

Abstract
This paper presents both a review of some recent results from our group and experimental groups, and some new theoretical results all of which are helping to form a more physically rigorous picture of the process of enzymatic catalysis. A common classical picture of enzymatic catalysis is the transition state tight binding model. Schwartz and Schramm1 have recently argued from both theoretical and experimental results that this picture is incorrect. We now investigate what the nature of barriers might be in enzymatic reactions, and what this viewpoint might imply for tunneling in a hydrogen transfer enzyme. For lactate dehydrogenase we conclude that the enzymes role in catalysis is at least partially to hunt through configuration space for those configurations that minimize chemical free energy barriers. Those configurations do not seem to be stable basins on the free energy surface, and in fact the overall free energy barrier to reaction may well largely be due to this stochastic hunt - both probabilistically and energetically. We suggest further computations to test this hypothesis.
Introduction
Enzymes manage to accomplish extraordinary chemical catalysis while maintaining exquisite preference for specific substrates and sensitivity to biological environment. The way in which enzymes perform such complex, and in some cases seemingly opposed goals remains a topic for heated debate after many decades of study. One of the earliest, and still most influential theories of enzymatic action is that of transition state binding. That is that the enzyme binds most strongly to the transition state as the reactants proceed to products, and this binding somehow releases energy to lower the free energy barrier to reaction1. There is even seeming experimental evidence for the correctness of this concept. Transition state geometries can be inferred from Kinetic isotope effect experiments2. This idea is closely coupled to that of electrostatic stabilization of transition states3. From these geometries one may construct quantum mechanical models of the transition state4, and from these models infer such properties as electrostatic potential at the van der Waals surface of the transition state5. Chemical intuition can then be designed to mimic these quantum mechanical properties, and these stable analogues of the transition state are some of the strongest enzymatic inhibitors in existence6. En face, this would seem to provide strong, if circumstantial, support for the transition state binding hypothesis. There is, however, almost equally strong experimental evidence that the concept is wrong.
Catalytic antibodies are designer enzymes7 which recognize a cognate ligand, much the same way the immune system designs antibodies to recognize invading threats. If binding to the transition state were the determining characteristic of enzymatic action, then an enzyme designed in this fashion to bind strongly to the transition state should be a superb catalyst. Though not for lack of effort, such designer enzymes have been fairly disappointing, achieving catalytic enhancements 10 to 15 orders of magnitude lower than that achieved by enzymes. It thus becomes clear that an important element designed into an enzyme by nature is missing in these artificial enzymes. Recent calculations have further demonstrated that there can be no tight binding. Transition path sampling8 with committor distribution analysis9 has shown for both lactate dehydrogenase (LDH) and for purine nucleoside phosphorylase (PNP) that residence time within the transition state region is extremely short - from one to 10 femtoseconds, clearly allowing no time for strong binding (or equilibration.)
We have thus determined the way in which an enzyme does not catalyze chemical reactions. The purpose of this paper is to begin to suggest a new view of the way in which it does. We will present some recent calculations which have appeared but which were not fully understood at the time for their deeper implications. Some of these calculations show that protein motion on a very fast timescale - picoseconds - are central to the chemical mechanism in both LDH and PNP. In other previously published work10, we demonstrated that protein conformations that create donor/acceptor (substrate/cofactor) distances which are small enough for such rapid “promoting vibrations” to be effective are quite rare. We will now finally present another piece of this puzzle to show that slow conformational rearrangements to stable potential basins in which donor/acceptor distances are small are transformations to high free energy states. This would imply that the free energy barrier in the case at hand is NOT a potential barrier, or at least not in total. We here suggest that for at least one hydrogen transfer enzyme, LDH, the overall free energy barrier to reaction is a probabilistic one, not a potential energy barrier. Thus, the role of the enzyme is changed from previous views. Originally the enzyme was thought to lower free energy barriers. In our previous published work we11 suggested that the role of the enzyme may be to make barriers thinner, and thus maximize tunneling. We now suggest that the dominant role of the enzyme may be to perform directed stochastic searches through conformation space that minimizes a barrier, and apparent free energies measured for reaction may largely be diagnostic of this probability distribution.
What this new concept implies regarding tunneling will then be addressed. Finally, we will conclude with further suggestions of calculations that will formalize the more speculative hypothesis raised in this article.
