

Abstract
Nuclear Factor (erythroid-derived 2)-like 2 (Nrf2) expression is deregulated in many cancers. Genetic and biochemical approaches coupled with functional assays in cultured cells were used to explore the consequence of Nrf2 repression. Nrf2 suppression by Keap1-directed ubiquitylation or expression of independent shRNA/siRNA sequences enhanced cellular ROS, Smad-dependent tumor cell motility, and growth in soft agar. Loss of Nrf2 was accompanied by concomitant Smad linker region/C-terminus phosphorylation, induction of the E-Cadherin transcriptional repressor Slug, and suppression of the cell-cell adhesion protein E-Cadherin. Ectopic expression of wildtype Nrf2, but not dominant negative Nrf2, suppressed the activity of a synthetic TGF-β1 responsive CAGA-directed luciferase reporter. shRNA knock-down of Nrf2 enhanced the activity of the synthetic CAGA-reporter, as well as the expression of the endogenous Smad target gene plasminogen activator inhibitor-1. Finally, we found that Nrf2/Smad3/Smad4 formed an immunoprecipitable nuclear complex. Thus, loss of Nrf2 increased R-Smad phosphorylation and R-Smad signaling, supporting the hypothesis that loss of Nrf2 in an oncogenic context-dependent manner can enhance cellular plasticity and motility, in part by using TGF-β/Smad signaling.
Keywords: Nrf2, TGF-β, Smad, Cadherin, motility, oncogenesis, ROS
Introduction
Nuclear Factor (erythroid-derived 2)-like 2 (Nrf2) is a transcription factor that mediates an adaptive response induced by heterodimerization at Antioxidant Response Elements (AREs) located in proximal promoters of target genes that detoxify oxidants and electrophiles (Venugopal and Jaiswal, 1998). This adaptive response represents the current paradigm describing Nrf2's primary physiological role.
The observation that Nrf2 null mice are viable and fertile (Chan et al., 1996) indicates that inducible ARE-dependent gene transcription is dispensable under homeostatic conditions. However, failure to induce Nrf2 and its target genes increases the susceptibility of Nrf2 null mice to chemical carcinogenesis (Ramos-Gomez et al., 2001).
Nrf2 expression is deregulated in human cancer. Nrf2 is elevated in some breast and lung cancers (Nioi and Nguyen, 2007; Padmanabhan et al., 2006; Singh et al., 2006). In non small cell lung cancer (NSCLC) Nrf2 overexpression can be a consequence of mutations in Keap1 that impede Nrf2 ubiquitination (Nioi and Nguyen, 2007; Padmanabhan et al., 2006; Singh et al., 2006) or a consequence of Nrf2 mutations that impair Nrf2's ability to bind to Keap1 (Shibata et al., 2008). Nrf2 overexpression in NSCLC is postulated to promote tumor growth because repression of Nrf2 expression in NSCLC xenografts has been shown to inhibit tumor proliferation (Singh et al., 2008).
As expected, given the complexity of tumorigenesis, there are tumors in which Nrf2 expression is suppressed. Stacy et al., (Stacy et al., 2006) found that approximately 7% of human head and neck squamous cell carcinomas expressed low levels of Nrf2, coincident with Keap1 overexpression (Stacy et al., 2006). An Oncomine bioinformatics analysis (www.oncomine.org) revealed that Nrf2 was downregulated in lung adenocarcinoma (Beer et al., 2002), ovarian serous adenocarcinoma, ovarian endometrioid adenocarcinoma, and clear cell adenocarcinoma (Hendrix et al., 2006; Welsh et al., 2001), as well as hepatocellular carcinoma (Chen et al., 2002; Wurmbach et al., 2007). Nrf2 and its target genes have been shown to be downregulated in human prostate tumors, in metastatic prostate cancer (Frohlich et al., 2008) and in the TRAMP mouse prostate tumor model (Frohlich et al., 2008; Yu et al., 2010). Nrf2 suppression can be a consequence of CUL3 overexpression (Loignon et al., 2009) or epigenetic events (Yu et al., 2010).
The consequences of Nrf2 deregulation in tumor etiology are just beginning to be explored. Exuberant ARE-directed gene expression in human head and neck cancer is associated with recurrence-free survival (Chung et al., 2004). Conversely, loss of ARE-directed gene expression, with subsequent elevation of reactive oxygen species has been shown to represent a primary oncogenic-associated stress that promotes proliferation and motility (Cao et al., 2009; Liu et al., 2004; Luo et al., 2009; Radisky et al., 2005; Sun et al., 1989; Szatrowski and Nathan, 1991; Weydert et al., 2006; Zhang et al., 2002). For example, loss of MnSOD results increased ROS levels, which in turn stimulates cyclins D1 and B1, making cells insensitive to anti-growth signals (Sarsour et al., 2008). Conversely, expression of MnSOD with subsequent suppression of ROS allows cells to respond to anti-proliferation signaling. Mates et al., (Mates et al., 2008) argue that the majority of ROS effects are a consequence of signaling pathway alterations rather than non-specific damage of macromolecules. ROS can function in an autocrine manner by reversibly altering the function of phosphatases and kinases (Kamata et al., 2005; Mates et al., 2008; Tonks, 2005).
The exuberant levels of ROS found in many tumor cells (Szatrowski and Nathan, 1991) can be a consequence of diminished expression of Nrf2-regulated genes involved in ROS detoxification (Hirayama et al., 2003; Kwak et al., 2001). Restoration of ROS to physiological levels by overexpression of Nrf2-regulated-antioxident proteins can lower ROS levels coincident with suppression of oncogenesis in such cancers as pancreatic, glioma and melanoma, (Chakraborty et al., 1992; Liu et al., 2004; Zhang et al., 2002).
Consistent with these observations, Coulouarn et al. (Coulouarn et al., 2008) found that loss of Nrf2 expression was associated with increased progression of human hepatocellular carcinomas. Based on data obtained with the TRAMP mouse model, Barve et al hypothesized that loss of Nrf2 contributes to high grade PIN and tumor formation (Barve et al., 2009). Repression of Nrf2 expression is part of a multi-gene signature that is predictive of TGF-β1-mediated invasiveness and metastasis. However, the biochemical pathways impacted by Nrf2 deregulation during tumorigenesis are not well understood.
In this report we show that genetic-mediated repression of Nrf2 in subpopulations of oncogenic cells grown in culture can increase their propensity for survival and motility. RNAi suppression of Nrf2 in an oncogenic Ras context was associated with increased ROS, increased survival and colony outgrowth. Cells expressing Nrf2 shRNA exhibit high levels of Smad phosphorylation in both the linker region and the C-terminus, elevated synthetic (CAGA) luciferase reporter activity, and elevated expression of endogenous PAI-1. We found that Nrf2/Smad3/Smad4 formed an immunoprecipitable nuclear complex and that wild type, but not dominant negative Nrf2 (Yu et al., 2000), suppressed CAGA-directed reporter activity. RNAi suppression of Nrf2 was associated with increased expression of Slug and subsequent repression of E-Cadherin. Functional studies revealed that loss of Nrf2 was accompanied by a high propensity for Smad-mediated motility. These results support a hypothesis that suppression of Nrf2 contributes to tumor cell plasticity and motility, in part through exuberant Smad-mediated signaling.
