

Abstract
Histone deacetylase (HDAC) inhibitors are emerging as effective therapies in the treatment of cancer, and the role of HDACs in the regulation of promoters is rapidly expanding. GRP78/BiP is a stress inducible endoplasmic reticulum (ER) chaperone with anti-apoptotic properties. We present here the mechanism for repression of the Grp78 promoter by histone deacetylase 1 (HDAC1). Our studies reveal that HDAC inhibitors specifically induce GRP78, and the induction level is amplified by ER stress. Through mutational analysis, we have identified the minimal Grp78 promoter and specific elements responsible for HDAC-mediated repression. We show the involvement of HDAC1 in the negative regulation of the Grp78 promoter not only by its induction in the presence of the HDAC inhibitors trichostatin A and MS-275, but also by exogenous overexpression and siRNA knockdown of specific HDACs. We present the results of chromatin immunoprecipitation analysis that reveals the binding of HDAC1 to the Grp78 promoter before but not after ER stress. Furthermore, overexpression of GRP78 confers resistance to HDAC inhibitor induced apoptosis in cancer cells and, conversely, suppression of GRP78 sensitizes them to HDAC inhibitor. These results define HDAC inhibitors as new agents that upregulate GRP78 without concomitantly inducing the ER or heat shock stress response, and suppression of GRP78 in tumors may provide a novel, adjunctive option to enhance anti-cancer therapies that utilize these compounds.
Keywords: histone deacetylase, HDAC inhibitor, GRP78/BiP, transcription, cancer
Introduction
Histone deacetylases (HDACs) represent a promising new target for drug development in cancer therapy. There are four classes of HDACs, which group the eleven known HDACs according to their homology with yeast deacetylases (1). Compounds that inhibit HDACs are currently being tested in clinical trials as primary or adjunctive anti-cancer agents. Most of the classic HDAC inhibitors result in global acetylation of histone and non-histone proteins, and changes in gene expression of approximately 2%-5% of target genes (1). The list of non-histone proteins known to be affected by differential acetylation is constantly expanding and it includes: E2F1, HSP90, p53, Rb and YY1, among others (2). The diversity of differentially acetylated proteins may in part explain why treatment of cancer cells with HDAC inhibitors leads to increased differentiation, growth arrest, and apoptosis. While HDAC inhibitors confer therapeutic benefits, they may inadvertently trigger additional events that render the cancer cells resistant to the treatment, such as induction of the pro-survival protein GRP78 (3).
GRP78, also referred to as BiP or HSPA5, is a stress-inducible chaperone located primarily in the endoplasmic reticulum (ER) (4, 5). When cells experience ER stress, such as ER Ca2+ efflux or accumulation of unfolded proteins in the ER, a signal transduction cascade is initiated that triggers the unfolded protein response (UPR) (6). The UPR mitigates ER stress by transiently arresting general translation, degrading misfolded proteins, and upregulating ER chaperones and folding enzymes. If these protective measures are insufficient to reduce ER stress, the UPR will activate apoptotic cell death. A major target of the UPR is the induction of GRP78, which assists in protein folding and assembly, targets misfolded proteins for degradation, binds ER Ca2+, and controls the activation of transmembrane ER stress sensors. As a major pro-survival arm of the UPR, GRP78 is emerging as a pivotal regulator of cancer growth and resistance (7, 8). GRP78 transcript and protein level is elevated in a wide variety of cancers, due to intrinsic factors such as altered glucose metabolism in cancer cells and extrinsic factors such as the abnormal pathological conditions of the tumor microenvironment that is known to trigger the UPR (9, 10). Through inactivation of the Grp78 allele in endogenous cancer mouse models, GRP78 is shown to be critical for tumor progression (11, 12). Further, utilizing overexpression or siRNA-mediated knockdown studies establish that GPR78 confers resistance to a variety of anti-cancer therapy, in tumor as well as tumor associated endothelial cells (7, 13-17).
The transcriptional activation of Grp78 is mediated primarily by highly conserved elements in its promoter referred to as the endoplasmic reticulum stress response element (ERSE), which serves as binding sites for a multitude of transcription factors, along with chromosomal modifications at the promoter region (18-20). The ERSE is evolutionarily conserved among eukaryotes and consists of 19 nucleotides characterized by a unique tripartite design. The three parts consist of: 1) a CCAAT motif that binds NF-Y; 2) a 9-nucleotide GC-rich domain that binds TF-II-I; and 3) a 5-nucleotide sequence that binds YY1 and ATF6. The typical mammalian Grp78 promoter contains three such ERSEs, and collectively they contribute to ER stress-induced transcriptional activation (18, 19).
Interestingly, the induction of GRP78 by an HDAC inhibitor was first discovered in normal rat brain tissue after prolonged treatment with valproic acid, a mood stabilizing and anti-convulsant drug later found to cause HDAC inhibition (3). However, the mechanism of induction of GRP78 by HDAC inhibitors is not known and its relevance in anti-cancer therapy has not been characterized. In this report we utilized a panel of cancer cell lines as well as xenograft tumor model to examine modulation of GRP78 expression by HDAC inhibitors. We report here our findings that characterize the specific mechanisms in HDACi-mediated transcriptional induction of GRP78 and the modulation of HDACi-induced apoptosis by GRP78, providing the proof-of-principle that adjunctive therapies targeting GRP78 could potentially sensitize cancer cells to HDAC inhibitor therapy.
Materials and Methods
Cell Lines and Drug Treatment Conditions
HCT116 and HT29 cell lines were provided by Dr. Robert Ladner, U87 and LN229 cell lines obtained from the American Tissue Culture Collection. The cells were propagated in DMEM supplemented with 10% fetal bovine serum, 100 units/mL penicillin, 0.1 mg/ml streptomycin at 37°C, and 5% CO2. Thapsigargin (Tg) and Trichostatin A (TSA) were obtained from Sigma-Aldrich. Tg was dissolved in DMSO at 1 mg/ml and added to cell culture at 300 nM final concentration. TSA was dissolved in DMSO at 300 mM and added to cell culture at 500 nM final concentration. MS-275, purchased from CalBiochem (La Jolla, CA), was dissolved in DMSO at 1 mM and added to cell culture at the concentrations indicated.
