

Abstract
β,γ-Fluoromethylene analogues of nucleotides are considered to be useful mimics of the natural substrates, but direct structural evidence defining their active site interactions has not been available, including the influence of the new chiral center introduced at the CHF carbon, as in β,γ-fluoromethylene-dGTP, which forms a active site complex with DNA polymerase β, a repair enzyme that plays an important role in base excision repair (BER) and oncogenesis. We report X-ray crystallographic results for a series of β,γ-CXY dGTP analogues, where X,Y = H, F, Cl, Br, and/or CH3. For all three monofluorinated analogues examined (CHF, 3/4; CCH3F, 13/14; CClF 15/16), a single CXF-diastereomer (3, 13, 15) is observed in the active site complex, with the CXF fluorine atom at a ~3 Å (bonding) distance to a guanidinium N of Arg183. In contrast, for the CHCl, CHBr and CHCH3 analogues, both diasteromers (6/7, 8/9, 10/11) populate the dGTP site in the enzyme complex about equally. The structures of the bound dichloro (5) and dimethyl (12) analogue complexes indicate little to no steric effect on the placement of the bound nucleotide backbone. The results suggest that introduction of a single fluorine atom at the β,γ-bridging carbon atom of these dNTP analogues enables a new, stereospecific interaction within the pre-organized active site complex that is unique to fluorine. The results also provide the first diverse structural dataset permitting an assessment of how closely this class of dNTP analogues mimics the conformation of the parent nucleotide within the active site complex.
INTRODUCTION
DNA polymerases are crucial to maintaining the fidelity of genetic information encoded into DNA, and failure to repair aberrant bases in damaged DNA strands is notoriously implicated in oncogenesis.1 During base excision repair (BER),2,3 DNA polymerase β (pol β) typically inserts a single deoxynucleoside triphosphate (dNTP) replacing an excised damaged or mismatched nucleoside residue with release of pyrophosphate. In the ongoing effort to elucidate mechanistic details of these processes, pol β, the smallest eukaryotic cellular DNA polymerase, has been the subject of extensive studies examining its key roles in BER3 and cancer1.
Designed modifications in the structures of natural dNTPs or NTPs can provide information on molecular interactions with nucleic acid polymerases.3–14 Often, such analogues are modified in their purine or pyrimidine bases or (deoxy)ribose moieties. However, changes in the triphosphate group4–8,10–13,15–18 are of particular interest because they involve the locus of chemical transformation catalyzed in the polymerase active site. Replacement of the Pα-O-Pβ bridging oxygen by a methylene carbon atom will prevent release of the pyrophosphate leaving group, whereas a Pβ-CXY-Pγ modification alters the leaving group properties depending on the nature of substituents X and Y. The introduction of these substituents may also enable entirely new bonding (or repulsive) active site interactions, not present with the natural nucleoside triphosphate, which if understood could be exploited to aid design of new inhibitors targeting DNA polymerases such as pol β.
Recently, we introduced a series of β,γ-CXY dGTPs to probe leaving group effects on pol β catalysis and fidelity.15–17 Unlike α, β–CXY dNTP analogues,18 these compounds are substrates of the polymerase, but release a substituted bisphosphonate in place of the natural pyrophosphate leaving group. Several β,γ-CXY nucleotide analogues were previously investigated in studies of DNA, viral RNA or RNA-directed DNA polymerases,4,5,7,10–13,19 but the structures of the putative complexes formed were not determined. The obtention of diastereomeric mixtures owing to the generation of a new chiral center when X ≠ Y in such analogues, and the resulting potential for a stereospecific interaction with the enzyme active site, was not addressed in these studies.10,11,13 We have presented X-ray crystallographic evidence that a β,γ-fluoromethylene-dGTP- primer-DNA ternary complex with pol β uniquely contained the (R)-CHF diastereomer (3), although the complex was obtained by exposing crystals of primer-DNA to a ~1:1 mixture (as confirmed by 19F NMR at high pH) of the R/S diastereomers (3, 4).15 A computer-based docking simulation using Autodock 319 was consistent with the experimental result, indicating the existence of a polar bonding interaction (3.1 Å) between one guanidininium nitrogen of Arg183 in the enzyme active site, and the bound (R)-CHF fluorine atom. The structures of complexes with the CH2 and CF2 dGTP analogues (1 and 2) were also determined, and demonstrated that the positions and conformations of 1–3 in the dGTP site of the complex were similar. No evidence was found for a steric or other energetically disfavoring interaction of the pro-(S) fluorine in 2 that could account for the presence of only one diastereomer in the ternary complex obtained from the 3/4 mixture.15
Here we report a systematic synthetic and structural investigation involving an extended series of halogenated and methylated β,γ-CXY dGTP analogues (1–16) exhibiting a range of stereoelectronic properties at the CXY group, with the goal of examining the uniqueness, scope and origin of the observed β,γ-CHF dGTP binding stereospecificity by DNA pol β. The results also provide the first diverse structural dataset permitting an assessment of how closely this class of dNTP analogues mimics the conformation of the parent nucleotide within the active site complex.
RESULTS AND DISCUSSION
Synthesis and purification of β,γ-CXY dGTP analogues
The previously unknown nucleotide analogues 10–14 were prepared by DCC-mediated conjugation in anhydrous DMSO of dGMP morpholidate6a,6c,20 with the tributylammonium salt of the appropriate methylene(bisphosphonic acid)15–17,21,22 (Scheme 1). The remaining analogues (1–9, 15, 16), prepared similarly, have been described previously.15–17 All compounds were purified by two-stage (SAX and C18) preparative HPLC to obtain samples free of nucleotide-like contaminants.15,18
Scheme 1.

