

Summary
Background
Plasmodium falciparum malaria infects 300–500 million people every year causing 1–2 million deaths annually. Evidence of a coagulation disorder, activation of endothelial cells (EC) and increase in inflammatory cytokines are often present in malaria.
Objectives
We have asked whether parasitized red blood cells (pRBC) interaction with EC induces Tissue Factor expression in vitro and in vivo. The potential of phosphatidylserine-containing pRBC to support the assembly of blood coagulation complexes was also investigated.
Results
We demonstrate that mature forms of pRBC induce functional expression of tissue factor (TF) by endothelial cells (EC) in vitro with productive assembly of the extrinsic Xnase complex and initiation of the coagulation cascade. Late stage pRBC also support the prothrombinase and intrinsic Xnase complex formation in vitro, and may function as activated platelets in the amplification phase of the blood coagulation. Notably, postmortem brain sections obtained from P. falciparum-infected children who died from Cerebral Malaria and other causes display a consistent staining for TF in the EC.
Conclusions
These findings place TF expression by endothelium and the amplification of the coagulation cascade by pRBC and/or activated platelets as potentially critical steps in the pathogenesis of malaria. Furthermore, it may allow investigators to test other therapeutic alternatives targeting TF or modulators of EC function in the treatment of malaria and/or its complications.
Keywords: endothelial cell, malaria, prothrombinase, platelets, Plasmodium falciparum, Tissue Factor
P. falciparum malaria manifests as a spectrum ranging from asymptomatic infection through mild, severe, and fatal disease. Uncomplicated malaria is an acute febrile illness characterized by fever, chills, and headache [1,2]. In some patients, particularly non-immune individuals, malaria infection can become complicated by severe anemia and cerebral malaria (CM)[1,2]. Clinically, the term "cerebral malaria" is used to describe a syndrome consisting of decreased consciousness not attributable to other causes in a patient with P. falciparum parasitemia [2,3]. These comatose patients often present with metabolic acidosis, hypoglycemia, and anemia. Recovery can be rapid and is usually complete, with a low incidence of long-term neurologic sequelae [2,3]. Although various hypotheses have been proposed, the mechanisms of CM pathogenesis remain incompletely understood [1,3]. Post mortem studies in children [4] have shown that this disease is a heterogeneous syndrome in which the sequestration of parasitized red blood cells (pRBC) in cerebral microvessels is a consistent finding [4–6]. Sequestration is mediated by various parasite-derived molecules located on the erythrocyte membrane. These molecules, collectively referred to as PfEMP-1 [7], interact in a synergistic manner with various endothelial cell surface receptors such as CD36 [8] and ICAM-1 [9]. Expression of these host receptors appears to be modulated, at least in part, by tumor necrosis factor alpha (TNF-α) a cytokine which concentration is increased in the plasma of malaria patients and that may play a role in the pathogenesis of the disease [10].
Sequestration has also been associated with localized and widespread activation of endothelial cells in mild and fatal cases of malaria [4–6]. A procoagulant state has been identified in these same patient populations, characterized by thrombocytopenia [11], hemostatic alterations [12], and microparticle production [13]. Hemostatic derangements may be contributing to the disease progression and organ failure observed in severe disease [12]. A systemic response characterized by increased levels of circulating cytokines and soluble adhesion molecules has also been observed in malaria in general and in cerebral malarial in particular [10].
No link has yet been described between sequestration, a coagulation disorder, endothelial cell activation, and the production of inflammatory cytokines, all of which have been separately described as features of severe malaria pathology. We hypothesized that parasitized red blood cells (pRBC) activate endothelial cells (EC) to express tissue factor (TF), a 47-kD protein that initiates the clotting cascade [14,15] and is increasingly recognized at the interface of blood coagulation and inflammation [16,17]. TF binds to coagulation FVIIa leading to FXa and FIXa formation with subsequent thrombin generation [18,19]. In this paper we describe three novel aspects related to malaria pathogenesis: i) interaction of pRBC with cultured microvascular endothelial cells (MVEC) in vitro is accompanied by TF expression by EC, ii) parasitized RBC supports the assembly of the multimolecular coagulation complex in vitro, leading to amplification of the coagulation cascade, and iii) we demonstrated the expression of TF in cerebral vascular endothelial cells of patients dying of cerebral malaria and patients with parasitemia who died of non-malarial causes. TF expression was not found in cerebral vascular tissue of subjects dying without malaria, suggesting that TF and subsequent hemostatic derangements may be contributing to malaria pathogenesis.
Materials and Methods
Culture of human dermal MVEC
MVEC were purchased from Clonetics (San Diego, CA) and grown in the presence of EBM-2 Plus as described [20] in detail in the Supplemental data.
Culture of P. falciparum parasites
Mycoplasma-free parasites (3D7) were thawed and initially grown in a 5% suspension of purified human O+ RBC in RPMI 1640 medium as described in detail in the Supplemental data.
TF expression by pRBC
Assembly of extrinsic Xnase in MVEC was carried out using a discontinuous assay as described in detail in the Supplemental data.
ELISA for TF
Human TF antigen was quantitated in triplicates with an enzyme-linked immunosorbent assay (ELISA) using Imubind Tissue Factor ELISA kit (American Diagnostica) as described in detail in the Supplemental data.
Reverse Transcriptase-PCR
Total RNA was isolated from MVEC cultures incubated with pRBC using TRIzol. RT-PCR was performed as described in detail in the Supplemental data.
Multimolecular coagulation assembly
Prothrombinase and intrinsic Xnase assemblies by pRBC using a discontinuous assay [21] as described in detail in the Supplemental data.
Pathology and IHC for TF
All patients included in this analysis met the standard clinical case definition of cerebral malaria during life [4]. The brain sections were fixed in buffered 10% neutral buffered formalin and embedded in paraffin casts before histologic sections were prepared. IHC was performed using the labeled polymer method as described in detail the Supplemental data.
Statistical analysis
Experiments were performed three or four times in triplicates or quadruplicates, and results are expressed as means ± SEM. Statistical significance was determined using Student`s t test or ANOVA (Bonferroni posttest comparison) using GraphPad Prism 3.0 software (GraphPad Software., Inc., San Diego, CA). Significance was set at p≤0.05.
Results
pRBC induce TF expression and extrinsic Xnase complex assembly by MVEC in vitro
To identify whether the interaction between mature pRBC (late-trophozoites and schizonts) and MVEC could induce TF expression, MVEC monolayers were incubated with 2% hematocrit of uninfected RBC or late-stage pRBC (0–50% parasitemia). FXa generation was measured as an estimate of TF expression. Figure 1A shows that FXa generation increased in a time- and parasitemia-dependent manner, reaching levels of ~40 pM with 50% parasitemia after 8 hrs. Unparasitized RBC and/or Hepes-AlbuMAX media alone induce negligible increase of FXa production. Control incubation of MVEC with TNF-α (10 ng/ml in AlbuMAX medium, 8 hrs) generated 206.5 ± 26 pM (not shown).
Figure 1. Parasitezed Red Blood Cells (pRBC) induce TF expression and productive assembly of the extrinsic Xnase complex in MVEC.