Results and Discussion
Cellular Plasticity and Motility
The Cullin3 adaptor protein, Keap1 directs E3-ubiquitin ligase mediated ubiquitylation and proteasome-dependent degradation of Nrf2 (Kobayashi et al., 2004). Keap1 overexpression has been shown to repress expression of Nrf2 and its target genes (Zhang et al., 2004). Keap1 overexpression has been observed in human cancer (Stacy et al., 2006), including metastatic hepatocellular carcinomas (www.oncomine.org)(Ye et al., 2003).
FLAG-tagged Keap1 or FLAG-tagged luciferase was stably expressed in HepG2 cells, as described in (Sekhar et al., 2003). As determined by quantitative image analysis, stable expression of FLAG/Keap1 reduced Nrf2 levels by 60% relative to expression of β-actin and compared to control cells stably expressing FLAG/luciferase (Figure 1, Panel A). Loss of Nrf2 produced a statistically significant decrease in the activity of a Nrf2-regulated ARE-directed reporter (Sekhar et al., 2003). GCLC, the catalytic subunit for glutamate cysteine ligase, the rate limiting enzyme in glutathione synthesis, is an Nrf2 target gene. Keap1-mediated repression of Nrf2 suppressed endogenous GCLC expression (Sekhar et al., 2003). Multidrug resistance-associated proteins (MRPs) are also Nrf2 target genes (Hayashi et al., 2003). Loss of Nrf2 decreased cellular MRP activity (Sekhar et al., 2003). These data indicate that overexpression of Keap1 repressed the expression of Nrf2 and several surrogate Nrf2 target genes.
Figure 1.

Overexpression of Keap1 in HepG2 cells drives down Nrf2 levels and is accompanied by increased colony forming ability, as well as morphological plasticity. A) Immunoblot of cells stably expressing FLAG/Luciferase or FLAG/Keap1; B) Colony forming ability; C) Cell proliferation. Cells expressing FLAG/Luciferase exhibited a doubling time of approximately 31 hrs. Cells expressing FLAG/Keap1 exhibited a doubling time of approximately 24 hrs; D) Light microscopic images (200 × magnification).
Cells that overexpressed Keap1 exhibited increased survival when plated as single cells (Figure 1). The cells changed to a spindle-like morphology with extended cellular pseudopodium characteristic of migrating cells (Greenburg and Hay, 1982), as assessed by phase microscopy (Figure 1D) or immunofluorescence (Supplemental Figure 1A). Changes in morphology were not observed in HepG2 cells stably expressing FLAG/luciferase, non-transformed HEK293 cells stably expressing FLAG/Keap1 (Hong et al., 2005) or non-transformed NMuMG cells that stably expressed FLAG/Keap1 (data not shown).
Overexpression of Keap1 in human colon Caco-2 carcinoma cells repressed Nrf2 and its target genes, resulting in cellular remodeling (Kusano et al., 2008). This change in cell morphology was due to Keap1-mediated inhibition of E-Cadherin recruitment to adherin junctions (Kusano et al., 2008). As HepG2 cells express low E-Cadherin levels due to loss of connexin 26 (Yano et al., 2001), we evaluated another marker of cellular remodeling, vimentin, the expression of which increases following loss of cell-cell adhesion (Zavadil and Bottinger, 2005). Overexpression of Keap1 was associated with upregulation of vimentin (Supplemental Figure 1B).
Keap1 also directs the ubiquitylation of PGAM5(Lo and Hannink, 2006), Fetal Alz-50 reactive clone (FAC1)(Strachan et al., 2005), IKK β (Lee et al., 2009), and p62 (Komatsu et al.). The changes in cellular morphology could be a direct consequence of loss of Nrf2 or a secondary consequence of increased degradation of the other substrates. Therefore use was made of human A549 non-small cell lung carcinoma cells, in which Keap1 contains a G to C mutation at residue 333 that impairs its ability to direct substrate ubiquitylation (Singh et al., 2006).
Several different non-overlapping shRNA/siRNA sequences were used to stably knockdown Nrf2 in A549 cells. Cells were transduced or transfected with retrovirus vector expressing Nrf2 shRNA, transfected with pSilencer vector expressing Nrf2 siRNA (Singh et al., 2008), or a pool consisting of 4 lentiviruses each expressing a different Nrf2 shRNA. Control cells were transduced or transfected with the appropriate control vectors. Drug selection of Nrf2 shRNA or siRNA expressing cells yielded large dense foci, which were isolated and expanded. In cells transduced with control non-silencing vector; only small discrete colonies were observed. Therefore, a polyclonal population stably expressing control non-silencing shRNA was obtained.
Stable expression of Nrf2 shRNA or siRNA repressed Nrf2 expression and suppressed the Nrf2 target gene GCLC (Figure 2A). Loss of GCLC was followed by a 70% decrease in intracellular glutathione (p <0.05; Student's t test; Supplemental Figure 2 A). As expected, suppression of GCLC and other Nrf2 target genes increased intracellular ROS, as determined by the 10 fold increase in C-400 fluorescence (p < 0.05 Student's t test; Supplemental Figure 2 B & C). Oxidative stress is known to activate AKT (Wang et al., 2000). Activated AKT accelerates mitochondrial ROS generation, resulting in a cycle of oxidant-induced PTEN inactivation and sustained AKT activation (Nogueira et al., 2008). Consistent with increased ROS observed in Supplemental Figure 2, immunoblotting revealed elevation of phospho-AKT, Cyclin D1 (Matsuzawa and Ichijo, 2008), and c-Jun in Nrf2 shRNA expressing cells compared to control (Supplemental Figure 3).
Figure 2.

RNAi suppression of Nrf2 increases soft agar colony forming ability and cell plasticity. A) Immunoblot of A549 cells stably expressing Nrf2 shRNA from a retrovirus vector (upper) or Nrf2 siRNA from a pSilencer vector (lower), or appropriate control shRNA/siRNA; B) A549 cells stably expressing Nrf2 shRNA from a retrovirus vector or Nrf2 siRNA from a pSilencer vector, or appropriate control shRNA/siRNA were inoculated into soft agar. Five days after inoculation, the numbers of single cells and multi-cellular colonies were quantitated by microscopy; C) Light microscopic images (400 × magnification) of A549 cells stably expressing Nrf2 shRNA or control non-silencing shRNA from a retrovirus vectors.
Cell proliferation was assessed by quantitating cell number as a function of time (Supplemental Figure 4). Stable expression of independent retrovirus-mediated Nrf2 shRNA (Supplemental Figure 4A) or lentivirus-mediated Nrf2 shRNA (Supplemental Figure 4B) did not significantly affect the rate of cell proliferation on a plastic substrate, compared to control, non-silencing shRNA (population doubling time of 19 hrs). Thus, the increased proliferation observed in cells that overexpress Keap1 cannot be attributed to loss of Nrf2.
Cells stably expressing independent Nrf2 shRNA from retrovirus or Nrf2 siRNA from a pSilencer vector (Singh et al., 2008) exhibited an anchorage-independent phenotype such that when plated as single cells in soft agar, they formed large colonies (p < 0.0001 Student's t test; Figure 2B).
shRNA containing cells exhibited a spindle-like morphology with extended cellular pseudopodium characteristic of migrating cells (Figure 2C). The change in morphology was not a consequence of transduction. A549 cells were transfected rather than transduced with the retrovirus shRNA vectors. Cells stably transfected with Nrf2 shRNA exhibited morphology similar to cells that were transduced with Nrf2 shRNA (data not shown).
