

Abstract
Cytidine triphosphate synthetases (CTPSs) synthesize CTP and regulate its intracellular concentration through direct interactions with the four ribonucleotide triphosphates. In particular, CTP product is a feedback inhibitor that competes with UTP substrate. Selected CTPS mutations that impart resistance to pyrimidine antimetabolite inhibitors also relieve CTP inhibition and cause a dramatic increase in intracellular CTP concentration, indicating that the drugs act by binding to the CTP inhibitory site. Resistance mutations map to a pocket that, although adjacent, does not coincide with the expected UTP binding site in apo Escherichia coli CTPS [EcCTPS; Endrizzi, J. A., et al. (2004) Biochemistry 43, 6447–6463], suggesting allosteric rather than competitive inhibition. Here, bound CTP and ADP were visualized in catalytically active EcCTPS crystals soaked in either ATP and UTP substrates or ADP and CTP products. The CTP cytosine ring resides in the pocket predicted by the resistance mutations, while the triphosphate moiety overlaps the putative UTP triphosphate binding site, explaining how CTP competes with UTP while CTP resistance mutations are acquired without loss of catalytic efficiency. Extensive complementarity and interaction networks at the interfacial binding sites provide the high specificity for pyrimidine triphosphates and mediate nucleotide-dependent tetramer formation. Overall, these results depict a novel product inhibition strategy in which shared substrate and product moieties bind to a single subsite while specificity is conferred by separate subsites. This arrangement allows for independent adaptation of UTP and CTP binding affinities while efficiently utilizing the enzyme surface.
Cytidine triphosphate (CTP) is synthesized de novo from uridine triphosphate (UTP), adenosine triphosphate (ATP), and glutamine by cytidine triphosphate synthetases (CTPSs,1 EC 6.4.3.2, 525–630 residues) (Figure 1a) (1–3; reviewed in ref 4). CTPSs catalyze three discrete reactions: MgATP-dependent phosphorylation of the UTP uracil O4 atom, glutamine hydrolysis to generate ammonia, and ammonia displacement of the uracil O4 phosphate (5–7). CTPSs also regulate intracellular CTP levels in response to the four ribonucleotide triphosphate concentrations (3, 8–16) and to protein phosphorylation in yeast (17) and likely other eukaryotes. In particular, substrate UTP and ATP binding induce oligomerization of inactive dimers to active tetramers, leading to positive cooperative behavior at physiological enzyme concentrations (Figure 1b) (3, 4, 11–13). Further, the product CTP provides negative feedback by acting as a competitive inhibitor of substrate UTP (3, 8, 9, 14), with a comparable Ki for CTP (110 μM) and Km for UTP (150 μM) (3) (Figure 1a). This particular interaction determines the upper limit for intracellular CTP, and Saccharomyces cerevisiae carrying CTPS mutants defective in product inhibition exhibit 16–20-fold increased levels (18).
Figure 1.
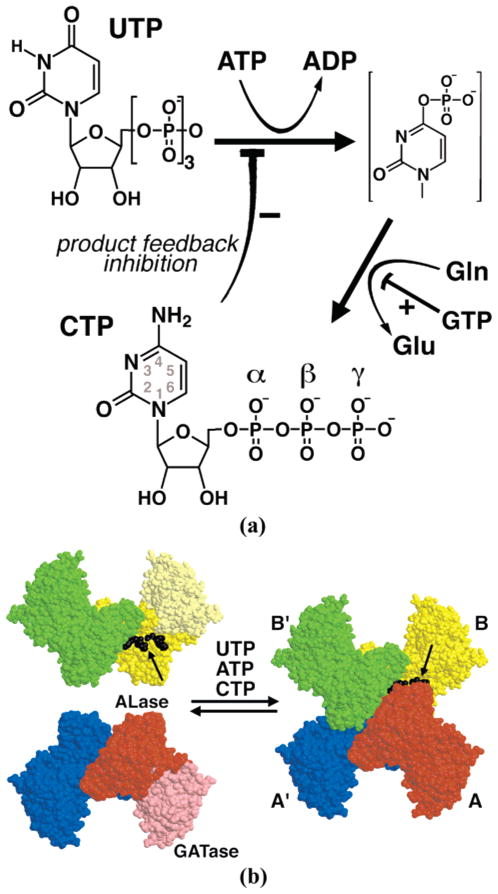
CTP synthesis and regulation by CTP synthetases. (a) CTPSs catalyze the amination of UTP to form CTP. The uracil O4 position is activated via ATP-dependent phosphorylation, and the resulting phosphate is displaced by ammonia generated by glutamine hydrolysis. CTPSs are regulated by all four nucleotide triphosphates: ATP and UTP promote oligomerization of inactive dimers to active tetramers (see panel b), GTP increases the kcat/Km of rate-limiting glutamine hydrolysis, and CTP is a negative feedback inhibitor that is competitive with UTP. (b) Space-filling representation of the EcCTPS tetramer of nearly identical subunits. Tetramerization of A–A′ (red and blue) and B–B′ (yellow and green) tightly associated dimers is mediated by the kinase ammonia ligase (ALase) domain (saturated yellow and red). GTP-activated glutamine hydrolysis is catalyzed by the glutamine amidotransferase (GATase) domain (light yellow and red). Tetramer formation is promoted by ATP and UTP substrates, as well as CTP product, binding in the CTP synthesis active site at the tetramer interfaces (black arrows). Bound ADP and CTP ligands at the B–A interface are also indicated (black).
Antimetabolite pyrimidine analogues cyclopentenylcytosine (CPEC) and 3-deazauridine (3-deazaU) are effective CTPS inhibitors that arrest cancer cell proliferation (18–24) and are toxic to yeast and bacteria (18, 25). Moreover, using these inhibitors to deplete the intracellular CTP concentration increases the efficacy of the anticancer/antiviral drugs cytosine arabinoside (26, 27), 2′,3′-dideoxy-3′-thia-cytidine (28), and 2′,2′-difluoro-2′-deoxycytidine (29). Trypanosoma brucei CTPS is a validated African sleeping sickness drug target (30), and malaria (31), giardiosis (32), chlamydia (33), and hemorrhagic fevers (34) are also potentially treatable using anti-CTPS therapies. However, spontaneous resistance to these drugs arises frequently through clustered CTPS gene mutations that release CTP feedback inhibition and increase intracellular CTP levels (Figures 2 and 6) (18, 25, 35, 36). These results define the CTPS role in regulating intracellular CTP as well as suggesting that the drugs act by binding to the CTP inhibitory site. Visualizing the structural mechanisms for CTP and CTP analogue inhibition will provide the basis for rational improvement of efficacy and resistance evasion of CTPS antagonists.
Figure 2.