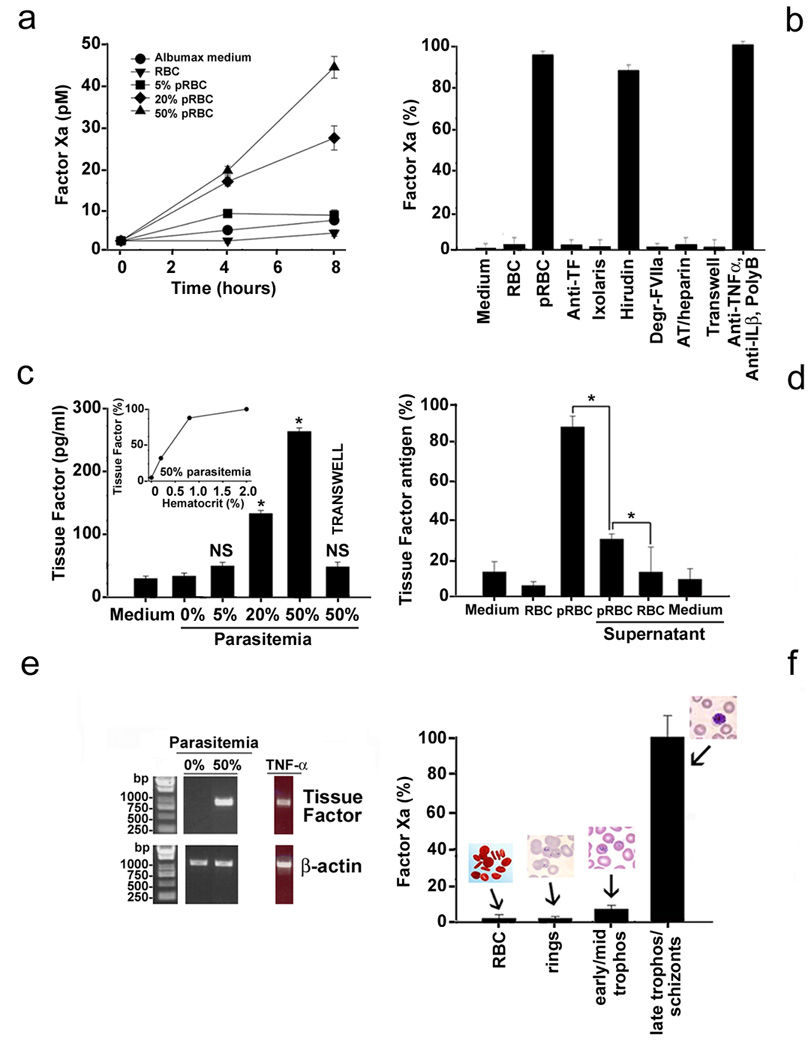
(A) Functional assays for TF. (B) Controls. Degr-FVIIa, catalytic site blocked FVIIa; AT/heparin, antithrombin/heparin; PolyB, polymyxin B. (C) Antigen detection for TF (ELISA). Inset: hematocrit dependence. (D) Effects of pRBC supernatant in TF expression (antigen detection). (E) Transcript detection for TF (RT-PCR). (F) Stage-specificity of TF expression (functional assay). For each data point, results are the mean ± SE from three or four independent experiments, or a typical experiment is shown (*, p<0.05; NS, non significant). Legends are described in detail in the Supplemental data.
A number of additional controls were performed to confirm whether FXa generation was specifically mediated by TF expression. Figure 1B shows that FXa generation was completely blunted by sheep anti-human polyclonal antibody against TF and by recombinant Ixolaris, a specific recombinant Tissue Factor Pathway Inhibitor (TFPI) from tick salivary gland [20]. No inhibition was attained with the specific thrombin inhibitor, hirudin. We then determined whether TF expression was associated with functional assembly of the extrinsic Xase complex. Figure 1B shows that FXa was not generated when FVIIa was replaced by dansyl-Glu-Gly-Arg-chloromethyl ketone-treated FVIIa (DEGR-FVIIa), a catalytic site-occupied FVIIa which interacts with TF but does not activate FX. Likewise, incubation of the supernatant with the specific FXa inhibitor — antithrombin/heparin — followed by addition of chromogenic substrate S-2222, was accompanied by 100% inhibition of chromogenic substrate hydrolysis. These experiments indicate that TF is specifically expressed on the surface of EC after incubation with pRBC, with productive assembly of the extrinsic Xnase complex leading to FXa production. Other controls were performed to rule out that TF expression was mediated by proinflammatory cytokines potentially released by activated MVEC or contaminating lipopolysaccharide (LPS). Figure 1B shows that a combination of anti-TNF-α and anti-interleukin-1β monoclonal antibodies (mAb) with polymyxin B did not affect pRBC-induced TF expression by MVEC. LPS contamination was below 0.05–0.025 EU/ml using the Limulus Ameboid Assay (data not shown). Figure 1B also shows that TF expression was not detected in experiments using Transwell plates, which do not allow physical contact between pRBC and MVEC but permit free exchange of macromolecules less than 1 µm between both compartments.
We confirmed the expression of TF using an enzyme-linked immunosorbent assay (ELISA) specific for human TF. Figure 1C shows that incubation of MVEC with 2% hematocrit (5–50% parasitemia) is accompanied by TF expression reaching concentrations as high as 271.8±12 pg/ml. Maximum TF expression was attained at 1–2% hematocrit, at 50% parasitemia (inset). In contrast, incubation of MVEC with Hepes-AlbuMAX medium or unparasitized RBC produced similar TF levels of ~35 pg/ml. When Transwell plates were used, TF detection was ~10% of control (6.3%, 8%, and 12% for each assay) under identical experimental conditions. In addition, Figure 1D shows that an increase of TF expression (~20% of control) was detected by ELISA when supernatants collected from pRBC (kept 6 h at 37°C, and centrifuged for 10 min at 0.1g) were added to fresh MVEC. TNF-α (10 ng/ml, diluted in AlbuMAX medium) induced 1,135 ± 155 pg/ml TF (not shown).
To determine whether increases in cell expression of TF were accompanied by changes in TF mRNA expression, MVEC were incubated with 2% hematocrit (0% or 50% parasitemia) or TNF-α (10 ng/ml) for 3 hours and mRNA was isolated for reverse transcriptase polymerase chain reactions (RT-PCR). Figure 1E shows that steady-state TF mRNA levels were undetectable when RBC or Hepes-AlbuMAX medium were used, but present when incubation was performed with pRBC or TNF-α. As a control, the identity of the band was checked by cloning and sequencing and it was found to be identical to human TF (gi 135666).
Temporal expression of specific genes has been reported during parasite development [22], and sequestration is associated with mature forms of pRBC [4–6]. Therefore, we examined whether TF expression was associated with a specific parasite developmental stage. Figure 1F shows that TF expression was detected after incubation of EC with late trophozoites/schizonts but was only marginally detectable with ring and early-mid trophozoite stages. Our results also demonstrate that pRBC did not inherently support the extrinsic Xnase assembly in the presence of FVIIa and FX, in contrast to lipidated TF (not shown).
pRBC support the assembly of the prothrombinase in vitro
Recently, it has been shown that pRBC expose phosphatidylserine on their surface as intracellular parasites mature to late trophozoite and schizont stages [23]. Because phosphatidylserine (PS) is critical for the assembly of multimolecular blood coagulation complexes [18,19], we attempted to determine whether late-stages pRBC support prothrombinase complex formation. In a first set of experiments, pRBC were incubated with FVa, FXa, and Ca2+ (prothrombinase) followed by addition of prothrombin to start reactions. Figure 2A shows that RBC did not induce thrombin generation. In contrast, 0.2% hematocrit at 5% parasitemia slightly increased thrombin formation, whereas at 50% parasitemia, the thrombin concentration reached 60 nM after 6 min. Figure 2B shows that 50% parasitemia consistently supported thrombin formation at hematocrits as low as 0.025%, and Figure 2C shows that thrombin production is exquisitely sensitive to FVa, a critical cofactor for prothrombinase assembly [18,19]. Figure 2D confirms that thrombin formation is mostly detected when mature, but not younger forms of pRBC are used in the experiments.
Figure 2. pRBC support assembly of prothrombinase and intrinsic Xnase complexes.
