

Abstract
Addictive drugs cause persistent restructuring of several neuronal cell types in the brain’s limbic regions thought to be responsible for long-term behavioral plasticity driving addiction. Although these structural changes are well documented in nucleus accumbens medium spiny neurons, little is known regarding the underlying molecular mechanisms. Additionally, it remains unclear whether structural plasticity and its synaptic concomitants drive addictive behaviors, or whether they reflect homeostatic compensations to the drug not related to addiction per se. Here, we discuss recent paradoxical data, which either support or oppose the hypothesis that drug-induced changes in dendritic spines drive addictive behavior. We define areas where future investigation can provide a more detailed picture of drug-induced synaptic reorganization, including ultrastructural, electrophysiological, and behavioral studies.
Keywords: dendritic spines, drug addiction, relapse, mesolimbic dopamine system, long-term potentiation (LTP), long-term depression (LTD), medium spiny neuron (MSN), α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA), N-methyl D-aspartate (NMDA), ΔFosB, cyclic AMP response element binding protein (CREB), nuclear factor kappaB (NFκB), and myocyte-enhancing factor 2 (MEF-2)
Introduction
Drug addiction is marked by long-lasting changes in behavior, such as craving and relapse. Correlated with these stable behavioral abnormalities is the persistent restructuring of many neuronal cell types in limbic regions of the brain. Two general types of structural plasticity have been observed: changes in the size of cell bodies [1] and changes in dendritic arborizations or spine morphology [2]. With respect to the latter, depending upon the class of addictive substance, nature of the drug administration paradigm (e.g., experimenter versus self-administered), and neuronal cell type examined, drugs of abuse can alter the complexity of dendritic branching, as well as the number and size of dendritic spines on neurons in several brain regions (Table 1). Correlative evidence suggests that certain morphological changes are important mediators of addictive behaviors. For example, morphine and cocaine alter the density of dendritic spines on medium spiny neurons (MSNs) in nucleus accumbens (NAc), a key brain reward region, to a greater extent in animals self-administering the drug, compared to animals given drug by the investigator, suggesting that volition may be important for key aspects of plasticity (reviewed in [3]). Additionally, cocaine-induced changes in NAc dendritic structure are tightly correlated with the induction of behavioral sensitization [4]: doses and drug administration paradigms that induce sensitization reliably increase dendritic spines and branching. Despite this evidence, however, the behavioral relevance of structural plasticity is still uncertain. Several recent studies using viral-mediated gene transfer and other methods to better understand the behavioral relevance and molecular basis of cocaine-induced changes in dendritic structure of MSNs have produced conflicting results, with two manuscripts supporting the hypothesis that cocaine-induced increases in dendritic spine density mediate behavioral sensitization and two others diametrically opposing it [5–8]. In this review, we discuss current paradoxical experimental data and formulate areas for future investigation. We detail key themes, starting with the types of synaptic plasticity induced by drugs of abuse and signaling pathways that mediate drug-induced structural plasticity, and progressing to more detailed discussions of spine morphometry and the functional role of actin reorganization in addiction.
Table 1.
Drug-induced changes in neuronal morphology
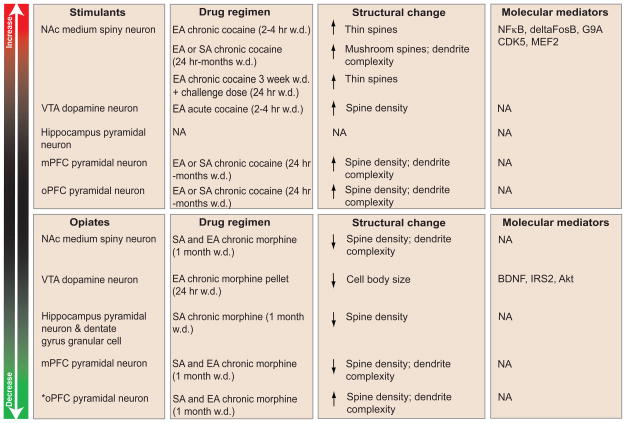 |
SA=self adminstered; EA=experimenter adminstered; w.d.=withdrawal; mPFC=medial prefrontal cortex; oPFC=orbital prefrontal cortex; NAc=nucleus accumbens; VTA=ventral tegmental area; BDNF=brain derived neurotrophic factor; IRS2=insulin receptor substrate 2; Akt=thymoma viral protooncogene; NFκB=nuclear factor kappa b; CDK5=cyclin dependent kinase 5; MEF2= myocyte enhancing factor 2; NA=not available.
oPFC is the only brain region studied that morphine increases spine density and complexity.
Structural plasticity induced by opiate and stimulant drugs of abuse
Drug-induced structural plasticity of dendrites was first described in 1997 (reviewed in [3, 9, 10]). Since then, numerous laboratories have shown that chronic administration of almost every drug of abuse induces structural plasticity in the brain’s reward circuitry. These studies have also correlated structural changes within specific brain regions to behavioral phenotypes associated with addiction. Since the original reports by Robinson and colleagues (reviewed in [3]), many researchers have added to this growing literature and have uncovered more subtle and drug class-specific effects on neuronal morphology. For example, opiates and stimulants regulate structural plasticity in the opposite direction. Opiates decrease the number and complexity of dendritic spines on NAc MSNs, medial prefrontal cortex (mPFC) and hippocampus pyramidal neurons, and also decrease the soma size of ventral tegmental area (VTA) dopaminergic neurons [1, 3, 11, 12]. To date, there is a single exception to these findings: chronic morphine increases spine number on orbitofrontal cortex (oPFC) pyramidal neurons [13]. In contrast to opiates, stimulants such as cocaine, amphetamine, and methylphenidate consistently increase dendritic complexity and spine density of NAc MSNs, VTA dopaminergic neurons, and mPFC pyramidal neurons [2, 8, 14–17]. From a behavioral perspective, morphine reduces spine density and dendritic complexity regardless of whether it is administered continuously to produce tolerance and dependence, or intermittently to maximize sensitization, whereas stimulant paradigms that increase spine density and complexity all use once to several times daily intermittent injections of the drug to induce drug sensitization [3, 9].
The opposite morphological changes induced in brain reward regions by opiates versus stimulants are paradoxical since the two drugs cause very similar behavioral phenotypes. Opiates and stimulants both induce locomotor activation acutely and locomotor as well as reward sensitization chronically [9]. They also both induce similar patterns of escalation of drug self-administration as well as a negative emotional state (dysphoria) during withdrawal [18]. Thus, if the opposite morphological changes induced by opiates and stimulants are important mediators of addiction, either they must have bidirectional properties, whereby a change from baseline in both directions produces the same behavioral phenotype, or there are key pieces of information regarding synaptic function that are not captured by measuring gross changes in dendritic spine density as this may be compensated for by a change in synaptic strength keeping total synaptic input per neuron constant [19]. For example, alcohol decreases neuronal complexity and density while consolidating pre-existing synapses [20], and it may be that opiates and stimulants produce similar effects on the size of the postsynaptic density (PSD) leading to the same net change in synaptic efficacy. It is also unclear whether chronic exposure to opiates or stimulants leads to similar electrophysiological changes at NAc synapses, as might be expected given the shared features of the addicted phenotype. Finally, we should consider that a drug-induced change in synaptic number and efficacy in one brain area may result in strengthening or weakening of connections with other brain areas, and may drive distinct aspects of addictive behaviors [21–23].
