDE (see below for expression). In contrast, there was no significant difference in the level of acetylated H3 in any of the 3 cell lines (see Figure 2C).
COX-1- and COX-2- mRNA expression in pancreatic cancer cells
Since different pancreatic cancers are known to overexpress as well as silence Cox-2, we further examined Cox-1 (Figure 3A) and Cox-2 expression (Figure 3B) in pancreatic cancer cell lines vs. non-neoplastic pancreatic cells using qRT-PCR. Surprisingly, 5 of 11 pancreatic cancer cell lines examined lacked expression of both COX1 and COX-2 RNA. All the non-neoplastic pancreatic fibroblast lines expressed COX-1 and COX-2. We also examined our Serial Analysis of Gene Expression (SAGE) data for COX-1 and COX-2 in normal pancreatic duct and 24 pancreatic cancers 42. There were no detectable SAGE tags for COX-1 in primary pancreatic duct whereas COX-2 tags were detectable (124 of ~1.5 million tags). Eleven pancreatic cancers did not have any COX-1 SAGE tags (out of >1 million tags sequenced per sample). Of the remaining 13 pancreatic cancers analyzed, only 9 had more than 5 COX-1 tags. COX-2 expression was more variable with several pancreatic cancers expressing high levels of COX-2 and 8 of 24 expressing little or no COX-2 (<6 tags per sample).
Figure 3.

(A) Expression level of Cox-1 was measured by qRT-PCR (mean ± SD (n=3), upper panel) and Western blot (lower panel). (B) Expression level of Cox-2 was measured by qRT-PCR (mean ± SD (n=3), upper panel) and Western blot (lower panel). For qRT-PCR, Ct values were normalized by GAPDH and calibrated by BxPC3 expression. For protein expression analysis by western blot, GAPDH (~36 kDa) was used as a reference protein. Note due to insufficient sample there is no protein data for CAF19.
Western blotting confirmed the RNA expression patterns (Figure 3A and 3B, lower panels). Bands of the expected size (~70 kDa) were consistent with qRT-PCR results, except that cell lines with low levels of RNA by qRT-PCR did not have detectable protein by Western.
Immunohistochemical analysis of pancreatic Cox-1 expression
To confirm the RNA expression patterns of Cox-1, we performed immunohistochemical labeling of primary pancreatic adenocarcinomas and corresponding non-neoplastic pancreatic tissues on TMAs. Recent evidence suggests that COX-1 is expressed in certain organs such as the collecting ducts in the kidney, astrocytes in the central nervous system, and endocrine cells in crypts of intestines3. We found these tissues (kidney, small intestine, and brain) expressed Cox-1 on our TMAs and served as an internal control. In pancreas tissues, positive labeling was clearly detectable in interstitial cells and in spindle shaped cells in pancreatic duct, but expression was usually undetectable in other pancreatic duct epithelial cells (Figure 4).
Figure 4.

Cox-1 expression in primary pancreatic tissues by immunohistochemistry:
(A) and (B): Cancers cells lacking Cox-1 labeling, with labeling in cancer associated fibroblasts (CAFs) and lymphocytes. (C). A cancer with Cox-1 labeling both in cancer cells and in CAFs. D. Normal pancreatic ductal epithelial cells do not show labeling with Cox-1 apart from a few strongly positive ductal endocrine cells (20× magnification).
Of 140 primary pancreatic cancers evaluated, only 14 cases (10%) showed diffuse immunolabeling for Cox-1 (labeling score ≥4; Figure 4B and 4C), whereas expression was only focal or completely absent throughout the tumor in the remaining score <4; Figure 4A). In contrast, cancer associated fibroblasts and stromal inflammatory cells displayed moderate to strong cox-1 labeling (Figure 4A and 4B).