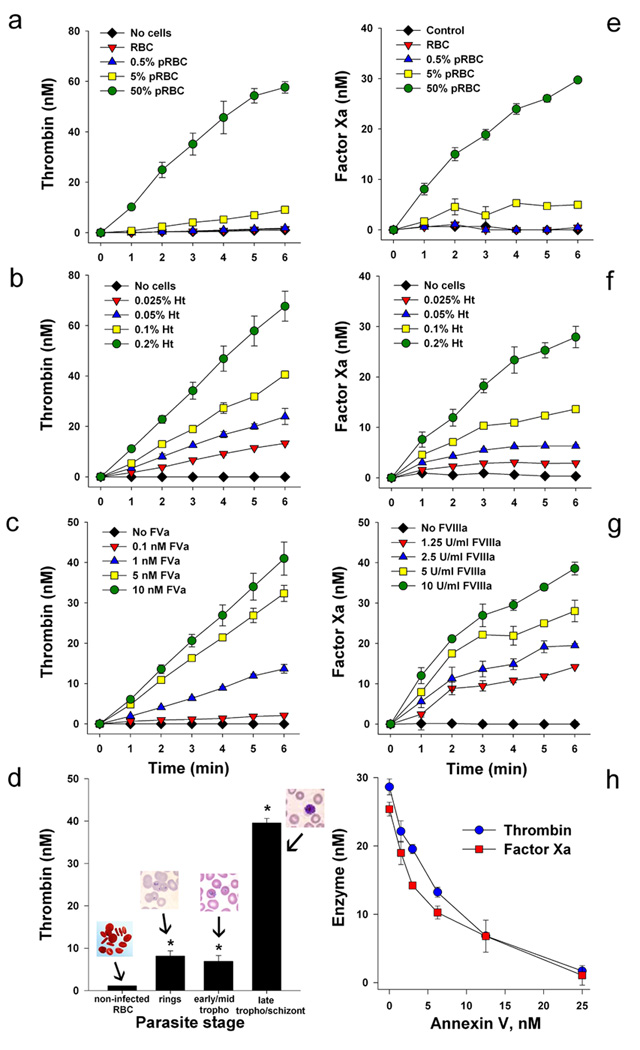
(A) pRBC support prothrombinase assembly. (B) Hematocrit-dependent prothrombinase assembly. (C) FVa-dependent prothrombinase assembly. (D) Stage specificity of pRBC-dependent prothrombinase assembly. Reactions were initiated by addition of human prothrombin, and thrombin formation was estimated using chromogenic substrate (S2238). (E) pRBC support intrinsic Xnase assembly. (F) Hematocrit-dependent intrinsic Xnase assembly. (G) FVIIIa-dependent intrinsic Xnase assembly. Reactions were initiated by addition of human FX, and FXa formation was estimated using chromogenic substrate (S2222) (H) Effects of annexin V in the prothrombinase and intrinsic Xnase assemblies by pRBC. For each data point, results are the mean ± SE from three independent experiments (*, p<0.05). Legends are described in detail in the Supplemental data.
pRBC support the assembly of the intrinsic Xnase in vitro
In a second set of experiments, pRBC (0.2% hematocrit, 0–50% parasitemia) was incubated with FVIIIa, FIXa, and Ca2+ (intrinsic Xnase) followed by addition of FX to initiate reactions. Figure 2E shows that pRBC induces FXa formation in a parasitemia-dependent manner, and Figure 2F shows that hematocrits as low as 0.025% are effective. The specificity of the intrinsic Xnase complex formation was estimated by increasing concentrations of FVIIIa, an indispensable cofactor in the intrinsic Xnase assembly [18,19]. We detected FXa production at FVIIIa concentrations as low as 1.25 U/ml (Figure 2G). Next, the specificity of prothrombinase and Xnase complexes were confirmed using annexin V, a specific PS binding protein [18,19]. Figure 2H depicts a dose-response inhibitory curve showing inhibition of thrombin and FXa formation with an IC50 around 2.5 nM; complete inhibition of enzyme formation was attained with 25 nM annexin V. Figures 2 thus show that PS is functionally expressed by pRBC.
TF expression in the EC from the brain of P. falciparum malaria patients
Brain slides from the frontal cortex of thirteen parasitemic children and 10 aparasitemic cases were used for immunohistochemistry (IHC) purposes using a specific mAb for TF and counterstaining with hematoxylin (see Methods). All parasitemic patients who met both clinical (in life) and pathological (at autopsy) case definition of CM were designated as “CM cases”. When another cause of death was identified in a parasitemic patient, they were designated as “parasitemic controls”. The patients who died without P falciparum infection in a malaria-free area were named “aparasitemic controls”. For the IHC assays, NovaRed dye was used to exclude any confounding effects from the brown malaria erythrocytic pigment (hemozoin) that is released by ruptured schizonts [1,2]. In some cases, polarized light that identifies hemozoin was employed to confirm that staining was associated with sequestration.
Figure 3 (A, C and E) shows three different CM cases with positive staining for TF in EC and associated with sequestration — hemozoin and/or pRBC were detectable using polarized light (Figure 3,B, D and F). Staining in EC was patchy and focal and found mostly in post capillary venules, or pre-capillary arterioles but not in capillaries. Figure 3G shows one CM case with intense TF expression in EC but no sequestration was observed using polarized light (Figure 3H). Figure 3I is a CM case showing TF staining of EC (arrow) and aggregates of extracellular hemozoin (open arrow), the latter indicating that schizonts have ruptured following sequestration (Figure 3J). Positive staining was present in the “parasitemic controls” with one exception. All “aparasitemic controls” were negative, with the exception of one Burkitt`s lymphoma case. Positive staining was not detected in mononuclear cells or in the subendothelial space, and was consistently not found in the absence of the primary antibody (not shown). Table 1 summarizes our findings.
Figure 3. TF staining in EC of the frontal cortex of CM cases.
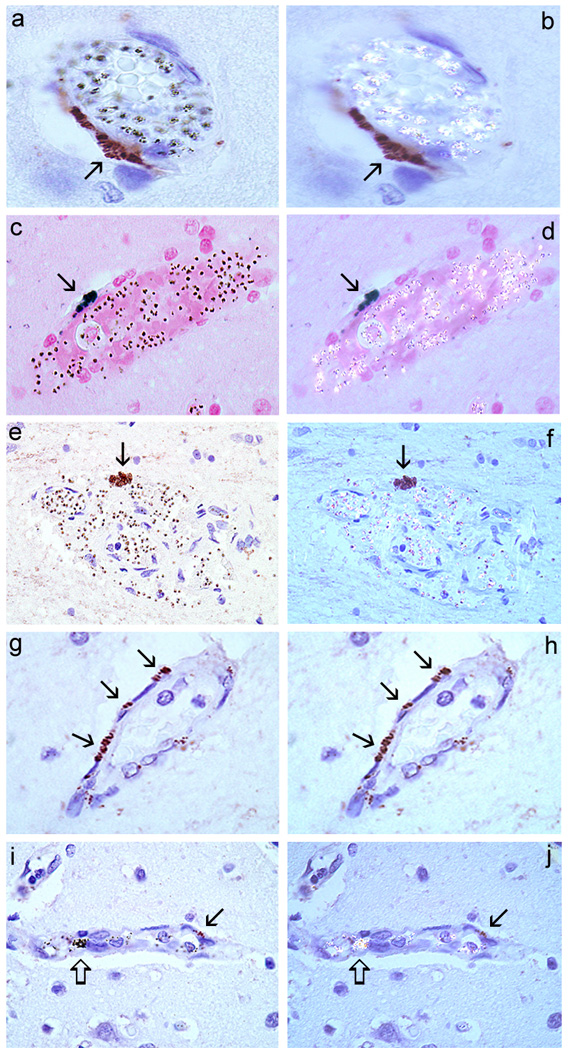
Case 75 (A and B), case 68 (C and D), case 74 (E and F), case 38 (G and H), and case 69 (I and J). (A), (C) and (E) show TF staining of EC (arrow) associated with sequestration only as confirmed by polarized light (B, D, and F) that detects hemozoin (malaria pigment). (G) Shows TF staining of EC but no sequestration (H). In (I) TF staining of EC (arrows) is shown in the presence of aggregated hemozoin (open arrows) (J). Staining was never detected for any of the slides in the absence of primary anti-TF antibodies. Counterstaining: Hematoxilin. Immunohistochemistry for TF is described in detail in the Supplemental data.