Immunoblots and Antibodies
Fifty micrograms of total cell lysate prepared in radioimmunoprecipitation assay buffer were processed for Western blot analysis as described (21). The antibodies against GRP78, CHOP, β-actin, GAPDH, HSP70, His, PDI, PARP (Santa Cruz Biotechnology, Inc.), caspase-7 (BD PharMingen), GRP94 (Stressgen) and FLAG (Sigma) were used per manufacturer’s recommendations. The secondary antibodies were coupled to horseradish peroxidase, and were detected by chemiluminescence using SuperSignal West substrate (Pierce). Each immunoblot was performed from 2 to 5 times.
Plasmid Construction
All luciferase reporters utilize the pGL3Basic vector backbone. For the construction of Grp78 promoter deletion mutants, the -169Luc plasmid was used as a template in a PCR reaction with the downstream primer 5’-ATCTCGAGGTCCAAGTCAGTGTAGTCACAGCCAGTA-3’ which contains an Xho1 site at the 3’ end. The following upstream primers were used, and introduced an Nhe1 site on the 5’ end of the fragment and an Xho1 site on the 3’ end: for -144Luc 5’-ATGCTAGCTTGGTGGCATGAACCAACCAGCG-3’; for -112Luc, 5’-ATGCTAGCGAGTAGCGAGTTCACCAATCGGAG-3’; for -79Luc, 5’-ATGCTAGCACGGGGCTGCGGGGAGGAT; and for -52Luc ATGCTAGCCGAGTCGGCGACCGGC. The PCR product was digested with Nhe1 and Xho1, purified, and ligated into pGL3Basic at the same sites to obtain the final plasmids. The ERSE mutants were generated by site-directed mutagenesis utilizing the following primers: ERSE2m, 5’-GAGGCCGCTTCTGATCGGCAGCG-3’ and ERSE1m, 5’-TGGCCGCTGGTCAGTTCATGCCAC-3’. For the construction of -112Luc ERSE1 mutants, the following primers were used in site-directed mutagenesis: mut 1, 5’CACtgATCGGAGGCCTCCACGACGG; mut2, 5’CACCAATCGGAtta CTCCACGACGG; mut 3, 5’CACCAATCGGAGGCCTaacaGACGG; mut 4, 5’CACCAATCGGAttaCTaacaGACGG. All plasmids were confirmed by sequencing in each direction.
Transient Transfection and Luciferase Assay
The Flag-tagged HDAC expression vectors and pGL191were kindly provided by Dr. Ed Seto (U Southern Florida). All transfections were carried out using Lipofectamine 2000 (Invitrogen) per the manufacturer’s guidelines. TSA, MS-275, or 100% DMSO was added to the culture medium 24 h after transfection, and cells were incubated at 37°C for an additional amount of time before harvesting with 50 μl of lysis buffer (Promega). Protein concentrations were determined using BPA reagent (Bio-Rad), and the relative luciferase activity was measured with firefly assay reagent (Promega) and a luminometer. Each transfection was repeated from 2 to 5 times.
Small Interfering RNA
The siRNA against Grp78 is 5’-ggagcgcauugauacuagadTdT-3’ and was previously described (22). The siRNAs against human HDAC1, 2 and 3 were purchased from Applied Biosystems. The control siRNA was purchased from Molecular Probes/Invitrogen and contains an alexa-fluor fluorescent tag to determine transfection efficiency. HeLa cells were grown to 80% confluence in 12-well dishes and transfected with 10 nM of either control siRNA or gene-specific siRNA, and with or without 0.2 μg of -112 Luc or pGL181sx reporter plasmids using Lipofectamine 2000 transfection reagent. The cells were harvested 48-72 h after transfection. The experiments were repeated 2 to 3 times.
RT-PCR Analysis
Total RNA was extracted using TRIZOL (Invitrogen) following the manufacturer’s instructions. First-strand cDNA was synthesized with the Superscript First-Strand Synthesis System for RT-PCR (Invitrogen). To detect human mRNA, PCR was performed using the following primers: Xbp-1 spliced 5’ GGTCTGCTGAGTCCGCAGCAGG-3’; GRP78, 5’-AACCATACATTCAAGTTGATATTGGAGGTG-3’; CHOP, 5’- GCAAGAGGTCCTGTCTTCAGATG-3’ HDAC1, 5’-GTTCTCCTGGTAGTGTATGC-3’; HDAC2, 5’-GTAATTCCACAGTTCTGACA-3’; HDAC3, 5’-CCGAAATGTTGCCCGCTGCTG-3’.
Chromatin Immunoprecipitation
Stably transfected HeLa cells grown to 80% confluence in 15 cm plates were subjected to chromatin immunoprecipitation (ChIP) assay using antibodies against HDAC1 (Upstate) as previously described (19). Purified DNA from the input and IP samples were subjected to quantitative PCR with SYBR green and the results were analyzed per manufacturer’s recommendations (Perkin-Elmer). The primers used were as follows: for Grp78, primer “c”, 5’-CATTGGTGGCCGTTAAGAATGACCAG (forward) and primer “d”, 5’-AGTATCGAGCGCGCCGTCGC (reverse), and RVprimer3 (primer “a”) 5’-CTAGCAAAATAGGCTGTCCC-3’ and GLprimer2 (primer “b”) 5’-GGAAGACGCCAAAAACATAAAG-3’.
Xenograft Mouse Models
Nude athymic mice were injected subcutaneously with 2×106 MDA-MB-435 human breast cancer cells in 0.1 ml cell culture medium. The injection of TSA at 500 μg/kg body weight or the solvent DMSO for 4 days started once palpable tumors were formed.