Synthesis of β,γ-methylenebis(phosphonate) dGTP analogues
When differently substituted methylenebisphosphonates are coupled to a nucleoside or deoxy nucleoside 5′-phosphate derivative, the prochiral CXY carbon becomes a new chiral center in the product NTP or dNTP analogue. McKenna and Eran23 reported that at alkaline pH, the 19F NMR spectrum of β,γ-CHF ATP6 exhibits two set of multiplets attributable to the generation of two diastereomers in the synthesis. However, subsequent work has tended to ignore this structural heterogeneity in unsymmetrically substituted β,γ-CXY nucleotide analogues.10,11,13 In our preliminary account of the present work, we reported that highly purified β,γ-CHF dGTP (i.e., 3/4) also displays two resolvable multiplets of close to equal intensity at pH 10 or higher, as shown in Fig. 1a. The spectra can be fitted to two independent resonances at δ = −218.61 and −218.77 ppm with coupling constants, 2JFH = 44.7, 2JFP = 55.9, 2JFP′ = 66.9 Hz (Fig. 1b) The diastereomeric components were not separable by C18 or SAX HPLC using the conditions applied for purification of the samples.
Figure 1.



a) 19F NMR of 3 and 4 in D2O at pH 10, δ −218.61 and −218.71 ppm; b) calculated individual 19F NMR (red) for the two diastereomers, ddd, 2JFH = 44.7, 2JFP = 55.9, 2JFP′ = 66.9 Hz, each superimposed on the experimental 19F NMR of 3 and 4; c) 19F NMR of mixture of 13 and 14 obtained synthetically, in D2O at pH 10, δ −176.51 and −176.49 ppm; d) Blue: actual NMR spectra of 13 and 14, red: predicted NMR for the two separate diastereomers; e) 19F NMR of 15 and 16 in D2O at pH 10, δ −136.49 and −136.51 ppm; f) Blue: actual NMR spectra of 3 and 4, red: predicted NMR for the two separate diastereomers. It should be noted that we are not yet able to assign the pairwise resonances to specific diastereomers.
We have now synthesized and investigated two new fluoromethylene analogues: β,γ-CCH3F dGTP (13/14) and β,γ-CClF dGTP (15/16). The 19F NMR spectrum of the β,γ-CCH3F dGTP analogue (13/14) in D2O at pH 10 also reveals two overlapping multiplets (Fig. 1c and 1d), with δ = −176.51, −176.49 (each tdd; 3JFH = 26, 2JFP = 62.5, 2JFP′ = 74 Hz) and confirms that both diastereomers are present in the purified product in a ratio close to 1:1. Similarly, the 19F NMR spectrum of 15/16 presents as a partially resolved pair (~1:1) of overlapping double doublets (Fig. 1e and 1f), δ = −136.49, −136.51, 2JFP = 66.5, 2JFP′ = 80.6 Hz (the difluoromethylene analogue 2 shows only a dd pattern as expected). The mixtures were used without any further attempt to separate their stereoisomeric components in subsequent co-crystallization experiments with DNA pol β.
X-ray diffraction crystallography of β,γ-CXY-dGTP human pol β-DNA ternary complexes
For protein crystallography, human pol β was overexpressed in E. coli and purified as described previously.24 The double-stranded DNA substrate consisted of a 16-mer template (5′-CCGACCGCGCATCAGC-3′), a complementary 9-mer primer (5′-GCTGATGCG-3′), and a 5-mer downstream oligonucleotide (5′-pGTCGG-3′), thus creating a two-nucleotide gap with annealed primer. Addition of ddCTP creates a one-nucleotide gapped product with a dideoxy-terminated primer and the remaining C in the gap. Crystals of the DNA-enzyme binary complex were first grown. The dGTP analogues were then soaked into the crystals, resulting in the ternary complex crystals used for crystallographic structure determination.
Well-diffracting single crystals were obtained from all the nucleotide-DNA pol β solutions, and the crystal structures were resolved at 1.90–2.15 Å. (Table 1; data for the monofluoro 3 complex were provided in our preliminary communication15). Comparison of these structures with those for the parent CH2 and CF2 dGTP analogues (1 and 2) along with the published structure of the ddCTP complex reveals that overlays of the deoxyribose-phosphobisphosphonate backbones of all the bound analogues are highly congruent, demonstrating that introduction of the bridging β,γ-methylene for the natural oxygen atom in a dNTP has little effect on the bound conformation. Even relatively bulky substituents such as bromine (8/9) or methyl (10–13) do not perturb the overall fit of the substrate in the active site dNTP binding region. For example, as shown in Fig. 2a, substitution of the β,γ-O in ddCTP (PDB ID 2FMP) with CCl2 (5) or C(CH3)2 (12) in the dGTP analogues is well tolerated structurally in the complex, with virtual superposition of the backbone moieties.
Table 1.
