

Abstract
Background and Objectives
Functional characterization of heterozygous TERC (telomerase RNA component) and TERT (telomerase reverse transcriptase) mutations found in autosomal dominant dyskeratosis congenita (DC) and aplastic anemia (AA) shows that telomerase function is defective and that this is associated with short telomeres. This leads to reduced cell longevity with maximal impact on tissues with high proliferate potential. The aim of this study was to establish the role of TERC in the pathophysiology of uncharacterized patients with AA with some features of DC.
Design and Methods
The TERC gene was screened for mutations by denaturing high performance liquid chromatography. To determine the functional significance of TERC mutations telomerase activity was assessed in an in vitro (TRAP) assay and telomere length of patients’ samples was determined using Southern blot analysis.
Results
This study led to the identification of four novel TERC mutations (G178A, C180T, Δ52-86 and G2C) and a recurrent TERC mutation (Δ110-113GACT).
Interpretation and Conclusions
Two of the de novo TERC mutations (G178A and C180T) found uniquely produce a clinical phenotype in the first generation, differing from previously published cases in which individuals in the first generation are usually asymptomatic. Curiously these mutations are located near the triple-helix domain of TERC. We also observed that the recurrent Δ110-113GACT can present with AA, myelodysplasia or leukemia. The Δ52-86 is associated with varied phenotypes including pulmonary disease (pulmonary fibrosis) as the first presentation. In summary, this study reports the functional characterization of several novel TERC mutations associated with varied hematologic and extra-hematologic presentations.
Keywords: aplastic anemia, de novo, dyskeratosis congenita, haplo-insufficiency, telomerase, TERC
Dyskeratosis congenita (DC), a rare bone marrow (BM) failure syndrome, has been linked to mutations in DKC1, TERC (telomerase RNA component) and TERT (telomerase reverse transcriptase).1-3 As these molecules associate to form the telomerase complex, it suggests that the various forms of DC are due to defective telomerase function, which results in loss of cell longevity in highly proliferative tissues.4
Heterozygous TERC mutations have been identified in a subset of patients with autosomal dominant DC, as well as other related BM failure syndromes, suggesting that disruption of the telomerase complex results in defective hematopoiesis.5 Previous reports have highlighted that the deletions and small base-pair substitutions in TERC impede telomerase function due to haplo-insufficiency.6-8 This occurs from either direct loss of catalytic activity or through dissociation of the telomerase complex itself. Subsequent studies showed that due to disease anticipation, heterogeneous inheritance patterns and variable penetrance, families with diverse clinical features can be linked to a TERC mutation.9 While there has been speculation as to the mechanism behind each defective telomerase complex, it has been suggested that disease anticipation occurs due to the gradual erosion of the telomeres in subsequent generations.10,11
In this paper, we report on the functional characterization of several novel TERC mutations, including a recurring 4bp pseudoknot deletion and two de novo substitutions associated with clinical features in the first generation. The latter observation is unique to these families and has not been observed with other TERC mutations published to date. We also observed that one of the mutations was primarily associated with pulmonary disease in some of the affected individuals. This study highlights that in addition to previously documented hematologic pathology, TERC mutations may present with disease features due to pathology in non-hematopoietic tissues.
Design and Methods
Screening of TERC in patients with AA and related syndromes
Clinical information was collected from many patients who have AA with features overlapping those of DC. These studies have been approved by the Local Research Ethics Committee and informed consent was gained in accordance with the Helsinki Declaration. TERC was screened by denaturing high performance liquid chromatography analysis and abnormal patterns were subjected to direct sequence analysis as previously described.9 Any mutations identified were confirmed by either sequencing the reverse strand or by re-amplification and restriction enzyme digestion using sites that are created or destroyed by the presence of the TERC mutation in question.