Apoptotic Assay
For mitochondrial membrane potential staining by the JC-1 assay, the Mitochondrial Permeability Transition Detection Kit (Immunochemistry, Bloomington, MN) was used per manufacturer’s protocol. Following wash with PBS, the cells were examined under a fluorescence microscope. Each assay was performed in triplicate.
Results
Histone Deacetylase Inhibitors Specifically Induce GRP78 but not Other Stress Response Pathways
GRP78 protein levels were examined by Western blots in a panel of cancer cell lines, which were either untreated or treated with 500 nM TSA, a pan-HDAC inhibitor, for 24 h. GRP78 induction was observed in varying levels in all five cancer cell lines tested which include the human colon cancer cell lines HCT116 (wt p53), and HT29 (p53 mutant), the human breast adenocarcinoma cell line MCF-7 and the human glioma cell lines LN229 and U87 (Fig. 1A). To test whether TSA induces GRP78 at transcript level, HCT116 cells were treated with TSA, and as positive control, treated with the 300 nM of the ER stress inducer Tg. As expected, Tg treatment results in upregulation of Grp78 transcript, as well as the transcript level of UPR targets CHOP and the spliced form of XBP-1 (Fig. 1B). In contrast, while TSA upregulated Grp78 mRNA level, CHOP and spliced XBP-1 mRNA levels were unaffected by TSA. To investigate the effect of TSA on tumor cells in vivo, nude mice with MDA/MB-435 xenografts were injected with TSA for 4 days. Western blot analysis of the tumor lysates shows induction of GRP78 but not the stress-inducible heat-shock protein HSP70 (Fig. 1C). The tumor microenvironment is subject to ER stress (10). To test whether ER stress modulates HDAC inhibitors induction of GRP78, HCT116 cells were pre-treated with increasing amounts of the class I HDAC-inhibitor MS-275 for 24 h, followed by Tg treatment. At 0.5 μM of MS-275, an additive effect was observed; whereas at 1.5 μM of MS-275, a synergistic effect was observed (Fig. 1D).
Figure 1. Induction of GRP78 by HDAC inhibitors in cancer cell lines and in xenograft models.
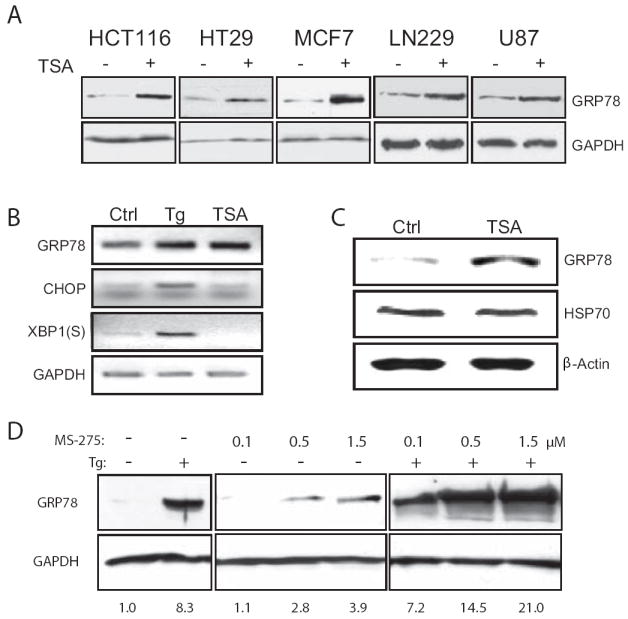
A, the cancer cell lines were either untreated (-) or treated (+) with TSA for 24 h and GRP78 protein levels were assessed by Western blot, with GAPDH levels serving as a loading control. B, HCT116 cells were treated with either TSA for 12 h or Tg for 4 h and the RNA was isolated and used in the RT-PCR reaction to determine the mRNA level of the UPR indicators Grp78, CHOP and the spliced (S) form of XBP-1. C, xenograft tumors were generated through subcutaneously injection of the human breast cancer cell line MDA-MB-435 in nude mice. Following DMSO (Ctrl) or TSA injection for 4 days, the tumors were harvested and Western blots performed to determine levels of GRP78 and HSP70, with β-actin as a loading control. D, HCT116 cells were either non-treated (-), treated with increasing concentrations of MS-275 for 24 h as indicated, or in combination with Tg for an additional 16 h. GRP78 levels were detected by Western blots with GAPDH as control. The fold increase of GRP78 under each condition after normalization against GAPDH is indicated below each lane.
Identification of the HDAC Inhibitor Response Elements in the Grp78 Promoter
Transcriptional induction of the rat Grp78 promoter primarily requires 170 base pair (bp) of the promoter sequence upstream of the TATA element (18). Within this region are three ERSEs, with ERSE1 being the most proximal to the TATA element (Fig. 2A). To map the location of the HDAC inhibitor response element, luciferase reporter plasmids -169Luc, -144Luc, -112Luc, -79 Luc containing three, two, one or no ERSE respectively, were constructed. Additionally, -52Luc, containing only the sequence immediately downstream of the TATA element to the transcription start site, was constructed. These were transfected into HeLa cells and 24 h later were either untreated, or treated with either TSA or MS-275. As shown in Fig. 2A, the -112Luc promoter was the minimal promoter fragment sufficiently inducible at 6- to 7.5-fold over control levels, and this was similar to the level of induction observed in the -144Luc and -169Luc plasmids. The -79Luc and -52Luc showed no response to HDAC inhibitor treatment. Additionally, the results obtained with TSA directly parallel to those obtained with MS275.
Figure 2. Mutational analysis to determine HDAC inhibitor response element in the Grp78 promoter.

A, luciferase reporter plasmids containing 5’ deletion or specific ERSE mutations of the rat Grp78 promoter as indicated were transiently transfected into HeLa cells, and luciferase activities were monitored 18 h after TSA or MS-275 treatment. For inactivation of ERSE, the CCAAT sequence was mutated. The luciferase activity for each construct in non-treated cells was set as 1, and the fold induction by TSA or MS-275 was plotted with standard deviations. B, the minimal HDAC inhibitor-inducible Grp78 promoter -112Luc was used as a template for successive mutation of putative transcription factor binding sites within the ERSE. The putative transcription factor binding sites are indicated. Following transfection and treatment with TSA or MS-275, the luciferase activities were determined. The fold induction was plotted with standard deviations.