Timescales and the reaction coordinate in the vicinity of the chemical barrier
As is by now well know, one principle reason study the of enzymatic catalysis is so complex is the wide range of timescales important in the process. Without belaboring the point, we refer without further comment to Figure 1. This range of timescales is due to the complexity of enzyme structure, and along with the difficulty highlighted by Figure 1, comes major philosophical questions as to the evolutionary design of enzymes. There seem to us to be three essential questions related to this issue of complexity: First, why are enzymes so large. Nature seems to rarely waste amino acids and ATP to join them, so the large size of the protein matrix of most enzymes would lead one to assume necessity for such a complex architecture. In some cases multimeric architecture can be ascribed to the need for physical stability in extreme environments, but even mesophiles seem to be quite large. The second question is why when there are many millions of primary sequences found in nature, there are only a relatively small number of basic fold designs (on the order of hundreds - many orders of magnitude less than the variability in sequence space.) The third and final question is simply why artificial enzyme design has been such a failure. As mentioned in the introduction, artificial enzymes are at least 6 and often 16 orders of magnitude less proficient than naturally occurring enzymes. If one takes catalytic antibodies as an example, clearly recognition of the transition state is not the sole design principle as work in creation of highly efficient biological catalysts.
Figure 1.
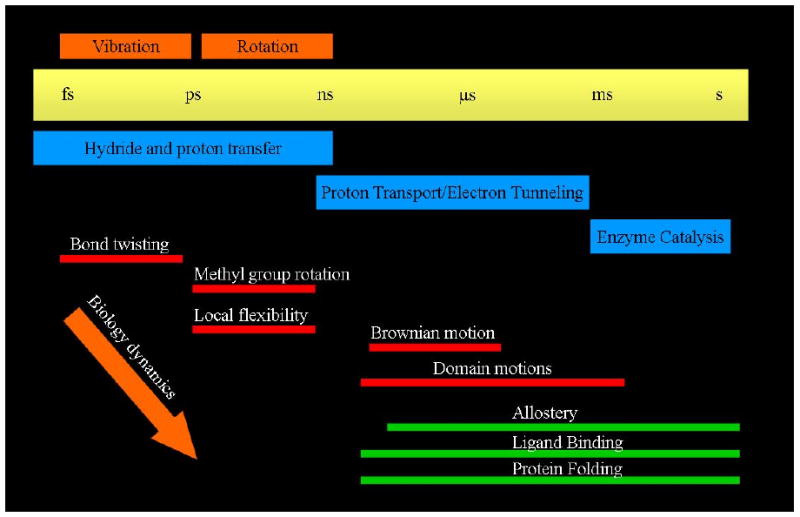
A graphical representation of the range of timescales on which functionally important events
A hint in our understanding of what this missing design principle may be, and a potential answer as well to the first two questions listed above came in our elucidation of the reaction coordinate of the hydride and proton transfer reaction catalyzed by lactate dehydrogenase. Using Transition Path Sampling we gathered a large ensemble of reactive trajectories for the chemical reaction in this enzyme13. These trajectories were then analyzed by committor distribution analysis to find a true chemical reaction coordinate. In addition to the chemical motions of a hydride and a proton in this reaction, there is a compression through the active site along with a subsequent relaxation on the distal side of the binding pocket. This is shown graphically for a monomer of human heart LDH in Figure 2. The need to create a protein architecture that allows energy to be channeled along what is not an obvious path (i.e. not along a particular helix for example) is an entirely reasonable explanation for the complexity of the enzyme. The corresponding lack of such design principle in artificial enzymes, for example catalytic antibodies, could well explain why even with superb recognition of the transition state, there is still poor catalytic proficiency.
Figure 2.