Neurophysiological relevance of drug-induced structural plasticity
Basic research into the relevance of dendritic spine changes in hippocampus and cerebral cortex indicates that the size and shape of individual spines correlates with forms of synaptic plasticity such as long-term potentiation (LTP) and long-term depression (LTD) [24, 25]. It is believed that stabilization of a transient, immature spine into a more permanent, functional spine occurs through an activity-dependent mechanism (reviewed in [26]). Stimulation protocols that induce LTD are associated with shrinkage or retraction of spines [27–29], whereas induction of LTP is associated with formation of new spines and enlargement of existing spines [27, 28, 30]. At a molecular level, it is believed that LTP and LTD initiate changes in signaling pathways, and in the synthesis and localization of cytoskeletal proteins, which alter polymerization of actin to affect spine maturation and stability and which either anchor or internalize α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA) glutamate receptors to produce a more functional spine (LTP) or retraction of an existing spine (LTD) [24, 26]. Upon stabilization, spines become mushroom-shaped, have larger postsynaptic densities [31], show increased surface expression of AMPA receptors, and persist for months [29, 32]. These changes reflect a highly stable cellular event that may be a plausible explanation for certain long-term behavioral changes associated with addiction.
Recent work in addiction models has indeed shown functional changes in NAc MSNs that are highly time-dependent and fluid during the addiction process (Figure 1). At early time-points after the last cocaine exposure, there is an increase in thin (more highly plastic) spines and synaptic depression [33, 34], which may represent an increased pool of silent synapses [35, 36]. Silent synapses contain N-methyl-D-aspartate (NMDA) glutamate receptors but few or no AMPA receptors, express relatively stable NMDA receptor-mediated excitatory postsynaptic currents, and are ideal substrates for LTP [36, 37]. Shortly after cocaine treatment, such silent synapses in NAc appear to express an increased proportion of NR2B-containing NMDA receptors [35], a finding consistent with these synapses being fairly new and immature [38, 39]. During the course of cocaine withdrawal, these recently formed spines appear to be highly transient and may retract or consolidate into mushroom-shaped spines [33], an event that is accompanied by an increase in surface expression of GluR2-lacking AMPA receptors and a potentiation of these glutamatergic synapses [40–42]. (GluR2-lacking AMPA receptors exhibit greater Ca2+ and overall conductance compared to GluR2-containing AMPA receptors.) Behaviorally, incubation of cocaine craving is seen during withdrawal from cocaine self-administration; this is characterized by a gradual and progressive increase in cocaine seeking and susceptibility to relapse, which may require these changes in the stoichiometry of synaptic AMPA receptors [42, 43]. However, behavioral studies using viral-mediated gene transfer show that overexpression of the AMPA GluR1 subunit paradoxically decreases behavioral sensitization to cocaine, highlighting the need for further research in this area [44]. Additional evidence shows that re-exposure to cocaine after either 14 or 30 days of withdrawal results in reduced spine head diameter [33], decreased surface expression of AMPA receptors [40], and depression of strength at these synapses [45]. During these transient changes in synapse structure and composition, there are also significant changes in activity of RhoGTPase signaling proteins required for actin polymerization, an effect that might be responsible for spine restructuring [46]. These data point to a complex interaction between spine head structure, electrophysiological properties of NAc MSNs, and addiction-related behavior. Given that many synaptic proteins can regulate these events, it will be important to identify the precise molecular networks involved in their regulation.
Figure 1. Model of addiction-related synaptic and structural plasticity.

Chronic exposure to cocaine results in a time–dependent and transient reorganization of α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA) and N-methyl-D-aspartate (NMDA) glutamate receptors at nucleus accumbens (NAc) medium spiny neuron (MSN) synapses, as well as structural changes in the spine head of NAc MSNs that correlate with distinct forms of synaptic plasticity. For example, chronic cocaine induces surface expression of NMDA receptors, silent synapse formation, and long-term depression (LTD) at early withdrawal time points. During more prolonged withdrawal, these synaptic changes reverse with the result being increased expression of surface AMPA receptors, a consolidation of the synapse into a mushroom-shaped spine, and long-term potentiation (LTP). These effects rapidly revert back again upon exposure to a challenge dose of cocaine leading to restructuring of the spine into thin spines and a depression of synaptic strength.
Mechanisms of opiate- and stimulant-induced structural plasticity
The functional relevance of structural plasticity in addiction models is complicated, as noted earlier, by the fact that morphine and cocaine have opposite effects on MSN spine density. Moreover, there is little direct examination of downstream drug actions to explain this dichotomy in structural plasticity. While there are several large-scale microarray studies examining changes in gene expression after psychostimulant administration, there is a relative paucity of such information available for opiates. Moreover, studies of gene expression changes in response to morphine or cocaine have used widely divergent time points, regimens, and doses, making direct comparisons impossible. Despite these caveats, it is clear that opiate and stimulant drugs of abuse regulate numerous genes that encode for cytoskeleton regulatory proteins. For example, in NAc, morphine decreases Homer 1 and PSD95 [47], scaffolding proteins associated with the postsynaptic cytoskeleton. Interestingly, cocaine similarly reduces these proteins in NAc [48–51]. Additionally, morphine decreases RhoA, Rac1, and Cdc42, small GTPases that regulate the actin cytoskeleton (see below) [47]. Activity of these GTPases and their downstream targets are reduced by cocaine as well [52]. These studies were not designed to directly compare morphine and cocaine regulation of structure-related genes, yet both drugs were found to induce many similar changes despite their opposite regulation of dendritic spines of NAc MSNs. This suggests that regulation of this pathway may serve as an initiator of plasticity; however, it does not explain the dichotomy between opiate- and stimulant-induced structural plasticity.
The fact that opiates and stimulants similarly induce many cytoskeleton regulatory genes may be attributed to their activation of similar transcriptional regulators, including the transcription factors, ΔFosB and cyclic AMP response element binding protein (CREB), in NAc [53–56] (Figure 2). ΔFosB is induced in NAc by virtually all classes of drugs of abuse [57] and enhances the rewarding effects of both morphine and cocaine [58, 59]. ΔFosB seems to account for roughly 25% of all genes regulated in NAc by chronic cocaine, including several genes associated with synaptic plasticity such as cofilin, actin-related protein-4 (ARP4), and activity-regulated cytoskeletal protein (Arc) [58, 60]. Furthermore, ΔFosB is both necessary and sufficient for cocaine-induced changes in dendritic spine density [7]. However, if both morphine and cocaine induce ΔFosB, and ΔFosB is a key mediator of enhanced spinogenesis, why does chronic morphine decrease NAc MSN spine density? One possibility is that ΔFosB regulates partly distinct subsets of genes in the context of morphine versus cocaine administration, depending on other transcriptional alterations involved, or that morphine induces other adaptations in NAc neurons that override the ΔFosB signal, which alone stimulates spinogenesis. Further studies are needed to address these and alternative explanations.