Crystallographic and refinement statistics
| Ligand (dGDPCxyP) | -Cl2- | -HCl- | -HBr- | -HCH3- | -(CH3)2- | -F(CH3)- | -FCl- |
| Compound | 5 | 6/7 | 8/9 | 10/11 | 12 | 13 | 16 |
| PDB ID | 3JPN | 3JPO | 3JPQ | 3JPP | 3JPR | 3JPS | 3JPT |
|
| |||||||
|
Data Collection
| |||||||
| Space Group | P21 | P21 | P21 | P21 | P21 | P21 | P21 |
| a (Å) | 50.65 | 50.46 | 50.68 | 50.70 | 50.63 | 50.57 | 50.66 |
| b (Å) | 80.11 | 79.99 | 80.18 | 80.11 | 80.11 | 79.86 | 80.12 |
| c (Å) | 55.45 | 55.43 | 55.56 | 55.55 | 55.33 | 55.55 | 55.45 |
| β (°) | 107.93 | 107.91 | 107.82 | 107.95 | 108.07 | 107.84 | 108.05 |
| dmin (Å) | 2.15 | 2.00 | 1.90 | 2.10 | 2.10 | 2.00 | 2.15 |
| Rmerge (%)a | 0.068 (0.272) | 0.071 (0.445) | 0.059 (0.264) | 0.073 (0.318) | 0.070 (0.304) | 0.055 (0.268) | 0.101 (0.412) |
| Completeness (%)b | 98.1 (95.3) | 99.4 (97.8) | 98.0 (96.3) | 99.8 (100) | 95.7 (93.8) | 95.2 (88.6) | 99.7 (98.3) |
| Unique Reflections | 22602 (2180) | 28269 (2766) | 32753 (3204) | 24695 (2441) | 23602 (2288) | 27208 (2479) | 22938 (2244) |
| Total Reflections | 82115 | 98958 | 120418 | 87669 | 87901 | 82991 | 82040 |
| I/s | 15.3 (4.17) | 13.9 (2.42) | 20.9 (4.74) | 16.0 (3.34) | 15.2 (3.76) | 21.2 (3.00) | 10.1 (2.54) |
|
| |||||||
|
Refinement
| |||||||
| r.m.s. Deviations | |||||||
| Bond angles (°) | 1.095 | 1.112 | 1.096 | 1.085 | 1.098 | 1.093 | 1.096 |
| Rwork (%)c | 18.71 | 19.36 | 19.34 | 19.76 | 20.07 | 19.10 | 19.35 |
| Rfree (%)d | 24.10 | 24.72 | 23.43 | 24.96 | 25.29 | 23.70 | 25.35 |
| Average B Factors (Å) | |||||||
| Protein | 24.71 | 28.58 | 21.70 | 28.44 | 30.45 | 25.08 | 25.28 |
| DNA | 35.80 | 38.90 | 34.17 | 40.49 | 40.68 | 38.29 | 36.18 |
| Analogue | 13.52 | 18.41 | 12.14 | 18.09 | 19.24 | 14.62 | 14.53 |
| Ramachandaran Analysise | |||||||
| Favored | 98.1 | 99.1 | 98.8 | 98.5 | 97.8 | 98.8 | 97.5 |
| Allowed | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
Rmerge=100 × ShSi|Ih,i−Ih| ShSiIh,j, where Ih is the mean intensity of symmetry related reflections Ih,j.
Numbers in the parentheses refer to the highest resolution shell of data.
Rwork = 100 × S||Fobs|−|Fcalc||/S|Fobs|
Rfree for a 10% subset of reflections.
As determined by Molprobity
Figure 2.
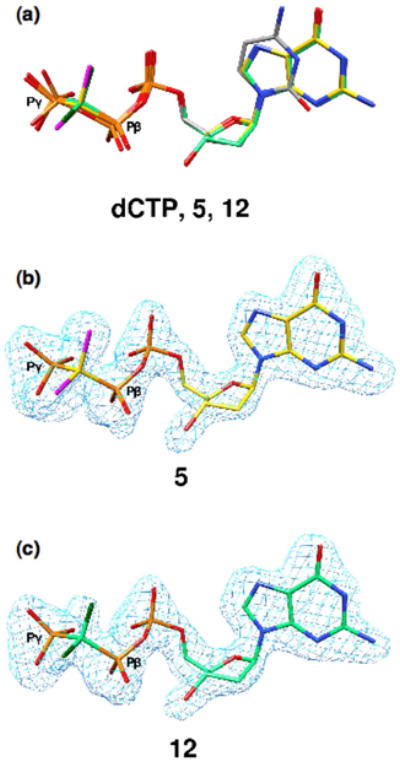
Congruence of dCTP and β,γ-CXY dGTP analogue sugar-phosphate backbones in ternary complexes with DNA-pol β. (a) The ternary complex of DNA-pol β with an incoming ddCTP (PDB ID 2FMP; gray carbons) is superimposed with the ternary complex obtained for the β,γ-CCl2 (5, yellow carbons, purple chlorines) and β,γ-C(CH3)2 (12, green carbons, dark green methyls) dGTP analogues. (b,c) Fo−Fc simulated annealing electron density omit maps (light blue) contoured at 4σ showing electron density for complex-bound 5 and 12.
Despite both stereoisomers being present in the solution used to soak the binary complex crystals, in the resulting ternary complexes from all three monofluoro analogues electron density is found only at the position proximal to an Arg183 nitrogen atom, indicating that only one diastereomer is present (i.e., (R)-CHF, 3; (R)-CCH3F, 13; (S)-CClF 16; Fig. 3). However, in the complexes obtained from the monochloro (6/7), monobromo (8/9) and monomethyl (10/11) analogues, the electron density map clearly shows population of the active site by both members of each diasteromer pair (Fig. 4). In the case of the monofluoro-analogues, a difference density map shows that there is electron density not accounted for when the opposite stereoisomer is considered (Figs. 3b and c, insets). This is most clearly seen in Figure 3c (inset) where (R)-CClF 15 was modeled. In this case, too few electrons are accounted for around the fluorine atom resulting in a positive difference density (green), and too many electrons are modeled in the position of the chlorine, resulting in negative density (red).
Figure 3.