Previously, it was determined that a 2 bp mutation of the CCAAT sequence site within the ERSE inactivates its transcriptional activity (18). To assess the contribution of the individual ERSEs, plasmids containing mutated CCAAT sequence of specific ERSEs were constructed (Fig. 2A). We observed that inactivation of ERSE2 in -169(ERSE2m) did not have any effect whereas inactivation of ERSE1 with intact ERSE2 and ERSE3 in -169(ERSE1m) substantially reduced induction of the Grp78 promoter by both TSA and MS-275. Similarly, inactivation of ERSE1 with intact ERSE2 in -144(ERSE1m) resulted in minimal induction by TSA or MS-275 (Fig. 2A). Collectively, these results establish that ERSE1 is essential for Grp78 induction by HDAC inhibitors, and that class 1 HDAC is likely to be responsible for suppression of the Grp78 promoter.
To determine the specific elements within the tripartite-structured ERSE1 responsible for the induction of the Grp78 promoter by HDAC inhibitors, the -112Luc plasmid was used as a template for creating plasmids containing mutations that prevented the binding of transcription factors with specific affinity for elements within ERSE1. The constructs mut1, mut2 and mut3 contained mutations in the NF-Y, TF-II-I/Sp and YY1/ATF6 binding sites, respectively (Fig. 2B). An additional mutant, mut 4, contained mutations in both the YY1/ATF6 and TFII-I/Sp binding sites. Transfections were carried out under similar conditions to Fig 2A. We observed that the CCAAT sequence representing NF-Y binding site is most important for HDAC inhibitors-induced transcription of the -112Luc promoter fragment, as its mutation (mut 1) abolished the promoter induction by both TSA and MS-275 (Fig. 2B). Mutation of the TF-II-1/Sp site (mut 2) was without effect and a 30% reduction of induction was observed for mutation of the YY1/ATF6 site (mut 3). Interestingly, when these sites were mutated in combination (mut 4), a 70% reduction was observed.
Grp78 Promoter Repression is Mediated by HDAC1
Our observation in transfection assays that MS-275, an HDAC inhibitor that only blocks HDAC1, -2 and -3 at the concentrations used in our experiments (23), is able to induce the Grp78 promoter similar to the level of TSA, provides the first hint that this class of HDACs may be responsible for Grp78 promoter repression. To identify the HDAC involved, HeLa cells were transfected with flag-tagged HDAC1, -2 and -3 expressing plasmids along with -169Luc, -112Luc, -79Luc and the luciferase activity were determined. As a control, the HDAC3-repressible luciferase reporter plasmid pGL191 (24) was analyzed in parallel. Prior to transfection, the expression level of each of the HDAC was determined to be similar by Western blot analysis (Fig. 3A insert). For each HDAC, varying doses were tested for effect on the promoter activity (Fig. 3A). For the Grp78 promoter, overexpression of HDAC1 at the higher doses substantially repressed both the -112Luc and -169Luc reporter activity. No change was seen in the -79Luc and pGL191 activity for HDAC1, and neither promoter responded to HDAC2 overexpression. The Gdf11 promoter (pGL191) was suppressed by overexpression of HDAC3 in a dosage dependent manner, as was expected (24), but the Grp78 promoter did not. These results show that HDAC1 is a repressor of the Grp78 promoter activity.
Figure 3. Identification of HDAC1 as a repressor of Grp78 promoter activity.

A, flag-tagged HDAC1, 2 and 3 expression plasmids at increasing amounts (μg) as indicated were transfected into HeLa cells along with either -79Luc, -112Luc, -169Luc or the HDAC3-repressed human GDF11 promoter reporter plasmid pGL191Luc. The transfected HDAC levels were determined by Western blot using anti-flag antibody (inset). The cells were harvested 72 h after transfection. The relative luciferase activity was plotted with standard deviations. B, HeLa cells were transfected with siRNA against HDACs 1, 2 or 3 and incubated for 48 h, harvested, and the RNA was isolated and subjected to quantitative RT-PCR to determine mRNA levels of each HDAC in comparison to control siRNA-treated cells. C, HeLa cells were transfected with siRNA against HDACs 1, 2 or 3, along with reporter plasmids -112Luc, -79Luc or pGL191Luc. The cells were harvested after 72 h and luciferase activity was determined. Fold induction in comparison to control siRNA is shown with standard deviations.
To confirm that HDAC1 is necessary for repression of the Grp78 promoter, siRNA oligos directed against human HDAC1, 2, or 3 were transfected them into HeLa cells along with the Grp78 promoter reporter plasmids. The pGL191 Gdf11 reporter was also transfected as a control. The siRNAs against HDAC 1, -2 and -3 sufficiently inhibited the expression their respective targets as determined by quantitative RT-PCR (Fig. 3B). In agreement with the data showing that overexpression of HDAC1 represses -112Luc, depletion of HDAC1 but not HDAC 2 or 3 increased the activity of the -112Luc. Additionally, the pGL191 Gdf11 promoter responded to the depletion of HDAC3 but not HDAC1 or 2, as expected (Fig. 3C). These findings identify HDAC1 as the primary HDAC responsible for transcriptional repression on the Grp78 promoter and it acts through the sequence spanning -112 and -79, which contains ERSE1.