PGE2 levels of pancreatic cell lines
Since Cox expression is important for the generation of prostanoids, we examined PGE2 levels in a panel of pancreatic cancer cell lines as well as control lines. As expected, pancreatic cancers lacking both Cox1 and Cox2 expression (e.g. A32-1, A38-5, Panc 2.5, AsPC1 and MiaPaca2) very low levels of PGE2 in their culture media (< 100pg/ml, Figure 5A). We suspect that these very low levels of PGE2 detectable in cancer cell lines lacking both cox-1 and cox-2 expression are due to the 1% fetal calf serum required to grow the cells. In contrast, the COX1 and COX-2-expressing cell line BxPC3 and the pancreatic cancer associated fibroblast line, CAF-19 had higher levels of PGE2 (282.25±23.69 pg/ml and 1140.69±89.57 pg/mL, respectively). Pancreatic cancer cell lines expressing either Cox1 or Cox2 or both (Figure 3, e.g. Panc215 and Capan-1) had higher levels of PGE2 overall in their culture media than cell lines without Cox expression (Figure 5A).
Figure 5.

(A) Prostaglandin E2 (PGE2) concentrations in conditioned media from pancreatic cell lines. (B) Pancreatic cancer cell proliferation in response to PGE2 by MTS colorimetric assay.
Effects of cyclooxygenase and lipoxygenase inhibitors on pancreatic cancer cells
We considered that sensitivity to cyclooxygenase and lipoxygenase inhibitors might vary by their COX gene expression status, but found no evidence for differences among 8 pancreatic cancer cell lines in response to COX and LOX inhibitors by their baseline Cox expression as measured by (I.C.50) determined using MTS assay (Supplemental Figure 3).
Effect of PGE2 on pancreatic cancer cell lines
Pancreatic cancer cell lines without COX expression (AsPC1 and MiaPaCa2) treated with PGE2 (1 μM and 10 μM) had significant increases in cell proliferation compared to untreated cells, whereas the COX-expressing line BxPC3 showed smaller but still significant increases in proliferation (Figure 5B).
Knockdown of the prostaglandin transporter MRP4
Since pancreatic cancer associated fibroblasts, such as CAF19, produce and release PGE2, we hypothesized that blocking the release of PGE2 from pancreatic cancer associated fibroblasts could be a selective strategy to therapeutically target pancreatic cancer cells that fail to produce PGE2 because of their lack of cyclooxygenases. The main prostaglandin transporter is MRP443. We therefore knocked down MRP4 in CAF19 cells. Using a Dharmacon Smartpool of MRP4 siRNAs we were able to knockdown MRP4 with 80–90% efficiency (Figure 6A). Cell culture media from untreated CAF19 cells had higher PGE2 levels than culture media from MRP4 knockdown fibroblasts (Figure 6B). To determine if fibroblast PGE2 contributed to cancer cell growth, we co-cultured 3 pancreatic cancer cell lines (AsPC1, BxPC3 and MiaPaCa2) with cancer associated fibroblasts with and without siRNA mediated knockdown of MRP4 (CAF19-KD and CAF19-control, respectively). Media from MRP4-knockdown CAFs had lower levels of PGE2 when co-cultured with the pancreatic cancer cell lines lacking cyclooxygenases (AsPC1 and MiaPaca2), but not for the cyclooxygenase-expressing line BxPC3 (Figure 6C). Moreover, when co-cultured with the knockdown CAFs, AsPC1 and MiaPaCa2 both showed a markedly slower growth (Figure 6D, dark grey bars) that was overcome by the addition of exogenous PGE2 in the culture media (Figure 6D, light grey bars).
Figure 6.

(A) Effect of MRP4 siRNA on MRP4 mRNA expression in CAF19 cells. (B) Effect of MRP4 knockdown on PGE2 concentrations. (C) PGE2 concentrations in pancreatic cancers and CAF co-cultures. ASPC1 and MiaPaca2 lack Cox expression while BxPC3 expresses Cox-1 and Cox-2. KD=MRP4 knockdown by siRNA in cancer associated fibroblasts. 1% DMEM containing 10mM PGE2 was used as a positive control. (D) Pancreatic cancer cell proliferation in response to co-culture with CAFs transfected or not with MRP4 siRNA.