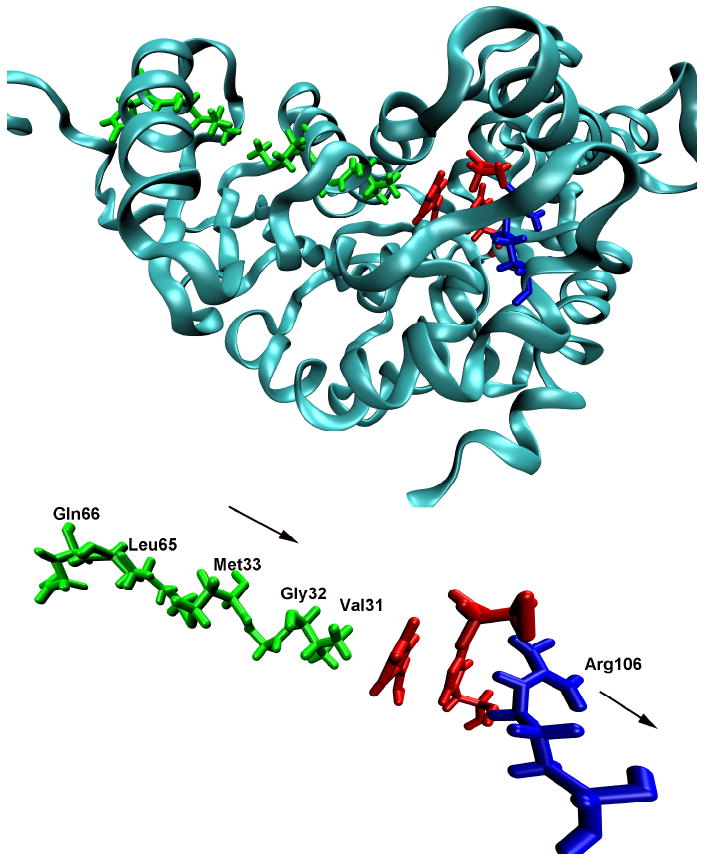
The “promoting vibration in human heart lactate dehydrogenase that was found to be an integral component of the reaction coordinate for chemistry.
Calculations, as demonstrated in Figure 3, have shown that the moment of transfer of the hydride comes at exactly the point of maximal compression of the promoting vibration14. We note that interestingly this is not always true of reaction in the enzyme from the thermophile Bacillus stearothermophillus, when dynamics are probed at mesophillic temperatures. This is another example of a lack of dynamic perfection when enzymes are asked to perform at temperatures other than that for which they were “designed” by evolution. That certainly has been noted by other groups15. This effect demonstrates the subtle detail in dynamics involved in the reactions of the respective enzymes. It is also worth noting that over the entire ensemble of calculations, committor distributions show that residence time in the transition state region is on the order of a femtosecond - far too short for any equilibrium. One example is shown in Figure 4. This feature has been suggested by Schramm and Schwartz1 to be a possibly general description of enzymatically catalyzed reactions.
Figure 3.

Hydride transfer from donor to cofactor comes at exactly the moment that the protein achieve maximal compression of the active site across the promoting vibration. This result is from the Bacillus enzyme in a case in which hydride and proton transfer is almost simultaneous, and in turn synchronized with the promoting vibration.
Figure 4.
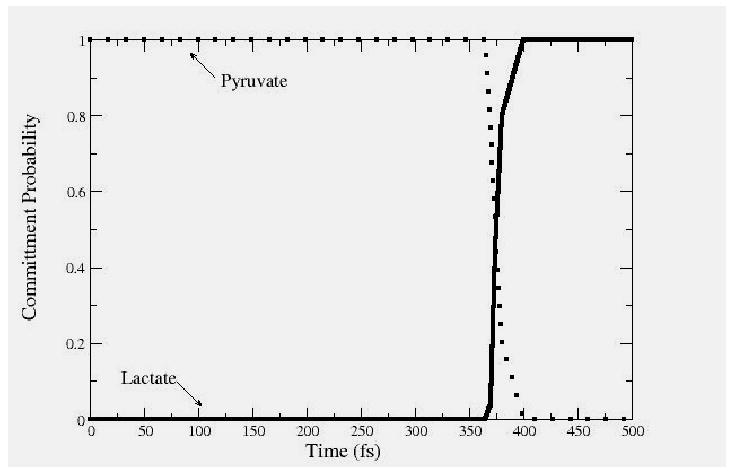
The committor distributions along a single reactive trajectory from the transition path ensemble for the reaction catalyzed by lactate dehydrogenase. The committor is calculated at each point along a trajectory by initiating new trajectories with momenta drawn from a Boltzmann distribution. The point of exactly equicommittor probability - .5 is defined as a transition state, and is a member of the stochastic separatrix.