SW480 colon carcinoma cells are deficient in TGF-β1 type II receptor function, functional Smad4, and E-Cadherin expression (Calonge and Massague, 1999; Muller et al., 2002). These cells exhibit a spindle-like morphology (Muller et al., 2002; Shiou et al., 2007). Forced expression of Smad4 in these cells restores expression of E-Cadherin in a TGF-β1 type II receptor-independent manner (Calonge and Massague, 1999; Muller et al., 2002) and the cells acquire an epithelioid morphology (Shiou et al., 2007). SW480 cells stably expressing Smad4 (Shiou et al., 2007) were transduced with either Nrf2 shRNA or control non-silencing vectors. Expression of Nrf2 shRNA reduced Nrf2 expression by 50%, as quantitated by immunoblotting. Loss of Nrf2 in SW480 cells expressing Smad4 was accompanied by a change in morphology: from epithelioid-like to spindle-like (Supplemental Figure 5). Taken all together, these results indicate that suppression of Nrf2 via overexpression of Keap1 in HepG2 cells or RNAi-mediated suppression of Nrf2 in A549 or SW480 cells was accompanied by morphological plasticity.
Interestingly, transient siRNA-mediated repression of Nrf2 in human lung cancer cell lines, including A549 cells, diminished proliferation and resistance to certain chemotherapeutic drugs (Homma et al., 2009; Ohta et al., 2008; Shibata et al., 2008). We hypothesize that the differences in proliferation observed by these investigators and the work reported here may be attributed to transient vs stable repression of Nrf2. Singh et al (Singh et al., 2008) stably expressed an Nrf2 siRNA vector in A549 and H460 cells. Knock down of Nrf2 in these cells was accompanied by a loss of proliferation and an inability to grow in soft agar. In addition, when these cells were grown as xenografts, tumor formation was inhibited (Singh et al., 2008). The differences in soft agar growth observed by Singh et al (Singh et al., 2008) and in the work presented here are not currently understood. Our study utilized cell culture models only and did not investigate xenograft growth.
A transwell migration assay was used to assess migration of A549 and HepG2 cells (Figure 3A &B). Migration through transwell inserts was assessed 24 hrs after inoculation. A549 cells stably expressing Nrf2 shRNA exhibited a 4 fold increase in migration compared to control. HepG2 cells expressing FLAG/Keap1 exhibited an 18 fold increase in migration compared to control HepG2 cells expressing FLAG/luciferase (Figure 3, p < 0.05; Student's t test). A 2-dimensional migration assay was used to assess migration. Loss of Nrf2 increased cellular motility (Figure 3, C & D). An area devoid of cells was created at time zero by scraping a monolayer of cells. The leading edges along the scraped area have almost coalesced 24 hrs after scraping cells expressing Nrf2 shRNA from lentivirus (Figure 3C), retrovirus (Supplemental Figure 6A), or Nrf2 siRNA from pSilencer (Supplemental Figure 6B), or overexpressing Keap1 (Figure 3D). Cells expressing control non-silencing shRNA/siRNA exhibited little migration. The observation that exposure of control cells to TGF-β (5 ng/ml) induced migration whereas addition of TGF-β plus the TGF-β receptor kinase inhibitor SB-431542 (10 μM) inhibited migration indicates that cells expressing non silencing shRNA retain the ability to migrate (Supplemental Figure 6C).
Figure 3.
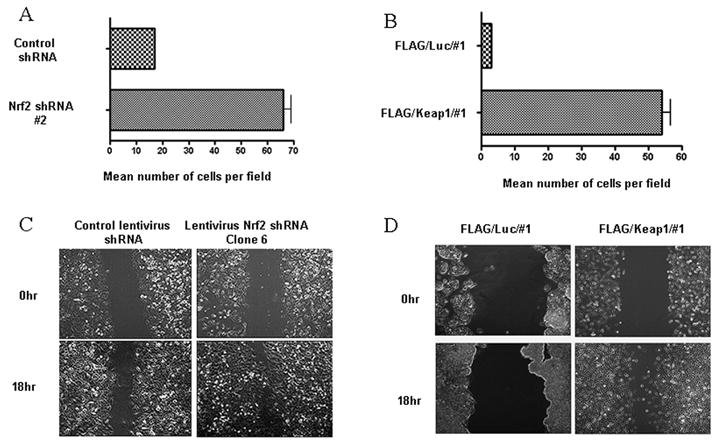
Loss of Nrf2 is accompanied by increased motility. A) & B) Transwell Migration Assay. The histogram illustrate the number of cells that penetrated the membrane 18 hrs after inoculation; C) & D) A scrape assay was used to assess migration. A) &C) A549 cells expressing Control or Nrf2 shRNA; B) & D) HepG2 cells stably expressing FLAG/luciferase or FLAG/Keap1.
SIS3 is a small molecule inhibitor of receptor-mediated phosphorylation of the Smad3 SSXS motif (Jinnin et al., 2006). SIS3 inhibits TGF-β receptor mediated association of phospho Smad3 with Smad4 and Smad-dependent transcriptional activation (Jinnin et al., 2006). SIS3 was found to inhibit phosphorylation of Smad2 and Smad3 in A549 cells (data not shown) and in Nrf2 shRNA expressing cells (Figure 4A). Use of SIS3 attenuated migration of cells stably expressing Nrf2 shRNA (Clone #2), as assessed using the 2-dimensional migration assay (Figure 4 B-F). In the same experiment cells stably expressing Nrf2 shRNA were exposed to 5 ng/ml TGF-β1 for 24 hrs in the absence or presence of SIS3. As a control for this experiment, cells stably expressing control shRNA were exposed to 5 ng/ml TGF-β1 for 24 hrs in the absence or presence of SIS3. Exposure to TGF-β1 enhanced cell migration, whereas SIS3 exposure attenuated TGF-β1-mediated migration (Supplemental Figure 7). These data support the conclusion that migration of Nrf2 shRNA expressing cells is Smad-dependent.
Figure 4.

Inhibition of Smad3 activity attenuates migration of A549 cells stably expressing Nrf2 shRNA. A) Nrf2 expressing shRNA cells were exposed to 10μM SIS3 for the indicated times. Cells were immunoprecipitated with antibody to pSmad2/3 and then immunoblotted with antibody to pSmad2/3; B-F Cells were subjected to the scrape assay. The assay was performed in the absence (Panel B, C, E) or presence of 10 μM SIS3 (Panels D & F). Migration was assessed 24 or 48 hrs after scraping.
We developed a novel methodology to interrogate cellular motility and plasticity. A microfabricated device, termed the μ-Taur, was designed to assay a cell's ability to migrate through narrow 3-dimensional channels of various dimensions. The device (see schematic in Supplemental Figure 8 and Supplemental Information) consists of a central channel into which cells are loaded. Three pairs of chambers, located to either side of the central loading channel contain arrays of posts that are manufactured to various dimensions. Figure 5A illustrates a central channel and two opposing chambers, and the inset shown in Figure 5B shows a higher magnification of the rows of posts and a single cell. The opposing chambers in Figure 5A have a series of posts with dimensions of 12 × 12 × 13μm (width × length × height). The inter-post gaps have the same dimensions as the posts. Each μ-Taur has two additional opposing chambers having interpost dimensions of 10 × 10 × 13 μm; and 8 × 8 × 13 μm (not shown). Thus, cells have to change from a cortical shape to an elongated shape in order to squeeze between two posts. Migration through the chambers was determined 16 hrs after loading. As shown in Figures 5A and B, A549 cells expressing control non-silencing shRNA could not progress through the first line of posts with interpost dimensions of either 12 × 12 × 13 μm or 8 × 8 × 13 μm. In contrast, cells expressing Nrf2 shRNA have migrated through 7 sets of 12 × 12 × 13 μm posts 16 hrs after loading (Figure 5C). Cell migration was slower when the interpost distances were 8 × 8 × 13 μm; the cells have migrated through only 3 sets of posts (Figure 5D). Please note that control cells loaded into the central loading channel were distributed throughout the loading channel and that some chambers had more cells in front of them than others. This is illustrated in the control samples by inspection of Figure 5A & 5B. These results lead to the hypothesis that repression of Nrf2 increases cellular plasticity.