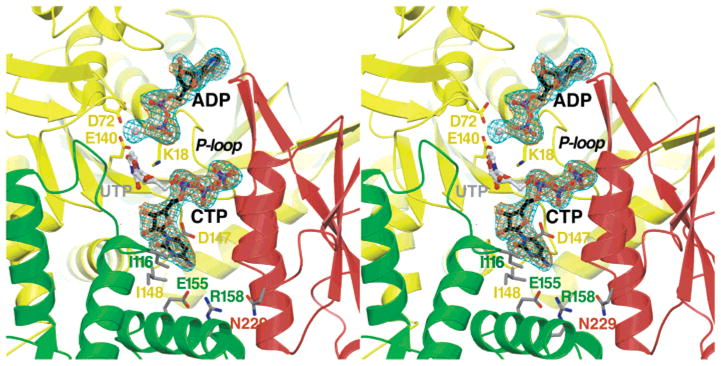
Crystallographic location of the CTP synthesis active site and the adenine and cytosine nucleotide binding sites at the EcCTPS tetramer interface. Fo − Fc omit difference density for ATP-, UTP-, and Mg2+-soaked crystals is shown (2.8 Å resolution, 4.5σ, cyan netting). The identities of the ADP and CTP ligands were confirmed by similar omit Fo − Fc difference density from ADP- and CTP-soaked crystals (2.8 Å resolution, 4.0σ, orange netting). Ribbons indicate secondary structure for the A (red), B (yellow), and B′ (green) EcCTPS subunits. The CTP and ADP positions from the refined 2AD5 model are enclosed by the electron density (black carbons). The hypothetical UTP positioning is also shown (4). Side chains for conserved catalytic residues K18, D72, and E140 identify the ALase catalytic site and the likely location of the UTP O4 atom for phosphorylation and/or amination, in analogy to the DTBS structure (38). Side chains at positions of drug resistance mutations (25, 35) that inhibit or abolish CTP feedback inhibition are also indicated (gray sticks; see Figure 6 for details).
Figure 6.
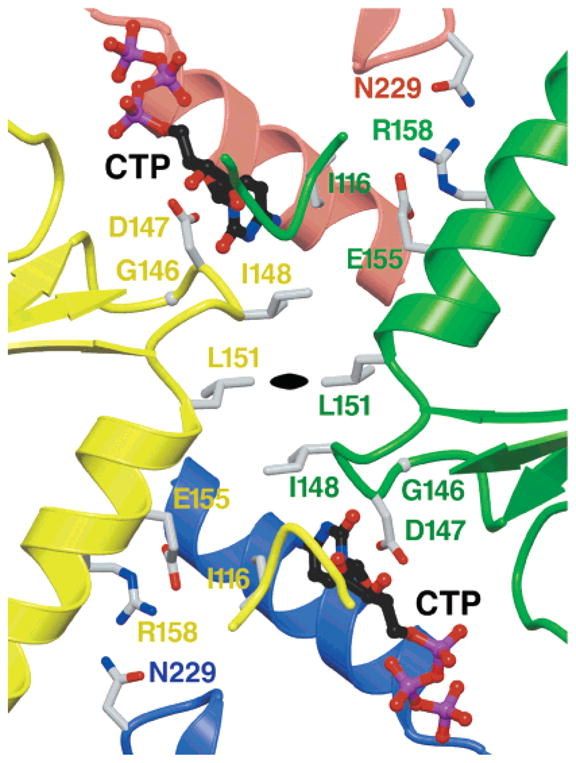
Proximity of CTP and drug resistance mutations to bound CTP and potential for allosteric interactions between bound CTPs. Ribbon diagram of the B–B′ crystallographic 2-fold interface, showing bound CTP (ball and stick with black carbon atoms), and side chains of residues that are replaced in CTP feedback inhibition-resistant CTPS mutants are shown as gray sticks. The associated subunits and residue labels are indicated by color (red for A, blue for A′, yellow for B, and green for B′). The black oval indicates the location of the 2-fold crystallographic symmetry axis, which relates subunits of the tightly bound dimer. The previously characterized CTP resistance substitutions are D147E [Chlamydia trachomatis (25)], V*116F, G146E, I148T, M*151I, R158H, and H*229K [hamster (35)], and E155K [hamster (35) and yeast (18)] (an asterisk denotes residues different from those of EcCTPS). Note the potential for substitutions at residues 148 and 151 to disrupt binding at both sites, and the potential for binding at one CTP site to influence binding at the two-fold-related site. Residues 155, 158, and 229 are not in direct contact with the bound CTP but could potentially interact with each other if the B–A′ and A–B′ interface distances were reduced by 1 Å.
Recently, we determined a prototypical CTPS structure, apo Escherichia coli CTPS, at 2.3 Å resolution (apo-EcCTPS, Protein Data Bank entry 1S1M) (4). Apo-EcCTPS is a nearly 222-symmetric homotetramer. Each monomer consists of an N-terminal ALase domain, which provides the oligomeric interfaces, and a C-terminal GATase domain (Figure 1b). The four kinase/ligase active site clefts where CTP is produced are assembled by highly conserved ALase domain surfaces from three different monomers, while GTP-regulated glutamine hydrolysis is carried out in the GATase domain glutaminase active site. In this P21212 crystal form, a crystallographic 2-fold axis defines the tight “dimer interface” (A–A′ or B–B′ interfaces2), while substrates bound in the kinase/ligase active sites mediate tetramer formation across the noncrystallographic dissociable “tetramer interface” (A–B′ and A′–B interfaces) (4).
Conservation of functionally and structurally important residues, which are ~20% identical with relatively few insertion or deletion differences between all members, suggests that EcCTPS is a viable model for understanding many aspects of CTPS function. This idea finds support in the structural similarity of EcCTPS and CTPS from Thermus thermophilus (37).
Previously, we used bioinformatic analysis to identify potential nucleotide binding sites (4). Structural relatedness of the ALase domain to the functionally related dethiobiotin synthetase (DTBS) provided predictions for the catalytic and ATP binding sites. The UTP site was deduced by modeling the uracil ring O4 position overlapping the analogous substrate oxygen position in the DTBS–DAPA–AlF3 complex (PDB entry 1BS1) (38), and inferring the UTP γ-phosphate position from that of a sulfate or iodine ion ligated by a conserved P-loop-like structure located at the tetramer interface (Figure 2).
Parsimonious interpretation of existing data suggested that the CTP inhibitory site and the UTP catalytic site were the same. CTP competitively inhibits UTP binding (3), is isosteric to UTP, and similarly induces tetramer formation (13). However, some selected drug resistance mutations that abolish CTP binding do not interfere with UTP binding, actually increase the kcat/Km (18), and are located away from the predicted uracil ring catalytic site (4). This behavior raises questions about how a single active site can both simultaneously recognize and discriminate between two highly similar ligands, and how mutations could severely affect the binding of one ligand but not the other.
To address the structural mechanisms of CTP inhibition, infer the binding sites of pyrimidine analogue anti-CTPS drugs, and understand the molecular basis for drug resistance, we determined the structures of two ADP/CTP CTPS complexes, obtained by soaking apo crystals in either substrates or products (Figure 2). The inhibited structures explain how some of the drug resistance mutations exert their effects, resolving the apparent paradox concerning UTP and CTP binding, and depict a novel mode of product inhibition in enzymes.
EXPERIMENTAL PROCEDURES
CTPS crystals in space group P21212 were grown in ammonium sulfate and Tris-HCl at pH 8.5 as previously described (4). The crystals were transferred to mother liquor containing either 30 mM UTP, 30 mM ATP, 30 mM GTP, and 5 mM MgCl2 (substrate-soaked) or 30 mM CTP, 30 mM ADP, and 30 mM GTP (product-soaked). After 20–40 min, the crystals were rapidly transferred to buffer containing 25% 2,4-methylpentanediol and flash-frozen in liquid nitrogen. Useable data to 2.8 Å resolution were collected for the substrate- and product-soaked crystals at SSRL beamline 9-2 and processed using MOSFLM/SCALA (39) with the assistance of Wedger and Scaler Elves (40) (Table 1).