| Patient | Age* | Sex†/Race‡ | Cause of death§ | Parasitemia‖ | Platelets¶ | TF staining in EC# |
|---|---|---|---|---|---|---|
| Malawian “Cerebral Malaria cases” (parasitemic patients, malarial coma) | ||||||
| 37 | 6 mo | M/B | Cerebral Malaria | 616,400 | 74,000 | 1 (9/286) |
| 38 | 7 yrs | M/B | Cerebral Malaria | 782,320 | 26,000 | 3 (121/267) |
| 68 | 13 yrs | M/B | Cerebral Malaria | 20,080 | 30,000 | 1 (21/267) |
| 69 | 39 mo | F/B | Cerebral Malaria | 81,840 | NA** | 1 (9/390) |
| 74 | 8 yrs | F/B | Cerebral Malaria | 280,000 | NA | 1 (3/259) |
| 75 | 12 yrs | M/B | Cerebral Malaria | 215,300 | 351,000 | 1 (33/299) |
| 78 | 15 mo | M/B | Cerebral Malaria | 637,000 | 73,000 | 1 (4/200) |
| 79 | 15 mo | M/B | Cerebral Malaria | 34,400 | 144,000 | 1 (1/196) |
| Malawian “parasitemic controls” (parasitemic patients, non-malarial coma) | ||||||
| 49 | 17 mo | M/B | Ruptured AVM | 1,943 | 276,000 | 3 (50/169) |
| 52 | 24 mo | M/B | Severe anemia | 319,287 | 87,000 | 1 (4/203) |
| 54 | 17 mo | M/B | Skull fractures | 96,520 | 195,000 | 0 (0/167) |
| 58 | 8 mo | M/B | Severe anemia | 788 | 469,000 | 1 (1/110) |
| 71 | 5 yrs | M/B | Subdural hematomas | 314 | 346,000 | 1 (1/183) |
| NIH “aparasitemic controls” | ||||||
| A90 | 9 yrs | F/W | Burkitt`s Lymphoma | NA | NA | 1 (4/266) |
| A83 | 9 yrs | M/B | Osteosarcoma | NA | NA | 0 (0/216) |
| A4 | 3 yrs | M/W | Burkitt`s Lymphoma | NA | NA | 0 (0/226) |
| A88 | 8 yrs | M/I | Burkitt`s Lymphoma | NA | NA | 0 (0/259) |
| A87 | 11 yrs | M/W | Osteogenic sarcoma | NA | NA | 0 (0/259) |
| Brazilian “aparasitemic controls” | ||||||
| A-08-6 | 6 yrs | F/B | Congenital cardiopathy | NA | NA | 0 (0/269) |
| A-08-14 | 6 yrs | F/B | Congenital cardiopathy | NA | NA | 0 (0/318) |
| A-02-12 | 7 yrs | F/B | Congenital cardiopathy | NA | NA | 0 (0/163) |
| A-02-8 | 7 yrs | F/B | Congenital cardiopathy | NA | NA | 0 (0/277) |
| Neural Tissue Array “aparasitemic controls”†† | ||||||
| TMA3001 | 54 yrs | NA | NA | NA | NA | 0 |
Age; mo (months); yrs (years).
Sex; M (male), F (Female).
Race; B (Black), W (White), I (Indian).
Cause of death; CM, cerebral malaria. AVM; arteriovenous malformation.
Parasitemia; parasites counts/µl at admission.
Platelets; platelet counts/µl at admission.
TF (Tissue Factor) staining in EC; 0, none; 1, mild; 2, moderate; and 3, severe immunoreactivity. [positive vessels/number of total vessels observed]. Vessel counting and scoring for TF performed by R.O.W.
NA; not available.
Frontal cortex obtained from neural tissue arrays TMA3001 (Chemicon Co.) was negative for all vessels examined.
Figure 4 shows the potential sources for TF expression in malaria (see Figure Legends).
Figure 4. Tissue Factor expression in Malaria.
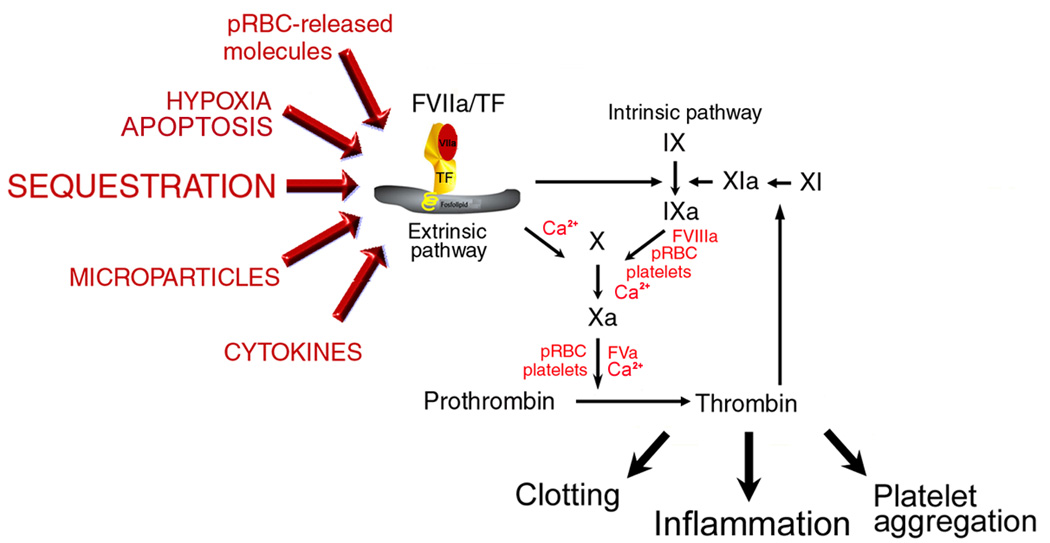
Normal, quiescent endothelium does not express TF in the absence of biologic stimulation [14–19]. Sequestration of pRBC, in addition to cytokines [10], microparticles [13], hypoxia [2,3], fibrin [5,6], apoptosis [54], and proinflammatory molecules released by pRBC [38–40] potentially contribute to TF expression in microvessels of the brain and in other vascular beds [69]. Monocytes may also express TF and/or produce TNF-α and thus contribute to the procoagulant tonus observed in the disease [12,27–33]. Mechanistically, TF/FVIIa complex activates FIX or FX (extrinsic Xase complex) generating respectively FIXa and FXa, in the presence of Ca2+. FXa, FVa, and prothrombin (prothrombinase complex) or Factor IXa, FVIIIa, and FX (intrinsic Xnase complex) assemble in the membrane of activated platelets and/or pRBC leading to amplification of blood coagulation, platelet aggregation [18,19], and inflammation [16,17]. The result is a convergence of signals leading to exacerbated TF expression that could sustain the coagulation-inflammatory cycle [16,17,47,52,67–68] in different vascular beds. Legends are described in detail in the Supplemental data.
Discussion
In patients with malaria, pRBC containing young, unpigmented parasites circulate in the peripheral blood for the first 18–24 h of the 48-h life cycle of P. falciparum [1,2]. pRBC containing the more mature, pigmented trophozoites and schizonts are rarely seen in the peripheral blood, because they are sequestered in the vascular beds of various organs [4–6] including the brain [4]. Although multiple pathogenic mechanisms have been suggested to explain the disease [10,24], including the “sequestration” [25] , and “cytokine” [26] hypotheses no unifying model has met with a consensus among investigators. However, four pathologic features have been described in severe malaria in general and in CM in particular: i) sequestration of mature forms of pRBC in different vascular beds [4–6]; ii) EC activation as detected by staining for adhesion molecules ICAM-1, VCAM-1, and E-selectin [6,10]; iii) an increase in the concentration of inflammatory cytokines such as TNF-α, IL-1β, IL-6, and others in plasma [10]; and iv) activation of the blood coagulation cascade [12, 27–33]. Collectively, these features suggest that pRBC and EC are involved with the pathogenesis of malaria, and may play an important role in the pro-coagulant state observed in the disease [12, 27–33].
Because blood coagulation in vivo is initiated by TF [14–19] we attempted to identify the mechanism by which pRBC might induce TF expression. Using three independent in vitro assays, we show that late-stage pRBC interact with cultured EC in vitro and induce TF expression with productive assembly of extrinsic Xnase complex, and initiation of the coagulation cascade. TF antigen and transcript were also detected by ELISA and RT-PCR, respectively. Functional expression of FXa was found in the pM range, was 3–4 times higher that the basal conditions (albumax or RBC), but ~ 5 times lower than the values obtained with TNF-α (10 ng/ml). These values were compatible with the levels of antigen detected by ELISA (~ 10 pg/well or ~ 0.5–1 pM TF, in 200 µl) and may also be due to the non-optimum binding of FX to apoptotic/activated EC [34,35].