Figure 5.

Morphological plasticity is increased in A549 cells stably expressing control or Nrf2 shRNA. A microfabricated μ-Taur device, was used to assay a cell's ability to migrate through narrow 3-diamensional channels of various dimensions 16 hrs after loading. A) Migration and plasticity of cells stably expressing control non-silencing shRNA in channels with 12 micron posts. Inset shows close ups of posts; B) Migration and plasticity of cells stably expressing control non-silencing shRNA in channels with 8 micron posts. Inset shows close ups of posts; C & D) Migration and plasticity of cells stably expressing Nrf2 shRNA in channels with 12 micron posts (C) or 8 micron posts (D).
We utilized a TGFβ1 ELISA assay (Biswas et al., 2007) to determine if the changes in motility were autocrine in origin. Cells stably expressing Nrf2 shRNA secreted slightly less TGF-β1 compared to control (p < 0.05. Student's t test; Supplemental Figure 9). Therefore, TGF-β1 autocrine effects could be ruled out.
Cell-Cell Adhesion
A migratory phenotype is often associated with loss of cell-cell adhesion (Yang and Weinberg, 2008). A549 cells expressing Nrf2 shRNA exhibited a loss of E-Cadherin expression, as determined by both immunofluorescence and immunoblotting (Figure 6A & B). The zinc finger protein Slug is one of several proteins that repress E-Cadherin expression (Peinado et al., 2007). Immunoblotting revealed that loss of Nrf2 was accompanied by increased expression of Slug (Figure 6C). We interpret these data to indicate that Slug expression in cells with repressed Nrf2 expression represses E-Cadherin.
Figure 6.

Loss of E-Cadherin is accompanied by increased expression of Slug. A) E-Cadherin immunofluorescence in control and Nrf2 shRNA containing A549 cells expressed from retrovirus; B) Immunoblotting for E-Cadherin in control and Nrf2 shRNA containing A549 cells expressed from retrovirus; C) Immunoblot of Slug expression in control and Nrf2 shRNA containing A549 cells expressed from retrovirus; D) Wild type A549 cells were transiently co-transfected with a luciferase reporter under the control of the human E-Cadherin promoter, a plasmid vector that expressed β-galactosidase, a vector that expressed human wild type Nrf2, or insertless pcDNA3.1. Luciferase/β-Gal activity was measured 72 hrs after transfection.
We found that transfection of a vector expressing human Nrf2 along with a luciferase reporter under the control of the human E-Cadherin promoter induced reporter activity (p < 0.05; Student's t test; Figure 6D). The E-Cadherin gene has not been reported to be regulated by an ARE in its promoter. Use of promoter analysis software did not reveal the presence of an ARE consensus sequence (data not shown). Thus, the data support the hypothesis that loss of Nrf2 impacts E-Cadherin expression indirectly.
Smad Phosphorylation and Transcriptional Responses
Thuault et al., (Thuault et al., 2006) have shown that Slug expression is positively regulated by HMGA2, which itself is induced by Smad2/3/4-dependent signaling. Smad-dependent signaling occurs in response to receptor-mediated phosphorylation of Smad2 and Smad3 at two serine residues at the C-terminus, with subsequent heterodimerization or heterotrimerization with Smad4 (Thuault et al., 2006). As shown in Figure 7A, nuclei isolated from cells expressing Nrf2 shRNA exhibit high levels of C-terminus Ser465/467 phospho-Smad2 and Ser423/425 phospho-Smad3 compared to control. Although the mechanism responsible for enhanced C-terminal phosphorylation of R-Smads has not been investigated in this study, one may postulate that the R-Smad dephosphorylation by cellular phosphatases may be inhibited as a consequence of elevated cellular ROS.
Figure 7.

Increased R-Smad phosphorylation and CAGA reporter activity in cells expressing Nrf2 shRNA. A) Immunoblot of phospho R-Smads in control and Nrf2 shRNA containing A549 cells expressed from retrovirus; B) Transient co-transfection of a luciferase reporter under control of a synthetic CAGA promoter and a reporter expressing Renilla luciferase into control and Nrf2 shRNA containing A549 cells expressed from retrovirus. Luciferase activity was measured 72 hrs after transfection; C) Immunoblot of PAI-1 expression in control and Nrf2 shRNA containing A549 cells expressed from retrovirus.
Phosphorylation of Ser245/250/255 in the linker region of Smad2 was also increased in Nrf2 shRNA containing cells. We interpret the presence of 2 immunoreactive bands in the pSer245/250/255 immunoblot to be a consequence of phosphorylation at both the linker region and C-terminus.
Matsuzaki et al., (Matsuzaki et al., 2009) have shown that simultaneous linker region and C-terminus phosphorylation correlate with increased malignant Smad signaling. As simultaneous linker region/C-terminus phosphorylation is not observed in non-transformed cells, we postulate that differential linker region/C-terminus phosphorylation may be the mechanism that distinguishes the differential results obtained in tumorigenic vs non-transformed cells. We found that transfection of a synthetic (CAGA) luciferase reporter (Mithani et al., 2004) into Nrf2 shRNA expressing cells yielded an activity that was 5 fold greater than that observed in control cells (Figure 7B, p < 0.001, Student's t test). The CAGA-LUC reporter activity reflects the presence of endogenous TGF-β present in the serum of the growth medium, as well as enhanced R-Smad phosphorylation. The type 1 plasminogen activator inhibitor (PAI-1) promoter contains a cis element responsive to a complex that includes phospho-Smad3 and Smad4 (Song et al., 1998). Therefore we interrogated the expression of endogenous PAI-1 in Nrf2 shRNA expressing cells. The immunoblot shown in Figure 7C indicates that PAI-1 expression is increased in Nrf2 repressed A549 cells.
Nrf2 belongs to the basic leucine zipper family of transcription factors (Moi et al., 1994) and has been shown to form heterodimers with c-Jun, JunB, and JunD, as well as ATF3 (Brown et al., 2008; Venugopal and Jaiswal, 1998). It has been shown that Smad3 binds to ATF3 (Kang et al., 2003) as well as the Jun family (Liberati et al., 1999). The MH1 domain of Smad3 binds directly to the leucine zipper domain of the bZip proteins ATF2 and c-Jun (Grinberg and Kerppola, 2003). Therefore, one may hypothesize that Smad3 may reside in a complex with the bZip leucine zipper protein Nrf2. Immunoprecipitation of endogenous Smad3 from A549 nuclear lysates followed by immunoblotting demonstrated the presence of Smad4 and Nrf2 in the immunoprecipitate (Figure 8A). FLAG/Smad3 and Nrf2 were either co-transfected or transfected alone into NMuMG cells. Co-transfection followed by immunoprecipitation of FLAG/Smad3 and immunoblotting for Nrf2 confirmed the association of Smad3 with Nrf2 (Figure 8B). We also interrogated H460 cells. Smad3 immunoprecipitation followed by immunoblotting of H460 nuclear lysate demonstrated the presence of Nrf2 in this complex.
Figure 8.