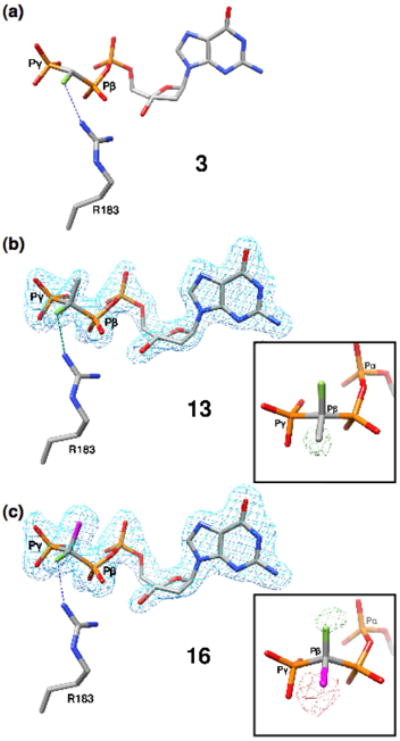
Structures of the monofluoro (β,γ-CXF) dGTP analogues in ternary DNA-pol β complexes. (a) Complex of DNA-pol β with an incoming (R)-β,γ-CHF-dGTP (3, PDB ID 2PXI). The fluorine atom is 3.1 Å from a guanidinium N of Arg183. (b) Only the (R)-isomer of the β,γ-CCH3F analogue 13 is observed in the pol β active site. The inset illustrates a Fo−Fc simulated annealing difference density map generated using the (S)-isomer and shows positive density (green, contoured at 3.2σ) in the vicinity of the CH3-group demonstrating that the CH3 cannot account for the observed density. (c) Similarly, the (S)-isomer of the β,γ-CClF (16) is the preferred stereoisomer bound to the pol β active site. The inset illustrates a difference density map generated using the (R)-isomer and shows positive (green, contoured at 3.2σ) and negative density (red, contoured at 4σ) in the vicinity of the chlorine and fluorine atoms, respectively, indicating that this isomer cannot account for the observed electron density.
Figure 4.
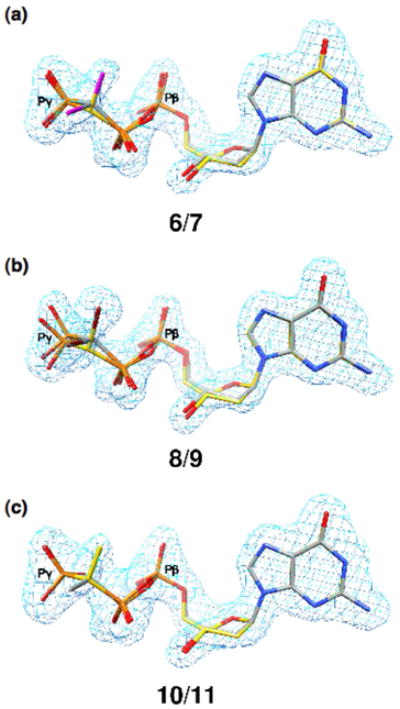
Structures of DNA-pol β ternary complexes with monochloro, monobromo and monomethyl β,γ-CXY dGTP analogues. The Fo−Fc simulated annealing electron density omit maps (light blue) contoured at 4σ show electron density for the corresponding β,γ-dGTP analogue. (a) Both stereoisomers of β,γ-CHCl-dGTP (6, 7) are observed in the ternary complex. The chlorine atoms are purple; the carbon atoms of the (R)-isomer (6) are yellow while those of the (S)-isomer (7) are gray. (b) Both stereoisomers of β,γ-CHBr-dGTP (8, 9) are observed in the ternary complex. The bromine atoms are dark red; the carbon atoms of the (R)-isomer (8) are yellow, while those of the (S)-isomer (9) are gray. (c) Both stereoisomers of β,γ-CHCH3-dGTP (10, 11) are observed in the ternary complex. The carbon atoms of the (R)-isomer (10) are gray while those of the (S)-isomer (11) are yellow.
Thus, for the entire set of CXY dGTP analogues where X ≠ Y, the structural data are consistent with preferred binding of one diastereomer only when Y is a fluorine atom, and that atom is always proximal to the active site Arg183. No stereospecificity is observed when the fluorine is replaced by a chloro, bromo or methyl substituent.
Stereospecific binding within the DNA pol β active site exclusively with the monofluorine analogues: evidence for a N-H···F-C ‘hydrogen bond’?
In our preliminary account,15 we suggested that in the absence of a dominant steric factor, asymmetric polarization induced by the F substituent presumably influences 3 vs. 4 binding specificity in some way. Assuming that the limit for detection of fluorine electron density at the disfavored position corresponds to a bound isomer ratio of roughly 1:4 or less, then a stereospecific interaction on the order of 1 kcal/mole would be sufficient. The fluorine atom in the 3 complex is located 3.1 Å from an Arg183 guanidinium N atom, raising the possibility that an unusual F···H bonding interaction contributes decisively to stabilizing the preferred stereoisomer within the highly preorganized enzyme active site complex. We did not exclude an alternative explanation (such as a directed polar effect of the C-F group acting on the effective charge vectors of the P-O anions, a small perturbation of the phosphophosphonate backbone confirmation, or a weak binding interaction of the relatively acidic25 CHF hydrogen with an active site water molecule). The latter explanations however do not appear to be consistent with persistence of stereospecificity for the fluoromethyl and fluorochloro analogues examined in the present study.
Fluorine-hydrogen bonds in HF are among the strongest known, but the existence of hydrogen bonds involving C-F groups and H donors such as NH or OH is controversial and has been debated vigorously for over a decade.9,26–28 The possibility of C-F interactions with amide or other groups is of particular interest due to the well-recognized importance of fluorine substitution in affecting the pharmacological properties of drugs.27 In a systematic search of a protein structure database recently carried out by Diderich and co-workers, several examples of “arginine fluorophilicity” were indentified,29 which provides support for such an interaction involving the arginine guanidinium as the source of the stereospecific binding found in this work.30,31 However, other factors, in particular spatial preorganization of the complex may play an important role as well in accounting for the phenomenon.
The data indicate that co-substitution in the fluoro analogues with an electron-donating (methyl) or withdrawing (chloro) group, which should respectively strengthen and weaken the C-F dipole, do not lead to loss of stereopreference in binding, although the F···H-N distance is changed from 3.1 Å in 3 to 3.2 Å in 13 and 3.5 Å in 16. This implies that the observable limit ratio of < 1:4 for the ‘wrong’ isomer may be better assigned to 16 and thus that the putative fluorine-NH interaction that stabilizes 3 and possibly 16 relative to their stereoisomers may somewhat exceed 1 kcal/mole.