Our results with the Transwell suggest that physical contact or close proximity between pRBC and EC is required for most TF expression — only ~ 10% of the response was observed with non-contact incubation. This is in agreement with previous reports showing that Transwell prevents pRBC-induced apoptosis [34] and EC activation [35]. On the other hand, we detected a small increase of ~ 20% in TF expression by ELISA using the supernatant obtained from pRBC that were kept at 37°C for 6 hrs. These findings are also in agreement with a recent report showing partial induction of ICAM-1 expression in EC by the supernatant of pRBC [36]. Therefore, molecules produced/released by pRBC [37–40] may contribute in part, although modestly, to TF expression by EC. At present, however, it is not known how pRBC induce TF expression but signaling triggered by its interaction with EC [41], or the molecules released by infected erythrocytes [38–40], and free radicals [42] may potentially contribute to this response. Also, the molecular mechanisms of TF gene expression triggered by pRBC remains unknown, but possibly involve NFkB, which is known to regulate TF expression in different cell types [14] and to be nuclear translocated in brain EC incubated in vitro with pRBC [36].
In a second set of experiments, the potential procoagulant activity of pRBC was investigated. Mature forms (late trophozoites and schizonts) of pRBC display PS [23], a negatively charged phospholipid expressed in activated platelets [18,19]. In the experiments described here, it was revealed that late-stage pRBC support productive assembly of both intrinsic Xnase and prothrombinase complexes with generation of FXa and thrombin, respectively. Both complexes were exquisitely sensitive to their respective cofactors FVa and FVIIIa and inhibited by annexin V, a PS-binding protein [18,19]. Assemblies were attained at a remarkable low hematocrit (e.g. 0.025 %) and parasitemia (e.g. 5%), indicating that minute amounts of pRBC presumably propagate the blood coagulation cascade in vivo. In fact, pRBC obtained directly from P. falciparum-infected patients shorten the clotting [43], and the recalcification time [44]. This indicates that pRBC are indeed procoagulant. Therefore, pRBC behaves functionally as activated platelets that, among other functions, operate as the limiting step in the amplification phase of the blood coagulation cascade [19]. These data are particularly important, because late-stage pRBC are associated with sequestration in the brain of CM patients [4–6]. It is plausible that initiation of the blood coagulation by endothelium TF may be amplified by pRBC and/or activated platelets at sites of sequestration. Therefore, a balance between pro- and anti-coagulant mechanisms may determine the pro-coagulant tonus in different vascular beds.
We have attempted to demonstrate whether sequestration in parasitemic children was associated with TF expression by brain EC. Accordingly, sections of the frontal cortex from children who died from CM were used for IHC assays using a specific mAB against TF. All except one of the malaria-infected cases have evidence of TF expression to a certain degree. In contrast, none of the aparasitemic patients displayed TF staining, with the exception of one of the three Burkitt`s Lymphoma cases, a condition where TF expression in the EC has been reported previously [45]. The presence of TF in the cerebral endothelium of children with severe malaria could serve to support activation of the coagulation cascade, a possibility corroborated by histopathologic findings of platelet accumulation [46] and fibrin deposition in the brain of some CM patients [2–6].
We cannot determine, however, whether TF was locally synthesized or was acquired from circulating particles via P-selectin/PSGL-1 interactions [47]. In some ECs, TF stained in patchy or granular structures that appear to be located on the cell surface, but it was not possible to determine whether TF was produced by ECs or was transferred to the EC surface from blood-borne microvesicles [47,48]. We did not detect positive staining for TF in mononuclear cells, but they cannot be excluded as a potential source of TF as described in other diseases [14–16,47,48]. In other slides, TF expression was found in the EC of vessels containing malaria pigment (hemozoin) but no sequestered parasites. Extra-erythrocytic hemoglobin is a marker of the schizont rupture that occurs once asexual replication is complete [22]. EC expression of TF was also found in the absence of hemozoin and pRBCs, suggesting that pRBC had been present, but had cleared, either as a result of the dynamic nature of sequestration or an effective anti-parasitic treatment. Our IHC studies also show variable degrees of TF staining in the five of six parasitemic children who died from non-malarial causes. The one ‘parasitemic control’ with no evidence of TF expression died from direct head trauma.
Therefore, TF expression appears to be a general feature of malaria, and not necessarily specific for CM. Consistent with this notion, widespread EC activation has been reported in uncomplicated malaria [6,49], a condition where sequestration [4–6,50], thrombocytopenia [11], and activation of blood coagulation [12] are generally observed. According to several reports [27–33], these non-comatose patients present none or few clinical manifestations compatible with a coagulation disorder such as bleeding but the plasma levels of coagulation factors are decreased to a moderate extend although not exhausted. These patients often present i) PT, PTT and fibrinogen often near the normal range, ii) low plasma levels of PC and PCI-1 and iii) increased levels of TAT, PAI-1 and D-dimers [12,27–33]. This laboratory and clinical profile therefore resembles "compensated" disseminated intravascular coagulation (DIC), a term that has been applied to atypical cases of DIC in which a continuous or intermittent slow rate of initiation of intravascular coagulation occur in vivo [51]. In theses cases, concomitant increase in coagulation factor synthesis may occur (e.g. fibrinogen), and therefore changes in coagulation tests may not be always remarkable [51]. However, it remains unclear why 1% of malaria patients develop CM, and why in 5–10% patients of severe malaria patients develop typical DIC [1,2] . It may be that blood coagulation, while normally homeostatic [16,52] may contribute to disease when it undergoes decompensation [16,53]. Of note, thrombocytopenia in malaria is associated with disease severity and is predictive of fatal outcome in humans [11]. Moreover, accumulation of platelets and monocytes in the vessels of the brain [46], apoptosis of EC [54] and formation of EC microparticles [13]—known to bear TF [55] and PS and to participate in inflammation [47]—have been specifically reported in CM but not in uncomplicated cases [13,46,54]. What catalyzes the transition from uncomplicated to complicated malaria, and the relative contribution of TF, activated platelets, activated/apoptotic EC to the pathogenesis of both conditions remains to be elucidated.
Our IHC studies show that some vessels contain sequestration and no TF staining. Perhaps, the tightly regulated mechanism of blood coagulation [14–19] may play a role in preventing an uncontrolled EC activation with widespread TF expression. Actually, in vivo TF staining in non-human primates or in human EC has been reported in only few studies [14,48,56,57], and it is remarkable that our cases consistently show positive staining and notably in some cases, at sites of sequestration. It is important to recognize that other events reported in malaria such as hypoxia [2], fibrin formation [5,6], and apoptosis [54]—and known to induce TF expression by EC in vitro [58–60]—may also play a proinflammatory role as may molecules released from ruptured pRBC [38–40] and monocyte-derived cytokines [10]. One of these, TNF-α, has been found in the P. falciparum patients plasma [12,61] which has been reported to induce TF expression and NF-κB activation in EC in vitro [62]. In addition, monocytes express TF in vitro after incubation with pRBC [37] suggesting that it could contribute to the pro-coagulant tonus observed in the disease [12,27–33]. It has also been reported that P. falciparum-infected placentas present macrophages that stain positive for TF [63], and display monocytes infiltrates which appear to be associated with complication in pregnancy in malaria [64]. Finally, the plasma level of the pro-inflammatory cytokine HMGB1 [65] is increased in severe cases of malaria [66]. Therefore, it is plausible to speculate that in malaria the positive feedback loop between clotting and inflammation [16,17,52,67–68], may ultimately contribute to neutrophil, leukocyte, and platelet interactions with the endothelium, resulting eventually in endothelial injury, increased vascular permeability, vesiculation and cellular apoptosis in different vascular beds [69].
The finding that TF is expressed in vivo may have implications for our understanding of the pathogenesis of malaria. Tissue factor is a structural member of the cytokine receptor family [16,52] which signify the expansion of the adaptive immune system in vertebrates, indicating a close connection of the coagulation pathways with the host response to infection [16,52]. TF is increasingly recognized as the interface of blood coagulation and inflammation [16,17,52,67,68], and reportedly plays a pivotal role in disease pathogenesis [70–74]. Our histological data to date suggest that expression of TF in cerebral vascular endothelium occurs in malaria, and our in vitro data indicate that this has the potential to activate the coagulation cascade and thus contribute to the severe disease. Therefore, malaria may provide a natural disease model for studying the interface between EC activation, inflammation and blood coagulation. Identification of TF as a potently critical mediator of malaria pathogenesis suggests that therapeutic agents targeted at TF and/or EC [75] might be useful as adjunct treatment in patients with severe disease [76].