Figure 2. Signaling pathways involved in addiction-related cytoskeleton reorganization.

Transcription factors, such as nuclear factor kappaB (NFκB), ΔFosB, cyclic AMP response element binding protein (CREB), and myocyte enhancing factor-2 (MEF2), play a role in regulating dendritic spines, and can be activated by a variety of signaling pathways. In addition to dopamine and opioid neurotransmitters, a key upstream signal may be brain-derived neurotrophic factor (BDNF) or other neurotrophins, which via receptor tyrosine kinases activate the Phosphoinositide 3-kinases (PI3K)-thymoma viral proto-oncogene (Akt), Ras-extracellular regulated kinase (ERK), and NFκB pathways, and ultimately regulate transcriptional activity and possibly control actin cytoskeletal dynamics through regulation of the Rho family of small GTPases (including Rac1 and p21-activated kinase (PAK1)). Activation of NFκB may occur additionally through a cytokine receptor mechanism to control spine plasticity, however, this remains speculative. Structural plasticity induced by psychostimulants can therefore result from manipulation of several signaling pathways that impinge upon actin assembly processes, with some of the changes mediated via altered gene expression. We hypothesize that the net effect of cocaine-induced activation of these fundamental signaling pathways are sensitized behavioral responses, although each pathway in isolation may produce distinct effects on addiction-like behavior and synaptic plasticity. PLCγ, phospholipase Cγ; IκK, inhibitory kappa kinase; IκB, nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor; TrKB, tyrosine receptor kinase B; Drd, dopamine receptor; LIMK, lim domain kinase; WASP, Wiskott-Aldrich Syndrome proteins; Cdk5, cyclin-dependent kinase-5.
In contrast to ΔFosB, CREB’s role in drug-induced structural plasticity is far more hypothetical. Despite the evidence that CREB induction in NAc mediates tolerance and dependence to morphine and cocaine reward (reviewed in [61]), there are few data examining whether CREB mediates structural changes following exposure to drugs of abuse. In several other brain areas, CREB induces spinogenesis [37, 62, 63], effects possibly mediated through transcriptional targets such as myocyte enhancing factor 2C (MEF2C) and brain-derived neurotrophic factor (BDNF), both of which are also involved in addiction-related plasticity [5, 64, 65]. CREB may also mediate plasticity through induction of microRNA, mir132, which was recently shown to induce neurite outgrowth of hippocampal neurons in culture, in part, by reducing levels of the GTPase p250GAP [66]. Given the large body of evidence implicating CREB’s role in structural plasticity in other neural circuits, a direct investigation of CREB’s role in mediating drug-induced structural plasticity in NAc is a top priority for future investigation. Here, too, however, there is the paradox that opiates and stimulants both induce CREB activity in NAc while inducing opposite effects on dendritic structure.
Molecular mechanisms mediating cocaine-induced structural plasticity
1. RhoGTPase signaling pathways regulate structural plasticity
Structural changes in the actin cytoskeleton are in large part governed by a family of small GTPases, namely, Rho, cell division cycle 42 (Cdc42), Ras, and Rac (see Figure 2). These small GTPases are activated by guanine nucleotide exchange factors (GEFs), such as Ras-guanine nucleotide releasing factor (RasGRF1/2), VAV, Kalirin 7, and Tiam1, all of which catalyze the exchange of GDP for GTP [67–71]. GEFs are themselves activated by numerous extracellular signals, including brain derived neurotrophic factor (BDNF) through a tyrosine receptor kinase (TRKB) mechanism, tumor growth factor-B (TGF-B), cell adhesion proteins (integrins), and NMDA glutamate receptors through an increase in Ca2+ and activation of Ca2+/calmodulin-dependent protein kinase-II (CAMKII) [71–74]. Binding of GTP activates these GTPases, which then leads to activation of downstream regulators of the actin cytoskeleton, including lim domain kinase (LIMK), Wiskott-Aldrich Syndrome proteins (WASPs), ARP, and WASP-family verprolin homologues (WAVEs) [75–77]. However, the detailed molecular steps through which these various proteins are regulated by extracellular signals, and in turn the mechanisms by which they regulate the generation, retraction, or reshaping of individual dendritic spines, remains poorly understood.
Recently, these small GTPases and their GEF activators have been investigated for their roles in drug-induced structural plasticity. Mice lacking the GEF Ras-GRF1 exhibit attenuated sensitivity to cocaine, while constitutive over-expression throughout the brain enhances drug sensitization and reward [78]. Furthermore, Ras-GRF1 appears to mediate expression of ΔFosB [78], which as noted earlier promotes spinogenesis on NAc MSNs [6, 7] Interestingly, chronic cocaine was recently shown to reduce levels of GTP-bound RhoA, presumably leading to decreases in downstream actin severing molecules such as LIMK and cofilin [52].
The active form of small GTPases is terminated by GTPase-activating proteins (GAPs), which enhance GTP hydrolysis and thus act as negative regulators of RhoGTPases. Although far less is known regarding the role of GAPs in addiction, one study demonstrated that mutations in RhoGAP18B convey an altered sensitivity for ethanol, nicotine, and cocaine in Drosophila [79]. These results highlight the need for much future research to define the regulation of RhoGTPases and their regulatory proteins upon exposure to cocaine or other addictive drugs.
2. Transcriptional regulators of structural plasticity
Although the precise molecular steps by which ΔFosB mediates cocaine-induced spine density changes on NAc MSNs remain unknown, several recent studies have characterized candidates genes downstream of ΔFosB that are likely to be involved in synaptic remodeling (see Figure 2). Using genome-wide analyses, ΔFosB has been shown to regulate several genes known to mediate spinogenesis [58]. One such target is cyclin dependent kinase 5 (Cdk5), which is induced by cocaine in NAc via ΔFosB [80] and known in other systems to regulate RhoGTPases. Local inhibition of Cdk5 prevents cocaine-induced spine proliferation in NAc [8]. One target for Cdk5 is MEF2: induction of Cdk5 phosphorylates and inhibits MEF2, which in turn increases dendritic spines on NAc MSNs [5]. Repression of MEF2 activity in response to cocaine may allow for transcription of cytoskeleton-associated genes, N-WASP and WAVEs, which have putative MEF binding sites in their proximal promoter regions. There is also evidence to suggest that one particular WAVE protein, WAVE1, regulates spine morphogenesis in a Cdk5-dependent manner [81, 82]. Thus, induction of Cdk5 by chronic cocaine via ΔFosB, could result in regulation of WAVE activity, while MEF2 may regulate its expression level to mediate longer-term changes involved in addiction. From a functional perspective, inhibition of Cdk5, or activation of MEF2, both of which would oppose cocaine’s effects on NAc dendritic spines, paradoxically enhances behavioral responses to cocaine [5, 83, 84]. These unexpected findings suggest that gross changes in overall spine density may not necessarily lead to sensitized drug responses per se, but may be a result of “homeostatic adaptations” to compensate for other changes caused by chronic cocaine exposure, such as a reduction in glutamatergic stimulation of MSNs by prefrontal cortical afferents [34, 85].