CONCLUSION
In conclusion, the stereoisomers 3, 13, and 16 are preferentially bound into ternary DNA-pol β complexes, conceivably due, at least in part, to a CXF-H bridge bond to Arg183. Introduction of a single fluorine atom at the bridging carbon atom of a dGTP methylenebis(phosphonate) analogue does not merely adjust the analogue pKa to more closely mimic the parent nucleotide,15,17,28 but also can result in stereospecific binding to an enzyme, determined by the CXF chirality. The introduction of these substituents thus enables entirely new active site interactions that must be taken into account in interpreting their use as enzyme probes, while offering a new factor to be considered for inhibitor design seeking to exploit DNA polymerases such as pol β as a drug targets.
EXPERIMENTAL SECTION
All reagents were purchased from Sigma-Aldrich except tetraisopropyl methylenebis(phosphonate) (TiPMBP) which was generously provided by Albright and Wilson Americas, Inc. The synthesis of 1–9, 15–1615,17 and the corresponding methylene(bisphosphonic acids)15, 22 has been described elsewhere. HPLC analytical and preparative separations were carried out using Varian ProStar 210 pumps and injector system equipped with a Shimadzu SPD-10A VP UV Vis detector operated at 266 nm with a standard cell pathlength (Shimadzu LC-8A pumps with a Shimadzu SPD-20A detector for 12–14), on: a) a Varian C-18 (ODS) Microsorb-MV 4.6 mm × 25 cm, 5 μm analytical column; b) a Dynamax C-18 21.4 mm × 25 cm, 5 μm preparative column; c) a Varian PureGel SAX 10 mm × 10 cm, 7 μm analytical column; or d) a Macherey-Nagel Nucleogel SAX 1000-10 25 mm × 15 cm preparative column. C-18 columns were eluted isocratically with 0.1 M TEAB containing 2% acetonitrile. SAX column elution conditions are given below. NMR spectra were measured on Bruker AM-360 or Varian Mercury 400 spectrometers. Chemical shifts (δ) are reported in parts per million (ppm) relative to internal residual CHCl3 in CDCl3 (δ 7.24, 1H), internal residual HDO in D2O (pH ~8, δ 4.8, 1H), external 85 % H3PO4 (δ 0.00, 31P, 1H decoupled) or external CFCl3 (δ 0.00, 19F). NMR spectra were simulated using the NUTSPro NMR Utility Transform software package from Acorn NMR, Inc. HRMS data were obtained at the UCR mass spectrometry facilities (Dr. Ron New). Elemental analysis of the methylene- and ethylidenebis(phosphonic acids) was performed by Galbraith Laboratories. HPLC, HRMS, and NMR spectra are collected in the Supporting Information.
Synthesis of tributylammonium salts of methylenebis(phosphonic acids)
The methylenebis(phosphonic acids)15, 22 were dissolved in 50% EtOH/H2O and placed in a conical flask. 1.5 equivalents of tributylamine were added dropwise and the solution kept at room temperature for 30 min. The solvent was removed and excess Bu3N coevaporated with ethanol. The residual tributylammonium salts were dried by coevaporation with DMF under vacuum and used without further characterization.
General procedure for synthesis of the β,γ-methylene-deoxyguanosine triphosphate analogues
1.1 eq. of dried 2′-deoxyguanosine 5′-phosphate, morpholidate (dGMP-Morph)15 was dissolved in freshly distilled anhydrous DMSO. In a separate flask, 4.4 eq. of the methylenebis(phosphonate) derivative (tributylammonium salt) was also dissolved in anhydrous DMSO. The latter solution was added slowly to the former, while monitoring by analytical HPLC (SAX ion exchange column, 0 – 100% 0.5 M TEAB buffer (pH = 8) gradient; or 0 – 50% 0.5 M LiCl gradient). After the reaction reached completion, the solvent was removed under reduced pressure. The yellowish oily residue was then dissolved in 1.5 mL of 0.5 M TEAB buffer and the desired product isolated by two-stage preparative HPLC; first using the SAX column (0 – 100% 0.5 M TEAB gradient) and then the C-18 column (0.1 N TEAB 4% CH3CN). The fractions containing the dGTP analogue product were collected, combined and lyophilized to obtain the TEA salt. The amount of nucleotide was found by determination of the concentration by UV absorption (λmax = 253 nm, ε262 = 13,700 M−1 cm−1 at pH = 832); yields reported are relatively low, but the two-stage HPLC purification procedure provides ultrapure samples of the dNTP analogues, free of detectable contaminating nucleotide or methylenebis(phosphonate). Following the general procedure, compounds 1–9, 15, and 16 were synthesized, purified and characterized according to our previously described procedures. 15,16
Synthesis of 2′-deoxyguanosine 5′-triphosphate β,γ-CHCH3 analogue, 10/11
Following the general procedure, 90 mg (0.215 mmol) of dried dGMP-Morph was reacted with the tributylammonium salt of 1,1-ethanediylbis(phosphonic acid) (0.859 mmol) in freshly distilled anhydrous DMSO. The crude product was separated from the reaction mixture and 10/11 isolated by prep. HPLC as a TEA salt (yield = 43 mg - 39%). 31P NMR: d −11.0 (d), 16.8 (d), 17.9 (dd); 1H NMR: d 8 (d), 6.25 (t), 4.8 (s), 4.3 (s), 4.2 (s), 3.8 (s), 2.75 (m), 2.4 (m), 2.2 (m), 1.4 (m); HRMS (ESI): calcd for C12H19N5O12P3−, [M−H]− 518.0249, found: 518.0246 m/z.