HDAC1 Differentially Occupies the Grp78 Promoter at the CCAAT Element in ERSE1
One mechanism for the repression of the Grp78 promoter by HDCA1 is that it binds to ERSE1 and inhibits acetylation of transcription factors and/or chromatin associated with the Grp78 promoter. When the Grp78 promoter is activated by Tg or TSA, this suppression is relieved through removal of HDAC1 binding to the promoter element. One explanation why CCAAT mutation of ERSE1 abolishes induction by HDAC inhibitors is that it is required for HDAC1 binding. To test these predictions, ChIP assays for HDAC1 were performed in HeLa cells stably transfected with -112Luc or mut1Luc, where the CCAAT sequence was mutated. Prior to immunoprecipitation with antibody against human HDAC1, the transfected cells were either untreated, or treated with TSA for 12 h or Tg for 4 h. The isolated DNA was amplified with primers matching pGL3 basic that either spanned the region encompassing the Grp78 promoter insert of the -112Luc and mut1Luc, (labeled “a” and “b”), or the sequence just upstream of ERSE3 and just downstream of the TATA box (labeled “c” and “d”), with the latter set of primers detecting HDAC1 binding to the endogenous Grp78 promoter in the transfected cells (Fig. 4A). We observed HDAC1 binding to the -112Luc promoter before but not after TSA or Tg treatment, and this binding is abolished by a mutation in the NF-Y binding site in mut1Luc (Fig. 4B). Concurrently, HDAC1 binds to the endogenous Grp78 promoter in both the -112Luc and the m1Luc stably transformed cells but binding was no longer detected after treatment with TSA or Tg (Fig. 4C).
Figure 4. The NF-Y binding site in ERSE1 is required for HDAC1 binding to the Grp78 promoter.

A, schematic drawing of the luciferase reporter constructs (-112 and mut 1 Luc) and the endogenous Grp78 promoter. The positions of the primer sets used for the ChIP assays are indicated. B, HeLa cells stably transfected with either -112Luc or mut1Luc were either non-treated (Ctrl) or treated with TSA for 12 h or Tg for 4 h and subjected to ChIP assay. Chromatin was immunoprecipitated with anti-HDAC1 antibody or IgG, and the purified DNA fraction was assayed by quantitative PCR with primers specific for detection of HDAC1 binding to plasmid DNA (a/b). Results are shown as percentage of input corrected by IgG, with standard error of the mean. C same as (B) except the primers (c/d) were used to detect HDAC1 binding to the endogenous Grp78 promoter in cells stably transfected with either -112Luc or mut 1 Luc.
Overexpression of GRP78 Confers Resistance to TSA-induced Apoptosis
TSA has been reported to induce apoptosis through the signaling cascade of Fas/FasL-mediated extrinsic and mitochondria-mediated intrinsic caspase pathways (25). To determine whether GRP78 overexpression would protect cells against TSA-induced apoptosis, His-tagged GRP78 expression vector was transfected into 293T cells. Apoptotic cells were identified using mitochondria membrane potential assay. As confirmed by Western blots, ectopic expression of GRP78 increased the total pool of GRP78 without affecting the expression level of other ER chaperones such as GRP94 and PDI (Fig. 5A). In cells transfected with the empty vector, over 90% of the cells entered apoptosis after TSA treatment (Fig. 5B and C). In cells transfected with increasing amounts of His-GRP78 expression vector, the percentage of cells in apoptosis decreased in a dose-dependent manner (Fig. 5B and C).
Figure 5. Overexpression of GRP78 protects 293T cells from TSA-induced apoptosis.
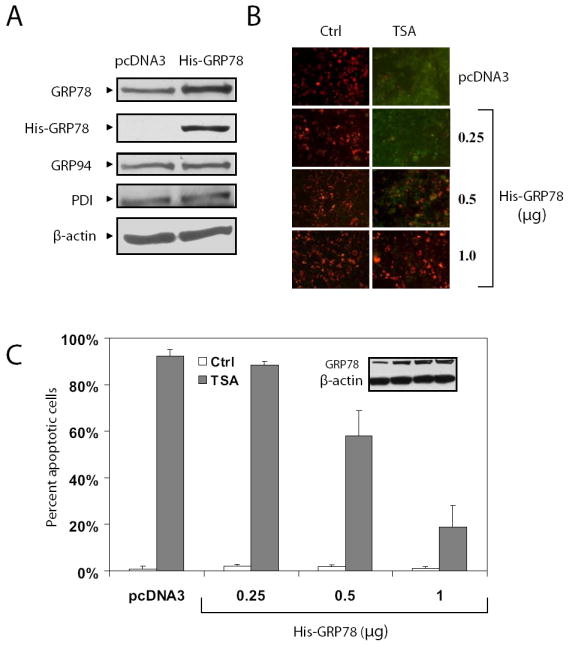
A, cell lysates prepared from 293T cells transfected with either 1.0 μg of a plasmid expressing His-tagged GRP78 or same amount of empty vector pcDNA3 were subjected to Western blot with anti-KDEL, anti-His, and anti-β-actin antibodies. The anti-KDEL antibody recognized the C-terminal KDEL motif of GRP78, GRP94 and protein disulphide isomerase (PDI). B, empty vector pcDNA3 or increasing amounts of the plasmid expressing His-GRP78 (0.25, 0.5 and 1.0 μg) as indicated were transfected to 293T cells. Empty vector was added to adjust the total amount of plasmids to be the same. Twenty-four h after transient transfection, the cells were treated with 500 nM TSA for 48 h and then subjected to mitochondrial membrane potential staining using the JC-1 assay, which detects cells at early stage of apoptosis. Red fluorescence indicates viable cells and green apoptotic cells. C, the percent of apoptotic cells under each condition in (B) was quantitated and plotted against the transfected amount of GRP78. The open and solid bars represent no treatment and TSA treatment respectively. The standard deviations are shown.