DISCUSSION
Using a global DNA methylation profiling strategy, we find that Cox-1 is epigenetically regulated in pancreatic cancers, and is not expressed in most normal pancreatic duct cells. Furthermore, as epigenetic silencing of Cox-2 can also occur during pancreatic cancer development 16, some pancreatic cancers evolve to lack both cyclooxygenases. Since stromal cells adjacent to infiltrating pancreatic cancers such as fibroblasts, endothelial cells 44 and inflammatory cells express high levels of Cox-1 and Cox-2, these cells are a likely source of prostaglandins for pancreatic cancer cells deficient in cyclooxygenases. Although pancreatic cancer cells lacking cyclooxygenases do not produce PGE2, they still proliferate in response to PGE2. Previous studies have identified cancers lacking Cox-2 expression, but to our knowledge no prior studies have recognized that cancer cells could be deficient in both Cox-1 and Cox-2.
Until recently, COX-1 had been reported to be the constitutive, and COX-2, the inducible cyclooxygenase. It is now clear that many tissues lack Cox-1 expression and based on our gene expression and immunohistochemical data it appears that pancreatic duct cells rely on Cox-2 rather than Cox-1 to produce prostaglandins 6, 45.
The lack of expression of Cox-1 in pancreatic duct may help explain epidemiological studies demonstrating that aspirin, which is primarily a Cox-1 inhibitor, does not prevent the development of pancreatic cancer, but does reduce colon cancer incidence and mortality(16).
We hypothesized that the lack of Cox expression in some pancreatic cancer cells may render them dependent on exogenous PGE2 such as from stromal fibroblasts. Prostaglandins are secreted by most cells, and act as autocrine- and paracrine-signaling molecules requiring controlled release, uptake, and metabolism to initiate and terminate signaling 7. The main efflux transporter of PGE2 is the multidrug resistance–associated protein (Mrp)-4 which exports PGE2 to the extracellular milieu) 46, 47. Exported PGE2 can bind to transmembrane prostaglandin receptors. The other main transmembrane prostaglandin transporter (PGT) carriesPGE2 into the cytoplasm. Interestingly, Mrp4 is overexpressed in colorectal and othercancers 48. In model systems, MRP4 knockout results in a pronounced reduction in extracellular PGE2, and Mrp4is inhibited by certain nonsteroidal anti-inflammatory drugs 46, 49. We find that stromal fibroblasts supply PGE2 to pancreatic cancer cells and blocking Mrp4-dependent PGE2 excretion from fibroblasts reduces the proliferation of pancreatic cancer cells lacking cyclooxygenases. Thus, inhibition of Mrp4 may represent a useful treatment strategy for pancreatic cancer cells deficient in cyclooxygenases. Inhibiting Mrp4 could be a more effective strategy for targeting the Cox pathway in pancreatic cancers since Cox inhibitors will not have any direct effect on pancreatic cancer cells lacking Cox expression. Cox inhibitors could still target stromal fibroblast Cox expression but since fibroblasts express both Cox-1 and Cox-2, non-selective Cox inhibitors would be required, with greater potential for systemic toxicity. The provision of prostaglandins by stromal fibroblasts may partly explain why Cox-2 inhibitors have not been shown to be effective in treating pancreatic cancers 50. It is not known if inhibiting PGE2 production by blocking Mrp4 would be a more targeted and therapeutically safer approach to blocking prostaglandin E2 effects. An alternative approach to inhibiting the cancer promoting effects of PGE2 would be to inhibit PGE2 receptors 51–53, but since there are 4 PGE2 receptors, EP1, EP2, EP3 and EP4, this might require blocking multiple receptors.
In summary, we find evidence for epigenetic regulation of Cox-1 in pancreatic cancers and demonstrate that most pancreatic cancers lack Cox-1 expression. Indeed, some pancreatic cancers are devoid of either Cox-1 or Cox-2 expression rendering dependent on exogenous sources of prostaglandins. Inhibiting the efflux of prostaglandins from stromal fibroblasts by blocking the prostaglandin transporter, MRP4, inhibits the proliferation of pancreatic cancer cells lacking cyclooxygenases.
Supplementary Material
Supplemental Table 1
Sequence information of primers used in MSP, BMS, and qRT-PCR
Supplemental Table 2
Genes silenced and significantly induced by >5-fold with combined 5-azacytidine and TSA in pancreatic cancer cell lines. (Fold change numbers are log2).