In a subsequent study, we examined another transcription factor, nuclear factor κB (NFκB). We found that cocaine induces NFκB activity in NAc and that the resulting activation of NFκB is necessary for cocaine-induced dendritic spine formation on MSNs [6]. As with the Cdk5-MEF2 pathway, ΔFosB is required for cocaine induction of NFκB subunits, indicating that ΔFosB regulates a larger program of altered gene expression that leads ultimately to spinogenesis of NAc MSNs. Interestingly, we also found that inhibition of the NFκB pathway inhibited behavioral responses to cocaine, in line with the prevailing hypothesis in the field that cocaine-induced increases in spine density mediate behavioral sensitization [6].
The paradoxical differences between the behavioral effects of Cdk5-MEF2 vs. the effects of NFκB, despite the fact that induction of both pathways is mediated via ΔFosB and increases dendritic spine density, highlight the complexity of these intracellular pathways and the importance of future research. Our hypothesis is that the net effect of cocaine is to induce, via ΔFosB, NAc spine density through multiple downstream targets (e.g., NFκB, Cdk5-MEF2, many others) and the net consequence is sensitized behavioral responses to cocaine. At the same time, however, an individual target pathway like Cdk5-MEF2 may in isolation elicit distinct behavioral effects via its own diverse downstream molecular consequences. Thus, it is crucial that future studies profile downstream molecular pathways for the many cocaine and ΔFosB targets to gain insight into specific contributions of each pathway to cocaine-induced spinogenesis and altered behavioral responses to cocaine. These discrepant results may also be explained by confounds associated with transgenic and knockout mice or viral overexpression systems. These models, which are critical in studying the molecular pathways involved in structural plasticity, can produce off-target gene effects and induce gene products at levels well beyond those seen after drug exposure. Finally, we must recognize that, by measuring total dendritic spine number only, we are losing vital information about whether these spines are forming active synapses and thus altering the flow of information through the circuit. With these caveats in mind, future studies are needed to examine more detailed changes in spine structure and composition and their presynaptic inputs (Box 1) as well as the electrophysiological consequences of these molecular manipulations in the context of drug-induced spine and synaptic plasticity (Box 2).
Box 1. Methods to quantify structural plasticity in NAc MSNs.
(A) The morphology and density of dendritic spines have been studied by several techniques, each with strengths and weaknesses. Golgi stains are inexpensive and relatively easy to perform. Viral-mediated expression of fluorescent proteins such as GFP allows the ability to probe intrinsic molecular pathways that govern structural plasticity. However, neither Golgi nor viral transfection allow for detailed 3-dimensional (3D) analysis of spine shape or number. The newer methodologies of diolistics (gene gun delivery of – most commonly – the carbocyanide dye DiI) and microinjection of fluorescent molecules such as Alexa Fluor dyes and Lucifer Yellow, in combination with high-resolution 3D confocal imaging, offer an unprecedented glimpse into the morphology of dendritic spines. (B) An example of microinjection (or cell loading) of NAc neurons with Lucifer Yellow imaged at 10X (lower panel), 40X (upper panel), and 100X (right panel). (C) By using transgenic mice that express GFP selectively in Drd2- or Drd1-expressing neurons (left panel), we can target diolistics or dye microinjections to study cell-type specific changes in morphology. (D) One advantage of microinjection is that it has been validated for use with NeuronStudio, a program to conduct automated 3D analysis of spine density and morphology, as well as unbiased classification of spines into thin, mushroom, stubby and other subtypes (http://www.mssm.edu/cnic/tools-ns.html). Similar systems exist for use with membrane bound dyes such as DiI [33]. (E) All light microscope-based methods have significant weaknesses compared to electron microscopy (EM). EM, the gold standard for visualizing synapses, exploits a unique feature of the synapse: postsynaptic densities (PSDs) are electron-dense and can be readily visualized. In addition, certain synaptic features such as multiple synaptic boutons (yellow bow) and perforated synapses (orange box) can only be visualized by EM. The size of PSDs provides a measure of synapse strength since PSD size is correlated with synaptic function and plasticity [91]. This level of information may be important in addiction models. For example, it is possible that a drug of abuse changes spine density without altering the functional output of the cell, either by consolidating existing synapses into fewer but stronger ones, or by creating new but silent synapses. Conversely, a drug-induced change in spine size or shape – and therefore function – may occur in the absence of a change in total spine number. To address these questions in future studies, we will need to directly compare opiate- and stimulant-induced structural plasticity of NAc and other neurons using light and electron microscopy, and 3D morphometric analysis of spine type, along with measuring the electrophysiological correlates of synaptic state. In addition, experiments using multi-photon microscopy combined with localized uncaging of caged glutamate, or stimulation of identified presynaptic nerve terminals with channel rhodopsins, are needed to directly test the function and efficacy of individual new spines. See Box 2 for a detailed description of these functional studies. Scale bar: 5 μm in (A), 1 μm in (E). In (D) blue, red, green indicate thin, mushroom, stubby type spines respectively. In (E) blue shading indicates axon, pink shading indicates spine, arrows point to PSDs.
Box 2. Quantifying synaptic strength at individual MSN synapses: why is this necessary?
An important priority in drug abuse research is to directly measure synaptic strength at individual spine synapses so that causal connections between structural spine changes and functional changes in synaptic transmission can be made. Currently, this can best be accomplished by combining multi-photon laser scanning microscopy to image individual spines with multi-photon laser uncaging of caged glutamate to activate the same individual spines [92, 93]. An additional important technical advance will be the ability to identify specific afferent inputs making synapses on individual spines, since drug-induced modifications of synaptic structure and function may differ depending on the input (e.g., hippocampal versus amygdala versus cortical inputs to NAc MSNs. An exciting but challenging method to accomplish this is to express light-activated channels, such as channel rhodopsins, in the synaptic terminals of specific afferent inputs. This could allow activation of visually identifiable, individual synapses in slice preparations while simultaneously imaging the spines upon which these synapses are made to record their individual responses to synaptically released glutamate. Finally, as emphasized in the text, the specific NAc cell type needs to be identified, since drug-induced structural and functional synaptic modifications likely differ between Drd1- and Drd2-expressing MSNs as well as for various types of interneurons in NAc.