Knockdown of GRP78 Induction Sensitizes Cancer Cells to TSA-induced Apoptosis
Since up-regulation of GRP78 is a potential mechanism for resistance to TSA-induced apoptosis, we determined whether suppression of GRP78 expression can sensitize cancer cells to TSA-mediated apoptosis. Annealed siRNA oligos against Grp78 (siGrp78) or a random control (siCtrl) were transfected into the breast carcinoma cell line MDA-MB-435 prior to treatment with 400 nM TSA for 24 h. Apoptotic cells were identified using mitochondria membrane potential assay. The MDA-MB-435 cells were resistant to this TSA treatment regimen as evidenced by the low percent (<10%) of apoptotic cells in cells treated with siCtrl (Fig. 6A). However, when GRP78 expression was knockdown by siGrp78, about 75% of the cells entered apoptosis (Fig. 5A). A similar experiment was carried out in colon carcinoma HCT116 cells, and PARP cleavage was used for detection of apoptosis. Prior to TSA treatment, no PARP cleavage was detected. In the cells where Grp78 was targeted by siGrp78, there was significant cleavage of PARP after TSA treatment compared to the cells transfected with siCtrl (Fig. 5B). Therefore, in 2 different cancer cell lines, using two different assays for detection of apoptosis, knockdown of GRP78 sensitizes the cancer cells to TSA-induced apoptosis.
Figure 6. Knockdown of GRP78 sensitizes cancer cells to HDAC inhibitors-mediated apoptosis.

A, MDA-MB-435 cells were transfected with siRNA against GRP78 (siGrp78) or a random control (siCtrl), and 48 h later, were either non-treated (-) or treated (+) with TSA for 24 h. The upper panel shows GRP78 levels in the transfected cells with β-actin as loading control. Lower panel shows the percent of apoptotic cells under each condition as determined by mitochondrial membrane potential staining using the JC-1 assay. B, HCT116 cells were subjected to the same conditions as (A) and the cell lysates were assayed for PARP cleavage by Western blot. The normal length (116 kDa) and apoptotic, cleaved forms (85 kDa) of PARP are indicated. GAPDH was used as a control.
Discussion
HDAC inhibitors represent a new class anti-cancer compounds with great therapeutic potential, with the HDAC inhibitor SAHA being the first HDAC inhibitor approved by the FDA for the treatment of cutaneous T-cell lymphoma (26). Here we show that in a wide variety of cancer cells ranging from colon, breast to brain tumor, HDAC inhibitors TSA and MS-275 induce the UPR target GRP78, a major anti-apoptotic protein representing the pro-survival arm of the UPR. Interestingly, the action of the HDAC inhibitors is distinct from ER stress inducers, which in addition to inducing GRP78 also induces the UPR signaling pathways, as exemplified by CHOP induction and generation of the spliced form of XBP-1 (6). The observation that HSP70, a major indicator of cytosolic stress, is not induced by HDAC inhibitors further suggests the upregulation of GRP78 by HDAC inhibitors is not a result of major cellular stress. In addition to cells in culture, GRP78 is induced by HDAC inhibitors in xenograft models, as well as biopsies from breast cancer patients undergoing TSA treatment (our unpublished results). Because of the clinical significance of HDAC inhibitors in cancer treatment and the emerging role of GRP78 as a major effector of drug resistance (7, 8), it is important to understand the how and which HDAC represses the Grp78 promoter and the functional consequence of GRP78 induction to HDAC inhibitors therapy in cancer cells.
Here we identified ERSE1, the most proximal ERSE to the TATA element of the Grp78 promoter, as the most critical for the HDAC inhibitor response, and within the ERSE tripartite structure the CCAAT motif as the essential control element. Other sequences within the ERSE may also contribute partially to the regulation and that await future investigation. The CCAAT sequence, which in the case of Grp78 promoter binds the transcription factor NF-Y (18, 19), are critical elements for HDAC inhibitor mediated induction of the GADD45, MDR1 and GTPase RhoB promoters, among others (1). Through the use of the class I HDAC inhibitor MS-275, overexpression and specific knockdown of individual HDACs, we identified HDAC1 as the key repressor of the Grp78 promoter. This is an important finding since while a multitude of activating transcription factors for the Grp78 promoter has been identified, including NF-Y, ATF6, YY1 and TF-II-1 (19), little is known on how the Grp78 promoter is maintained at low basal level in non-stressed cells. The discovery here HDAC1 binds the Grp78 promoter constitutively under normal culture conditions but departs from the promoter when it is activated provides the first evidence that acetylation inhibition is a novel mechanism for suppression of this multifunctional protein. Interestingly, it has been reported that HDAC1 regulates itself by self-mediated repression, utilizing a NF-Y site along with a distil Sp-1 site to its own promoter (27).
While the precise mechanism whereby HDAC1 regulates the Grp78 promoter awaits further investigation, possible candidates for HDAC1 include the chromatin and transcription factors associated with the Grp78 promoter. Our mutational analysis of the ERSE1 shows that the NF-Y binding site is required for HDAC1 binding to the Grp78 promoter, suggesting that NF-Y mediates binding of HDAC1, which through protein protein interaction, exerts its effect on the transcription factors and chromatin that associates with the Grp78 promoter. Consistent with this notion, NF-Y, Sp proteins and YY1 have been reported to be targets for HDAC modifications (28-30). As for the chromatin, recently, it has been determined through single molecule footprinting that the 350 bp region upstream of the transcription initiation site spanning the three ERSEs of the Grp78 promoter is constitutively depleted of nucleosomes, however, nucleosome footprints are detected immediately downstream of the transcription initiation site, as well as further upstream of promoter (28). These chromatin structures, as well as the ERSE binding factors, are prime targets of HDAC1 modification although this remains to be determined.
GRP78 promotes tumor growth through enhancing cell proliferation and tumor angiogenesis, and at the same time, inhibits stress-induced apoptosis (11). The ability of GRP78 to block apoptosis includes its binding and inactivation of pro-apoptotic components such as BIK and caspase-7 that locate to the ER, as well as suppression of induction of CHOP, which mediates the apoptotic arm of the UPR (13, 14). Recently, GRP78 is also identified as a regulator of autophagy due to its role in maintaining ER integrity and homeostasis (31). The potent pro-survival property of GRP78 explains why it is found to confers drug resistance to a wide variety of cancer cells, as well as dormant cancer cells and blood vessels associated with tumor (7, 8, 15-17). Anti-angiogenesis therapy, while promising as an anti-cancer therapy, inadvertently induces GRP78 in the treated tumor due to nutrient and oxygen deprivation, leading to the prediction that GRP78 induction by these agents may result in drug resistance (9). Here we show that GRP78 overexpression leads to resistance of TSA induced apoptosis. Our results with siRNA against GRP78 show that lowering GRP78 levels significantly increases the apoptotic effects of the HDAC inhibitor TSA. Compounds and agents that can specifically block GRP78 expression and/or its activity have been reported and are being evaluated (8, 32-37). Their use as an adjunct to HDAC-inhibitor therapy to reduce resistance warrants vigorous investigation. Conversely, agents that induce GRP78 have been reported to be neuroprotective (38). That may explain the therapeutic benefits of valporic acid and suggest that other HDAC inhibitors that upregulate GRP78 expression or activity may be potential agents for organ or tissue protection during stress.