Table 1.
Data Collection and Refinement Statisticsa
| apob | UTP/ATP | CTP/ADP | |
|---|---|---|---|
| cell dimensions (Å) | |||
| a | 165.5 | 163.27 | 163.10 |
| b | 106.81 | 106.38 | 107.77 |
| c | 130.03 | 130.47 | 130.67 |
| wavelength (Å) | 1.08 | 0.975 | 0.975 |
| resolution (Å) (outer bin) | 2.3 (2.34–2.3) | 2.8 (2.95–2.8) | 2.85 (3.02–2.85) |
| Rsym (%) | 7.7 (38) | 8.4 (31) | 7.8 (38) |
| coverage (%) | 99.2 (99.1) | 98.9 (95.1) | 99.4 (99.1) |
| multiplicity (outer bin) | 3.7 (3.6) | 3.3 (3.0) | 3.7 (3.1) |
| I/SD (outer bin) | 23.8 (2.1) | 8.5 (2.6) | 10.5 (3.2) |
| refinement | |||
| resolution range (Å) (outer bin) | 20–2.3 (2.34–2.30) | 20–2.8 (2.95–2.8) | |
| R-work (%) | 21.4 (36.0) | 20.2 (31) | |
| R-free (%) | 28.1 (38.0) | 27.7 (40) | |
| no. of reflections | |||
| working | 97650 | 53781 | |
| free | 4051 | 1932 | |
| no. of atoms | 8992 | 8694 | |
| protein | 8334 | 8328 | |
| solvent | 532 | 252 | |
| ligands | |||
| Mg2+ | 4 | 2 52 (CTP) 54 (ADP) |
|
| rmsd from ideality | |||
| bonds (Å) | 0.009 | 0.007 | |
| angles (deg) | 1.95 | 1.93 | |
| B-factors (Å2) | 3.4 | 2.0 | |
| average B-factor (Å2) | |||
| protein | 59.4 | 60.3 | |
| solvent | 65.8 | 66.2 | |
| Mg2+ | 4 | 39.3 42.7 (CTP) 73.8 (ADP) |
|
Data were collected at SSRL beamline 7-1 using a MAR imaging plate. The data were integrated by MOSFLM and merged with SCALA (39) assisted by Wedger Elves (40). The merging R values were based on intensities for all of the data calculated by SCALA. R-free and R-factor values were calculated by TNT release 5F (41) using the following scaling parameters: Ksol = 0.75, Bsol = 121, K = 1.33. Anisotropic thermal corrections were also used: B11 = −10.17, B22 = 19.57, and B33 = −9.40. Model deviations from ideal geometry were calculated by TNT using Engh and Huber parameters (59) and the BCORRELS library (42). Average model B-factors were calculated with EDPDB (44).
From PDB entry 1S1M (4).
Refinement was carried out using TNT Release 5F with anisotropic temperature factor scaling (41, 42). The apo-EcCTPS 1S1M model lacking solvent or ligand molecules was used as a starting model. After one round of refinement, the CTP and ADP positions were unambiguously visualized in Fo − Fc and 2Fo − Fc maps (Figure 2). However, no GTP density was discernible in preliminary or refined maps. Close inspection indicated a rearrangement in residues 227–233, and the formation of a new disulfide bond between Cys2613 and Cys268 which are adjacent but reduced in the apo structure. The structure converged after several more rounds of model building with O (43) and TNT refinement. The final models were generally similar to the 1S1M structure (main chain rmsd = 0.62 Å). The structure described here is that of the ATP/UTP-soaked crystals which had generally more well-defined CTP density. It contains 1050 of 1090 residues, two CTP and two ADP molecules, two Mg ions, and 252 solvent molecules, and has R-factor and R-free values of 0.201 and 0.277, respectively, with good geometry (Table 1). This structure was deposited in the Protein Data Bank as entry 2AD5.
Structural comparisons and accompanying model calculations were performed utilizing EDPDB (44). Figures were generated using MOLSCRIPT (45) or BOBSCRIPT (46) rendered with RASTER3D (47). Although the active site structures of the two unique monomers in the asymmetric unit are quite similar (rmsd = 0.83 Å for 372 atoms of residues within 8 Å of the bound CTP), the ligand densities were somewhat clearer for monomer B and all figures depict this active site (Figure 2).
The kinetic constants for ATP and 2′-deoxyATP (dATP) in glutamine-dependent CTP synthesis were determined using uncleaved N-terminally His6-tagged EcCTPS protein. The PCR-amplified gene was cloned into NdeI- and XhoI-cleaved pET28b(+), and the protein was overexpressed in B21(DE3) cells and purified to homogeneity using a single metal chelate chromatographic step. Assay for CTP formation was carried out in triplicate as previously described (12) with 100 nM recombinant His6-EcCTPS, following the change in absorbance at 291 nm in 60 mM Na-HEPES (pH 8.0), 10 mM MgCl2, 1 mM UTP, 0.2 mM GTP, 5 mM Gln, and 30, 50, 100, 200, 400, 600, and 1000 μM ATP or dATP at 37°C. Apparent S0.5, Vmax, and Hill coefficient values were obtained by manual fitting to the Hill equation using Excel. For His6-tagged EcCTPS, the specific activity and kinetic constants for ATP were comparable to those of the native enzyme purified from E. coli (12) (H. Kim, unpublished results).
RESULTS
Data from the product- and substrate-soaked crystals yielded identical electron density maps in the vicinity of the CTP synthesis active sites, suggesting that they both depict the product complexes (Figure 2). Thus, the crystals are catalytically active and competent to carry out both the phosphorylation and ammonia ligase reactions. The crystallization mother liquor containing ~0.8 M ammonium sulfate at pH 8.5 likely provided ammonia for the reaction in lieu of Gln hydrolysis, which is readily utilized by CTPSs [KM-(NH3) ~ 2 mM (2, 48)].
Difference electron density features in both substrate- and product-soaked crystals indicated that ADP was bound in the putative ATP binding site, since density for the γ-phosphate was absent (Figure 2). Relative to the P-loop (residues 15–20), the ADP ribose and phosphate moieties bind in the essentially same positions as in DTBS complexes (38), but the adenine ring is shifted ~1 Å deeper into its binding pocket in EcCTPS coincident with a 3–4 Å lateral displacement in the ATP-binding “lid”.
The adenine ring packs into a pit created by the loops of residues 17–21 and 238–247 and the side chains of Ile20 and Arg211 (Figure 3). The specificity for adenine ring N1 and N6 atoms is provided by main chain hydrogen bonds from the amide of Val241 and carbonyl oxygen of Lys239, respectively. As in other similar kinases, such as APS kinase [PDB entry 1M7G (49)], a conserved arginine, Arg211, stacks against the adenine, and is stabilized by a salt bridge with Asp240.
Figure 3.