Synthesis of 2′-deoxyguanosine 5′-triphosphate β,γ-C(CH3) analogue, 12
Following the general procedure, 60 mg (0.144 mmol) of dried dGMP-Morph was reacted with the tributylammonium salt of 2,2-propanediylbis(phosphonic acid) (0.634 mmol) in freshly distilled anhydrous DMSO. The product 12 was isolated as the TEA salt (yield = 15 mg - 20%). 31P NMR: d −11.0 (d), 19.5 (m), 23 (d); 1H NMR: d 8 (d), 6.25 (t), 4.2 (s), 4.1 (m), 2.75 (m), 2.4 (m); HRMS (ESI): calcd for C13H21N5O12P3−, [M−H]− 532.0405, found: 532.0408 m/z.
Synthesis of 2′-deoxyguanosine 5′-triphosphate β,γ-CCH3F analogue, 13/14
Following the general procedure, 80 mg (0.192 mmol) of dried dGMP-Morph was reacted with the tributylammonium salt of (1-fluoro-1,1-ethanediyl)bis(phosphonic acid) (0.769 mmol) in freshly distilled anhydrous DMSO. The product 13/14 was obtained as above (yield = 13 mg - 13%). 31P NMR: d −11.0 (d), 10 (m), 11.5 (dd); 1H NMR: d 8 (d), 6.25 (t), 4.8 (s), 4.2 (s), 4.1 (m), 2.75 (m), 2.5 (m), 1.7 (dt); 19F NMR: d −176.5 (m); HRMS (ESI): calcd for C12H18FN5O12P3−, [M−H]− 536.0154, found: 536.0149 m/z.
Crystallization of the pol β ternary complexes
Human DNA polymerase β was over-expressed in E. coli and purified.24 The DNA substrate consisted of a 16-mer template, a complementary 9-mer primer strand, and a 5-mer downstream oligonucleotide. The annealed 9-mer primer creates a two-nucleotide gap. The sequence of the downstream oligonucleotide was 5′-GTCGG–3′ and the 5′-terminus was phosphorylated. The template sequence was 5′-CCGACCGCGCATCAGC–3′ and the primer sequence was 5′-GCTGATGCG-3′. Oligonucleotides were dissolved in 20 mM MgCl2 in 100 mM Tris/HCl, pH 7.5. Each set of template, primer, and downstream oligonucleotides were mixed in a 1:1:1 ratio and annealed using a PCR thermocycler by heating for 10 min at 90 °C and cooling to 4 °C (1 °C min-1) resulting in 1 mM gapped duplex DNA. This solution was mixed with an equal volume of pol β (15 mg/ml in 20 mM Bis-Tris, pH 7.0) at 4 °C, the mixture warmed to 35 °C and then gradually cooled to 4 °C. A four-fold excess of 2′,3′-dideoxycytosine 5′-triphosphate (ddCTP) was added to obtain a 1-nucleotide gap complex with a dideoxy primer terminus. Pol β-DNA complexes were crystallized by sitting drop vapor diffusion. The crystallization buffer for binary complexes (1-nucleotide gap) contained 16% PEG-3350, 350 mM sodium acetate, and 50 mM imidazole, pH 7.5. Drops were incubated at 18 °C and streak seeded after 1 day. Crystals grew in approximately 2 to 4 days after seeding. The binary DNA complex crystals were soaked in artificial mother liquor with 200 mM MgCl2, 90 mM sodium acetate, 4–6 mM of the analogue, 20% PEG-3350, and 12% ethylene glycol resulting in crystals of ternary complexes.
Data collection and structure determination
Data were collected on the ternary complex crystals at 100K on a Saturn92 CCD detector system mounted on a MicroMax-007HF (Rigaku Corporation) rotating anode generator. Data were integrated and reduced with HKL2000 software.33 Ternary substrate complex structures were determined by molecular replacement with a previously determined structure of pol β complexed with one-nucleotide gapped DNA and a complementary incoming ddCTP (PDB accession 2FMP).34 The crystal structures have similar lattices and are sufficiently isomorphous to refine directly using CNS35 and manual model building using O. The crystallographic images were prepared in Chimera.36 The parameters and topology files for the analogs were prepared using the program XPOL2D.37
Supplementary Material
Acknowledgments
We thank Dr. Kym Faull and Dr. Ron New for assistance with HRMS analysis and J.M. Krahn for his help in preparing the analogue parameters and topology files for structure determination. This research was supported by NIH Grant 5-U19-CA105010 and in part by Research Project Numbers Z01 ES050158-12 and Z01 ES050161-12 (S. H. Wilson) in the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences.