Supplementary Material
Acknowledgments
We thank Drs. Thomas E. Wellems, Robert W. Gwadz, and Kathryn Zoon (NIAID/NIH) for encouragement and support. We express our thanks to Drs. Thomas E. Wellems and José Marcos C. Ribeiro for helpful discussions and critical reading of the manuscript. We acknowledge Drs. David Kleiner and Stephen M. Hewitt from the Laboratory of Pathology (NCI/NIH) for providing slides containing frontal cortex from malaria-free patients. We are thankful to Larry Faucette (NIAID/NIH) for expertise in the immunohistochemistry assays. We appreciate the cooperation of parents of the children studied, and the care provided by the nursing and laboratory staff within the Paediatric Department of the College of Medicine, University of Malawi. This study received funding from the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health, USA; from NIAID contract to SoBran, Inc., and from The Wellcome Trust, UK. The autopsy study is jointly funded by NIH RO1 grant AI 34969 to Dr. TE Taylor and a Wellcome Trust Research Leave Fellowship grant 058390 to Dr. ME Molyneux. The authors acknowledge Brenda Rae Marshall (NIAID/NIH) for correction of English style and editorial assistance.
Footnotes
Authors` Contributions
I. Francischetti, K. Seydel, R. Monteiro, J. Ward, study design, writing the paper, performing research, data analysis, vital reagents; C. Erexson, performing research; A. Noronha, G. Ostera; vital reagents; S. Kamiza, data analysis; R. Whitten, M. Molyneux, T. Taylor, data analysis, writing the paper, vital reagents.
References
- 1.Miller LH, Baruch DI, Marsh K, Doumbo OK. The pathogenic basis of malaria. Nature. 2002;415:673–679. doi: 10.1038/415673a. [DOI] [PubMed] [Google Scholar]
- 2.White NJ, Breman JG. Malaria and babesiosis: diseases caused by red blood cell parasites. In: Kasper DL, Braunwald E, Fauci AS, Hauser SL, Longo DL, Jameson LH, editors. Harrison`s Principles of Internal Medicine. Vol 1. New York, NY: McGraw Hill; 2005. pp. 1218–1233. [Google Scholar]
- 3.Newton CR, Taylor TE, Whitten RO. Pathophysiology of fatal falciparum malaria in African. Am J Trop Med Hyg. 1998;58:673–683. doi: 10.4269/ajtmh.1998.58.673. [DOI] [PubMed] [Google Scholar]
- 4.Taylor TE, Fu WJ, Carr RA, Whitten RO, Mueller JS, Fosiko NG, Lewallen S, Liomba NG, Molyneux ME. Differentiating the pathologies of cerebral malaria by postmortem parasite counts. Nat Med. 2004;10:143–145. doi: 10.1038/nm986. [DOI] [PubMed] [Google Scholar]
- 5.Pongponratn E, Turner GD, Day NP, Phu NH, Simpson JA, Stepniewska K, Mai NT, Viriyavejakul P, Looareesuwan S, Hien TT, Ferguson DJ, White NJ. An ultrastructural study of the brain in fatal Plasmodium falciparum malaria. Am J Trop Med Hyg. 2003;69:345–349. [PubMed] [Google Scholar]
- 6.Turner GD, Ly VC, Nguyen TH, Tran TH, Nguyen HP, Bethell D, Wyllie S, Louwrier K, Fox SB, Gatter KC, Day NP, Tran TH, White NJ, Berendt AR. Systemic endothelial activation occurs in both mild and severe malaria. Correlating dermal microvascular endothelial cellphenotype and soluble cell adhesion molecules with disease severity. Am J Pathol. 1998;152:1477–1487. [PMC free article] [PubMed] [Google Scholar]
- 7.Su XZ, Heatwole VM, Wertheimer SP, Guinet F, Herrfeldt JA, Peterson DS, Ravetch JA, Wellems TE. The large diverse gene family var encodes proteins involved in cytoadherence and antigenic variation of Plasmodium falciparum-infected erythrocytes. Cell. 1995;82:89–100. doi: 10.1016/0092-8674(95)90055-1. [DOI] [PubMed] [Google Scholar]
- 8.Baruch DI, Gormley JA, Ma C, Howard RJ, Pasloske BL. Plasmodium falciparum erythrocyte membrane protein 1 is a parasitized erythrocyte receptor for adherence to CD36, thrombospondin, and intercellular adhesion molecule 1. Proc Natl Acad Sci USA. 1996;93:3497–3502. doi: 10.1073/pnas.93.8.3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berendt AR, Simmons DL, Tansey J, Newbold CI, Marsh K. Intercellular adhesion molecule-1 is an endothelial cell adhesion receptor for Plasmodium falciparum. Nature. 1989;341:57–59. doi: 10.1038/341057a0. [DOI] [PubMed] [Google Scholar]
- 10.Clark IA, Alleva LM, Mills AC, Cowden WB. Pathogenesis of malaria and clinically similar conditions. Clin Microbiol Rev. 2004;17:509–539. doi: 10.1128/CMR.17.3.509-539.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gerardin P, Rogier C, Ka AS, Jouvencel P, Brousse V, Imbert P. Prognostic value of thrombocytopenia in African children with falciparum malaria. Am J Trop Med Hyg. 2002;66:686. doi: 10.4269/ajtmh.2002.66.686. [DOI] [PubMed] [Google Scholar]
- 12.Hemmer CJ, Kern P, Holst FG, Radtke KP, Egbring R, Bierhaus A, Nawroth PP, Dietrich M. Activation of the host response in human Plasmodium falciparum malaria: relation of parasitemia to tumor necrosis factor/cachectin, thrombin-antithrombin III, and protein C levels. Am J Med. 1991;91:37–44. doi: 10.1016/0002-9343(91)90071-5. [DOI] [PubMed] [Google Scholar]
- 13.Combes V, Taylor TE, Juhan-Vague I, Mege JL, Mwenechanya J, Tembo M, Grau GE, Molyneux ME. Circulating endothelial microparticles in Malawian children with severe falciparum malaria complicated with coma. J Am Med Assn. 2004;291:2542–2544. doi: 10.1001/jama.291.21.2542-b. [DOI] [PubMed] [Google Scholar]
- 14.Broze GJ., Jr . The tissue factor pathway of coagulation. In: Loscalzo J, Schafer AI, editors. Thrombosis and Hemorrhage. Baltimore, MD: Williams & Wilkins; 1998. pp. 77–104. [Google Scholar]
- 15.Giesen PL, Rauch U, Bohrmann B, Kling D, Roque M, Fallon JT, Badimon JJ, Himber J, Riederer MA, Nemerson Y. Blood-borne tissue factor: another view of thrombosis. Proc NatlAcad Sci U S A. 1999;96:2311–2315. doi: 10.1073/pnas.96.5.2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ruf W. Protease-activated receptor signaling in the regulation of inflammation. Crit Care Med. 2004;32:S287–S292. doi: 10.1097/01.ccm.0000126364.46191.12. [DOI] [PubMed] [Google Scholar]
- 17.Coughlin SR. Protease-activated receptors in hemostasis, thrombosis and vascular biology. J Thromb Haemost. 2005;3:1800–1814. doi: 10.1111/j.1538-7836.2005.01377.x. [DOI] [PubMed] [Google Scholar]
- 18.Mann KG, Butenas S, Brummel K. The dynamics of thrombin formation. Arterioscler Thromb Vasc Biol. 2003;23:17–25. doi: 10.1161/01.atv.0000046238.23903.fc. [DOI] [PubMed] [Google Scholar]
- 19.Monroe DM, Hoffman M, Roberts HR. Platelets and thrombin generation. Arterioscler Thromb Vasc Biol. 2002;22:1381–1389. doi: 10.1161/01.atv.0000031340.68494.34. [DOI] [PubMed] [Google Scholar]
- 20.Francischetti IM, Valenzuela JG, Andersen JF, Mather TN, Ribeiro JM. Ixolaris, a novelrecombinant tissue factor pathway inhibitor (TFPI) from the salivary gland of the tick, Ixodes scapularis: identification of factor X and factor Xa as scaffolds for the inhibition of factor VIIa/tissue factor complex. Blood. 2002;99:3602–3612. doi: 10.1182/blood-2001-12-0237. [DOI] [PubMed] [Google Scholar]