Adenine nucleotide recognition. The ADP binding site at the 2AD5 model A–B subunit interface is shown. The bound ADP is indicated with black carbon atoms. Residue subunit identities are indicated by color (see Figure 1). The α- and β-phosphates are bound by the P-loop structure of residues 15–20. The ribose ring packs between I20 and A182(A), providing a basis for ATP-induced tetramer formation. Water recognition of O4′ provides sugar specificity; however, no hydrogen bonds form with the 2′- and 3′-hydroxyl groups, and ATP and dATP are equally good substrates (see Results). The adenine ring packs into a pocket formed by the binding “lid” of residues 238–244 and the R211 side chain. Specific hydrogen bonds from lid residues O239 and N241 provide the specificity for adenine. The identities of G17, K18, G19, D72, E140, R211, and D240 are conserved in 41 of 43 of the CTPS COG0504 sequences (4; http://www.ncbi.nlm.nih.gov/COG/old/aln/COG0504.aln), while D303 is conserved in 37 of 43.
The ADP ribose ring is sandwiched between the Ile20 side chain and Ala182(A)4 from the adjacent noncrystallographically-related “dissociable” subunit (Figure 3). This direct contact combined with the extensive interactions of loop residues 176–187 of the adjacent subunit with ATP binding site residues provides the structural basis for ATP-induced tetramer formation. In addition, the ribose O4′ atom is bound via a water-mediated hydrogen bond to Ser15 carbonyl and Arg211 guanidinium moieties. The ribose 2′- and 3′-hydroxyls have no direct protein contacts, although they are proximal to Asp303 and Lys306. However, 2′-hydroxyl recognition is apparently unimportant since ATP and dATP were equally effective cosubstrates with UTP. The apparent S0.5, Vmax, and Hill coefficient values for ATP (140 ± 30 μM, 2.9 ± 0.1 s−1, and 1.4 ± 0.3, respectively) were essentially identical to those for dATP (120 ± 20 μM, 4.2 ± 0.4 s−1, 1.4 ± 0.2, respectively). However, ddATP was not an efficient substrate (data not shown).
The diphosphate moiety is bound by the P-loop via a network of main chain and side chain hydrogen bonds (Figure 3). The β-phosphate is ligated by amide hydrogen bonds with residues 15, 17, and 18, while the α-phosphate is ligated by residue 19 and 20 amides. A Mg2+ ion also bridges the β-phosphate and putative catalytic residues Glu140 and Asp72 in the ATP/UTP-soaked crystals.
The CTP binding site location was defined by unambiguous pyrimidine nucleotide triphosphate density at the interface between three subunits (Figure 2), and binding was accompanied by only minor structural changes. Compared to apo-EcCTPS, the main chain rmsd for the entire tetramer is 0.62 Å, with individual domain main chain rmsd values ranging from 0.4 to 0.5 Å. Relative to the B subunit, the ALase domains of the A and B′ subunits rotated 1.7° and 1.1°, respectively, and shifted their centers of mass 0.6 and 0.9 Å, respectively, bringing the B and B′ subunits 0.31 Å closer together. A 1.1° hinging of the B subunit ALase and GATase domains was also observed.
More significant changes proximal to the CTP-binding site were observed, with main chain shifts, side chain rotations, and increased protein ordering (Figure 4a). B-Factors were 17 Å2 lower for residues within 5 Å of the bound CTP, after correcting for the ~0.6 Å2 rms difference between the product complex and apo-EcCTPS. The cytosine and ribose are accommodated by an “induced fit” protein rearrangement (Figure 4a,b). A Glu149 side chain rotamer switch, rotations of Asp147 (25°) and Ile148 (40°) side chains, and an inward shift of Ile116(B′) create the cytosine ring-binding pocket, while the Gln114(B′) side chain is rotated to both provide space and pack against the ribose ring. Main chain amides from α-helical loop residues 148 and 149 donate hydrogen bonds that recognize the unique pattern of cytosine O2 and N3 acceptors, while the Asp147 carboxylate provides favorable stacking interactions for the electron-deficient pyrimidine ring. The N4 exocyclic atom is not directly contacted, explaining inhibitor tolerance to some substitutions at this position (50), but a water-mediated hydrogen bond to the Gly146 carbonyl oxygen provides potential recognition of N4. The cytosine C5 and C6 atoms are proximal to Phe227-(A); however, they do not form a tight interaction, and the side chain electron density is not clearly defined (data not shown).
Figure 4.
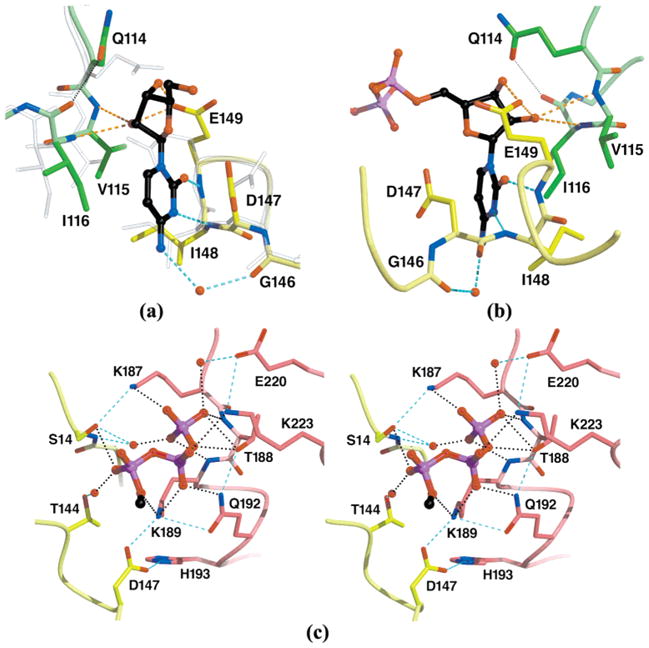
Atomic details of CTP recognition by EcCTPS. The CTP binding site at the model 2AD5 A–B–B′ subunit interface is shown. The bound CTP is indicated with black carbon atoms. Residue subunit identities are indicated by color (see Figure 2). (a) Cytidine recognition by the B and B′ subunits. The triphosphate moiety has been removed for clarity. Subunit B′ rotated 1.1°, and its center of mass shifted 0.3 Å closer to subunit B; side chains D147, E149, and Q114(B′) rotate to accommodate CTP binding (compare CTP complex opaque colored atoms to translucent apo-EcCTPS 1S1M atoms). The cytosine ring is bound via two protein-mediated hydrogen bond with N147 and N148 and one water-mediated hydrogen bond to O146. The ring is sandwiched in a pocket formed by D147, I148, V115(B′), and I116(B′). The identities of Q114, G146, D147, I148, and E149 are conserved in >95% of the CTPS sequences. (b) Ribose recognition. This view is rotated approximately 150° about the vertical axis from the view in panel a. D149 packs against the ribose ring accepting two hydrogen bonds from the 2′- and 3′-hydroxyls. The 2′-hydroxyl is additionally recognized by main chain hydrogen bonding with I116(B′). (c) A–B cross-subunit recognition of the CTP triphosphate. The cytidine nucleoside has been removed for clarity. The triphosphate moiety cements a bridge between the A and B subunits, suggesting a basis for CTP-induced oligomerization. Extensive γ-phosphate interactions are mediated by a P-loop-like structure formed by residues 187–189(A). Note the extensive network of ligand-dependent side chain–side chain hydrogen bonds that would amplify binding specificity. The rotation of D147 induced by cytosine ring binding (see panel a) promotes intersubunit contacts with K189(A) and H193(A). S14, T144, D147, K187, T188, K189, Q192, K223, are conserved in 41 of 43 CTPS sequences (4; http://www.ncbi.nlm.nih.gov/COG/old/aln/COG0504.aln), while H193 is conserved in 37 of 43.