3-D volcano plots of pancreatic cell line gene expression. The left panel (A) displays gene expression (log2 fold change) between pancreatic cancer lines vs. non-neoplastic cells (X-axis) and the p-value (Y-axis) derived from a 2-way ANOVA model. In right panel is the differential gene expression (log2 fold change) between 5-aza-dC/TSA treated pancreatic cancers vs. untreated pancreatic cancers is shown in a Z-axis and represented by color in the scale bar. The vertical green lines represent a 5 fold change between treated and untreated cells and the horizontal green lines represent the p< 0.001 significance threshold set to compensate for multiple testing errors. Based on these filters only 10 genes (including PTGS1 and TFPI2) were identified as epigenetically silenced and having epigenetic silencing in pancreatic cancers compared to non-neoplastic pancreatic cells
PTGS2 (Cox-2) expression by Affymetrix exon array. Each cell line was treated with 5Aza-dC (red), TSA (blue), the combination (green) or mock-treated (purple). Each dot represents one sample and each cell line is connected by a solid line. The box plot shows the variance among each type of cell lines. Signal intensity is plotted in log2 scale.
Correlation between COX inhibitor or LOX inhibitor and relative expression level of either of COXs is represented in four scatter plots. Cell viability after each COX and LOX inhibitor treatment was determined by MTS assay. I.C.50 was determined as shown in material and methods. R value was obtained by Pearson’s correlation model. Each data point represents one cell line and labeled above X-axis. X-axis is the relative COXs mRNA level (log10 scale). The level of BxPC3 as a calibrator is one. Y-axis is I.C.50 (μM) of each inhibitor.
Acknowledgments
Grant support: Supported by the NCI grants Specialized Programs of Research Excellence in Gastrointestinal Malignancies (CA62924), R01CA120432, the Jimmy V foundation, the Michael Rolfe Foundation.
References
- 1.Smith WL, Garavito RM, DeWitt DL. Prostaglandin endoperoxide H synthases (cyclooxygenases)-1 and -2. J Biol Chem. 1996;271:33157–60. doi: 10.1074/jbc.271.52.33157. [DOI] [PubMed] [Google Scholar]
- 2.Parente L, Perretti M. Advances in the pathophysiology of constitutive and inducible cyclooxygenases: two enzymes in the spotlight. Biochem Pharmacol. 2003;65:153–9. doi: 10.1016/s0006-2952(02)01422-3. [DOI] [PubMed] [Google Scholar]
- 3.Zidar N, Odar K, Glavac D, Jerse M, Zupanc T, Stajer D. Cyclooxygenase in normal human tissues - is COX-1 really a constitutive isoform, and COX-2 an inducible isoform? J Cell Mol Med. 2008 doi: 10.1111/j.1582-4934.2008.00430.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Molina MA, Sitja-Arnau M, Lemoine MG, Frazier ML, Sinicrope FA. Increased cyclooxygenase-2 expression in human pancreatic carcinomas and cell lines: growth inhibition by nonsteroidal anti-inflammatory drugs. Cancer Res. 1999;59:4356–62. [PubMed] [Google Scholar]
- 5.Tucker ON, Dannenberg AJ, Yang EK, et al. Cyclooxygenase-2 expression is up-regulated in human pancreatic cancer. Cancer Res. 1999;59:987–90. [PubMed] [Google Scholar]
- 6.Matsubayashi H, Infante JR, Winter J, et al. Tumor COX-2 expression and prognosis of patients with resectable pancreatic cancer. Cancer Biol Ther. 2007;6:1569–75. doi: 10.4161/cbt.6.10.4711. [DOI] [PubMed] [Google Scholar]
- 7.Wang D, Mann JR, DuBois RN. The role of prostaglandins and other eicosanoids in the gastrointestinal tract. Gastroenterology. 2005;128:1445–61. doi: 10.1053/j.gastro.2004.09.080. [DOI] [PubMed] [Google Scholar]