- 21.Monteiro RQ, Rezaie AR, Ribeiro JM, Francischetti IM. Ixolaris: a factor Xa heparin-binding exosite inhibitor. Biochem J. 2005;387:871–877. doi: 10.1042/BJ20041738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bozdech Z, Llinas M, Pulliam BL, Wong ED, Zhu J, DeRisi JL. The transcriptome of the intraerythrocytic developmental cycle of Plasmodium falciparum. PLoS Biol. 2003;1:85–100. doi: 10.1371/journal.pbio.0000005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eda S, Sherman IW. Cytoadherence of malaria-infected red blood cells involves exposure of phosphatidylserine. Cell Physiol Biochem. 2002;12:373–384. doi: 10.1159/000067908. [DOI] [PubMed] [Google Scholar]
- 24.Hunt NH, Grau GE. Cytokines: accelerators and brakes in the pathogenesis of cerebral malaria. Trends Immunol. 2003;24:491–499. doi: 10.1016/s1471-4906(03)00229-1. [DOI] [PubMed] [Google Scholar]
- 25.Berendt AR, Tumer GD, Newbold CI. Cerebral malaria: the sequestration hypothesis. Parasitol Today. 1994;10:412–414. doi: 10.1016/0169-4758(94)90238-0. [DOI] [PubMed] [Google Scholar]
- 26.Clark IA, Rockett KA. The cytokine theory of human cerebral malaria. Parasitol Today. 1994;10:410–412. doi: 10.1016/0169-4758(94)90237-2. [DOI] [PubMed] [Google Scholar]
- 27.Dennis LH, Eichelberger JW, Inman MM, Conrad ME. Depletion of coagulation factors in drug-resistant Plasmodium falciparum malaria. Blood. 1967;29:713–721. [PubMed] [Google Scholar]
- 28.O`Leary S, Barr CF, Wellde BT, Conrad ME. Experimental infection with Plasmodium falciparum in Aotus Monkeys. III-The development of Disseminated Intravascular Coagulation. Am J Trop Med Hyg. 1972;21:282–287. doi: 10.4269/ajtmh.1972.21.282. [DOI] [PubMed] [Google Scholar]
- 29.Horstmann RD, Dietrich M. Haemostatic alterations in malaria correlate to parasitaemia. Blut. 1985;51:329–335. doi: 10.1007/BF00320043. [DOI] [PubMed] [Google Scholar]
- 30.Pukrittayakamee S, White NJ, Clemens R, Chittamas S, Karges HE, Desakorn V, Looareesuwan S, Bunnag D. Activation of the coagulation cascade in falciparum malaria. Trans R Soc Trop Med Hyg. 1989;83:762–766. doi: 10.1016/0035-9203(89)90321-0. [DOI] [PubMed] [Google Scholar]
- 31.Clemens R, Pramoolsinsap C, Lorenz R, Pukrittayakamee S, Bock HL, White NJ. Activation of the coagulation cascade in severe falciparum malaria through the intrinsic pathway. Br J Haematol. 1994;87:100–105. doi: 10.1111/j.1365-2141.1994.tb04877.x. [DOI] [PubMed] [Google Scholar]
- 32.Mohanty D, Ghosh K, Nandwani SK, Shetty S, Phillips C, Rizvi S, Parmar BD. Fibrinolysis, inhibitors of blood coagulation, and monocyte derived coagulant activity in acute malaria. Am J Hematol. 1997;54:23–29. doi: 10.1002/(sici)1096-8652(199701)54:1<23::aid-ajh4>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 33.Holst FG, Hemmer CJ, Foth C, Seitz R, Egbring R, Dietrich M. Low levels of fibrin-stabilizing factor (factor XIII) in human Plasmodium falciparum malaria: correlation with clinical severity. Am J Trop Med Hyg. 1999;60:99–104. doi: 10.4269/ajtmh.1999.60.99. [DOI] [PubMed] [Google Scholar]
- 34.Pino P, Vouldoukis I, Kolb JP, Mahmoudi N, Desportes-Livage I, Bricaire F, Danis M, Dugas B, Mazier D. Plasmodium falciparum-infected erythrocyte adhesion induces caspase activation and apoptosis in human endothelial cells. J Infect Dis. 2003;187:1283–1290. doi: 10.1086/373992. [DOI] [PubMed] [Google Scholar]
- 35.Viebig NK, Wulbrand U, Forster R, Andrews KT, Lanzer M, Knolle PA. Direct activation of human endothelial cells by Plasmodium falciparum-infected erythrocytes. Infect Immun. 2005;73:3271–3277. doi: 10.1128/IAI.73.6.3271-3277.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tripathi AK, Sullivan DJ, Stins MF. Plasmodium falciparum-infected erythrocytes increase intercellular adhesion molecule 1 expression on brain endothelium through NF-kappaB. Infect Immun. 2006;74:3262–3270. doi: 10.1128/IAI.01625-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pernod G, Polack B, Peyron F, Luisy A, Kolodie L, Ambroise-Thomas P, Santoro F. Monocyte tissue factor expression induced by Plasmodium falciparum-infected erythrocytes. Thromb Haemost. 1992;68:111–114. [PubMed] [Google Scholar]
- 38.Schofield L, Novakovic S, Gerold P, Schwarz RT, McConville MJ, Tachado SD. Glycosylphosphatidylinositol toxin of plasmodium up-regulates intercellular adhesion molecule-1, vascular cell adhesion molecule-1, and E-selectin expression in vascular endothelial cells and increases leukocyte and parasite cytoadherence via tyrosine kinase-dependent signal transduction. J Immunol. 1996;156:1886–1996. [PubMed] [Google Scholar]
- 39.Krishnegowda G, Hajjar AM, Zhu J, Douglass EJ, Uematsu S, Akira S, Woods AS, Gowda DC. Induction of proinflammatory responses in macrophages by the glycosylphosphatidylinositols of Plasmodium falciparum: cell signaling receptors, glycosylphosphatidylinositol (GPI) structural requirement, and regulation of GPI activity. J Biol Chem. 2005;280:8606–8616. doi: 10.1074/jbc.M413541200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jaramillo M, Plante I, Ouellet N, Vandal K, Tessier PA, Olivier M. Hemozoin-inducible proinflammatory events in vivo: potential role in malaria infection. J Immunol. 2004;172:3101–3110. doi: 10.4049/jimmunol.172.5.3101. [DOI] [PubMed] [Google Scholar]
- 41.Yipp BG, Robbins SM, Resek ME, Baruch DI, Looareesuwan S, Ho M. Src-family kinase signaling modulates the adhesion of Plasmodium falciparum on human microvascular endothelium under flow. Blood. 2003;101:2850–2857. doi: 10.1182/blood-2002-09-2841. [DOI] [PubMed] [Google Scholar]
- 42.Taoufiq Z, Pino P, Dugas N, Conti M, Tefit M, Mazier D, Vouldoukis I. Transient supplementation of superoxide dismutase protects endothelial cells against Plasmodium falciparum-induced oxidative stress. Mol Biochem Parasitol. 2006 doi: 10.1016/j.molbiopara.2006.07.008. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 43.Udeinya IJ, Miller LH. Plasmodium falciparum: effect of infected erythrocytes on clotting time of plasma. Am J Trop Med Hyg. 1987;37:246–249. doi: 10.4269/ajtmh.1987.37.246. [DOI] [PubMed] [Google Scholar]
- 44.Mohanty D, Marwaha N, Ghosh K, Chauhan AP, Shah S, Sharma S, Das KC. Vascular occlusion and disseminated intravascular coagulation in falciparum malaria. Br Med J. 1985;290:115–116. doi: 10.1136/bmj.290.6462.115-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sase T, Wada H, Yamaguchi M, Ogawa S, Kamikura Y, Nishikawa M, Kaneko T, Abe Y, Nishioka J, Nobori T, Shiku H. Haemostatic abnormalities and thrombotic disorders in malignant lymphoma. Thromb Haemost. 2005;93:153–159. doi: 10.1160/TH04-04-0260. [DOI] [PubMed] [Google Scholar]
- 46.Grau GE, Mackenzie CD, Carr RA, Redard M, Pizzolato G, Allasia C, Cataldo C, Taylor TE, Molyneux ME. Platelet accumulation in brain microvessels in fatal pediatric cerebral malaria. J Infect Dis. 2003;187:461–466. doi: 10.1086/367960. [DOI] [PubMed] [Google Scholar]