Nrf2 is found in a complex with Smad3 and Smad4 and suppresses the activity of a luciferase reporter under control of a synthetic CAGA promoter. A) Immunoprecipitation of endogenous Smad3 from A549 cell lysates followed by immunoblotting for Smad4 and Nrf2. B) FLAG/Smad3 and Nrf2 were either co-transfected or transfected alone into NMuMG cells. FLAG/Smad3 was then immunoprecipitated and immunoblotted for Nrf2. Endogenous Smad3 was immunoprecipitated from H460 cells and then immunoblotted for Nrf2; C) A549 cells were treated with 2 ng/ml TGF beta and solubilized. Cell protein was subjected to GST or GST/Nrf2 pulldown and immunoblotted to pSmad2/3 antibody; D) Wild type A549 cells were transiently co-transfected with a luciferase reporter under control of a synthetic CAGA reporter, a vector expressing β-galactosidase, vectors expressing Smad3, wild type human Nrf2, dominant negative Nrf2, or insertless pcDNA3.1. Reporter activity was determined 72 hrs after transfection.
A549 cells were treated with 2 ng/ml of TGF-β, washed and solubilized in cell lysis buffer (please see Supplemental Methods). GST or GST/Nrf2, immobilized on GSH-agarose beads (Brown et al., 2008), was used to capture soluble protein. Captured protein was immunoblotted using antibody to C-terminus phospho-Smad2/3 (Figure 8C). We observed immunoreactive protein of approximately 55 kDa and 60 kDA (arrows Figure 8C). We interpret these phospho proteins to be pSmad3 and pSmad2 respectively. We also observed an immunoreactive phospho protein that migrated just above pSmad2, which we interpret to be Smad2 phosphorylated in both the linker region and the C-terminus.
The observation that Smad3, Smad4, and Nrf2 resided in a complex prompted us to determine if Nrf2 impacted Smad-mediated transcription. A549 cells were co-transfected with vectors expressing a synthetic (CAGA) luciferase reporter, Renilla luciferase reporter, Smad3 and wild type or dominant negative Nrf2 (Yu et al., 2000) (Figure 8D). CAGA-LUC reporter activity (Mithani et al., 2004) was inhibited 70% when wildtype but not dominant negative Nrf2 was ectopically overexpressed (p = 0.001, Student's t test). Transfection of Smad3 increased CAGA-LUC reporter activity 2.6 fold (p = 0.002, Student's t test). However, co-transfection of wildtype Nrf2 but not dominant negative Nrf2 (amino acids 399-589(Yu et al., 2000)) inhibited Smad3-mediated transcription (p = 0.004, Student's t test). These data show that Nrf2 negatively impacts transcription mediated by R-Smad/Smad4.
Dominant negative Nrf2, which contains the CNC homology region and the DNA binding leucine zipper motif but is missing the Neh4 and Neh5 transactivation domains, was not able to function antagonistically. The Neh4 and Neh5 domains of Nrf2 recruit p300/CBP (Katoh et al., 2001). In the nucleus the Smad3/Smad4 oligomers also recruit the coactivators p300/CBP. This interaction results in the acetylation of Lys 378 of Smad3 and promotion of transcriptional activity (Inoue et al., 2007). A competition between DNA-bound Smad3 complexes and Nrf2/Smad complexes for CBP/p300 is one possibility that may contribute to the results presented herein. Another possibility relates to the work of Verrecchia et al. (Verrecchia et al., 2001). They have shown that Smad3/Jun proteins form immunoprecipitable ‘off-DNA’ complexes that down regulate gene transcription driven by Smad binding sequences. Testing of these hypotheses is beyond the scope of the current investigation.
In summary, the data shown herein demonstrate that loss of Nrf2 increases cellular motility of tumor cells. TGF-β signaling represents one important regulator of cell motility and as discussed in Safina et al., (Safina et al., 2009) involves R-Smads, Smad4, and PI3 Kinase, as well as p38. Because Valcourt et al., have shown that TGF-β/R-Smads/Co-Smad signaling is intimately involved in cellular processes that enhance tumor cell motility, we focused on this pathway. Supplemental Figure 10 illustrates the signaling pathways impacted by loss of Nrf2.
Our data support a new and novel dimension to the Nrf2 paradigm. We have used immunoprecipitation assays to show that Nrf2 resides in a complex with Smad3 and Smad4. Ectopic expression of wild type Nrf2, but not dominant negative Nrf2, dampened the activity of a synthetic CAGA-directed luciferase reporter. Conversely, shRNA knock down of Nrf2 in tumor cells enhanced the activity of the synthetic CAGA-directed reporter, as well as enhanced expression of the endogenous Smad target genes PAI-1 and Slug, while suppressing E-Cadherin luciferase reporter activity, compared to non-silencing control shRNA. Consistent with these observations, transient ectopic expression of Nrf2 enhanced activity of an E-Cadherin reporter. Complementing these observations, we found that E-Cadherin expression was suppressed in tumor cells expressing Nrf2 shRNA. Functional assays revealed that RNAi-mediated knock down of Nrf2 in cell culture was accompanied by increased tumor cell motility. Thus, in the context of oncogenesis, these data lead to the hypothesis that suppression of Nrf2 expression in tumor cells can increase cell plasticity and motility, due in part to malignant Smad signaling.
Materials and Methods
Cells, Plasmids, and Antibodies
HepG2 cells, characterized by dominant-acting N-Ras (Huber and Thorgeirsson, 1987), were maintained in DMEM. A549 cells, characterized by an activating K-Ras mutation (Valenzuela and Groffen, 1986), were maintained in RPMI 1640 medium. SW480 cells express activating K-Ras were maintained as described (Shiou et al., 2007). The cDNA KIAA0132 (human Keap1) was a gift from Dr. Takahiro Nagase and was subcloned into pCMV-Tag (Stratagene). The vector pCMV-Tag places a FLAG epitope NH2 terminus to Keap1. pCMV-Tag/luciferase was obtained from Stratagene. pcDNA/V5mNrf2 was a graciousgift from Dr. M. McMahon. The CAGA-LUC reporter was used as described in (Mithani et al., 2004). GST and GST/Nrf2 are described in (Brown et al., 2008). The NFE2L2 (Nrf2) pSM2 Retroviral shRNAmir clone V2HS_64255 (Sense sequence CCATTGATGTTTCTGATCT) and the target set Nrf2 pGIPZ lentiviral shRNAmir (Sense sequences GCTGCTCAGAATTGCAGAA; CTGAGTTACAGTGTCTTAA; CTCCTACTGTGATGTGAAA; CAGTTGACAGTGAACTCAT), and corresponding non-silencing controls were obtained from Open Biosystems. pSilencer (Applied Biosystems) was used to express Nrf2 siRNA (sense sequence 5′GATCCGTAAGAAGCCAGATGTTAATTCAAGAGACATTCTTCGGTCTACAATTTTTT TTGGAAA-3′) or appropriate controls as described in (Singh et al., 2008).
Antibodies were obtained as follows: Nrf2 (Santa Cruz: Cat# sc-13032; Abcam: Cat #ab62352), GCLC (Neomarkers: Cat # P1697), β-actin (Sigma: Cat #A5441). The following antibodies were obtained from Cell Signaling: pAKT (Cat #9275S), AKT (Cat #9272), E-Cadherin (Cat #610404), pSmad2 (ser245 Cat #3104S, Ser 465 Cat #3101S), Smad2 (Cat #3103), pSmad3 (Ser 423 Cat #9520S), Smad3 (Cat #9523S), Smad 4 (Cat #9515), cyclin D (Cat #2926), and Lamin A/C (Cat #2032). The following antibodies were obtained from Santa Cruz: PAI-1 (#sc8979), SLUG (#sc-15391), c Jun ((#822), Keap1 (#sc15246), pSmad2/3 (Ser423/425) (#sc-11769) and GAPDH (#sc32233).