- 8.Thun MJ, Namboodiri MM, Heath CW. Aspirin use and reduced risk of fatal colon cancer. N Engl J Med. 1991;325:1593–6. doi: 10.1056/NEJM199112053252301. [DOI] [PubMed] [Google Scholar]
- 9.Hawk ET, Levin B. Colorectal Cancer Prevention. J Clin Oncol. 2005;23:378–91. doi: 10.1200/JCO.2005.08.097. [DOI] [PubMed] [Google Scholar]
- 10.Giardiello FM, Hamilton SR, Krush AJ, et al. Treatment of Colonic and Rectal Adenomas with Sulindac in Familial Adenomatous Polyposis. N Engl J Med. 1993;328:1313–6. doi: 10.1056/NEJM199305063281805. [DOI] [PubMed] [Google Scholar]
- 11.Labayle D, Fischer D, Vielh P, et al. Sulindac causes regression of rectal polyps in familial adenomatous polyposis. Gastroenterology. 1991;101:635–9. doi: 10.1016/0016-5085(91)90519-q. [DOI] [PubMed] [Google Scholar]
- 12.Nugent KP, Farmer KC, Spigelman AD, Williams CB, Phillips RK. Randomized controlled trial of the effect of sulindac on duodenal and rectal polyposis and cell proliferation in patients with familial adenomatous polyposis. Br J Surg. 1993;80:1618–9. doi: 10.1002/bjs.1800801244. [DOI] [PubMed] [Google Scholar]
- 13.Steinbach G, Lynch PM, Phillips RKS, et al. The Effect of Celecoxib, a Cyclooxygenase-2 Inhibitor, in Familial Adenomatous Polyposis. N Engl J Med. 2000;342:1946–52. doi: 10.1056/NEJM200006293422603. [DOI] [PubMed] [Google Scholar]
- 14.Jacobs EJ, Connell CJ, Rodriguez C, Patel AV, Calle EE, Thun MJ. Aspirin use and pancreatic cancer mortality in a large United States cohort. J Natl Cancer Inst. 2004;96:524–8. doi: 10.1093/jnci/djh084. [DOI] [PubMed] [Google Scholar]
- 15.Schernhammer ES, Kang JH, Chan AT, et al. A prospective study of aspirin use and the risk of pancreatic cancer in women. J Natl Cancer Inst. 2004;96:22–8. doi: 10.1093/jnci/djh001. [DOI] [PubMed] [Google Scholar]
- 16.Sato N, Maehara N, Goggins M. Gene expression profiling of tumor-stromal interactions between pancreatic cancer cells and stromal fibroblasts. Cancer Res. 2004;64:6950–6. doi: 10.1158/0008-5472.CAN-04-0677. [DOI] [PubMed] [Google Scholar]
- 17.El-Rayes BF, Zalupski MM, Shields AF, et al. A phase II study of celecoxib, gemcitabine, and cisplatin in advanced pancreatic cancer. Invest New Drugs. 2005;23:583–90. doi: 10.1007/s10637-005-1028-z. [DOI] [PubMed] [Google Scholar]
- 18.Cruz-Correa M, Hylind LM, Romans KE, Booker SV, Giardiello FM. Long-term treatment with sulindac in familial adenomatous polyposis: a prospective cohort study. Gastroenterology. 2002;122:641–5. doi: 10.1053/gast.2002.31890. [DOI] [PubMed] [Google Scholar]
- 19.Giardiello FM, Hamilton SR, Krush AJ, et al. Treatment of colonic and rectal adenomas with sulindac in familial adenomatous polyposis. N Engl J Med. 1993;328:1313–6. doi: 10.1056/NEJM199305063281805. [DOI] [PubMed] [Google Scholar]
- 20.Phillips RK, Wallace MH, Lynch PM, et al. A randomised, double blind, placebo controlled study of celecoxib, a selective cyclooxygenase 2 inhibitor, on duodenal polyposis in familial adenomatous polyposis. Gut. 2002;50:857–60. doi: 10.1136/gut.50.6.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Young SD, Whissell M, Noble JC, Cano PO, Lopez PG, Germond CJ. Phase II clinical trial results involving treatment with low-dose daily oral cyclophosphamide, weekly vinblastine, and rofecoxib in patients with advanced solid tumors. Clin Cancer Res. 2006;12:3092–8. doi: 10.1158/1078-0432.CCR-05-2255. [DOI] [PubMed] [Google Scholar]