- 47.Furie B, Furie BC. Role of platelet P-selectin and microparticle PSGL-1 in thrombus formation. Trends Mol Med. 2004;10:171–178. doi: 10.1016/j.molmed.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 48.Lupu C, Westmuckett AD, Peer G, Ivanciu L, Zhu H, Taylor FB, Jr, Lupu F. Tissue factor-dependent coagulation is preferentially up-regulated within arterial branching areas in a baboon model of Escherichia coli sepsis. Am J Pathol. 2005;167:1161–1172. doi: 10.1016/S0002-9440(10)61204-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Garcia F, Cebrian M, Dgedge M, Casademont J, Bedini JL, Neves O, Filella X, Cinta Cid M, Corachan M, Grau JM. Endothelial cell activation in muscle biopsy samples is related to clinical severity in human cerebral malaria. J Infect Dis. 1999:475–483. doi: 10.1086/314598. [DOI] [PubMed] [Google Scholar]
- 50.Dondorp AM, Desakorn V, Pongtavornpinyo W, Sahassananda D, Silamut K, Chotivanich K, Newton PN, Pitisuttithum P, Smithyman AM, White NJ, Day NP. Estimation of the total parasite biomass in acute falciparum malaria from plasma PfHRP2. PLoS Med. 2005;2:788–797. doi: 10.1371/journal.pmed.0020204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mammen EF. Disseminated intravascular coagulation (DIC) Clin. Lab. Sci. 2000;13:239–245. [PubMed] [Google Scholar]
- 52.Opal SM, Esmon CT. Bench-to-bedside review: Functional relationship between coagulation and the innate immune response and their respective role in the pathogenesis of sepsis. Crit Care. 2003;7:22–38. doi: 10.1186/cc1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Taylor FB., Jr Staging of the pathophysiologic responses of the primate microvasculature to Escherichia coli and endotoxin: examination of the elements of the compensated response and their links to the corresponding uncompensated lethal variants. Crit Care. 2001;29:S78–S89. doi: 10.1097/00003246-200107001-00026. [DOI] [PubMed] [Google Scholar]
- 54.Hemmer CJ, Lehr HA, Westphal K, Unverricht M, Kratzius M, Reisinger EC. Plasmodium falciparum malaria: reduction of endothelial cell apoptosis in vitro. Infect Immun. 2005;73:1764–1770. doi: 10.1128/IAI.73.3.1764-1770.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jin M, Drwal G, Bourgeois T, Saltz J, Wu HM. Distinct protein features of plasma microparticle. Proteomics. 2004;5:1940–1952. doi: 10.1002/pmic.200401057. [DOI] [PubMed] [Google Scholar]
- 56.Drake TA, Cheng J, Chang A, Taylor FB. Expression of tissue factor, thrombomodulin, and E-selectinin baboons with lethal Escherichia coli sepsis. Am J Pathol. 1996;5:1458–1470. [PMC free article] [PubMed] [Google Scholar]
- 57.Solovey A, Gui L, Key NS, Hebbel RP. Tissue factor expression by endothelial cells in sickle cell anemia. J Clin Invest. 1998;101:1899–1904. doi: 10.1172/JCI1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yan S, Pinsky DJ, Stern DM. A pathway leading to hypoxia-induced vascular fibrin deposition. Sem Thromb Hemost. 2000;26:479–483. doi: 10.1055/s-2000-13203. [DOI] [PubMed] [Google Scholar]
- 59.Contrino J, Goralnick S, Qi J, Hair G, Rickles FR, Kreutzer DL. Fibrin induction of tissue factor expression in human vascular endothelial cells. Circulation. 1997;96:605–613. [PubMed] [Google Scholar]
- 60.Bombeli T, Karsan A, Tait JF, Harlan JM. Apoptotic vascular endothelial cells become procoagulant. Blood. 1997;89:2429–2442. [PubMed] [Google Scholar]
- 61.Kwiatkowski D. Tumor necrosis factor, fever, and fatality in falciparum malaria. Immunol Lett. 1990;25:213–216. doi: 10.1016/0165-2478(90)90117-9. [DOI] [PubMed] [Google Scholar]
- 62.Bierhaus A, Hemmer CJ, Mackman N, Kutob R, Ziegler R, Dietrich M, Nawroth PP. Antiparasitic treatment of patients with P. falciparum malaria reduces the ability of patient serum to induce tissue factor by decreasing NF-kappa B activation. Thromb Haemost. 1995;73:39–48. [PubMed] [Google Scholar]
- 63.Imamura T, Sugiyama T, Cuevas LE, Makunde R, Nakamura S. Expression of tissue factor, the clotting initiator, on macrophages in Plasmodium falciparum-infected placentas. J Infect Dis. 2002;186:436–440. doi: 10.1086/341507. [DOI] [PubMed] [Google Scholar]
- 64.Rogerson SJ, Pollina E, Getachew A, Tadesse E, Lema VM, Molyneux ME. Placental monocyte infiltrates in response to Plasmodium falciparum malaria infection and their association with adverse pregnancy outcomes. Am J Trop Med Hyg. 2000;68:115–119. [PubMed] [Google Scholar]
- 65.Fiuza C, Bustin M, Talwar S, Tropea M, Gerstenberger E, Shelhamer JH, Suffredini AF. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood. 2003;101:2652–2660. doi: 10.1182/blood-2002-05-1300. [DOI] [PubMed] [Google Scholar]
- 66.Alleva LM, Yang H, Tracey KJ, Clark IA. High mobility group box 1 (HMGB1) protein: possible amplification signal in the pathogenesis of falciparum malaria. Trans R Soc Trop Med Hyg. 2005;99:171–174. doi: 10.1016/j.trstmh.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 67.Liu Y, Pelekanakis K, Woolkalis MJ. Thrombin and tumor necrosis factor α synergistically stimulate tissue factor expression in human endothelial cells. Regulation through c-Fos and c-Jun. J Biol Chem. 2004;270:36142–36147. doi: 10.1074/jbc.M405039200. [DOI] [PubMed] [Google Scholar]
- 68.Hezi-Yamit A, Wong PW, Bien-Ly N, Komuves LG, Prasad KS, Phillips DR, Sinha U. Synergistic induction of tissue factor by coagulation factor Xa and TNF: evidence for involvement of negative regulatory signaling cascades. Proc Natl Acad Sci USA. 2005;23(102):12077–11282. doi: 10.1073/pnas.0504526102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Seydel KB, Milner DA, Jr, Kamiza SB, Molyneux ME, Taylor TE. The distribution and intensity of parasite sequestration in comatose Malawian children. J Infect Dis. 2006;194:208–205. doi: 10.1086/505078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Charo IF, Taubman MB. Chemokines in the pathogenesis of vascular disease. Circ Res. 2004;95:858–866. doi: 10.1161/01.RES.0000146672.10582.17. [DOI] [PubMed] [Google Scholar]
- 71.Mackman N. Role of tissue factor in hemostasis, thrombosis, and vascular development. Arterioscler Thromb Vasc Biol. 2004;24:1015–1022. doi: 10.1161/01.ATV.0000130465.23430.74. [DOI] [PubMed] [Google Scholar]
- 72.Hebbel RP, Osarogiagbon R, Kaul D. The endothelial biology of sickle cell disease: inflammation and a chronic vasculopathy. Microcirculation. 2004;11:129–151. [PubMed] [Google Scholar]
- 73.Geisbert TW, Hensley LE, Jahrling PB, Larsen T, Geisbert JB, Paragas J, Young HA, Fredeking TM, Rote WE, Vlasuk GP. Treatment of Ebola virus infection with a recombinant inhibitor of factor VIIa/tissue factor: a study in rhesus monkeys. Lancet. 2003;362:1953–1958. doi: 10.1016/S0140-6736(03)15012-X. [DOI] [PubMed] [Google Scholar]
- 74.Ruf W. Emerging roles of tissue factor in viral hemorrhagic fever. Trends Immunol. 2004;25:461–464. doi: 10.1016/j.it.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 75.Kendrick BJ, Gray AG, Pickworth A, Watters MP. Drotrecogin alfa (activated) in severe falciparum malaria. Anaesthesia. 2006;61:899–902. doi: 10.1111/j.1365-2044.2006.04752.x. [DOI] [PubMed] [Google Scholar]
- 76.Wellems TE, Miller LH. Two worlds of malaria. N Engl J Med. 2003;1349:1496–1498. doi: 10.1056/NEJMp038127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.