Acknowledgments
We thank Dr Edward Seto for the generous gift of reagents and Dr. Robert Ladner and for cell lines and helpful discussions.
Grant Support: NCI grants CA027607 and CA111700 (ASL).
Abbreviations
- ChIP
chromatin immunoprecipitation
- ER
endoplasmic reticulum
- ERSE
endoplasmic reticulum stress response element
- HDAC
histone deacetylase
- HDAC1
histone deacetylase 1
- Tg
thapsigargin
- TSA
trichostatin A
- UPR
unfolded protein response
References
- 1.Glozak M, Seto E. Histone deacetylases and cancer. Oncogene. 2007;26:5420–32. doi: 10.1038/sj.onc.1210610. [DOI] [PubMed] [Google Scholar]
- 2.Yang X, Seto E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene. 2007;26:5310–8. doi: 10.1038/sj.onc.1210599. [DOI] [PubMed] [Google Scholar]
- 3.Wang J, Bown C, Young L. Differential display PCR reveals novel targets for the mood-stabilizing drug valproate including the molecular chaperone GRP78. Mol Pharmacol. 1999;55:521–7. [PubMed] [Google Scholar]
- 4.Lee AS. The glucose-regulated proteins: stress induction and clinical applications. Trends Biochem Sci. 2001;26:504–10. doi: 10.1016/s0968-0004(01)01908-9. [DOI] [PubMed] [Google Scholar]
- 5.Hendershot LM. The ER function BiP is a master regulator of ER function. Mt Sinai J Med. 2004;71:289–97. [PubMed] [Google Scholar]
- 6.Zhang K, Kaufman R. The unfolded protein response: a stress signaling pathway critical for health and disease. Neurology. 2006;66:S102–9. doi: 10.1212/01.wnl.0000192306.98198.ec. [DOI] [PubMed] [Google Scholar]
- 7.Li J, Lee A. Stress induction of GRP78/BiP and its role in cancer. Curr Mol Med. 2006;6:45–54. doi: 10.2174/156652406775574523. [DOI] [PubMed] [Google Scholar]
- 8.Lee A. GRP78 induction in cancer: therapeutic and prognostic implications. Cancer Res. 2007;67:3496–9. doi: 10.1158/0008-5472.CAN-07-0325. [DOI] [PubMed] [Google Scholar]
- 9.Dong D, Ko B, Baumeister P, et al. Vascular targeting and antiangiogenesis agents induce drug resistance effector GRP78 within the tumor microenvironment. Cancer Res. 2005;65:5785–91. doi: 10.1158/0008-5472.CAN-05-0754. [DOI] [PubMed] [Google Scholar]
- 10.Bi M, Naczki C, Koritzinsky M, et al. ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. EMBO J. 2005;24:3470–81. doi: 10.1038/sj.emboj.7600777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dong D, Ni M, Li J, et al. Critical role of the stress chaperone GRP78/BiP in tumor proliferation, survival, and tumor angiogenesis in transgene-induced mammary tumor development. Cancer Res. 2008;68:498–505. doi: 10.1158/0008-5472.CAN-07-2950. [DOI] [PubMed] [Google Scholar]
- 12.Fu Y, Wey S, Wang M, et al. Pten null prostate tumorigenesis and AKT activation are blocked by targeted knockout of the stress response chaperone GRP78/BiP in prostate epithelium. Proc Natl Acad Sci USA. 2008;105:19443–48. doi: 10.1073/pnas.0807691105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reddy R, Mao C, Baumeister P, Austin R, Kaufman R, Lee A. Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: role of ATP binding site in suppression of caspase-7 activation. J Biol Chem. 2003;278:20915–24. doi: 10.1074/jbc.M212328200. [DOI] [PubMed] [Google Scholar]
- 14.Fu Y, Li J, Lee A. GRP78/BiP inhibits endoplasmic reticulum BIK and protects human breast cancer cells against estrogen starvation-induced apoptosis. Cancer Res. 2007;67:3734–40. doi: 10.1158/0008-5472.CAN-06-4594. [DOI] [PubMed] [Google Scholar]
- 15.Pyrko P, Schonthal A, Hofman F, Chen T, Lee A. The unfolded protein response regulator GRP78/BiP as a novel target for increasing chemosensitivity in malignant gliomas. Cancer Res. 2007;67:9809–16. doi: 10.1158/0008-5472.CAN-07-0625. [DOI] [PubMed] [Google Scholar]
- 16.Virrey JJ, Dong D, Stiles C, et al. Stress chaperone GRP78/BiP confers chemoresistance to tumor-associated endothelial cells. Mol Cancer Res. 2008;6:1268–75. doi: 10.1158/1541-7786.MCR-08-0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ranganathan AC, Zhang L, Adam AP, Aguirre-Ghiso JA. Functional coupling of p38-induced up-regulation of BiP and activation of RNA-dependent protein kinase-like endoplasmic reticulum kinase to drug resistance of dormant carcinoma cells. Cancer Res. 2006;66:1702–11. doi: 10.1158/0008-5472.CAN-05-3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roy B, Lee AS. The mammalian endoplasmic reticulum stress response element consists of an evolutionarily conserved tripartite structure and interacts with a novel stress-inducible complex. Nucleic Acids Res. 1999;27:1437–43. doi: 10.1093/nar/27.6.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baumeister P, Luo S, Skarnes W, et al. Endoplasmic reticulum stress induction of the Grp78/BiP promoter: activating mechanisms mediated by YY1 and its interactive chromatin modifiers. Mol Cell Biol. 2005;25:4529–40. doi: 10.1128/MCB.25.11.4529-4540.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoshida H, Haze K, Yanagi H, Yura T, Mori K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J Biol Chem. 1998;273:33741–9. doi: 10.1074/jbc.273.50.33741. [DOI] [PubMed] [Google Scholar]