The CTP ribose packs against residues 114–116 (B′) and Glu149. Gln114(B′) vacates the space occupied by ribose, simultaneously capping the main chain carbonyl of Ile116-(B′) (Figure 4a, b). The Glu149 side chain switches from capping its own main chain amide in apo-EcCTPS to accept hydrogen bonds from both the ribose 2′- and 3′-hydroxyl groups. Val115(B′) and Ile116(B′) main chain amide donate additional hydrogen bonds to the 2′-hydroxyl, with main chain atoms 114–117(B′) shifting 0.9–1.0 Å toward the ribose (Figures 4a, b and 5). This trivalent 2′-hydroxyl recognition explains the preference for CTP over dCTP as an inhibitor (50).
Figure 5.
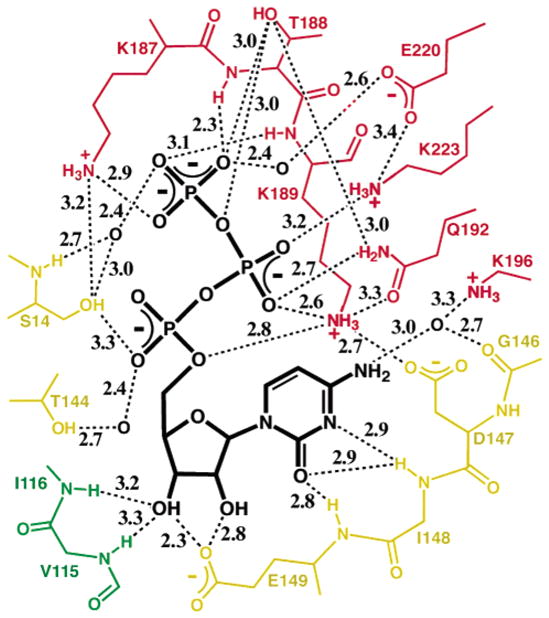
Hydrogen bond summary of bound CTP recognition at the A–B–B′ interface. Potential hydrogen bonds were suggested by distances between nitrogen and/or oxygen atoms that are ≤3.5 Å with appropriate geometry, and are indicated with the stippled lines along with the distances in angstroms. The associated subunits are indicated by color (yellow for B, red for A, and green for B′).
The triphosphate moiety overlaps the predicted UTP triphosphate binding site (4), providing the structural basis for CTP binding competition with UTP and the absolute triphosphate requirement for cytosine ribonucleotide inhibition (50) (Figure 2). The CTP triphosphate makes extensive protein contacts via a network of polar interactions, provided primarily by the noncrystallographically-related “dissociable” subunit (Figures 4c and 5). The γ-phosphate is effectively pinned by six hydrogen bonds from the Lys187(A) and Thr188(A) side chains and the Thr188(A) and Lys189(A) main chain amides, as well as water-mediated interactions with Ser14 and Glu220(A). Lys189(A), Gln192(A), and Lys223(A) donate side chain hydrogen bonds to the β-phosphate. Within the nucleoside-binding reference subunit, the α-phosphate is recognized directly by the Ser14 side chain and via water by the Thr144 side chain. The critical role of the Lys187 side chain–γ-phosphate interaction in UTP bindng is underscored by the inability of Lys187Ala EcCTPS to perform Gln- or NH3-dependent CTP synthesis (51).
In addition to the individual interactions, a number of interdependent protein–protein contacts cooperatively increase the specificity for CTP. Prominently, Thr188(A), Gln192(A), Lys189(A), and Asp147 co-align for phosphate recognition (Figures 4c and 5). This interlocking recognition network also provides an energetic basis for CTP-dependent tetramerization (13) in several ways (Figure 1b). First, the parsing of phosphate and nucleoside contacts to different subunits allows CTP, and presumably UTP, to act as “mortar” to cement the tetramer contacts. Second, phosphate-directed ordering of Lys189(A) and cytosine ring-directed repositioning of Asp147 generate an intersubunit salt bridge network that includes His193(A), while Lys187(A) is repositioned to hydrogen bond with Ser14 (Figures 4c and 5). These interactions are absent in the apo structure. Interestingly, Lys187Ala EcCTPS is not defective in ATP/UTP-dependent tetramerization (51), perhaps because oligomerization is still driven by ATP binding (11, 12). Neutralization of clustered positive charges at the interface by bound triphosphate may also contribute to oligomerization, since Lys187Ala EcCTPS tends to form tetramers in the absence of nucleotides (51).
DISCUSSION
Although feedback inhibition is a common theme in unbranched biosynthetic pathways, relatively few enzymes appear to be primarily product-regulated. The simplest mode entails product binding in a substrate like pose, with product dissociation limiting the maximum flux through the enzyme. Indeed, E. coli chorismate lyase (52), adenylosuccinate synthase (53), brain hexokinase I (54), and Thermoanaerobacter tencongensis HGPRT (55) are inhibited in this manner. Alternatively, second allosteric product binding sites may be present, as in diguanylate cyclase (56), but if substrates are structurally similar to products, substrate inhibition may ensue.
CTPSs have evolved a hybrid strategy for distinguishing between UTP and CTP. The overlapping regions of the product feedback inhibitory and substrate sites recognize a common feature in both substrates, the triphosphate moiety. The feasibility of UTP sharing the CTP triphosphate binding subsite is supported by the lack of obvious alternative phosphate binding sites and by the ease with which the uracil ring can be placed in the putative catalytic site (Figure 2), by rotating the nucleoside ~120° about the O5′–Pα–O3α–Pβ torsion angle, combined with further adjustments about the α, β, and γ torsion angles, while maintaining the entire set of protein–triphosphate interactions. In pantothenate kinase [PDB entries 1ESM and 1ESN (57)], the adenine rings of the ATP substrate and coenzyme A downstream product inhibitor also occupy widely separated pockets while their phosphate positions overlap. The novel aspect for CTPS is utilization of alternative binding modes to distinguish its remarkably similar substrate and product in contiguous enzyme surfaces.
The observed CTP positioning, along with the expected overlapping UTP substrate pose, explains at once how CTP competes for UTP binding and promotes tetramer formation, while mutations can abolish the CTP binding without reducing UTP affinity. The physical spanning of the dissociable tetramer interface by the ligand binding sites provides the basis for substrate- and product-induced tetramer formation. The extensive interdependent contacts, particularly with the γ-phosphate, provide an explanation for the absolute requirement for triphosphate substrate and regulator, as well as the requirement for conversion of noninhibitory CPEC and 3deazaU in vivo to the inhibitory nucleoside triphosphates (22, 50, 58). The thermodynamic linkage between substrate binding and the assembly of competent CTP synthesis sites clearly explains the apparent positive cooperativity of ATP and UTP at subsaturating substrate concentrations, and supports the idea that CTPSs are regulated in part by an association–dissociation mechanism (3, 11, 12). In addition, the product-inhibited enzyme is still in the “active” tetrameric form, and is conceivably poised to receive substrates, potentially allowing for a faster response to changing metabolic needs for CTP. Indeed, at physiological enzyme concentrations, a significant lag in activity is observed if EcCTPS is not preincubated with UTP or ATP (12), and at low enzyme concentrations, CTP stimulates activity, presumably by promoting tetramerization (3, 13). The ability to exchange the pyrimidine ring between sites without shifting the triphosphate suggests the possibility that CTP might bind the inhibitory site immediately after synthesis and prior to dissociating from the enzyme.