Telomere length measurement
Telomere length was measured as previously described.12 A linear regression line was calculated for telomere length against age in unaffected siblings and spouses in families in which DKC1 mutations have been characterized. This value was then used to determine the age-adjusted telomere length of affected individuals by expressing the difference between the observed length and the predicted telomere length from the linear regression line (Δtel) as previously described.13
Micro-satellite analysis
Paternity was assessed through the analysis of ten short tandem repeat (STR) loci (FGA, VWA, TH01, D13S1358, D16S539, D2S1338, D8S1179, D21S11, D18S51 and D19S433) provided within the AmpFlSTR SGM Plus PCR amplification kit (Applied Biosystems). The tetranucloetide STR loci were amplified in a single polymerase chain reaction (PCR), separated on a 3130xl Genetic Analyzer and visualised using GeneMapper software (Applied Biosystems).
TERC plasmid constructs and mutagenesis
Wild type (WT) TERC and TERT plasmids were constructed as previously described.7 TERC mutations were produced by a two-stage PCR approach7 or by using the QuikChange site-directed mutagenesis kit (Stratagene, CA, USA). Complementary overlapping primers were designed (Table 1) for each mutation and were added to 1x reaction buffer, 10 ng of WT TERC, 2% dNTP mix, 6% QuikSolution mix and 1.25 units of PfuTurbo DNA polymerase. Each reaction was denatured at 95°C for 30secs, cycled 14 times at 95°C for 30 secs, 55°C for 1 min, and 68°C for 9 min and completed with an extension cycle for 7 min at 68°C. Competent cells were transformed with DpnI-treated DNA following the manufacturer’s instructions. Resulting colonies from each transformation were selected for plasmid DNA extraction (Qiagen, CA, USA) and the TERC sequence of each construct was verified.
Table 1.
Primers used during TERC plasmid mutagenesis
| TERC mutation 1 | Sequence 2 |
|---|---|
| c.180C→T (C180T) | caaacaaaaaatgtTagctgctggcccgt acgggccagcagctAacattttttgtttg |
| c.112C→T (C112T) complement to c.178G?A |
gtttttctcgctgaTtttcagcgggc gcccgctgaaaAtcagcgagaaaaac |
| c.110G→A (G110A) complement to c.180C?T |
gtttttctcgctAactttcagcgggc gcccgctgaaagtTagcgagaaaaac |
| c.2G→C (G2C) | gcagcgcaccgCgttgcggagggt accctccgcaacGcggtgcgctgc |
Nucleotide number in TERC gene where G of the initial RNA sequence GGG is depicted as number 1
Primer sequence 5′→3′ with the forward primer above the reverse primer. The mutated base in each primer is highlighted in bold.
Telomerase repeat amplification protocol (TRAP) analysis in transfected WI-38 VA13 cells
WI-38 VA13 cells were transfected and split into 1x CHAPS buffer for TRAP analysis and 1x lysis buffer for luciferase analysis as previously described.7 TRAP lysates were analyzed by using the TRAPese telomerase detection kit (Intergen, NY, USA).7 The TRAP percentages shown are approximate values derived from comparing serial dilutions from WT samples. The luciferase lysates were analyzed for transcription efficiency using the Renilla luciferase assay protocol (Promega, Southampton, UK) as previously described.7
Results
Identification of de novo autosomal dominant TERC mutations
The index case of family A presented at 2 years old with abdominal pain and easy bruising. He also had mild developmental retardation with delayed speech attributed to a hearing impairment secondary to recurring otitis media, for which he underwent grommet insertion. Investigations demonstrated severe BM hypocellularity. He had a normal 46XY karyotype and normal chromosomal breakage scores with DNA cross-linking agents. His parents were asymptomatic so he was diagnosed as having AA (Figure 1A; Table 2). The index case of family B also presented with features of AA. He had avascular necrosis of the hips, patches of hypo-pigmentation on his back and gray hair from the age of 13 years old, but no other signs of DC. He responded poorly to immunosuppressive treatment (anti-lymphocyte globulin (ALG) and cyclosporine) but has had some response to oxymetholone. His parents were clinically normal (Figure 1A; Table 2). TERC screening found two novel heterozygous substitutions c.178G→A (G178A) and c.180C→T (C180T) in the index cases of family A and B, respectively (Figure 1A). Both these mutations are in the P3 pseudoknot stem of TERC. Subsequent analysis of the TERC gene in both sets of parents showed that they were normal with respect to these mutations. Family histories from both cases found no evidence of non-paternity. Analysis of ten STR loci in the two families showed consistent segregation of alleles. This indicated a relative chance of paternity of over 99.999% in both cases, providing virtual proof of the declared paternity. We, therefore, concluded that the TERC mutation arose as a de novo event in both of these cases. G178A was also detected in the buccal smear of the index case indicating that this mutation is constitutional. These mutations were not identified in 484 healthy subjects (67 of mixed ethnicity, 147 Caucasian, 170 West African and 100 Turkish) or in any other reported TERC screens.8,9,14-16
Figure 1.