One may rightfully ask if the short residence time in the transition state region is a characteristic of hydrogen transfer reactions. Because of their chemical simplicity, the phenomena may not be universal. While such a claim is difficult to make, we can examine a very different reaction - that catalyzed by purine nucleoside phosphorylase. The chemistry is shown below in Figure 5. In fact similar application of transition path sampling shows that while this reaction is far more complex, residence time in the transition state region is quite short - on the order of 10 femtoseconds. A standard committor from our computations is shown in Figure 6. While residence time is still quite short the structure of the committor distribution makes it apparent that the chemistry is far more complex than that found in lactate dehydrogenase. Analysis of the reactive trajectories gathered in the transition path ensemble reveal that the chemistry is due not to the attacking nucleophile, but rather, the glycosidic bond is first broken through the aid of a promoting vibration in the protein. This promoting vibration in the protein polarizes this bond to form a good leaving group. After the scission of this bond, further protein motion moves the ribosyl group down to the rigidly held phosphate group. This finally allows formation of the ribose 1 phosphate product. Thus protein motions is central to reaction, there is not time for equilibrium in the transition state region, and the mechanism of chemistry is completely distinct from the expected organic solution phase SN2 reaction. The reaction is in no way dependent on motion of the nucleophile, but rather on motion of the protein which in two steps first polarizes the glycosidic bond to allow facile leaving group formation, and in a second step rotates the ribosyl group down towards a firmly locked phosphate.
Figure 5.
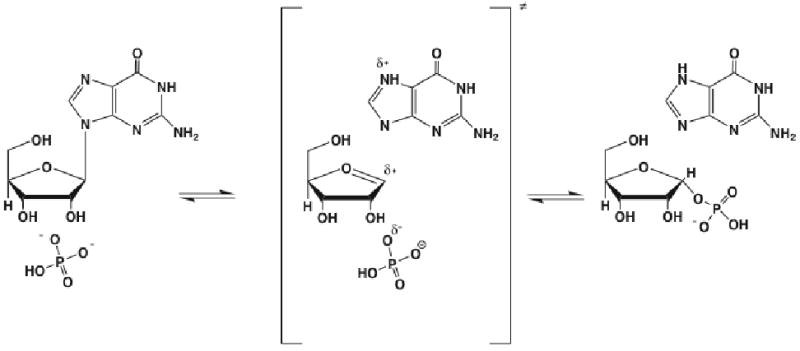
The chemical reaction catalyzed by purine nucleoside phosphorylase. It this were solution phase chemistry, one would suppose that this is an SN2 reaction with phosphate the attacking nucleophile. Our results show that the enzymatic reaction proceeds by a completely difference chemical mechanism.
Figure 6.

An example of a committor distribution for the reaction catalyzed by PNP. While the reaction is far more complex, residence in the transition state region is still quite short, not allowing for the formation of any equilibrium with the transition state. In addition, the mechanism is not that of the corresponding organic solution phase reaction, but again critically involves a promoting vibration in the protein matrix of the enzyme.
The question we must now address is given the extremely short time of barrier passage, and the fact that the promoting vibrations which allow the passage are on the order of picoseconds, why does the enzyme take milliseconds to turnover. It is often the case that the enzyme is limited by product release, but it is also surely the case that the chemical reaction is not in general complete in picoseconds, with the enzyme simply waiting so many more orders of magnitude of time to release the already formed product. Thus the issue is not why is the enzyme so fast, but rather, why is it so slow. Understanding this mystery will begin to elucidate the nature of the barrier to reaction in enzymatic catalysis.
Conformational fluctuations and catalysis
It has certainly been known for many years that fluctuations in protein conformation on time scales far slower than picoseconds effects protein function. This idea was pioneered by Frauenfelder in his studies of hemeprotein ligand binding16. More recently groups such as Hammes Schiffer's17 and Brooks'18 using partially phenomenological models have been able to reproduce some rate constants of enzymatically catalyzed reactions. The question is how these slower motions relate to promoting vibrations, and eventually to catalysis.
The most useful picture of protein motion is obtained through a hierarchical model of the potential energy surface. In this picture, proteins have rapid conformational fluctuations in “basins.”19 Transformations between basins which involve greater potential barriers are concomitantly less frequent. One may imagine higher level groupings of basins into superbasins, etc. A schematic view of this model is shown in Figure 7. Here, A, B, and C are individual basins with rapid internal transformations between local internal minima. Transformations between the basins are significantly slower activated events. The hierarchical nature of this view is shown schematically by the right panel in Figure 7. These ideas are of value in a purely pedagogical sense, but do little to explain the function of enzymes until one can find such conformational fluctuations on a real surface. The actual potential energy surface of a folded protein is expected to be highly complex, and it is also expected that not all conformations are directly relevant to catalysis. What is needed then is a “reporter,” or in the words of transition path sampling, an order parameter which allows identification of those conformations central to catalysis.