To differentiate isosteric substrate and product pyrimidine rings, an additional pocket, lined by residues 115, 116, and 146–148 and far from the expected kinase/ligase catalytic site, specifically recognizes the cytosine and ribose portions of the inhibitor. The bidentate hydrogen bonds donated by the Ile148 and Glu149 main chain amides distinguish the acceptor–acceptor–donor (A–A–D) pattern of cytosine from the A–D–A pattern of uracil, possibly supported by a water-mediated O146–N4 interaction. Presumably, recognition of the 2-oxo-4-hydroxypyridine ring of 3-deazaU ring relies more heavily on the latter contact.
The cytosine nucleoside positioning is in agreement with the locations of CTP inhibition-releasing mutations at positions 116, 146–148, and 151 (Figure 6). Not surprisingly, none of the substitutions occur in the triphosphate subsite, which would affect UTP binding and CTP synthesis. Instead, these substitutions are predicted to specifically disrupt CTP binding. For example, the Asp147Glu substitution would be unable to stack on the cytosine ring and form the intersubunit salt bridge with Lys189 and His193 (Figure 4c); the Gly146Glu substitution would destabilize the glycine left-hand turn and disrupt the loop of residues 143–147, and the Ile116Phe substitution would incur a steric conflict with bound CTP (Figure 4a, b). Interestingly, Ile148 and Leu151 participate in the B′–B′ interface and potentially provide a direct interaction path between dimer interface-related CTP sites, suggesting both a multiplying effect of position 148 and 151 substitutions on CTP binding and the potential for allosteric communication of CTP sites with each other or with the interfacial UTP or ATP sites. Indeed, given the proximity of the segments of each of the four protein chains, binding CTP at even a single site could conceivably influence interactions of all of the subunits.
While the CTP complex structure presented here provides straightforward rationales for some of the resistance mutations, positions 155, 158, and 229 do not directly contact the bound CTP and it is not apparent how the associated resistance substitutions exert their effects (Figure 6). These sites line a loosely associated and sequence-conserved interface formed by the B, B′, and A subunits. The lack of intimate contacts and the disorder of residues near the CTP site, such as Phe227 in the apo or CTP-inhibited forms described above, may indicate a yet-unidentified conformational change that tightens the interface upon CTP binding in solution but is not allowed in the crystals.
The hybrid strategy used by CTPSs for differentiation of substrate and product allows for greater evolutionary adaptability without requiring a separate product regulatory site. Affinity and specificity for substrates and products can be fine-tuned by adaptive mutation or protein modification independently, allowing responses to intracellular CTP concentrations without compromising catalytic efficiency. For CTPS, this property also makes cytidine analogues less attractive as anti-CTPS drugs since resistance to them can be acquired while maintaining CTP synthesis activity. A further limitation, in agreement with inhibitor studies (50), is the suggestion from the CTP complex structure that modifications to improve cytidine triphosphate analogue binding would be limited to cytosine N4, with size limits to C5 and C6. Finally, selected resistances to some pyrimidine compounds, such as 5-fluorouracil (36) which is not a CTPS substrate (50), may not involve altered drug–enzyme interactions but rather selection for increased intracellular CTP levels that inhibit their uptake and incorporation.
Acknowledgments
We thank Profs. Irwin Segel and Michael Toney (University of California) for helpful discussions. Protein purifications, home-source data collections, and all computations were carried out in the W. M. Keck Protein Expression and X-ray Crystallographic Facilities at the University of California. Synchrotron data were obtained at the Stanford Synchrotron Radiation Laboratory, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences.
Footnotes
This work was funded by UC Systemwide Biotechnology Training Program Grant 2002–07 and the National Institutes of Health, General Medical Sciences (Grant GM63109).
The coordinates have been deposited in the Protein Data Bank as entry 2AD5.
Abbreviations: CTPS, cytidine triphosphate synthetase; CPEC, cyclopentenylcytosine; 3deazaU, 3-deazauridine; EcCTPS, Escherichia coli cytidine triphosphate synthetase; ALase, ammonia ligase reaction, i.e., the formation of a carbon–nitrogen bond by displacement of a phosphorylated oxygen; GATase, glutamine amidotransferase, which refers to a conserved glutamine hydrolysis domain that provides ammonia for a number of diverse enzyme-catalyzed reactions; DTBS, E. coli dethiobiotin synthetase (BioD) enzyme; rmsd, root-mean-square difference in position between atom sets, in angstroms.
The asymmetric unit of the EcCTPS P21212 crystal contains subunits A and B. Symmetry chains in the functional tetramer are denoted by a prime; for example, the dyad-related subunit of chain B is B′.
In the text, specific protein residues are indicated by their three-letter residue type and residue number. In figures and legends, the one-letter abbreviation is used to reduce label size.
For residues that are donated by other subunits, the chain designation is in parentheses, after the residue designations described in footnote 3. For example, lysine 187 from the symmetry mate of the B subunit is denoted Lys187(B′).
References
- 1.Liebermann I. Enzymatic amination of uridine triphosphate to cytidine triphosphate. J Biol Chem. 1956;222:765–775. [PubMed] [Google Scholar]
- 2.Charkraborty KP, Hurlbert RB. Role of glutamine in the biosynthesis of cytidine nucleotides in Escherichia coli. Biochim Biophys Acta. 1961;47:607–609. doi: 10.1016/0006-3002(61)90563-7. [DOI] [PubMed] [Google Scholar]
- 3.Long CW, Pardee AB. Cytidine triphosphate synthetase of Escherichia coli B. I. Purification and kinetics. J Biol Chem. 1967;242:4715–4721. [PubMed] [Google Scholar]
- 4.Endrizzi JA, Kim H, Anderson PM, Baldwin EP. Crystal structure of Escherichia coli cytidine triphosphate synthetase, a nucleotide-regulated glutamine amidotransferase/ATP-dependent amidoligase fusion protein and homologue of anticancer and antiparasitic drug targets. Biochemistry. 2004;43:6447–6463. doi: 10.1021/bi0496945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levitzki A, Koshland DE., Jr Cytidine triphosphate synthetase. Covalent intermediates and mechanisms of action. Biochemistry. 1971;10:3365–3371. doi: 10.1021/bi00794a008. [DOI] [PubMed] [Google Scholar]