- 22.Anderson KE, Johnson TW, Lazovich D, Folsom AR. Association between nonsteroidal anti-inflammatory drug use and the incidence of pancreatic cancer. J Natl Cancer Inst. 2002;94:1168–71. doi: 10.1093/jnci/94.15.1168. [DOI] [PubMed] [Google Scholar]
- 23.Tong WG, Ding XZ, Witt RC, Adrian TE. Lipoxygenase inhibitors attenuate growth of human pancreatic cancer xenografts and induce apoptosis through the mitochondrial pathway. Mol Cancer Ther. 2002;1:929–35. [PubMed] [Google Scholar]
- 24.Hennig R, Ding XZ, Tong WG, et al. 5-Lipoxygenase and leukotriene B(4) receptor are expressed in human pancreatic cancers but not in pancreatic ducts in normal tissue. Am J Pathol. 2002;161:421–8. doi: 10.1016/S0002-9440(10)64198-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Niijima M, Yamaguchi T, Ishihara T, et al. Immunohistochemical analysis and in situ hybridization of cyclooxygenase-2 expression in intraductal papillary-mucinous tumors of the pancreas. Cancer. 2002;94:1565–73. doi: 10.1002/cncr.10358. [DOI] [PubMed] [Google Scholar]
- 26.Funahashi H, Satake M, Dawson D, et al. Delayed progression of pancreatic intraepithelial neoplasia in a conditional Kras(G12D) mouse model by a selective cyclooxygenase-2 inhibitor. Cancer Res. 2007;67:7068–71. doi: 10.1158/0008-5472.CAN-07-0970. Epub 2007 Jul 24. [DOI] [PubMed] [Google Scholar]
- 27.Cipollone F, Toniato E, Martinotti S, et al. A polymorphism in the cyclooxygenase 2 gene as an inherited protective factor against myocardial infarction and stroke. Jama. 2004;291:2221–8. doi: 10.1001/jama.291.18.2221. [DOI] [PubMed] [Google Scholar]
- 28.Brosens LAA, Iacobuzio-Donahue C, Keller JJ, Hustinx SR, Carvalho R, Morsink FH, Hylind L, Offerhaus GJ, Giardiello FM, Goggins M. Increased Cyclooxygenase-2 Expression in Duodenal compared to colonic tissues in Familial Adenomatous Polyposis andrelationship to the -765G>C COX-2 polymorphism. Clin Cancer Res. 2005;11:4090–6. doi: 10.1158/1078-0432.CCR-04-2379. [DOI] [PubMed] [Google Scholar]
- 29.Loftin CD, Tiano HF, Langenbach R. Phenotypes of the COX-deficient mice indicate physiological and pathophysiological roles for COX-1 and COX-2. Prostaglandins Other Lipid Mediat. 2002;68–69:177–85. doi: 10.1016/s0090-6980(02)00028-x. [DOI] [PubMed] [Google Scholar]
- 30.Ethridge RT, Chung DH, Slogoff M, et al. Cyclooxygenase-2 gene disruption attenuates the severity of acute pancreatitis and pancreatitis-associated lung injury. Gastroenterology. 2002;123:1311–22. doi: 10.1053/gast.2002.35951. [DOI] [PubMed] [Google Scholar]
- 31.Koliopanos A, Friess H, Kleeff J, Roggo A, Zimmermann A, Buchler MW. Cyclooxygenase 2 expression in chronic pancreatitis: correlation with stage of the disease and diabetes mellitus. Digestion. 2001;64:240–7. doi: 10.1159/000048868. [DOI] [PubMed] [Google Scholar]
- 32.Walter K, Omura N, Hong SM, Griffith M, Goggins M. Pancreatic cancer associated fibroblasts display normal allelotypes. Cancer Biol Ther. 2008;7:882–8. doi: 10.4161/cbt.7.6.5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walter K, Omura N, Hong SM, et al. Overexpression of Smoothened activates the Sonic Hedgehog signaling pathway in pancreatic cancer associated fibroblasts. Clin Cancer Res. 2010 doi: 10.1158/1078-0432.CCR-09-1913. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ueki T, Toyota M, Sohn T, et al. Hypermethylation of multiple genes in pancreatic adenocarcinoma. Cancer Res. 2000;60:1835–9. [PubMed] [Google Scholar]