Footnotes
Supporting Information Available. Characterization (HPLC detail and separation traces, HRMS spectra, NMR spectra) data for 10–14; crystallographic data for the ternary complexes of the analogues with DNA-pol β (PDB ID, 2PXI, 3JPN, 3JPO, 3JPQ, 3JPP, 3JPR, 3JPS, 3JPT. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Albertella MR, Lau A, O’Connor MJ. DNA Repair. 2005;4:583–593. doi: 10.1016/j.dnarep.2005.01.005. [DOI] [PubMed] [Google Scholar]; (b) Sweasy JB, Lauper JM, Eckert KA. Radiat Res. 2006;166:693–714. doi: 10.1667/RR0706.1. [DOI] [PubMed] [Google Scholar]; (c) Lang T, Dalal S, Chikova A, DiMaio D, Sweasy JB. Mol Cell Biol. 2007;27:5587–5596. doi: 10.1128/MCB.01883-06. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Bergoglio V, Canitrot Y, Hogarth L, Minto L, Howell SB, Cazaux C, Hoffmann JS. Oncogene. 2001;20:6181–6187. doi: 10.1038/sj.onc.1204743. [DOI] [PubMed] [Google Scholar]; (e) Bergoglio V, Pillaire MJ, Lacroix-Triki M, Raynaud-Messina B, Canitrot Y, Bieth A, Gares M, Wright M, Delsol G, Loeb LA, Cazaux C, Hoffmann JS. Cancer Res. 2002;62:3511–3514. [PubMed] [Google Scholar]; (f) Loeb LA, Monnat RJ., Jr Nat Rev Genet. 2008;9:594–604. doi: 10.1038/nrg2345. [DOI] [PubMed] [Google Scholar]; (g) Dalal S, Hile S, Eckert KA, Sun K-w, Starcevic D, Sweasy JB. Biochemistry. 2005;44:15664–15673. doi: 10.1021/bi051179z. [DOI] [PubMed] [Google Scholar]; (h) Iwanaga A, Ouchida M, Miyazaki K, Hori K, Mukai T. Mutat Res. 1999;435:121–128. doi: 10.1016/s0921-8777(99)00036-1. [DOI] [PubMed] [Google Scholar]; (i) Starcevic D, Dalal S, Sweasy JB. Cell Cycle. 2004;3:998–1001. [PubMed] [Google Scholar]; (j) Sweasy JB, Lang T, DiMaio D. Cell Cycle. 2006;5:250–259. doi: 10.4161/cc.5.3.2414. [DOI] [PubMed] [Google Scholar]; (k) Sweasy JB, Lang T, Starcevic D, Sun KW, Lai CC, DiMaio D, Dalal S. Proc Nat Acad Sci U S A. 2005;102:14350–14355. doi: 10.1073/pnas.0505166102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barnes DE, Lindahl T. Ann Rev Gen. 2004;38:445–476. doi: 10.1146/annurev.genet.38.072902.092448. [DOI] [PubMed] [Google Scholar]
- 3.Beard WA, Wilson SH. Chem Rev. 2006;106:361–382. doi: 10.1021/cr0404904. [DOI] [PubMed] [Google Scholar]
- 4.Alexandrova LA, Skoblov AY, Jasko MV, Victorova LS, Krayevsky AA. Nucl Acids Res. 1998;26:778–786. doi: 10.1093/nar/26.3.778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arabshahi L, Khan NN, Butler M, Noonan T, Brown NC, Wright GE. Biochemistry. 1990;29:6820–6826. doi: 10.1021/bi00481a010. [DOI] [PubMed] [Google Scholar]
- 6.(a) Blackburn GM, Kent DE, Kolkmann F. J Chem Soc, Chem Commun. 1981:1188–1190. [Google Scholar]; (b) McKenna CE, Leswara ND, Shen PD. Fed Proc. 1982;41:860. [Google Scholar]; (c) Blackburn GM, Kent DE, Kolkmann F. J Chem Soc Perkin Trans 1 (1972–1999) 1984:1119–1125. [Google Scholar]
- 7.Hamilton CJ, Roberts SM, Shipitsin A. Chem Commun (Cambridge) 1998;1087–:1088. [Google Scholar]
- 8.Kashemirov BA, Roze CN, McKenna CE. Phos Sulf Sil Rel Elem. 2002;177:2275. [Google Scholar]
- 9.Kim TW, Delaney JC, Essigmann JM, Kool ET. Proc Nat Acad Sci US. 2005;102:15803–15808. doi: 10.1073/pnas.0505113102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krayevsky A, Arzumanov A, Shirokova E, Dyatkina N, Victorova L, Jasko M, Alexandrova L. Nucleosides Nucleotides. 1998;17:681–693. doi: 10.1080/07328319808005209. [DOI] [PubMed] [Google Scholar]
- 11.Martynov BI, Shirokova EA, Jasko MV, Victorova LS, Krayevsky AA. FEBS Letters. 1997;410:423–427. doi: 10.1016/s0014-5793(97)00577-2. [DOI] [PubMed] [Google Scholar]
- 12.McKenna CE, Kashemirov BA, Roze CN. Bioorg Chem. 2002;30:383–395. doi: 10.1016/s0045-2068(02)00521-7. [DOI] [PubMed] [Google Scholar]
- 13.Shipitsin AV, Victorova LS, Shirokova EA, Dyatkina NB, Goryunova LE, Beabealashvilli RS, Hamilton CJ, Roberts SM, Krayevsky A. J Chem Soc Perkin Trans 1. 1999;1039–:1050. doi: 10.1080/15257779908041639. [DOI] [PubMed] [Google Scholar]
- 14.Victorova L, Sosunov V, Skoblov A, Shipytsin A, Krayevsky A. FEBS Letters. 1999;453:6–10. doi: 10.1016/s0014-5793(99)00615-8. [DOI] [PubMed] [Google Scholar]
- 15.McKenna CE, Kashemirov BA, Upton TG, Batra VK, Goodman MF, Pedersen LC, Beard WA, Wilson SH. J Am Chem Soc. 2007;129:15412–15413. doi: 10.1021/ja072127v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sucato CA, Upton TG, Kashemirov BA, Batra VK, Martinek V, Xiang Y, Beard WA, Pedersen LC, Wilson SH, McKenna CE, Florian J, Warshel A, Goodman MF. Biochemistry. 2007;46:461–471. doi: 10.1021/bi061517b. [DOI] [PubMed] [Google Scholar]
- 17.Sucato CA, Upton TG, Osuna J, Oertell K, Kashemirov BA, Beard WA, Wilson SH, McKenna CE, Florian J, Warshel A, Goodman MF. Biochemistry. 2008;47:870–879. doi: 10.1021/bi7014162. [DOI] [PubMed] [Google Scholar]