- 6.von der Saal W, Anderson PM, Villafranca JJ. Mechanistic investigations of Escherichia coli cytidine-5′-triphosphate synthetase. Detection of an intermediate by positional isotope exchange experiments. J Biol Chem. 1985;260:14993–14997. [PubMed] [Google Scholar]
- 7.Lewis DA, Villafranca JJ. Investigation of the mechanism of CTP synthetase using rapid quench and isotope partitioning methods. Biochemistry. 1989;28:8454–8459. doi: 10.1021/bi00447a027. [DOI] [PubMed] [Google Scholar]
- 8.Aronow B, Ullman B. In situ regulation of mammalian CTP synthetase by allosteric inhibition. J Biol Chem. 1987;262:5106–5112. [PubMed] [Google Scholar]
- 9.Kizaki H, Ohsaka F, Sakurada T. CTP synthetase from Ehrlich ascites tumor cells. Subunit stoichiometry and regulation of activity. Biochim Biophys Acta. 1985;829:34–43. doi: 10.1016/0167-4838(85)90065-2. [DOI] [PubMed] [Google Scholar]
- 10.Levitzki A, Koshland DE., Jr Role of an allosteric effector. Guanosine triphosphate activation in cytosine triphosphate synthetase. Biochemistry. 1972;11:241–246. doi: 10.1021/bi00752a015. [DOI] [PubMed] [Google Scholar]
- 11.Levitzki A, Koshland DE., Jr Ligand-induced dimer-to-tetramer transformation in cytosine triphosphate synthetase. Biochemistry. 1972;11:247–253. doi: 10.1021/bi00752a016. [DOI] [PubMed] [Google Scholar]
- 12.Anderson PM. CTP synthetase from Escherichia coli: An improved purification procedure and characterization of hysteretic and enzyme concentration effects on kinetic properties. Biochemistry. 1983;22:3285–3292. doi: 10.1021/bi00282a038. [DOI] [PubMed] [Google Scholar]
- 13.Pappas A, Yang WL, Park TS, Carman GM. Nucleotide-dependent tetramerization of CTP synthetase from Saccharomyces cerevisiae. J Biol Chem. 1998;273:15954–15960. doi: 10.1074/jbc.273.26.15954. [DOI] [PubMed] [Google Scholar]
- 14.Yang WL, McDonough VM, Ozier-Kalogeropoulos O, Adeline MT, Flocco MT, Carman GM. Purification and characterization of CTP synthetase, the product of the URA7 gene in Saccharomyces cerevisiae. Biochemistry. 1994;33:10785–10793. doi: 10.1021/bi00201a028. [DOI] [PubMed] [Google Scholar]
- 15.Hurlbert RB, Kammen HO. Formation of cytidine nucleotides from uridine nucleotides by soluble mammalian enzymes: Requirements for glutamine and guanosine nucleotides. J Biol Chem. 1960;235:443–449. [Google Scholar]
- 16.Weinfeld H, Savage CR, Jr, McPartland RP. CTP synthetase of bovine calf liver. Methods Enzymol. 1978;51:84–90. doi: 10.1016/s0076-6879(78)51015-x. [DOI] [PubMed] [Google Scholar]
- 17.Carman GM, Kersting MC. Phospholipid synthesis in yeast: Regulation by phosphorylation. Biochem Cell Biol. 2004;82:62–70. doi: 10.1139/o03-064. [DOI] [PubMed] [Google Scholar]
- 18.Ostrander DB, O’Brien DJ, Gorman JA, Carman GM. Effect of CTP synthetase regulation by CTP on phospholipid synthesis in Saccharomyces cerevisiae. J Biol Chem. 1998;273:18992–19001. doi: 10.1074/jbc.273.30.18992. [DOI] [PubMed] [Google Scholar]
- 19.Lim MI, Moyer JD, Cysyk RI, Marquez VE. Cyclopentenyluridine and cyclopentenylcytidine analogues as inhibitors of uridine-cytidine kinase. J Med Chem. 1984;27:1536–1538. doi: 10.1021/jm00378a002. [DOI] [PubMed] [Google Scholar]
- 20.Gharehbaghi K, Zhen W, Fritzer-Szekeres M, Szekeres T, Jayaram HN. Studies on the antitumor activity and biochemical actions of cyclopentenyl cytosine against human colon carcinoma HT-29 in vitro and in vivo. Life Sci. 1999;64:103–112. doi: 10.1016/s0024-3205(98)00540-2. [DOI] [PubMed] [Google Scholar]
- 21.Verschuur AC, van Gennip AH, Brinkman J, Voute PA, van Kuilenburg AB. Cyclopentenyl cytosine induces apoptosis and secondary necrosis in a T-lymphoblastic leukemic cell-line. Adv Exp Med Biol. 2000;486:319–325. doi: 10.1007/0-306-46843-3_62. [DOI] [PubMed] [Google Scholar]
- 22.Wang MC, Bloch A. Studies on the mode of action of 3 deazapyrimidines. 1 Metabolism of 3-deazauridine and 3-deazacytidine in microbial and tumor cells. Biochem Pharmacol. 1972;21:1063–1073. doi: 10.1016/0006-2952(72)90100-1. [DOI] [PubMed] [Google Scholar]
- 23.McPartland RP, Wang MC, Bloch A, Weinfeld H. Cytidine 5′-triphosphate synthetase as a target for inhibition by the antitumor agent 3-deazauridine. Cancer Res. 1974;34:3107–3111. [PubMed] [Google Scholar]
- 24.Brockman RW, Shaddix SC, Williams M, Nelson JA, Rose LM, Schabel FM., Jr The mechanism of action of 3-deazauridine in tumor cells sensitive and resistant to arabinosylcytosine. Ann NY Acad Sci. 1975;255:501–521. doi: 10.1111/j.1749-6632.1975.tb29254.x. [DOI] [PubMed] [Google Scholar]
- 25.Wylie JL, Wang LL, Tipples G, McClarty G. A single point mutation in CTP synthetase of Chlamydia trachomatis confers resistance to cyclopentenyl cytosine. J Biol Chem. 1996;271:15393–15400. doi: 10.1074/jbc.271.26.15393. [DOI] [PubMed] [Google Scholar]
- 26.Bierau J, Van Gennip AH, Leen R, Helleman J, Caron HN, Van Kuilenburg AB. Cyclopentenyl cytosine primes SK-N-BE(2)c neuroblastoma cells for cytarabine toxicity. Int J Cancer. 2003;103:387–392. doi: 10.1002/ijc.10858. [DOI] [PubMed] [Google Scholar]
- 27.Verschuur AC, Van Gennip AH, Leen R, Voute PA, Brinkman J, Van Kuilenburg AB. Cyclopentenyl cytosine increases the phosphorylation and incorporation into DNA of 1-β-D-arabinofuranosyl cytosine in a human T-lymphoblastic cell line. Int J Cancer. 2002;98:616–623. doi: 10.1002/ijc.10211. [DOI] [PubMed] [Google Scholar]
- 28.Dereuddre-Bosquet N, Roy B, Routledge K, Clayette P, Foucault G, Lepoivre M. Inhibitors of CTP biosynthesis potentiate the anti-human immunodeficiency virus type 1 activity of 3TC in activated peripheral blood mononuclear cells. Antiviral Res. 2004;61:67–70. doi: 10.1016/j.antiviral.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 29.Verschuur AC, Van Gennip AH, Leen R, Van Kuilenburg AB. Increased cytotoxicity of 2′,2′-difluoro-2′-deoxycytidine in human leukemic cell-lines after a preincubation with cyclopentenyl cytosine. Nucleosides, Nucleotides Nucleic Acids. 2004;23:517–521. doi: 10.1081/NCN-200027733. [DOI] [PubMed] [Google Scholar]