3. Cell-type specificity of structural plasticity
NAc MSNs exist in two major subtypes, predominantly containing either Drd1 or Drd2 dopamine receptors. The intracellular pathways downstream of the receptors differ greatly, and thus the molecular pathways governing neuronal structure may differ accordingly. Although the induction of dendritic spines after repeated treatment with psychostimulants occurs in both Drd1- and Drd2-expressing MSNs, the long-term stability of new spines appears to be greater in Drd1 neurons. These observations support the idea that intracellular signaling pathways downstream of Drd1 may mediate longer-term stabilization of spines than in Drd2 neurons [17, 86]. Indeed, the persistence of increased dendritic spines in Drd1-containing MSNs highly correlates with the persistent induction of ΔFosB in Drd1 MSNs and sensitized behavioral response to chronic drug exposure [87, 88]. Thus, it is possible that morphine and cocaine regulate distinct intracellular cascades in Drd1 and Drd2 MSNs. A key question therefore is whether different drugs of abuse differentially regulate neuronal structure through selective regulation of gene expression in these distinct NAc MSNs. This is a crucial consideration as these two populations are implicated in distinct aspects of NAc function, still incompletely defined, including different contributions to cocaine’s behavioral effects. For example, selective knockout of dopamine and cAMP-regulated phosphoprotein of 32 kDa (DARPP-32) from Drd1 versus Drd2 cells exerts opposite effects on cocaine-induced locomotion [89]. Furthermore, a selective knockout of glucocorticoid receptor from Drd1 neurons reduced motivation for cocaine and suppressed intake along a wide range of doses [90]. The ability to now use more sensitive methodologies for probing molecular changes in Drd1 and Drd2 MSNs (Box 1) will help us to understand how molecular changes occurring in these neuronal cell types may lead to distinct changes in neuronal structure in response to different classes of drugs of abuse, and how these changes influence addictive behaviors.
Conclusions
Drug-induced structural plasticity is one of the more replicable and enduring changes associated with addiction models. Numerous correlative studies, and a few functional studies, provide convincing evidence that these neuroadaptations are critical in mediating behavioral sensitization to cocaine. However, there are also several functional reports that argue that drug-induced spine plasticity is an epi-phenomenon unrelated to sensitization. It is clear that more work is necessary to fully understand the involvement of synaptic and structural plasticity in addictive behaviors. At this stage, it is premature to argue definitively for either side, as most published studies rely on measurements of total dendritic spine density, ignoring numerous features of spine plasticity (see Box 1). Throughout this review, we have outlined key areas for future investigation, summarized in Table 2, which are needed to clarify the paradoxical experimental data and help explain the role of dendritic spine plasticity in addiction. Future studies using multi-photon and electron microscopy will be needed to compare the effects of opiate and stimulant drugs of abuse on detailed structural properties of excitatory synapses, such as number of docked versus reserve pool presynaptic vesicles, PSD and active zone length, and spine head density and volume. This will help answer the question of whether the paradoxical differences observed in total dendritic spine density after morphine and cocaine do indeed reflect differences in synapse number and strength. Additionally, due to the transient nature of many electrophysiological changes, we need far more detailed time-course information of dendritic plasticity, of LTD/LTP, and of insertion or internalization of glutamate receptors induced by opiates and stimulants that might reflect particular behavioral features of addiction. To establish causality, we will then need to determine how each of these functional and structural changes affects addictive-like behavior. This last point is particularly important and will require an integration of several techniques. First, a molecular pathway is identified as being regulated by drugs of abuse and downstream target genes profiled for any relevant structural plasticity-related genes. Then, by using viral-mediated gene transfer, expression of shRNAs, or inducible genetic mutant mice to manipulate these molecular pathways, it will be possible to determine their specific roles in electrophysiological, structural, and behavioral changes following chronic drug administration. Finally, all of these studies must be considered on a cell-type and brain region-specific basis for a meaningful understanding of the precise mechanisms of brain pathology in addiction.
Table 2.
Outstanding questions
|
Box 1.

Acknowledgments
Preparation of this review was supported by grants from the National Institute on Drug Abuse
Glossary List of Terms
- Addiction-related behavior
This is most often studied by use of drug self-administration paradigms, including acquisition and maintenance of self-administration, withdrawal and extinction, as well as reinstatement (relapse)
- Stimulant treatment regimens
This includes experimenter- or self-administered cocaine amphetamine, or nicotine at a given dose and frequency for a given duration of time. Animals are then analyzed at varying times after the last drug dose
- Opiate treatment paradigms
This includes experimenter- or self-administered morphine, heroin, or other opiate drugs of abuse at a given dose and frequency for a given duration of time. Animals are then analyzed at varying times after the last drug dose
- Brain reward regions
These include midbrain dopaminergic neurons in the ventral tegmental area, and the limbic regions to which these neurons project, including the nucleus accumbens (ventral striatum) amygdala, hippocampus, and several regions of prefrontal cortex (e.g., medial, orbitofrontal, etc.)
- Glutamate receptors
The major ionotropic glutamate receptors in brain are named for specific agonists, α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA) and N-methyl-D-aspartate (NMDA)
- Dopamine receptor
Two major types of dopamine receptors are expressed in nucleus accumbens, containing either Drd1 or Drd2 receptors, which differ in their post-receptor signaling mechanisms. Drd1 receptors are Gs-coupled and stimulate adenylyl cyclase, while Drd2 receptors are Gi/o-coupled and inhibit adenylyl cyclase, activate inwardly rectifying K+ channels, and inhibit voltage-gated Ca2+ channels. Both receptors can also regulate extracellular signal regulated kinase (ERK) cascades
- RhoGTPases
These small G proteins play a central role in regulation of the actin cytoskelelton, thought to be integral in the growth and retraction of dendritic spines. They are activated by guanine nucleotide exchange factors (GEFs) and inhibited by GTPase-activating proteins (GAPs)
- Transcription factors