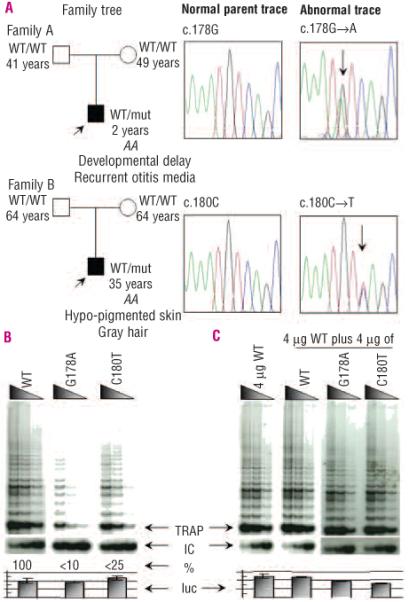
TERC mutation identification and analysis for Families A and B. A. Sequence traces depicting the normal and abnormal heterozygous TERC traces of G178A and C180T for families A and B, respectively. Age (years), TERC allele status (WT: as published TERC sequence; mut: nucleotide substitution as indicated in the sequence trace) and relevant family clinical history are indicated in the family trees in which the index cases (black squares) are indicated by an arrow and the parents appear asymptomatic (white squares or circles). B. Reconstituted TRAP assays showed that G178A and C180T reduce in vitro telomerase activity to less than 10% and 25% of WT control levels, respectively. C. In mixing experiments, activity did not drop below 50% of WT activity, suggesting no dominant negative effect for these two de novo heterozygous TERC mutations. The arrows denote the start of the TRAP ladder, the corresponding internal control (IC), the amount of activity in relationship to the WT TRAP control (%) and the levels of luciferase (luc) to control for the corresponding TRAP experiment. Serial dilutions of each transfected lysate were assayed as described in the methods section.
Table 2.
Hematologic data (at their last clinic visit) from affected cases with TERC mutations
|
Family and
TERC mutation |
Age
(years) |
Hb
(g/dL) |
MCV
(fL) |
HbF
(%) |
PLT
(×109/L) |
WCC
(×109/L) |
Neut
(×109/L) |
|---|---|---|---|---|---|---|---|
| A: G178A | 5 | 10.7 | 97.6 | 5.7 | 29 | 6.6 | 2.1 |
| B: C180T1 | 35 | 13.9 | 117 | – | 37 | 2.8 | 1.5 |
| C: ΔD110-113GACT | 17 | 11.7 | 104 | 3.3 | 90 | 3.4 | 1.6 |
| D: ΔD53-872 | 26 | 10.8 | 125 | 2.9 | 35 | 3.6 | 2.1 |
| E: G2C | 27 | 8.3 | 117 | 3.0 | 35 | 2.4 | 0.9 |
| Normal ranges | |||||||
| child | 2-6 yrs |
12.5±1.5 | 81±6 | <1 | 345±145 | 10.0±5.0 | 15.0±3.0 |
| adult | |||||||
| males | 13.5±1.5 | 92±9 | <1 | 280±130 | 7.0±3.0 | 4.5±2.5 | |
| females | 15.0 ± 2.0 | ||||||
Hb: hemoglobin; HbF: fetal Hb; MCV: mean corpuscular volume; Neut: neutrophil; PLT: platelets; WCC: white cell count
on oxymetholone
sister of the index case.