- 30.Hofer A, Steverding D, Chabes A, Brun R, Thelander L. Trypanosoma brucei CTP synthetase: A target for the treatment of African sleeping sickness. Proc Natl Acad Sci USA. 2001;98:6412–6416. doi: 10.1073/pnas.111139498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hendriks EF, O’Sullivan WJ, Stewart TS. Molecular cloning and characterization of the Plasmodium falciparum cytidine triphosphate synthetase gene. Biochim Biophys Acta. 1998;1399:213–218. doi: 10.1016/s0167-4781(98)00108-0. [DOI] [PubMed] [Google Scholar]
- 32.Jimenez BM, O’Sullivan WJ. CTP synthetase and enzymes of pyrimidine ribonucleotide metabolism in Giardia intestinalis. Int J Parasitol. 1994;24:713–718. doi: 10.1016/0020-7519(94)90125-2. [DOI] [PubMed] [Google Scholar]
- 33.Tipples G, McClarty G. Cloning and expression of the Chlamydia trachomatis gene for CTP synthetase. J Biol Chem. 1995;270:7908–7914. doi: 10.1074/jbc.270.14.7908. [DOI] [PubMed] [Google Scholar]
- 34.Andrei G, De Clercq E. Molecular approaches for the treatment of hemorrhagic fever virus infections. Antiviral Res. 1993;22:45–75. doi: 10.1016/0166-3542(93)90085-w. [DOI] [PubMed] [Google Scholar]
- 35.Whelan J, Phear G, Yamauchi M, Meuth M. Clustered base substitutions in CTP synthetase conferring drug resistance in Chinese hamster ovary cells. Nat Genet. 1993;3:317–322. doi: 10.1038/ng0493-317. [DOI] [PubMed] [Google Scholar]
- 36.Aronow B, Ullman B. Mammalian mutants genetically altered in CTP synthetase activity. Adv Exp Med Biol. 1986;195(Part B):263–269. doi: 10.1007/978-1-4684-1248-2_42. [DOI] [PubMed] [Google Scholar]
- 37.Goto M, Omi R, Nakagawa N, Miyahara I, Hirotsu K. Crystal structures of CTP synthetase reveal ATP, UTP, and glutamine binding sites. Structure. 2004;12:1413–1423. doi: 10.1016/j.str.2004.05.013. [DOI] [PubMed] [Google Scholar]
- 38.Kack H, Sandmark J, Gibson KJ, Schneider G, Lindqvist Y. Crystal structure of two quaternary complexes of dethiobiotin synthetase, enzyme-MgADP-AlF3-diaminopelar-gonic acid and enzyme-MgADP-dethiobiotin-phosphate; implications for catalysis. Protein Sci. 1998;7:2560–2566. doi: 10.1002/pro.5560071209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Collaborative Computational Project Number 4. The CCP4 suite of programs for protein crystallography. Acta Crystallogr. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 40.Holton J, Alber T. Automated protein crystal structure determination using ELVES. Proc Natl Acad Sci USA. 2004;101:1537–1542. doi: 10.1073/pnas.0306241101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tronrud DE. TNT refinement package. Methods Enzymol. 1997;277:306–319. doi: 10.1016/s0076-6879(97)77017-4. [DOI] [PubMed] [Google Scholar]
- 42.Tronrud D. Knowledge-based B-factor restraints for the refinement of proteins. J Appl Crystallogr. 1996;29:100–104. [Google Scholar]
- 43.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. 1991;A47(Part 2):110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 44.Zhang XJ, Matthews BW. EDPDB: A multi-functional tool for protein structure analysis. J Appl Crystallogr. 1995;28:624–630. [Google Scholar]
- 45.Kraulis PJ. MOLSCRIPT: A Program to Produce Both Detailed and Schematic Plots of Protein Structures. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 46.Esnouf RM. An extensively modified version of MolScript that includes greatly enhanced coloring capabilities. J Mol Graphics Modell. 1997;15:132–134. 112–113. doi: 10.1016/S1093-3263(97)00021-1. [DOI] [PubMed] [Google Scholar]
- 47.Merritt EA, Bacon DJ. Raster3D: Photorealistic Molecular Graphics. Methods Enzymol. 1997;277:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
- 48.Iyengar A, Bearne SL. Aspartate-107 and leucine-109 facilitate efficient coupling of glutamine hydrolysis to CTP synthesis by Escherichia coli CTP synthase. Biochem J. 2003;369:497–507. doi: 10.1042/BJ20021110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lansdon EB, Segel IH, Fisher AJ. Ligand-induced structural changes in adenosine 5′-phosphosulfate kinase from Penicillium chrysogenum. Biochemistry. 2002;41:13672–13680. doi: 10.1021/bi026556b. [DOI] [PubMed] [Google Scholar]
- 50.Scheit KH, Linke HJ. Substrate specificity of CTP synthetase from Escherichia coli. Eur J Biochem. 1982;126:57–60. doi: 10.1111/j.1432-1033.1982.tb06745.x. [DOI] [PubMed] [Google Scholar]
- 51.Simard D, Hewitt KA, Lunn F, Iyengar A, Bearne SL. Limited proteolysis of Escherichia coli cytidine 5′-triphosphate synthase. Identification of residues required for CTP formation and GTP-dependent activation of glutamine hydrolysis. Eur J Biochem. 2003;270:2195–2206. doi: 10.1046/j.1432-1033.2003.03588.x. [DOI] [PubMed] [Google Scholar]
- 52.Gallagher DT, Mayhew M, Holden MJ, Howard A, Kim KJ, Vilker VL. The crystal structure of chorismate lyase shows a new fold and a tightly retained product. Proteins. 2001;44:304–311. doi: 10.1002/prot.1095. [DOI] [PubMed] [Google Scholar]
- 53.Iancu CV, Borza T, Fromm HJ, Honzatko RB. Feedback inhibition and product complexes of recombinant mouse muscle adenylosuccinate synthetase. J Biol Chem. 2002;277:40536–40543. doi: 10.1074/jbc.M204952200. [DOI] [PubMed] [Google Scholar]
- 54.Aleshin AE, Zeng C, Bourenkov GP, Bartunik HD, Fromm HJ, Honzatko RB. The mechanism of regulation of hexokinase: New insights from the crystal structure of recombinant human brain hexokinase complexed with glucose and glucose-6-phosphate. Structure. 1998;6:39–50. doi: 10.1016/s0969-2126(98)00006-9. [DOI] [PubMed] [Google Scholar]
- 55.Chen Q, Liang Y, Su X, Gu X, Zheng X, Luo M. Alternative IMP binding in feedback inhibition of hypoxanthine phosphoribosyltransferase from Thermoanaerobacter tengcongensis. J Mol Biol. 2005;348:1199–1210. doi: 10.1016/j.jmb.2005.02.064. [DOI] [PubMed] [Google Scholar]
- 56.Chan C, Paul R, Samoray D, Amiot NC, Giese B, Jenal U, Schirmer T. Structural basis of activity and allosteric control of diguanylate cyclase. Proc Natl Acad Sci USA. 2004;101:7084–7089. doi: 10.1073/pnas.0406134101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yun M, Park CG, Kim JY, Rock CO, Jackowski S, Park HW. Structural basis for the feedback regulation of Escherichia coli pantothenate kinase by coenzyme A. J Biol Chem. 2000;275:28093–28099. doi: 10.1074/jbc.M003190200. [DOI] [PubMed] [Google Scholar]
- 58.McPartland RP, Weinfeld H. Cytidine 5′-triphosphate synthetase of calf liver. Size, polymerization, and reaction stoichiometry. J Biol Chem. 1976;251:4372–4378. [PubMed] [Google Scholar]
- 59.Engh R, Huber R. Accurate bond and angle parameters for X-ray protein structure refinement. Acta Crystallogr. 1991;A47:392–400. [Google Scholar]