Figure 7.

A hierarchical view of conformational fluctuations in a protein. The left panel shows a hypothetical idealized 1 dimensional potential energy surface, while the right emphasizes the groupings of conformations in a hierarchy.
In the case of LDH, we know that the promoting vibration, part of the reaction coordinate will not be effective unless the donor and acceptor, the NAD+ cofactor and the substrate, are close enough together that the small amplitude promoting vibration can be effective. A simple MD search of the surface will not yield transformations between basins, and so we began our hunt for conformational basins using Monte Carlo searches of the structure. We showed some years ago, that conformations that resulted in close donor acceptor distances were quite rare10. To perform a more systematic examination of these conformational fluctuations, we are currently employing Langevin dynamics to search for potential minima via quench of the structures after a certain time. We accelerate the search by constraining donor acceptor to be relatively short. During the quench process we allow full minimization. Again, many conformations show relaxation away from a close donor acceptor distance. A small minority of conformations retain the close donor acceptor distance. In order to understand the dynamics we interrogate the minimum potential energy path between two such conformations. A representative sample is given in Figure 8. This is accomplished using the string method of van den Eijnden and E20. While there is significant roughness in the complex potential energy surface, clearly the two potential basins exist at about 0.75 and .75 in RMSD units. The potential barrier between them is quite high - about 80 kcal/mol in one direction and 50 in the other. That having been said, the physically relevant quantity is the free energy for the transformation. This is the quantity that will determine the rate of conformational fluctuation between the two states. Calculation of the free energy is not trivial and a “reaction coordinate is not obvious. We perform the calculation in two separate steps. First, using the structures found along the minimum potential (the so called zero temperature string) we locate the free energy minimum path using a finite temperature string. To insure convergence of the free energy value, we then apply WHAM sampling to the average structure at each node in the free energy string. The resulting free energy plot for the transformation between the two configurations shown in Figure 8 is given in Figure 9.
Figure 8.

The minimum potential energy path between two conformations of small donor acceptor distance.
Figure 9.

The free energy map corresponding to the conformational transformation shown in Figure 8.
At first glance, this result is strikingly odd the free energy minimum is actually between the two potential minima. We have repeated this for the other small donor acceptor distance conformations we have located, and in all cases similar results obtain in our preliminary calculations. Recall that the small donor acceptor distance conformations are very rare; then this free energy is an indication of the difficulty of achieving this conformation.
The nature of the free energy barrier to catalysis
The overall rate of the chemical reaction catalyzed by an enzyme is determined by the overall free energy of catalysis. This is not a physical statement it is a definitional one. The free energy is defined as that number which when multiplied by the inverse temperature β and then exponentiated yields the experimental rate. This number has been both measured21 and predicted theoretically22. The result for LDH is always about 20 kcal/mol. A quick glance at Figure 9 shows that to go from the free energy minimum to the structure that will allow the promoting vibration to be effective requires between 10 and 20 kcal per mol. This is in no way a definitive statement regarding the nature of catalysis, but it is a striking result. Taken at face value, it would seem to indicate that the vast majority of free energy needed for the catalytic reaction in the enzyme is that needed to hunt through configuration space to locate one of the reactive conformations. At that point, the promoting vibration is enough to carry the reaction to completion in what may well be essentially a barrierless process. The compression of a promoting vibration will both lower and thin barriers. This is a vastly different picture of catalysis that any standard view. It also has deep implications for the possibility of tunneling in an enzyme such as LDH. It must be stated that no one has ever claimed tunneling for LDH, but the striking similarity in both chemistry and structure of this enzyme to alcohol dehydrogenase suggests that analogy is at least not a wasted effort.
One of the great mysteries of tunneling in ADH is why there is no significant primary kinetic isotope effect. In the past we have argued that tunneling may be present, but the known mathematical effect of the promoting vibration can mask the primary KIE23. This fact remains true. The presence of the promoting vibration, however, does not prove the existence of tunneling, it simply rationalizes why if tunneling is present there is no major sign of it in the primary KIE. An alternative explanation could be that there simply is very little tunneling. The function of the enzyme is not to maximize tunneling, but to limit the search through configuration space to efficiently find ones in which both tunneling and activated barrier passage are maximized. The free energy is employed in this picture not for barrier passage, but rather for barrier elimination.