These are proteins that bind to specific DNA sequences (called response elements) within responsive genes and thereby increase or decrease the rate at which those genes are transcribed. Examples of transcription factors that regulate dendritic spines are: ΔFosB (a Fos family protein), cyclic AMP response element binding protein (CREB), nuclear factor κB (NFκB), and myocyte-enhancing factor-2 (MEF2)
- Protein kinases
Several protein kinases, enzymes that phosphorylate other proteins to regulate their function, have been implicated in the control of dendritic spine formation, including Ca2+/calmodulin-dependent protein kinase-II (CaMKII), cyclin-dependent kinase-5 (Cdk5), p21-activated kinase (PAK1), and lim domain kinase (LIMK), among many others
- Actin-related proteins
The actin cytoskeleton is regulated by a large number of proteins, however, the detailed role of each in ultimately growing or retracting a spine, or altering a spine’s size and shape, remain incompletely understood. Examples include actin-related proteins (ARPs), Wiskott-Aldrich Syndrome proteins (WASPs), WASP-family verprolin homologues (WAVEs), and cofilin, among many others
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sklair-Tavron L, et al. Chronic morphine induces visible changes in the morphology of mesolimbic dopamine neurons. Proc Natl Acad Sci U S A. 1996;93(20):11202–7. doi: 10.1073/pnas.93.20.11202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Robinson TE, Kolb B. Persistent structural modifications in nucleus accumbens and prefrontal cortex neurons produced by previous experience with amphetamine. J Neurosci. 1997;17(21):8491–7. doi: 10.1523/JNEUROSCI.17-21-08491.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robinson TE, Kolb B. Structural plasticity associated with exposure to drugs of abuse. Neuropharmacology. 2004;47(Suppl 1):33–46. doi: 10.1016/j.neuropharm.2004.06.025. [DOI] [PubMed] [Google Scholar]
- 4.Li Y, Acerbo MJ, Robinson TE. The induction of behavioural sensitization is associated with cocaine-induced structural plasticity in the core (but not shell) of the nucleus accumbens. Eur J Neurosci. 2004;20(6):1647–54. doi: 10.1111/j.1460-9568.2004.03612.x. [DOI] [PubMed] [Google Scholar]
- 5.Pulipparacharuvil S, et al. Cocaine regulates MEF2 to control synaptic and behavioral plasticity. Neuron. 2008;59(4):621–33. doi: 10.1016/j.neuron.2008.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Russo SJ, et al. Nuclear factor kappa B signaling regulates neuronal morphology and cocaine reward. J Neurosci. 2009;29(11):3529–37. doi: 10.1523/JNEUROSCI.6173-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maze I, et al. Essential role of the histone methyltransferase G9a in cocaine-induced plasticity. Science. 327(5962):213–6. doi: 10.1126/science.1179438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Norrholm SD, et al. Cocaine-induced proliferation of dendritic spines in nucleus accumbens is dependent on the activity of cyclin-dependent kinase-5. Neuroscience. 2003;116(1):19–22. doi: 10.1016/s0306-4522(02)00560-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Russo SJ, et al. Neurotrophic factors and structural plasticity in addiction. Neuropharmacology. 2009;56(Suppl 1):73–82. doi: 10.1016/j.neuropharm.2008.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dietz DM, et al. Molecular mechanisms of psychostimulant-induced structural plasticity. Pharmacopsychiatry. 2009;(42 Suppl 1):S69–78. doi: 10.1055/s-0029-1202847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nestler EJ. Molecular mechanisms of drug addiction. J Neurosci. 1992;12(7):2439–50. doi: 10.1523/JNEUROSCI.12-07-02439.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Russo SJ, et al. IRS2-Akt pathway in midbrain dopamine neurons regulates behavioral and cellular responses to opiates. Nat Neurosci. 2007;10(1):93–9. doi: 10.1038/nn1812. [DOI] [PubMed] [Google Scholar]
- 13.Robinson TE, et al. Widespread but regionally specific effects of experimenter- versus self-administered morphine on dendritic spines in the nucleus accumbens, hippocampus, and neocortex of adult rats. Synapse. 2002;46(4):271–9. doi: 10.1002/syn.10146. [DOI] [PubMed] [Google Scholar]
- 14.Robinson TE, et al. Cocaine self-administration alters the morphology of dendrites and dendritic spines in the nucleus accumbens and neocortex. Synapse. 2001;39(3):257–66. doi: 10.1002/1098-2396(20010301)39:3<257::AID-SYN1007>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 15.Robinson TE, Kolb B. Alterations in the morphology of dendrites and dendritic spines in the nucleus accumbens and prefrontal cortex following repeated treatment with amphetamine or cocaine. Eur J Neurosci. 1999;11(5):1598–604. doi: 10.1046/j.1460-9568.1999.00576.x. [DOI] [PubMed] [Google Scholar]
- 16.Sarti F, et al. Acute cocaine exposure alters spine density and long-term potentiation in the ventral tegmental area. Eur J Neurosci. 2007;26(3):749–56. doi: 10.1111/j.1460-9568.2007.05689.x. [DOI] [PubMed] [Google Scholar]
- 17.Lee KW, et al. Cocaine-induced dendritic spine formation in D1 and D2 dopamine receptor-containing medium spiny neurons in nucleus accumbens. Proc Natl Acad Sci U S A. 2006;103(9):3399–404. doi: 10.1073/pnas.0511244103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koob GF, Le Moal M. Plasticity of reward neurocircuitry and the ‘dark side’ of drug addiction. Nat Neurosci. 2005;8(11):1442–4. doi: 10.1038/nn1105-1442. [DOI] [PubMed] [Google Scholar]
- 19.Zito K, et al. Induction of spine growth and synapse formation by regulation of the spine actin cytoskeleton. Neuron. 2004;44(2):321–34. doi: 10.1016/j.neuron.2004.09.022. [DOI] [PubMed] [Google Scholar]
- 20.Hamilton GF, Whitcher LT, Klintsova AY. Postnatal binge-like alcohol exposure decreases dendritic complexity while increasing the density of mature spines in mPFC Layer II/III pyramidal neurons. Synapse. 2009;64(2):127–135. doi: 10.1002/syn.20711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luscher C, Bellone C. Cocaine-evoked synaptic plasticity: a key to addiction? Nat Neurosci. 2008;11(7):737–8. doi: 10.1038/nn0708-737. [DOI] [PubMed] [Google Scholar]
- 22.Ikemoto S. Dopamine reward circuitry: two projection systems from the ventral midbrain to the nucleus accumbens-olfactory tubercle complex. Brain Res Rev. 2007;56(1):27–78. doi: 10.1016/j.brainresrev.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Belin D, Everitt BJ. Cocaine seeking habits depend upon dopamine-dependent serial connectivity linking the ventral with the dorsal striatum. Neuron. 2008;57(3):432–41. doi: 10.1016/j.neuron.2007.12.019. [DOI] [PubMed] [Google Scholar]
- 24.Bourne J, Harris KM. Do thin spines learn to be mushroom spines that remember? Curr Opin Neurobiol. 2007;17(3):381–6. doi: 10.1016/j.conb.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 25.Carlisle HJ, Kennedy MB. Spine architecture and synaptic plasticity. Trends Neurosci. 2005;28(4):182–7. doi: 10.1016/j.tins.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 26.Tada T, Sheng M. Molecular mechanisms of dendritic spine morphogenesis. Curr Opin Neurobiol. 2006;16(1):95–101. doi: 10.1016/j.conb.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 27.Nagerl UV, et al. Bidirectional activity-dependent morphological plasticity in hippocampal neurons. Neuron. 2004;44(5):759–67. doi: 10.1016/j.neuron.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 28.Okamoto K, et al. Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat Neurosci. 2004;7(10):1104–12. doi: 10.1038/nn1311. [DOI] [PubMed] [Google Scholar]