- 35.Sato N, Fukushima N, Maitra A, et al. Discovery of novel targets for aberrant methylation in pancreatic carcinoma using high-throughput microarrays. Cancer Res. 2003;63:3735–42. [PubMed] [Google Scholar]
- 36.Sugimoto T, Koizumi T, Sudo T, et al. Correlative expression of cyclooxygenase-1 (Cox-1) and human epidermal growth factor receptor type-2 (Her-2) in endometrial cancer. Kobe J Med Sci. 2007;53:177–87. [PubMed] [Google Scholar]
- 37.Omura M, Li C-P, Li A, et al. Genome-wide profiling of methylated promoters in pancreatic adenocarcinoma. Cancer Biol Ther. 2008;7:1146–56. doi: 10.4161/cbt.7.7.6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vincent A, Perrais M, Desseyn JL, Aubert JP, Pigny P, Van Seuningen I. Epigenetic regulation (DNA methylation, histone modifications) of the 11p15 mucin genes (MUC2, MUC5AC, MUC5B, MUC6) in epithelial cancer cells. Oncogene. 2007;26:6566. doi: 10.1038/sj.onc.1210479. [DOI] [PubMed] [Google Scholar]
- 39.Infante JR, Matsubayashi H, Sato N, Tonascia J, Klein AP, Riall TA, Yeo C, Iacobuzio-Donahue C, Goggins M. Peritumoral fibroblast SPARC expression and patient outcome with resectable pancreatic adenocarcinoma. J Clin Onc. 2007;25:319–25. doi: 10.1200/JCO.2006.07.8824. [DOI] [PubMed] [Google Scholar]
- 40.Salaria SN, Illei P, Sharma R, et al. Palladin is Overexpressed in the Non-Neoplastic Stroma of Infiltrating Ductal Adenocarcinomas of the Pancreas, but is only Rarely Overexpressed in Neoplastic Cells. Cancer Biol Ther. 2007;6:324–8. doi: 10.4161/cbt.6.3.3904. Epub 2007 Mar 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sato N, Parker AR, Fukushima N, et al. Epigenetic inactivation of TFPI-2 as a common mechanism associated with growth and invasion of pancreatic ductal adenocarcinoma. Oncogene. 2005;24:850–8. doi: 10.1038/sj.onc.1208050. [DOI] [PubMed] [Google Scholar]
- 42.Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–6. doi: 10.1126/science.1164368. Epub 2008 Sep 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin ZP, Zhu YL, Johnson DR, et al. Disruption of cAMP and prostaglandin E2 transport by multidrug resistance protein 4 deficiency alters cAMP-mediated signaling and nociceptive response. Mol Pharmacol. 2008;73:243–51. doi: 10.1124/mol.107.039594. Epub 2007 Oct 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Raut CP, Nawrocki S, Lashinger LM, et al. Celecoxib inhibits angiogenesis by inducing endothelial cell apoptosis in human pancreatic tumor xenografts. Cancer Biol Ther. 2004;3:1217–24. doi: 10.4161/cbt.3.12.1221. Epub 2004 Dec 9. [DOI] [PubMed] [Google Scholar]
- 45.Langenbach R, Loftin CD, Lee C, Tiano H. Cyclooxygenase-deficient mice. A summary of their characteristics and susceptibilities to inflammation and carcinogenesis. Ann N Y Acad Sci. 1999;889:52–61. doi: 10.1111/j.1749-6632.1999.tb08723.x. [DOI] [PubMed] [Google Scholar]
- 46.Reid G, Wielinga P, Zelcer N, et al. The human multidrug resistance protein MRP4 functions as a prostaglandin efflux transporter and is inhibited by nonsteroidal antiinflammatory drugs. Proc Natl Acad Sci U S A. 2003;100:9244–9. doi: 10.1073/pnas.1033060100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rius M, Thon WF, Keppler D, Nies AT. Prostanoid transport by multidrug resistance protein 4 (MRP4/ABCC4) localized in tissues of the human urogenital tract. J Urol. 2005;174:2409–14. doi: 10.1097/01.ju.0000180411.03808.cb. [DOI] [PubMed] [Google Scholar]