μTaur device
The device is made through standard photolithographic techniques with two layer deposition of SU8-2010 photo resist (Whitesides et al., 2001). Cells were loaded into the device and the device placed into in an incubator for approximately 3 hrs. The device was then placed in an Axiovert 200M Zeiss microscope which maintained a temperature of 37°C. For detailed information please consult Supplemental Information.
The information concerning the methods for other experiments is provided in Supplemental Information.
Supplementary Material
Acknowledgments
This research was supported in part by NIH Grants RO1CA104590, RO1CA115556, RO1CA069457, R01DK052334, RO1CA095195, RO1CA113519, P50CA090949 (Project# 4), U01AI061223, the Vanderbilt-Ingram Cancer Center grant P30CA68485, and the Vanderbilt Institute for Integrative Biosystems Research and Education.
Footnotes
There are no conflicts of interest.
References
- Barve A, Khor TO, Nair S, Reuhl K, Suh N, Reddy B, et al. Gamma-tocopherol-enriched mixed tocopherol diet inhibits prostate carcinogenesis in TRAMP mice. Int J Cancer. 2009;124:1693–9. doi: 10.1002/ijc.24106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beer DG, Kardia SL, Huang CC, Giordano TJ, Levin AM, Misek DE, et al. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med. 2002;8:816–24. doi: 10.1038/nm733. [DOI] [PubMed] [Google Scholar]
- Biswas S, Guix M, Rinehart C, Dugger TC, Chytil A, Moses HL, et al. Inhibition of TGF-beta with neutralizing antibodies prevents radiation-induced acceleration of metastatic cancer progression. J Clin Invest. 2007;117:1305–13. doi: 10.1172/JCI30740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SL, Sekhar KR, Rachakonda G, Sasi S, Freeman ML. Activating transcription factor 3 is a novel repressor of the nuclear factor erythroid-derived 2-related factor 2 (Nrf2)-regulated stress pathway. Cancer Res. 2008;68:364–8. doi: 10.1158/0008-5472.CAN-07-2170. [DOI] [PubMed] [Google Scholar]
- Calonge MJ, Massague J. Smad4/DPC4 silencing and hyperactive Ras jointly disrupt transforming growth factor-beta antiproliferative responses in colon cancer cells. J Biol Chem. 1999;274:33637–43. doi: 10.1074/jbc.274.47.33637. [DOI] [PubMed] [Google Scholar]
- Cao J, Schulte J, Knight A, Leslie NR, Zagozdzon A, Bronson R, et al. Prdx1 inhibits tumorigenesis via regulating PTEN/AKT activity. Embo J. 2009;28:1505–17. doi: 10.1038/emboj.2009.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty AK, Ueda M, Mishima Y, Ichihashi M. Intracellular glutathione and its metabolizing enzyme activities in a metastatic variant melanoma cell line. Melanoma Res. 1992;2:315–9. doi: 10.1097/00008390-199212000-00004. [DOI] [PubMed] [Google Scholar]
- Chan K, Lu R, Chang JC, Kan YW. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc Natl Acad Sci U S A. 1996;93:13943–8. doi: 10.1073/pnas.93.24.13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Cheung ST, So S, Fan ST, Barry C, Higgins J, et al. Gene expression patterns in human liver cancers. Mol Biol Cell. 2002;13:1929–39. doi: 10.1091/mbc.02-02-0023.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CH, Parker JS, Karaca G, Wu J, Funkhouser WK, Moore D, et al. Molecular classification of head and neck squamous cell carcinomas using patterns of gene expression. Cancer Cell. 2004;5:489–500. doi: 10.1016/s1535-6108(04)00112-6. [DOI] [PubMed] [Google Scholar]
- Coulouarn C, Factor VM, Thorgeirsson SS. Transforming growth factor-beta gene expression signature in mouse hepatocytes predicts clinical outcome in human cancer. Hepatology. 2008;47:2059–67. doi: 10.1002/hep.22283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohlich DA, McCabe MT, Arnold RS, Day ML. The role of Nrf2 in increased reactive oxygen species and DNA damage in prostate tumorigenesis. Oncogene. 2008;27:4353–62. doi: 10.1038/onc.2008.79. [DOI] [PubMed] [Google Scholar]
- Greenburg G, Hay ED. Epithelia suspended in collagen gels can lose polarity and express characteristics of migrating mesenchymal cells. J Cell Biol. 1982;95:333–9. doi: 10.1083/jcb.95.1.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinberg AV, Kerppola T. Both Max and TFE3 cooperate with Smad proteins to bind the plasminogen activator inhibitor-1 promoter, but they have opposite effects on transcriptional activity. J Biol Chem. 2003;278:11227–36. doi: 10.1074/jbc.M211734200. [DOI] [PubMed] [Google Scholar]
- Hayashi A, Suzuki H, Itoh K, Yamamoto M, Sugiyama Y. Transcription factor Nrf2 is required for the constitutive and inducible expression of multidrug resistance-associated protein 1 in mouse embryo fibroblasts. Biochem Biophys Res Commun. 2003;310:824–9. doi: 10.1016/j.bbrc.2003.09.086. [DOI] [PubMed] [Google Scholar]
- Hendrix ND, Wu R, Kuick R, Schwartz DR, Fearon ER, Cho KR. Fibroblast growth factor 9 has oncogenic activity and is a downstream target of Wnt signaling in ovarian endometrioid adenocarcinomas. Cancer Res. 2006;66:1354–62. doi: 10.1158/0008-5472.CAN-05-3694. [DOI] [PubMed] [Google Scholar]
- Hirayama A, Yoh K, Nagase S, Ueda A, Itoh K, Morito N, et al. EPR imaging of reducing activity in Nrf2 transcriptional factor-deficient mice. Free Radic Biol Med. 2003;34:1236–42. doi: 10.1016/s0891-5849(03)00073-x. [DOI] [PubMed] [Google Scholar]
- Homma S, Ishii Y, Morishima Y, Yamadori T, Matsuno Y, Haraguchi N, et al. Nrf2 Enhances Cell Proliferation and Resistance to Anticancer Drugs in Human Lung Cancer. Clin Cancer Res. 2009 doi: 10.1158/1078-0432.CCR-08-2822. [DOI] [PubMed] [Google Scholar]
- Hong F, Sekhar KR, Freeman ML, Liebler DC. Specific patterns of electrophile adduction trigger Keap1 ubiquitination and Nrf2 activation. J Biol Chem. 2005;280:31768–75. doi: 10.1074/jbc.M503346200. [DOI] [PubMed] [Google Scholar]
- Huber BE, Thorgeirsson SS. Analysis of c-myc expression in a human hepatoma cell line. Cancer Res. 1987;47:3414–20. [PubMed] [Google Scholar]
- Inoue Y, Itoh Y, Abe K, Okamoto T, Daitoku H, Fukamizu A, et al. Smad3 is acetylated by p300/CBP to regulate its transactivation activity. Oncogene. 2007;26:500–8. doi: 10.1038/sj.onc.1209826. [DOI] [PubMed] [Google Scholar]
- Jinnin M, Ihn H, Tamaki K. Characterization of SIS3, a novel specific inhibitor of Smad3, and its effect on transforming growth factor-beta1-induced extracellular matrix expression. Mol Pharmacol. 2006;69:597–607. doi: 10.1124/mol.105.017483. [DOI] [PubMed] [Google Scholar]
- Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120:649–61. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- Kang Y, Chen CR, Massague J. A self-enabling TGFbeta response coupled to stress signaling: Smad engages stress response factor ATF3 for Id1 repression in epithelial cells. Mol Cell. 2003;11:915–26. doi: 10.1016/s1097-2765(03)00109-6. [DOI] [PubMed] [Google Scholar]
- Katoh Y, Itoh K, Yoshida E, Miyagishi M, Fukamizu A, Yamamoto M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells. 2001;6:857–68. doi: 10.1046/j.1365-2443.2001.00469.x. [DOI] [PubMed] [Google Scholar]
- Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24:7130–9. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- Kusano Y, Horie S, Shibata T, Satsu H, Shimizu M, Hitomi E, et al. Keap1 regulates the constitutive expression of GST A1 during differentiation of Caco-2 cells. Biochemistry. 2008;47:6169–77. doi: 10.1021/bi800199z. [DOI] [PubMed] [Google Scholar]
- Kwak MK, Itoh K, Yamamoto M, Sutter TR, Kensler TW. Role of transcription factor Nrf2 in the induction of hepatic phase 2 and antioxidative enzymes in vivo by the cancer chemoprotective agent, 3H-1, 2-dimethiole-3-thione. Mol Med. 2001;7:135–45. [PMC free article] [PubMed] [Google Scholar]
- Lee DF, Kuo HP, Liu M, Chou CK, Xia W, Du Y, et al. KEAP1 E3 ligase-mediated downregulation of NF-kappaB signaling by targeting IKKbeta. Mol Cell. 2009;36:131–40. doi: 10.1016/j.molcel.2009.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberati NT, Datto MB, Frederick JP, Shen X, Wong C, Rougier-Chapman EM, et al. Smads bind directly to the Jun family of AP-1 transcription factors. Proc Natl Acad Sci U S A. 1999;96:4844–9. doi: 10.1073/pnas.96.9.4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Hinkhouse MM, Sun W, Weydert CJ, Ritchie JM, Oberley LW, et al. Redox regulation of pancreatic cancer cell growth: role of glutathione peroxidase in the suppression of the malignant phenotype. Hum Gene Ther. 2004;15:239–50. doi: 10.1089/104303404322886093. [DOI] [PubMed] [Google Scholar]
- Lo SC, Hannink M. PGAM5, a Bcl-XL-interacting protein, is a novel substrate for the redox-regulated Keap1-dependent ubiquitin ligase complex. J Biol Chem. 2006;281:37893–903. doi: 10.1074/jbc.M606539200. [DOI] [PubMed] [Google Scholar]
- Loignon M, Miao W, Hu L, Bier A, Bismar TA, Scrivens PJ, et al. Cul3 overexpression depletes Nrf2 in breast cancer and is associated with sensitivity to carcinogens, to oxidative stress, and to chemotherapy. Mol Cancer Ther. 2009 doi: 10.1158/1535-7163.MCT-08-1186. [DOI] [PubMed] [Google Scholar]
- Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–37. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mates JM, Segura JA, Alonso FJ, Marquez J. Intracellular redox status and oxidative stress: implications for cell proliferation, apoptosis, and carcinogenesis. Arch Toxicol. 2008;82:273–99. doi: 10.1007/s00204-008-0304-z. [DOI] [PubMed] [Google Scholar]
- Matsuzaki K, Kitano C, Murata M, Sekimoto G, Yoshida K, Uemura Y, et al. Smad2 and Smad3 phosphorylated at both linker and COOH-terminal regions transmit malignant TGF-beta signal in later stages of human colorectal cancer. Cancer Res. 2009;69:5321–30. doi: 10.1158/0008-5472.CAN-08-4203. [DOI] [PubMed] [Google Scholar]
- Matsuzawa A, Ichijo H. Redox control of cell fate by MAP kinase: physiological roles of ASK1-MAP kinase pathway in stress signaling. Biochim Biophys Acta. 2008;1780:1325–36. doi: 10.1016/j.bbagen.2007.12.011. [DOI] [PubMed] [Google Scholar]
- Mithani SK, Balch GC, Shiou SR, Whitehead RH, Datta PK, Beauchamp RD. Smad3 has a critical role in TGF-beta-mediated growth inhibition and apoptosis in colonic epithelial cells. J Surg Res. 2004;117:296–305. doi: 10.1016/S0022-4804(03)00335-4. [DOI] [PubMed] [Google Scholar]
- Moi P, Chan K, Asunis I, Cao A, Kan YW. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc Natl Acad Sci U S A. 1994;91:9926–30. doi: 10.1073/pnas.91.21.9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller N, Reinacher-Schick A, Baldus S, van Hengel J, Berx G, Baar A, et al. Smad4 induces the tumor suppressor E-cadherin and P-cadherin in colon carcinoma cells. Oncogene. 2002;21:6049–58. doi: 10.1038/sj.onc.1205766. [DOI] [PubMed] [Google Scholar]
- Nioi P, Nguyen T. A mutation of Keap1 found in breast cancer impairs its ability to repress Nrf2 activity. Biochem Biophys Res Commun. 2007;362:816–21. doi: 10.1016/j.bbrc.2007.08.051. [DOI] [PubMed] [Google Scholar]
- Nogueira V, Park Y, Chen CC, Xu PZ, Chen ML, Tonic I, et al. Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell. 2008;14:458–70. doi: 10.1016/j.ccr.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta T, Iijima K, Miyamoto M, Nakahara I, Tanaka H, Ohtsuji M, et al. Loss of Keap1 function activates Nrf2 and provides advantages for lung cancer cell growth. Cancer Res. 2008;68:1303–9. doi: 10.1158/0008-5472.CAN-07-5003. [DOI] [PubMed] [Google Scholar]
- Padmanabhan B, Tong KI, Ohta T, Nakamura Y, Scharlock M, Ohtsuji M, et al. Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol Cell. 2006;21:689–700. doi: 10.1016/j.molcel.2006.01.013. [DOI] [PubMed] [Google Scholar]
- Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7:415–28. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- Radisky DC, Levy DD, Littlepage LE, Liu H, Nelson CM, Fata JE, et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature. 2005;436:123–7. doi: 10.1038/nature03688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Gomez M, Kwak MK, Dolan PM, Itoh K, Yamamoto M, Talalay P, et al. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc Natl Acad Sci U S A. 2001;98:3410–5. doi: 10.1073/pnas.051618798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safina AF, Varga AE, Bianchi A, Zheng Q, Kunnev D, Liang P, et al. Ras alters epithelial-mesenchymal transition in response to TGFbeta by reducing actin fibers and cell-matrix adhesion. Cell Cycle. 2009;8:284–98. doi: 10.4161/cc.8.2.7590. [DOI] [PubMed] [Google Scholar]
- Sarsour EH, Venkataraman S, Kalen AL, Oberley LW, Goswami PC. Manganese superoxide dismutase activity regulates transitions between quiescent and proliferative growth. Aging Cell. 2008;7:405–17. doi: 10.1111/j.1474-9726.2008.00384.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekhar KR, Crooks PA, Sonar VN, Friedman DB, Chan JY, Meredith MJ, et al. NADPH oxidase activity is essential for Keap1/Nrf2-mediated induction of GCLC in response to 2-indol-3-yl-methylenequinuclidin-3-ols. Cancer Res. 2003;63:5636–45. [PubMed] [Google Scholar]