It is important to note that there are undoubtedly cases where there is a true chemical barrier to surmount. An example may be found in cases where there is a large primary kinetic isotope effect which seems clearly diagnostic of deep barrier tunneling14.
This paper as part of a “symposium in print” has presented what is undoubtedly a radical view of enzymatic catalysis. Far more effort is needed before this view can be accepted as correct, even in a limited case of a single enzyme. First, more configurations which achieve small transfer distances must be located. The free energy of transformation between these conformations must be computed. Finally, and critically, using appropriate QM/MM techniques, the chemical barriers to reaction must be computed as a function along these conformational rearrangements. Experimental confirmation of this picture is a bit more complex. The best tools available would seem to be a combination of mutagenesis and crosslinking to prevent the conformational shifts we have discussed, or make them more energetically costly. The prediction would then be that rates should fall in exponential relation to the shift in conformational free energy.
Acknowledgments
We gratefully acknowledge the support of the National Institutes of Health, NIGMS through grant GM068036, and the Chemistry division of the National Science Foundation through grant CHE-0714118. SDS thanks Prof. Vern Schramm for constantly stimulating discussions on this and many other topics.
References
- 1.Schwartz SD, Schramm VL. Nature Chem Bio. in press. [Google Scholar]
- 2.Pauling LC. Molecular architecture and biological reactions. Chem & Eng News. 1946;24:1375–1377. [Google Scholar]
- 3.Kline PC, Schramm VL. Purine nucleoside phosphorylase. Catalytic mechanism and transition-state analysis of the arsenolysis reaction. Biochemistry. 1993;32:13212–13219. doi: 10.1021/bi00211a033. [DOI] [PubMed] [Google Scholar]
- 4.Warshel A, Sharma PK, Kato M, Xiang Y, Liu H, Olsson MHM. Electrostatic Basis for Enzyme Catalysis. Chem Rev. 2006;106:3210–3235. doi: 10.1021/cr0503106. [DOI] [PubMed] [Google Scholar]
- 5.Schramm VL. Enzymatic transition state theory and transition state analogue design. J Biol Chem. 2007;282:28297–28300. doi: 10.1074/jbc.R700018200. [DOI] [PubMed] [Google Scholar]
- 6.Braunheim BB, Miles RW, Schramm VL, Schwartz SD. Prediction of Inhibitor Binding Free Energies by Quantum Neural Networks. Nucleoside Analogues binding to Trypanosomal Nucleoside Hydrolase. Biochemistry. 1999;38:16076–16083. doi: 10.1021/bi990830t. [DOI] [PubMed] [Google Scholar]
- 7.Murkin AS, Clinch K, Mason JM, Tyler PC, Schramm VL. Immucillins in custom catalytic-site cavities. Bioorg Med Chem Lett. 2008;18:5900–5903. doi: 10.1016/j.bmcl.2008.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu Y, Yamamoto N, Janda KD. Catalytic antibodies: hapten design strategies and screening methods. Bioorg Med Chem. 2004;12:5247–5268. doi: 10.1016/j.bmc.2004.03.077. [DOI] [PubMed] [Google Scholar]
- 9.Bolhuis PG, Chandler D, Dellago C, Geissler PL. Transition path sampling: Throwing ropes over rough mountain passes in the dark. Annu Rev Phys Chem. 2002;53:291–318. doi: 10.1146/annurev.physchem.53.082301.113146. [DOI] [PubMed] [Google Scholar]; Transition path sampling is described as a way to probe, in an unbiased fashion, the nature of barrier crossing in condensed phase reactions.