- 29.Zuo Y, et al. Development of long-term dendritic spine stability in diverse regions of cerebral cortex. Neuron. 2005;46(2):181–9. doi: 10.1016/j.neuron.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 30.Matsuzaki M, et al. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429(6993):761–6. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harris KM, Jensen FE, Tsao B. Three-dimensional structure of dendritic spines and synapses in rat hippocampus (CA1) at postnatal day 15 and adult ages: implications for the maturation of synaptic physiology and long-term potentiation. J Neurosci. 1992;12(7):2685–705. doi: 10.1523/JNEUROSCI.12-07-02685.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holtmaat AJ, et al. Transient and persistent dendritic spines in the neocortex in vivo. Neuron. 2005;45(2):279–91. doi: 10.1016/j.neuron.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 33.Shen HW, et al. Altered dendritic spine plasticity in cocaine-withdrawn rats. J Neurosci. 2009;29(9):2876–84. doi: 10.1523/JNEUROSCI.5638-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thomas MJ, et al. Long-term depression in the nucleus accumbens: a neural correlate of behavioral sensitization to cocaine. Nat Neurosci. 2001;4(12):1217–23. doi: 10.1038/nn757. [DOI] [PubMed] [Google Scholar]
- 35.Huang YH, et al. In vivo cocaine experience generates silent synapses. Neuron. 2009;63(1):40–7. doi: 10.1016/j.neuron.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Malenka RC, Nicoll RA. Long-term potentiation--a decade of progress? Science. 1999;285(5435):1870–4. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- 37.Marie H, et al. Generation of silent synapses by acute in vivo expression of CaMKIV and CREB. Neuron. 2005;45(5):741–52. doi: 10.1016/j.neuron.2005.01.039. [DOI] [PubMed] [Google Scholar]
- 38.Sheng M, et al. Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature. 1994;368(6467):144–7. doi: 10.1038/368144a0. [DOI] [PubMed] [Google Scholar]
- 39.Elias GM, et al. Differential trafficking of AMPA and NMDA receptors by SAP102 and PSD-95 underlies synapse development. Proc Natl Acad Sci U S A. 2008;105(52):20953–8. doi: 10.1073/pnas.0811025106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boudreau AC, et al. Cell surface AMPA receptors in the rat nucleus accumbens increase during cocaine withdrawal but internalize after cocaine challenge in association with altered activation of mitogen-activated protein kinases. J Neurosci. 2007;27(39):10621–35. doi: 10.1523/JNEUROSCI.2163-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boudreau AC, Wolf ME. Behavioral sensitization to cocaine is associated with increased AMPA receptor surface expression in the nucleus accumbens. J Neurosci. 2005;25(40):9144–51. doi: 10.1523/JNEUROSCI.2252-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Conrad KL, et al. Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature. 2008;454(7200):118–21. doi: 10.1038/nature06995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Anderson SM, et al. CaMKII: a biochemical bridge linking accumbens dopamine and glutamate systems in cocaine seeking. Nat Neurosci. 2008;11(3):344–53. doi: 10.1038/nn2054. [DOI] [PubMed] [Google Scholar]
- 44.Bachtell RK, et al. Role of GluR1 expression in nucleus accumbens neurons in cocaine sensitization and cocaine-seeking behavior. Eur J Neurosci. 2008;27(9):2229–40. doi: 10.1111/j.1460-9568.2008.06199.x. [DOI] [PubMed] [Google Scholar]
- 45.Kourrich S, et al. Cocaine experience controls bidirectional synaptic plasticity in the nucleus accumbens. J Neurosci. 2007;27(30):7921–8. doi: 10.1523/JNEUROSCI.1859-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Toda S, et al. Cocaine increases actin cycling: effects in the reinstatement model of drug seeking. J Neurosci. 2006;26(5):1579–87. doi: 10.1523/JNEUROSCI.4132-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Spijker S, et al. Morphine exposure and abstinence define specific stages of gene expression in the rat nucleus accumbens. FASEB J. 2004:03–0612fje. doi: 10.1096/fj.03-0612fje. [DOI] [PubMed] [Google Scholar]
- 48.Roche KW. The expanding role of PSD-95: a new link to addiction. Trends in Neurosciences. 2004;27(12):699–700. doi: 10.1016/j.tins.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 49.Szumlinski KK, et al. Homer Isoforms Differentially Regulate Cocaine-Induced Neuroplasticity. Neuropsychopharmacology. 2005;31(4):768–777. doi: 10.1038/sj.npp.1300890. [DOI] [PubMed] [Google Scholar]
- 50.Yao WD, et al. Identification of PSD-95 as a Regulator of Dopamine-Mediated Synaptic and Behavioral Plasticity. Neuron. 2004;41(4):625–638. doi: 10.1016/s0896-6273(04)00048-0. [DOI] [PubMed] [Google Scholar]
- 51.Heiman M, et al. A Translational Profiling Approach for the Molecular Characterization of CNS Cell Types. Cell. 2008;135(4):738–748. doi: 10.1016/j.cell.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim WY, et al. Cocaine regulates ezrin-radixin-moesin proteins and RhoA signaling in the nucleus accumbens. Neuroscience. 2009;163(2):501–505. doi: 10.1016/j.neuroscience.2009.06.067. [DOI] [PubMed] [Google Scholar]
- 53.Hope BT, et al. Induction of a long-lasting AP-1 complex composed of altered Fos-like proteins in brain by chronic cocaine and other chronic treatments. Neuron. 1994;13(5):1235–44. doi: 10.1016/0896-6273(94)90061-2. [DOI] [PubMed] [Google Scholar]
- 54.Alibhai IN, et al. Regulation of fosB and DeltafosB mRNA expression: in vivo and in vitro studies. Brain Res. 2007;1143:22–33. doi: 10.1016/j.brainres.2007.01.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shaw-Lutchman TZ, et al. Regional and cellular mapping of cAMP response element-mediated transcription during naltrexone-precipitated morphine withdrawal. J Neurosci. 2002;22(9):3663–72. doi: 10.1523/JNEUROSCI.22-09-03663.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shaw-Lutchman TZ, et al. Regulation of CRE-mediated transcription in mouse brain by amphetamine. Synapse. 2003;48(1):10–7. doi: 10.1002/syn.10172. [DOI] [PubMed] [Google Scholar]
- 57.Perrotti LI, et al. Distinct patterns of DeltaFosB induction in brain by drugs of abuse. Synapse. 2008;62(5):358–69. doi: 10.1002/syn.20500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McClung CA, Nestler EJ. Regulation of gene expression and cocaine reward by CREB and [Delta]FosB. Nat Neurosci. 2003;6(11):1208–1215. doi: 10.1038/nn1143. [DOI] [PubMed] [Google Scholar]
- 59.Zachariou V, et al. An essential role for [Delta]FosB in the nucleus accumbens in morphine action. Nat Neurosci. 2006;9(2):205–211. doi: 10.1038/nn1636. [DOI] [PubMed] [Google Scholar]