De novo autosomal dominant TERC mutations reduce telomerase activity by haplo-insufficiency
Telomere length measurements established that the two index cases had reduced Δtel values of −2.16 and −4.51 for G178A and C180T, respectively (Figure 2). These results placed the index case of both family A and B below the 90% deviation which suggests that these telomere lengths are below the 5th percentile when compared to the normal control panel. The telomere lengths of the parents of both families were located within the 68% percentile or higher which is between the 50th and 68th percentile when compared to the normal control panel (Figure 2).
Figure 2.
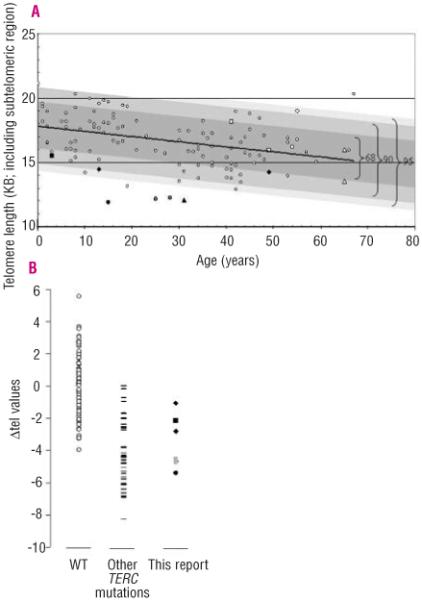
Telomere lengths of investigated families compared to those of normal controls. A. Normal subjects are indicated by small white circles. The different families are represented as follows: G178A affected and normal parents, black and white squares; C180T affected and normal parents, black and white triangles; 110-113delGACT affected, black circle; 53-87del affected and normal parent, black and white diamonds; G2C affected and asymptomatic sister and mother, gray and white circles respectively. The line of best fit through this normal range is shown as a black line which corresponds to the equation Y=17.821 – 0.0407X. Deviation from the best-fit-line is highlighted as a dark gray box for 68%, a lighter gray box for 90% and the palest grey box for 95%. Therefore the index cases lie on or below the 90% deviation range, which represents the 5th percentile when compared to the panel of normal controls. B. The Δtel values from normal subjects from panel A (n=112) are represented on a linear graph and compared to the telomere lengths from previously reported patients with TERC mutations (n=38; black lines) and the patients from this report (n=7). The patients in this report have similar Δtel values to those of other patients with TERC mutations. Other labels are as in panel A.
The functional consequences of these two novel TERC substitutions were investigated by telomerase reconstitution analysis in telomerase-negative cells. The telomerase activity was found to be reduced to approximately 10% and 25% of WT control levels for G178A and C180T, respectively (Figure 1B). Furthermore, no significant evidence of a dominant negative effect was found when either TERC mutation was mixed in equal concentration with WT TERC (Figure 1C). These data, therefore, show that the presence of either novel substitution is capable of reducing telomerase activity via haplo-insufficiency and leads to prematurely reduced telomere lengths.
Additional functional characterization of de novo TERC mutations shows that the primary sequence is as critical as the secondary structure for normal activity
In previous investigations of TERC stem mutations, the reduction of telomerase activity was restored when the stem structure was reconstituted.7,8,15 When we investigated the two de novo TERC stem mutations, we observed that the two mutations acted in a different manner (Figure 3). G178A had significantly reduced telomerase activity compared to WT controls (Figure 1B, Figure 3), while telomerase activity was close to WT levels when either the complementary stem mutation c.112C→T (C112T) was present alone (Stem in Figure 3) or when both of these mutations were present in cis, restoring the stem structure (Double in Figure 3). For C180T, the presence of the original mutation, the stem mutation (c.110G→A (G110A)) or both mutations in cis (Double in Figure 3) showed reduced telomerase activity in comparison to WT controls. This suggests that the primary sequence at positions G178 and C180 is as critical as the intact secondary stem structure at this location.