- 18.Upton TG, Kashemirov BA, McKenna CE, Goodman MF, Prakash GKS, Kultyshev R, Batra VK, Shock DD, Pedersen LC, Beard WA, Wilson SH. Org Lett. 2009;11:1883–1886. doi: 10.1021/ol701755k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boyle NA, Fagan P, Brooks JL, Prhavc M, Lambert J, Cook PD. Nucleosides Nucleotides Nucleic Acids. 2005;24:1651–1664. doi: 10.1080/15257770500267055. [DOI] [PubMed] [Google Scholar]
- 20.Moffatt JG, Khorana HG. J Am Chem Soc. 1961;83:649–658. [Google Scholar]
- 21.(a) Grabenstetter RJ, Quimby OT, Flautt TJ. J Phys Chem. 1967;71:4194–4202. [Google Scholar]; Kabachnik MI. Dokl Akad Nauk. 1967;177:582. [Google Scholar]; (b) Aboujaoude EE, Lietje S, Collignon N, Teulade MP, Savignac P. Tetrahedron Lett. 1985;26:4435–4438. [Google Scholar]; (c) Martynov BI, Sokolov VB, Aksinenko AY, Goreva TV, Epishina TA, Pushin AN. Russ Chem Bull. 1998;47:1983–1984. [Google Scholar]
- 22.Upton TG. PhD Dissertation. Univ. Southern Calif; 2008. Note that 19F NMR 2JFH and 2JFPvalues for 3/4 given in this reference were inadvertently reversed, leading to a similar simulated multiplet15 to that presented here, but with reversed assignment of the second outermost peaks. The CFH 1H NMR peak is obscured by HDO in the nucleotide analogue spectra preventing observation of the fluorine splitting, however our assignment is consistent with the CFH 1H value of 44–48 Hz determined for 2JFH in the ethyl, isopropyl and trimethylsilyl esters of the parent bisphosphonate25. [Google Scholar]
- 23.McKenna CE, Harutunian V. FASEB J. 1988;2 [Google Scholar]
- 24.Beard WA, Wilson SH. Meth Enzymol. 1995;262:98–107. doi: 10.1016/0076-6879(95)62013-3. [DOI] [PubMed] [Google Scholar]
- 25.McKenna CE, Shen PD. J Org Chem. 1981;46:4573–4576. [Google Scholar]
- 26.(a) Wang X, Houk KN. Chem Commun (Cambridge) 1998;2631–:2632. [Google Scholar]; (b) Hof F, Scofield DM, Schweizer WB, Diederich F. Angew Chem, Int Ed. 2004;43:5056–5059. doi: 10.1002/anie.200460781. [DOI] [PubMed] [Google Scholar]; (c) Howard JAK, Hoy VJ, O’Hagan D, Smith GT. Tetrahedron. 1996;52:12613–12622. [Google Scholar]; (d) Mecozzi S. Abstract MEDI-467. 230th ACS National Meeting; Washington, DC. Aug. 28–Sept. 1, 2005. [Google Scholar]; (e) Mecozzi S, Hoang KC, Martin O. Abstract FLUO-047. 226th ACS National Meeting; New York. Sept. 7–11, 2003. [Google Scholar]; (f) Morgenthaler M, Aebi JD, Gruninger F, Mona D, Wagner B, Kansy M, Diederich F. J Fluorine Chem. 2008;129:852–865. [Google Scholar]; (g) Brammer L, Bruton EA, Sherwood P. Cryst Growth Des. 2001;1:277–290. [Google Scholar]; (h) Carosati E, Sciabola S, Cruciani G. J Med Chem. 2004;47:5114–5125. doi: 10.1021/jm0498349. [DOI] [PubMed] [Google Scholar]; (i) Romanenko VD, Kukhar VP. Chem Rev (Washington, DC, U S) 2006;106:3868–3935. doi: 10.1021/cr051000q. [DOI] [PubMed] [Google Scholar]; (j) Schneider HJ. Angew Chem, Int Ed Engl. 48:3924–3977. doi: 10.1002/anie.200802947. [DOI] [PubMed] [Google Scholar]; (k) Smart BE. J Fluorine Chem. 2001;109:3–11. [Google Scholar]; (l) Woo LWL, Fischer DS, Sharland CM, Trusselle M, Foster PA, Chander SK, Di F, Anna, Supuran CT, De S, Giuseppina, Purohit A, Reed MJ, Potter BVL. Mol Cancer Ther. 2008;7:2435–2444. doi: 10.1158/1535-7163.MCT-08-0195. [DOI] [PubMed] [Google Scholar]
- 27.O’Hagan D, Rzepa HS. Chem Commun (Cambridge) 1997:645–652. [Google Scholar]
- 28.Berkowitz DB, Bose M, Pfannenstiel TJ, Doukov T. J Org Chem. 2000;65:4498–4508. doi: 10.1021/jo000220v. [DOI] [PubMed] [Google Scholar]
- 29.Mueller K, Faeh C, Diederich F. Science. 2007;317:1881–1886. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- 30.For an example of aprotic cation (K+, Ag+) stabilization by 6 C-F groups in a macrocage complex, see: Takemura H, Kon N, Yasutake M, Kariyazono H, Shinmyozu T, Inazu T. Angew Chem Int Ed. 1999;38:959–961. doi: 10.1002/(SICI)1521-3773(19990401)38:7<959::AID-ANIE959>3.0.CO;2-8.Somewhat greater stabilization of NH4+ was also observed.
- 31.Lu YX, Wang Y, Xu ZJ, Yan XH, Luo XM, Jiang HL, Zhu WL. J Phys Chem B. 2009;113:12615–12621. doi: 10.1021/jp906352e. [DOI] [PubMed] [Google Scholar]
- 32.Dawson RMC, Elliot Daphne C, Elliot William H, Jones Kenneth M. Data for Biochemical Research. Oxford University Press; New York: 1986. [Google Scholar]
- 33.Otwinowski Z, Minor W. Meth Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 34.Sawaya MR, Prasad R, Wilson SH, Kraut J, Pelletier H. Biochemistry. 1997;36:11205–11215. doi: 10.1021/bi9703812. [DOI] [PubMed] [Google Scholar]
- 35.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Acta Crystallogr Sect D: Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 36.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 37.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Acta Crystallogr, Sect A: Found Crystallogr. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.