- 21.Lane D, Harlow E. Two different viral transforming proteins bind the same host tumour antigen. Nature. 1982;298:517. doi: 10.1038/298517a0. [DOI] [PubMed] [Google Scholar]
- 22.Tsutsumi S, Namba T, Tanaka K, et al. Celecoxib upregulates endoplasmic reticulum chaperones that inhibit celecoxib-induced apoptosis in human gastric cells. Oncogene. 2006;25:1018–29. doi: 10.1038/sj.onc.1209139. [DOI] [PubMed] [Google Scholar]
- 23.Suzuki T, Ando T, Tsuchiya K, et al. Synthesis and histone deacetylase inhibitory activity of new benzamide derivatives. J Med Chem. 1999;42:3001–3. doi: 10.1021/jm980565u. [DOI] [PubMed] [Google Scholar]
- 24.Zhang X, Wharton W, Yuan Z, Tsai S, Olashaw N, Seto E. Activation of the growth-differentiation factor 11 gene by the histone deacetylase (HDAC) inhibitor trichostatin A and repression by HDAC3. Mol Cell Biol. 2004;24:5106–18. doi: 10.1128/MCB.24.12.5106-5118.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim HR, Kim EJ, Yang SH, et al. Trichostatin A induces apoptosis in lung cancer cells via simultaneous activation of the death receptor-mediated and mitochondrial pathway? Exp Mol Med. 2006;38:616–24. doi: 10.1038/emm.2006.73. [DOI] [PubMed] [Google Scholar]
- 26.Marks P. Discovery and development of SAHA as an anticancer agent. Oncogene. 2007;26:1351–6. doi: 10.1038/sj.onc.1210204. [DOI] [PubMed] [Google Scholar]
- 27.Schuettengruber B, Simboeck E, Khier H, Seiser C. Autoregulation of mouse histone deacetylase 1 expression. Mol Cell Biol. 2003;23:6993–7004. doi: 10.1128/MCB.23.19.6993-7004.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gal-Yam E, Jeong S, Tanay A, Egger G, Lee A, Jones P. Constitutive nucleosome depletion and ordered factor assembly at the GRP78 promoter revealed by single molecule footprinting. PLoS Genet. 2006;2:e160. doi: 10.1371/journal.pgen.0020160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park SH, Lee SR, Kim BC, et al. Transcriptional regulation of the transforming growth factor beta type II receptor gene by histone acetyltransferase and deacetylase is mediated by NF-Y in human breast cancer cells. J Biol Chem. 2002;277:5168–74. doi: 10.1074/jbc.M106451200. [DOI] [PubMed] [Google Scholar]
- 30.Won J, Yim J, Kim TK. Sp1 and Sp3 recruit histone deacetylase to repress transcription of human telomerase reverse transcriptase (hTERT) promoter in normal human somatic cells. J Biol Chem. 2002;277:38230–8. doi: 10.1074/jbc.M206064200. [DOI] [PubMed] [Google Scholar]
- 31.Li J, Ni M, Lee B, Barron E, Hinton DR, Lee AS. The unfolded protein response regulator GRP78/BiP is required for endoplasmic reticulum integrity and stress-induced autophagy in mammalian cells. Cell Death Differ. 2008;15:1460–71. doi: 10.1038/cdd.2008.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davidson DJ, Haskell C, Majest S, et al. Kringle 5 of human plasminogen induces apoptosis of endothelial and tumor cells through surface-expressed glucose-regulated protein 78. Cancer Res. 2005;65:4663–72. doi: 10.1158/0008-5472.CAN-04-3426. [DOI] [PubMed] [Google Scholar]
- 33.Zhou Y, Lee A. Mechanism for the suppression of the mammalian stress response by genistein, an anticancer phytoestrogen from soy. J Natl Cancer Inst. 1998;90:381–8. doi: 10.1093/jnci/90.5.381. [DOI] [PubMed] [Google Scholar]
- 34.Deng WG, Ruan KH, Du M, Saunders MA, Wu KK. Aspirin and salicylate bind to immunoglobulin heavy chain binding protein (BiP) and inhibit its ATPase activity in human fibroblasts. FASEB J. 2001;15:2463–70. doi: 10.1096/fj.01-0259com. [DOI] [PubMed] [Google Scholar]
- 35.Park HR, Tomida A, Sato S, et al. Effect on tumor cells of blocking survival response to glucose deprivation. J Natl Cancer Inst. 2004;96:1300–10. doi: 10.1093/jnci/djh243. [DOI] [PubMed] [Google Scholar]
- 36.Park HR, Ryoo IJ, Choo SJ, et al. Glucose-deprived HT-29 human colon carcinoma cells are sensitive to verrucosidin as a GRP78 down-regulator. Toxicology. 2007;229:253–61. doi: 10.1016/j.tox.2006.11.049. [DOI] [PubMed] [Google Scholar]
- 37.Paton AW, Beddoe T, Thorpe CM, et al. AB5 subtilase cytotoxin inactivates the endoplasmic reticulum chaperone BiP. Nature. 2006;443:548–52. doi: 10.1038/nature05124. [DOI] [PubMed] [Google Scholar]
- 38.Kudo T, Kanemoto S, Hara H, et al. A molecular chaperone inducer protects neurons from ER stress. Cell Death Differ. 2008;15:364–75. doi: 10.1038/sj.cdd.4402276. [DOI] [PubMed] [Google Scholar]