Figure 3.
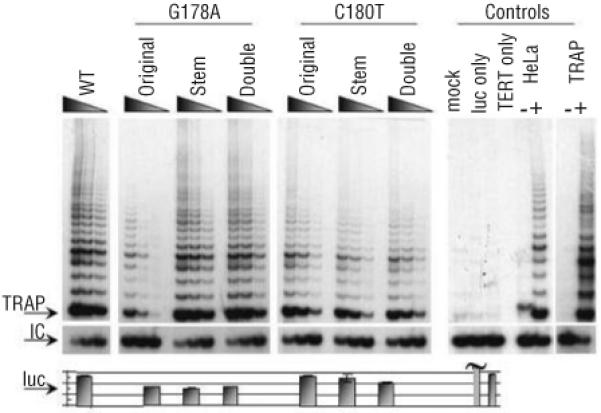
Reconstituted telomerase activity of disrupted and restored pseudoknot helices. Original represents the TERC mutation found in the index case, Stem represents the mutation formed on the corresponding stem opposite the original mutation and Double represents the TERC RNA when both the original and stem mutation are present together in the same molecule, reconstituting the stem but with the mutated primary sequence. Other labels are as in Figure 1.
Other novel TERC mutations in patients with diverse clinical presentations
In family C, a girl presented at 6 years old with chronic iron deficiency anemia and dysphagia. Her BM was markedly hypocellular with no evidence of myelodysplasia (MDS) associated with peripheral cytopenia (Table 2). This patient had congenital deafness, learning difficulties, short stature, scoliosis, dental abnormalities and dental caries. She had normal nails but spider naevi on her neck. Her family history is noteworthy: two of her four brothers had congenital deafness and schizophrenia, one of whom had AA and died from carcinoma of the tongue. A third brother died following a BM transplant for transforming MDS. Her fourth brother has normal hearing, a normal blood count and is not schizophrenic. Her mother was also deaf and her father died of acute myeloid leukemia (AML) (Figure 4). Fanconi anemia was excluded in the index case by diepoxybutane stress testing and this family was given the diagnosis of familial MDS/AML.
Figure 4.
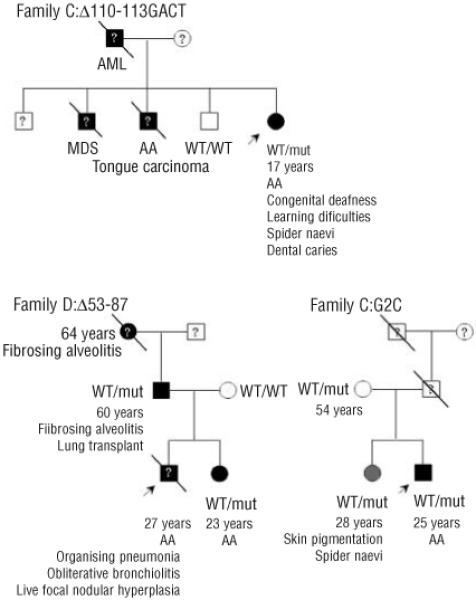
Clinical history and inheritance of TERC mutations in families C, D and E. The arrow denotes the index case and a line through the person represented indicates that the person has died. Black squares represent an AA/DC phenotype while gray circles represent borderline phenotypes. White squares and circles indicate asymptomatic people. ? denotes that a sample was not available for TERC mutation screening. Other labels are as in Figure 1.
As this patient had been fostered, it was not possible to perform genetic analysis on all family members apart from her fourth brother. The index case was found to be heterozygous for a c.110-113delGACT deletion located within the conserved helix P3 stem domain of the TERC pseudoknot. This TERC mutation has not been identified among healthy individuals including the fourth asymptomatic brother but has been observed in another family with autosomal dominant DC.17 The family was described to have non-severe AA, elphin appearance and significantly reduced telomerase activity via haplo-insufficiency.6,7,18 The index case of family C has a reduced Δtel value of −5.32, finding that is complementary to the results of the TRAP analysis (Figure 5A). When compared to the normal telomere length range, this index case is located well below the 95% deviation range which suggests that this telomere length is within the 1st and 5th percentile when compared to the normal control panel (Figure 2).