- Shibata T, Ohta T, Tong KI, Kokubu A, Odogawa R, Tsuta K, et al. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc Natl Acad Sci U S A. 2008;105:13568–73. doi: 10.1073/pnas.0806268105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiou SR, Singh AB, Moorthy K, Datta PK, Washington MK, Beauchamp RD, et al. Smad4 regulates claudin-1 expression in a transforming growth factor-beta-independent manner in colon cancer cells. Cancer Res. 2007;67:1571–9. doi: 10.1158/0008-5472.CAN-06-1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Boldin-Adamsky S, Thimmulappa RK, Rath SK, Ashush H, Coulter J, et al. RNAi-mediated silencing of nuclear factor erythroid-2-related factor 2 gene expression in non-small cell lung cancer inhibits tumor growth and increases efficacy of chemotherapy. Cancer Res. 2008;68:7975–84. doi: 10.1158/0008-5472.CAN-08-1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Misra V, Thimmulappa RK, Lee H, Ames S, Hoque MO, et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006;3:e420. doi: 10.1371/journal.pmed.0030420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song CZ, Siok TE, Gelehrter TD. Smad4/DPC4 and Smad3 mediate transforming growth factor-beta (TGF-beta) signaling through direct binding to a novel TGF-beta-responsive element in the human plasminogen activator inhibitor-1 promoter. J Biol Chem. 1998;273:29287–90. doi: 10.1074/jbc.273.45.29287. [DOI] [PubMed] [Google Scholar]
- Stacy DR, Ely K, Massion PP, Yarbrough WG, Hallahan DE, Sekhar KR, et al. Increased expression of nuclear factor E2 p45-related factor 2 (NRF2) in head and neck squamous cell carcinomas. Head Neck. 2006;28:813–8. doi: 10.1002/hed.20430. [DOI] [PubMed] [Google Scholar]
- Strachan GD, Ostrow LA, Jordan-Sciutto KL. Expression of the fetal Alz-50 clone 1 protein induces apoptotic cell death. Biochem Biophys Res Commun. 2005;336:490–5. doi: 10.1016/j.bbrc.2005.08.127. [DOI] [PubMed] [Google Scholar]
- Sun Y, Oberley LW, Elwell JH, Sierra-Rivera E. Antioxidant enzyme activities in normal and transformed mouse liver cells. Int J Cancer. 1989;44:1028–33. doi: 10.1002/ijc.2910440615. [DOI] [PubMed] [Google Scholar]
- Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991;51:794–8. [PubMed] [Google Scholar]
- Thuault S, Valcourt U, Petersen M, Manfioletti G, Heldin CH, Moustakas A. Transforming growth factor-beta employs HMGA2 to elicit epithelial-mesenchymal transition. J Cell Biol. 2006;174:175–83. doi: 10.1083/jcb.200512110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonks NK. Redox redux: revisiting PTPs and the control of cell signaling. Cell. 2005;121:667–70. doi: 10.1016/j.cell.2005.05.016. [DOI] [PubMed] [Google Scholar]
- Valenzuela DM, Groffen J. Four human carcinoma cell lines with novel mutations in position 12 of c-K-ras oncogene. Nucleic Acids Res. 1986;14:843–52. doi: 10.1093/nar/14.2.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venugopal R, Jaiswal AK. Nrf2 and Nrf1 in association with Jun proteins regulate antioxidant response element-mediated expression and coordinated induction of genes encoding detoxifying enzymes. Oncogene. 1998;17:3145–56. doi: 10.1038/sj.onc.1202237. [DOI] [PubMed] [Google Scholar]
- Verrecchia F, Vindevoghel L, Lechleider RJ, Uitto J, Roberts AB, Mauviel A. Smad3/AP-1 interactions control transcriptional responses to TGF-beta in a promoter-specific manner. Oncogene. 2001;20:3332–40. doi: 10.1038/sj.onc.1204448. [DOI] [PubMed] [Google Scholar]
- Wang X, McCullough KD, Franke TF, Holbrook NJ. Epidermal growth factor receptor-dependent Akt activation by oxidative stress enhances cell survival. J Biol Chem. 2000;275:14624–31. doi: 10.1074/jbc.275.19.14624. [DOI] [PubMed] [Google Scholar]
- Welsh JB, Zarrinkar PP, Sapinoso LM, Kern SG, Behling CA, Monk BJ, et al. Analysis of gene expression profiles in normal and neoplastic ovarian tissue samples identifies candidate molecular markers of epithelial ovarian cancer. Proc Natl Acad Sci U S A. 2001;98:1176–81. doi: 10.1073/pnas.98.3.1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weydert CJ, Waugh TA, Ritchie JM, Iyer KS, Smith JL, Li L, et al. Overexpression of manganese or copper-zinc superoxide dismutase inhibits breast cancer growth. Free Radic Biol Med. 2006;41:226–37. doi: 10.1016/j.freeradbiomed.2006.03.015. [DOI] [PubMed] [Google Scholar]
- Whitesides GM, Ostuni E, Takayama S, Jiang X, Ingber DE. Soft lithography in biology and biochemistry. Annu Rev Biomed Eng. 2001;3:335–73. doi: 10.1146/annurev.bioeng.3.1.335. [DOI] [PubMed] [Google Scholar]
- Wurmbach E, Chen YB, Khitrov G, Zhang W, Roayaie S, Schwartz M, et al. Genome-wide molecular profiles of HCV-induced dysplasia and hepatocellular carcinoma. Hepatology. 2007;45:938–47. doi: 10.1002/hep.21622. [DOI] [PubMed] [Google Scholar]
- Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14:818–29. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- Yano T, Hernandez-Blazquez FJ, Omori Y, Yamasaki H. Reduction of malignant phenotype of HEPG2 cell is associated with the expression of connexin 26 but not connexin 32. Carcinogenesis. 2001;22:1593–600. doi: 10.1093/carcin/22.10.1593. [DOI] [PubMed] [Google Scholar]
- Ye QH, Qin LX, Forgues M, He P, Kim JW, Peng AC, et al. Predicting hepatitis B virus-positive metastatic hepatocellular carcinomas using gene expression profiling and supervised machine learning. Nat Med. 2003;9:416–23. doi: 10.1038/nm843. [DOI] [PubMed] [Google Scholar]
- Yu R, Chen C, Mo YY, Hebbar V, Owuor ED, Tan TH, et al. Activation of mitogen-activated protein kinase pathways induces antioxidant response element-mediated gene expression via a Nrf2-dependent mechanism. J Biol Chem. 2000;275:39907–13. doi: 10.1074/jbc.M004037200. [DOI] [PubMed] [Google Scholar]
- Yu S, Khor TO, Cheung KL, Li W, Wu TY, Huang Y, et al. Nrf2 expression is regulated by epigenetic mechanisms in prostate cancer of TRAMP mice. PLoS One. 2010;5:e8579. doi: 10.1371/journal.pone.0008579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavadil J, Bottinger EP. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene. 2005;24:5764–74. doi: 10.1038/sj.onc.1208927. [DOI] [PubMed] [Google Scholar]
- Zhang DD, Lo SC, Cross JV, Templeton DJ, Hannink M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol. 2004;24:10941–53. doi: 10.1128/MCB.24.24.10941-10953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zhao W, Zhang HJ, Domann FE, Oberley LW. Overexpression of copper zinc superoxide dismutase suppresses human glioma cell growth. Cancer Res. 2002;62:1205–12. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.