- Dellago C, Bolhuis PG, Chandler D. Efficient transition path sampling: application to Lennard-Jones cluster rearrangements. J Chem Phys. 1998;108:9236–9245. [Google Scholar]
- 10.Saen-Oon S, Quaytman S, Schramm VL, Schwartz SD. Atomic Detail of Chemical Transformation at the transition state of an enzymatic reaction. PNAS. 2008;105:16543–16548. doi: 10.1073/pnas.0808413105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basner JE, Schwartz SD. How Enzyme Dynamics Helps Catalyze a Chemical Reaction in Atomic Detail: A Transition Path Sampling Study. JACS. 2005;127:13822–13831. doi: 10.1021/ja043320h. [DOI] [PubMed] [Google Scholar]
- 11.Pineda JRET, Schwartz SD. Protein Dynamics and Catalysis – The Problems of Transition State Theory and the Subtlety of Dynamic Control. Philosophical Transactions of the Royal Society. 2006;361:1433–1438. doi: 10.1098/rstb.2006.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Antoniou D, Caratzoulas S, Kalyanaraman C, Mincer JS, Schwartz SD. Barrier passage and protein dynamics in enzymatically catalyzed reactions. European Journal of Biochemistry. 2002;269:3103–3112. doi: 10.1046/j.1432-1033.2002.03021.x. [DOI] [PubMed] [Google Scholar]
- 13.Quaytman S, Schwartz SD. The reaction coordinate of an enzymatic reaction: TPS studies of lactate dehydrogenase. PNAS USA. 2007;104:12253–12258. doi: 10.1073/pnas.0704304104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Quaytman S, Schwartz SD. Comparison Studies of the Human Heart and Bacillus stearothermophilus Lactate Dehydrogenase by Transition Path Sampling. J Phys Chem A. 2009;113:1892–1897. doi: 10.1021/jp804874p. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meyer MP, Tomchick DR, Klinman JP. Enzyme structure and dynamics affect hydrogen tunneling: the impact of a remote side chain (I553) in soybean lipoxygenase-1. Proc Natl Acad Sci U S A. 2008 Jan 29;105(4):1146–51. doi: 10.1073/pnas.0710643105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frauenfelder H, McMahon BH, Austin RH, Chu K, Groves JT. The role of structure, energy landscape, dynamics, and allostery in the enzymatic function of myoglobin. Proc Natl Acad Sci USA. 2001;98:2370–2374. doi: 10.1073/pnas.041614298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hammes-Schiffer S, Benkovic SJ. Relating protein motion to catalysis. Annu Rev Biochem. 2006;75:519–541. doi: 10.1146/annurev.biochem.75.103004.142800. [DOI] [PubMed] [Google Scholar]
- 18.Conformational substates modulate hydride transfer in dihydrofolate reductase. Thorpe IF, Brooks CL., 3rd J Am Chem Soc. 2005;127:12997–3006. doi: 10.1021/ja053558l. [DOI] [PubMed] [Google Scholar]
- 19.Becker OM, Karplus M. The Topology of Multidimensional Potential Energy Surfaces: Theory and Application to Peptide Structure and Kinetics. J Chem Phys. 1997;106:1495–1517. [Google Scholar]
- Despa Mark F, Fernandez A, Stephen Berry R, Levy Y, Jortner J. Interbasin Motion Approach to Conformationally Constrained Peptides. J Chem Phys. 2003;118:5673–5682. [Google Scholar]
- 20.E W, Ren W, Vanden-Eijnden E. String method for the study of rare events. Physical Review B: Condensed Matter and Materials Physics. 2002;66:052301/1–052301/4. [Google Scholar]
- E W, Ren W, Vanden-Eijnden E. Finite Temperature String Method for the Study of Rare Events. Journal of Physical Chemistry B. 2005;109:6688–6693. doi: 10.1021/jp0455430. [DOI] [PubMed] [Google Scholar]
- 21.Menon MP, Hunter FR, Miller S. Kinetic Studies on Human Lactate Dehydrogenase Isoenzyme-Catalyzed Lactate-to-Pyruvate Reaction. J Prot Chem. 1987;6:413–429. [Google Scholar]
- 22.Ferrer S, Tunon I, Marti S, Moliner V, Garcia-Viloca M, Gonzalez-Lafont A, Lluch JM. A Theoretical Analysis of Rate Constants and Kinetic Isotope Effects Corresponding to Different Reactant Valleys in Lactate Dehydrogenase. J Am Chem Soc. 2006;128:16851–16863. doi: 10.1021/ja0653977. [DOI] [PubMed] [Google Scholar]
- 23.Antoniou D, Schwartz SD. Internal Enzyme Motions as a Source of Catalytic Activity: Rate Promoting Vibrations and Hydrogen Tunneling. J Phys Chem. 2001;B105:5553–5558. [Google Scholar]