Figure 5.
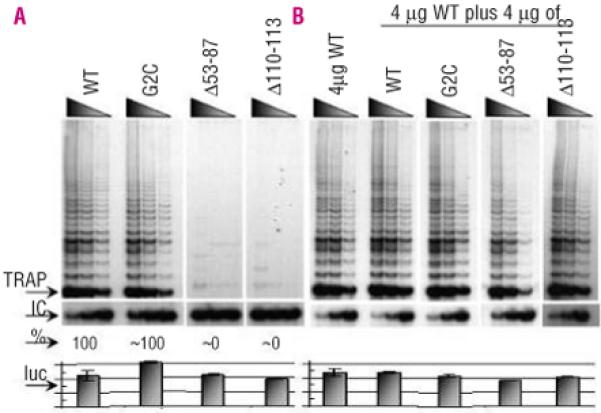
Reconstituted telomerase activity of other TERC mutations studied in vitro. A. Reconstituted TRAP assays show that 53-87del and 110-113delGACT mutations reduce in vitro telomerase activity to near 0% of that of WT controls while the G2C mutation appears to have near normal activity. B. In mixing experiments, there was no observed drop below 50% WT activity, suggesting no dominant negative effect for these three heterozygous TERC mutations. Other labels are as in Figure 1.
The index case in Family D presented with AA in childhood. His blood count was maintained with oxymetholone for several years and he was diagnosed as having constitutional AA. Eventually he became refractory to oxymetholone and underwent reduced intensity matched unrelated BM transplant at the age of 25. This was associated with complications (recurrent gastrointestinal bleeding, ascites, infections and pulmonary complications) and he died 2 years after the transplant. Post-mortem examination showed an abnormal liver with focal nodular hyperplasia and oesophageal varices. Pulmonary histology showed cryptogenic organizing pneumonia and obliterative bronchiolitis. The patient’s father had cryptogenic fibrosing alveolitis for which he had a successful heart/lung transplantation. The patient’s paternal grandmother had a history of anaemia and was diagnosed with fibrosing alveolitis which was treated unsuccessfully with steroids and other immuno-suppressive agents. She was described as having dry skin and died at the age of 64 from respiratory failure. The index case’s younger sister also developed AA (Figure 4; Table 2). The 35nt index case’s sister and father were found to have a novel heterozygous deletion spanning nucleotides 53 to 87 of TERC, while his mother was normal. This deletion removes the highly conserved alignment region of the RNA template as well as the P2a.1 stem of the pseudoknot. TRAP analysis of this deletion showed that telomerase activity was abolished in comparison to that in normal controls with no evidence of a dominant negative effect on WT TERC (Figure 5). Due to the nature of this deletion, it is not possible to determine whether the loss of telomerase activity is due to the inability of the resulting complex to align itself on the telomere and/or the loss of the P2a.1 stem. Telomere length data suggest that this deletion is capable of reducing telomerase function in vivo as the father has c̷tel values of −1.60 and the sister has Δtel values of −2.82 in comparison to the normal population (within 68% and 90% deviation, respectively, which relates to the 5th percentile, in Figure 2).
The index case from family E presented with non-severe AA at the age of 25 years (Table 2). He had no obvious clinical features suggestive of DC but was put forward for a TERC screen in an attempt to rule out this diagnosis since he had failed to respond to immuno-suppressive therapy. His blood count responsed to oxymetholone treatment. His sister and mother were both asymptomatic with normal blood counts although his sister had some patches of skin hyper-pigmentation and spider naevi. His father died at 51 years old from a myocardial infarct (Figure 4).