- 60.Renthal W, et al. Genome-wide analysis of chromatin regulation by cocaine reveals a role for sirtuins. Neuron. 2009;62(3):335–48. doi: 10.1016/j.neuron.2009.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Carlezon WA, Jr, Duman RS, Nestler EJ. The many faces of CREB. Trends Neurosci. 2005;28(8):436–45. doi: 10.1016/j.tins.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 62.Murphy DD, Segal M. Morphological plasticity of dendritic spines in central neurons is mediated by activation of cAMP response element binding protein. Proc Natl Acad Sci U S A. 1997;94(4):1482–7. doi: 10.1073/pnas.94.4.1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Seigo S, et al. Opposing functions of CREB and MKK1 synergistically regulate the geometry of dendritic spines in visual cortex. The Journal of Comparative Neurology. 2007;503(5):605–617. doi: 10.1002/cne.21424. [DOI] [PubMed] [Google Scholar]
- 64.Graham DL, et al. Dynamic BDNF activity in nucleus accumbens with cocaine use increases self-administration and relapse. Nat Neurosci. 2007;10(8):1029–37. doi: 10.1038/nn1929. [DOI] [PubMed] [Google Scholar]
- 65.Pu L, Liu QS, Poo MM. BDNF-dependent synaptic sensitization in midbrain dopamine neurons after cocaine withdrawal. Nat Neurosci. 2006;9(5):605–7. doi: 10.1038/nn1687. [DOI] [PubMed] [Google Scholar]
- 66.Vo N, et al. A cAMP-response element binding protein-induced microRNA regulates neuronal morphogenesis. Proc Natl Acad Sci U S A. 2005;102(45):16426–31. doi: 10.1073/pnas.0508448102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Abe K, et al. Vav2 is an activator of Cdc42, Rac1, and RhoA. J Biol Chem. 2000;275(14):10141–9. doi: 10.1074/jbc.275.14.10141. [DOI] [PubMed] [Google Scholar]
- 68.Farnsworth CL, et al. Calcium activation of Ras mediated by neuronal exchange factor Ras-GRF. Nature. 1995;376(6540):524–7. doi: 10.1038/376524a0. [DOI] [PubMed] [Google Scholar]
- 69.Krapivinsky G, et al. The NMDA receptor is coupled to the ERK pathway by a direct interaction between NR2B and RasGRF1. Neuron. 2003;40(4):775–84. doi: 10.1016/s0896-6273(03)00645-7. [DOI] [PubMed] [Google Scholar]
- 70.Penzes P, et al. Rapid induction of dendritic spine morphogenesis by trans-synaptic ephrinB-EphB receptor activation of the Rho-GEF kalirin. Neuron. 2003;37(2):263–74. doi: 10.1016/s0896-6273(02)01168-6. [DOI] [PubMed] [Google Scholar]
- 71.Tolias KF, et al. The Rac1-GEF Tiam1 couples the NMDA receptor to the activity-dependent development of dendritic arbors and spines. Neuron. 2005;45(4):525–38. doi: 10.1016/j.neuron.2005.01.024. [DOI] [PubMed] [Google Scholar]
- 72.Edlund S, et al. Transforming growth factor-beta-induced mobilization of actin cytoskeleton requires signaling by small GTPases Cdc42 and RhoA. Mol Biol Cell. 2002;13(3):902–14. doi: 10.1091/mbc.01-08-0398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang JQ, et al. Glutamate signaling to Ras-MAPK in striatal neurons: mechanisms for inducible gene expression and plasticity. Mol Neurobiol. 2004;29(1):1–14. doi: 10.1385/MN:29:1:01. [DOI] [PubMed] [Google Scholar]
- 74.Yuan XB, et al. Signalling and crosstalk of Rho GTPases in mediating axon guidance. Nat Cell Biol. 2003;5(1):38–45. doi: 10.1038/ncb895. [DOI] [PubMed] [Google Scholar]
- 75.Machesky LM, et al. Scar, a WASp-related protein, activates nucleation of actin filaments by the Arp2/3 complex. Proc Natl Acad Sci U S A. 1999;96(7):3739–44. doi: 10.1073/pnas.96.7.3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Miki H, et al. Induction of filopodium formation by a WASP-related actin-depolymerizing protein N-WASP. Nature. 1998;391(6662):93–6. doi: 10.1038/34208. [DOI] [PubMed] [Google Scholar]
- 77.Miki H, Suetsugu S, Takenawa T. WAVE, a novel WASP-family protein involved in actin reorganization induced by Rac. EMBO J. 1998;17(23):6932–41. doi: 10.1093/emboj/17.23.6932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fasano S, et al. Ras-Guanine Nucleotide-Releasing Factor 1 (Ras-GRF1) Controls Activation of Extracellular Signal-Regulated Kinase (ERK) Signaling in the Striatum and Long-Term Behavioral Responses to Cocaine. Biol Psychiatry. 2009 doi: 10.1016/j.biopsych.2009.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rothenfluh A, et al. Distinct behavioral responses to ethanol are regulated by alternate RhoGAP18B isoforms. Cell. 2006;127(1):199–211. doi: 10.1016/j.cell.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 80.Kumar A, et al. Chromatin remodeling is a key mechanism underlying cocaine-induced plasticity in striatum. Neuron. 2005;48(2):303–14. doi: 10.1016/j.neuron.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 81.Kim Y, et al. Phosphorylation of WAVE1 regulates actin polymerization and dendritic spine morphology. Nature. 2006;442(7104):814–7. doi: 10.1038/nature04976. [DOI] [PubMed] [Google Scholar]
- 82.Sung JY, et al. WAVE1 controls neuronal activity-induced mitochondrial distribution in dendritic spines. Proc Natl Acad Sci U S A. 2008;105(8):3112–6. doi: 10.1073/pnas.0712180105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Benavides DR, et al. Cdk5 modulates cocaine reward, motivation, and striatal neuron excitability. J Neurosci. 2007;27(47):12967–76. doi: 10.1523/JNEUROSCI.4061-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bibb JA, et al. Effects of chronic exposure to cocaine are regulated by the neuronal protein Cdk5. Nature. 2001;410(6826):376–80. doi: 10.1038/35066591. [DOI] [PubMed] [Google Scholar]
- 85.Berglind WJ, et al. A single intra-PFC infusion of BDNF prevents cocaine-induced alterations in extracellular glutamate within the nucleus accumbens. J Neurosci. 2009;29(12):3715–9. doi: 10.1523/JNEUROSCI.5457-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kim Y, et al. Methylphenidate-induced dendritic spine formation and DeltaFosB expression in nucleus accumbens. Proc Natl Acad Sci U S A. 2009;106(8):2915–20. doi: 10.1073/pnas.0813179106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hope BT, et al. Induction of a long-lasting AP-1 complex composed of altered Fos-like proteins in brain by chronic cocaine and other chronic treatments. Neuron. 1994;13(5):1235–1244. doi: 10.1016/0896-6273(94)90061-2. [DOI] [PubMed] [Google Scholar]
- 88.Nestler EJ. Review. Transcriptional mechanisms of addiction: role of DeltaFosB. Philos Trans R Soc Lond B Biol Sci. 2008;363(1507):3245–55. doi: 10.1098/rstb.2008.0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bateup HS, et al. Distinct populations of medium spiny neurons differentially regulate striatal motor behaviors. Proc Natl Acad Sci U S A. doi: 10.1073/pnas.1009874107. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ambroggi F, et al. Stress and addiction: glucocorticoid receptor in dopaminoceptive neurons facilitates cocaine seeking. Nat Neurosci. 2009;12(3):247–249. doi: 10.1038/nn.2282. [DOI] [PubMed] [Google Scholar]
- 91.Lisman JE, Raghavachari S, Tsien RW. The sequence of events that underlie quantal transmission at central glutamatergic synapses. Nat Rev Neurosci. 2007;8(8):597–609. doi: 10.1038/nrn2191. [DOI] [PubMed] [Google Scholar]
- 92.Steiner P, et al. Destabilization of the postsynaptic density by PSD-95 serine 73 phosphorylation inhibits spine growth and synaptic plasticity. Neuron. 2008;60(5):788–802. doi: 10.1016/j.neuron.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kantevari S, et al. Two-color, two-photon uncaging of glutamate and GABA. Nat Methods. 7(2):123–5. doi: 10.1038/nmeth.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]