- 48.Maubon N, Le Vee M, Fossati L, et al. Analysis of drug transporter expression in human intestinal Caco-2 cells by real-time PCR. Fundam Clin Pharmacol. 2007;21:659–63. doi: 10.1111/j.1472-8206.2007.00550.x. [DOI] [PubMed] [Google Scholar]
- 49.Lin ZP, Zhu YL, Johnson DR, et al. Disruption of cAMP and prostaglandin E2 transport by multidrug resistance protein 4 deficiency alters cAMP-mediated signaling and nociceptive response. Mol Pharmacol. 2008;73:243–51. doi: 10.1124/mol.107.039594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jimeno A, Amador ML, Kulesza P, et al. Assessment of celecoxib pharmacodynamics in pancreatic cancer. Mol Cancer Ther. 2006;5:3240–7. doi: 10.1158/1535-7163.MCT-06-0565. [DOI] [PubMed] [Google Scholar]
- 51.Zheng Y, Ritzenthaler JD, Sun X, Roman J, Han S. Prostaglandin E2 stimulates human lung carcinoma cell growth through induction of integrin-linked kinase: the involvement of EP4 and Sp1. Cancer Res. 2009;69:896–904. doi: 10.1158/0008-5472.CAN-08-2677. Epub 2009 Jan 27. [DOI] [PubMed] [Google Scholar]
- 52.Subbaramaiah K, Hudis C, Chang SH, Hla T, Dannenberg AJ. EP2 and EP4 receptors regulate aromatase expression in human adipocytes and breast cancer cells. Evidence of a BRCA1 and p300 exchange. J Biol Chem. 2008;283:3433–44. doi: 10.1074/jbc.M705409200. Epub 2007 Dec 14. [DOI] [PubMed] [Google Scholar]
- 53.Fulton AM, Ma X, Kundu N. Targeting prostaglandin E EP receptors to inhibit metastasis. Cancer Res. 2006;66:9794–7. doi: 10.1158/0008-5472.CAN-06-2067. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1
Sequence information of primers used in MSP, BMS, and qRT-PCR
Supplemental Table 2
Genes silenced and significantly induced by >5-fold with combined 5-azacytidine and TSA in pancreatic cancer cell lines. (Fold change numbers are log2).
3-D volcano plots of pancreatic cell line gene expression. The left panel (A) displays gene expression (log2 fold change) between pancreatic cancer lines vs. non-neoplastic cells (X-axis) and the p-value (Y-axis) derived from a 2-way ANOVA model. In right panel is the differential gene expression (log2 fold change) between 5-aza-dC/TSA treated pancreatic cancers vs. untreated pancreatic cancers is shown in a Z-axis and represented by color in the scale bar. The vertical green lines represent a 5 fold change between treated and untreated cells and the horizontal green lines represent the p< 0.001 significance threshold set to compensate for multiple testing errors. Based on these filters only 10 genes (including PTGS1 and TFPI2) were identified as epigenetically silenced and having epigenetic silencing in pancreatic cancers compared to non-neoplastic pancreatic cells
PTGS2 (Cox-2) expression by Affymetrix exon array. Each cell line was treated with 5Aza-dC (red), TSA (blue), the combination (green) or mock-treated (purple). Each dot represents one sample and each cell line is connected by a solid line. The box plot shows the variance among each type of cell lines. Signal intensity is plotted in log2 scale.
Correlation between COX inhibitor or LOX inhibitor and relative expression level of either of COXs is represented in four scatter plots. Cell viability after each COX and LOX inhibitor treatment was determined by MTS assay. I.C.50 was determined as shown in material and methods. R value was obtained by Pearson’s correlation model. Each data point represents one cell line and labeled above X-axis. X-axis is the relative COXs mRNA level (log10 scale). The level of BxPC3 as a calibrator is one. Y-axis is I.C.50 (μM) of each inhibitor.