TERC screening showed that the index case, mother and sister were heterozygous for a novel c.2G→C (G2C) substitution. Telomere length analysis in family E showed that only the two siblings have short telomeres that are below the 5th percentile when compared to the normal control panel (Figure 2). TRAP analysis revealed that this substitution had apparent WT activity when compared to normal controls, with no evidence of a dominant negative effect (Figure 5). The TRAP data suggest that the G2C mutation is a rare polymorphism although the possibility that it could act as a disease risk factor cannot be completely ruled out. In this respect, it is intriguing that the asymptomatic sister, like the index case, has very short telomeres.
Discussion
We describe the functional characterization of four novel heterozygous mutations in TERC: G178A, C180T, 53-87del, G2C and a recurrent TERC mutation (110-113delGACT). G178A and C180T are the first reported de novo TERC mutations that result in a definitive pathophysiology of AA with indications of additional characteristics of DC (Figure 1A). As with previously reported TERC mutations identified in AA/DC families, telomerase functional analysis showed that both of these substitutions were capable of reducing telomerase activity in reconstituted telomerase assays (Figure 1B). The surprising aspect of these results however was the unique observation that these mutations produced a phenotype in the first generation. This feature has not been previously described in TERC families, in which disease anticipation has been observed.10 One possible explanation of this new phenomenon was that these particular TERC mutations exerted a dominant-negative effect on telomerase activity. This theory was rejected since further analysis found that the defective telomerase induced by these substitutions appeared to result from haplo-insufficiency in these experiments (Figure 1C). Therefore the severity of clinical phenotype in the first generation in these two case studies is not easily explained. One possible explanation is that although telomerase activity can be reconstituted in vitro, these mutations still have some dominant negative effect on telomere maintenance in vivo. Further studies are required to determine whether this is the case.
The G178A and C180T mutations, along with the previously reported 96-97delCT, GC107-8AG, 110-113delGACT, C116T, and A117C, lie close to each other both in the primary sequence and the secondary structure of TERC. This stem region has over 80% shared conservation with other TERC RNA species, as defined by mutagenesis and phylogenetic analysis, and is stabilized by a triple helix that surrounds the two stems (Figure 6).18-20 Although the mutations are contained within the minimal pseudoknot domain, they appear to function in different ways. One report suggests that the P3 helix, and not the intraloop base-pairings, is essential for telomerase activity, where the sequence of the J2b/3 loop rather than its base-pairing ability within the loop region is required for activity.21 As these two substitutions localize to an area of the pseudoknot domain that involves a proposed triple helix structure, it is possible that the physical location of these novel substitutions disrupts molecular switching in a different way to that of previously reported TERC mutations (Figure 6).
Figure 6.
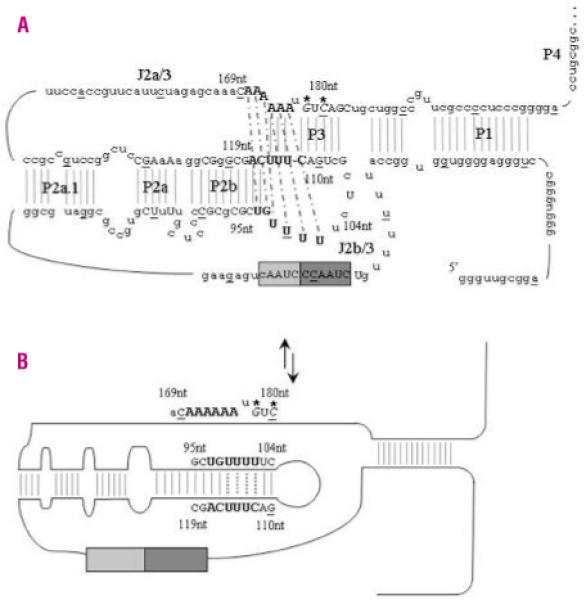
TERC pseudoknot structure and proposed molecular switch model. A. The secondary structure of the TERC pseudoknot has been elucidated by functional analysis. Every tenth base of the 451bp molecule is underlined and <80% vertebrate nucleotide conservation is denoted by upper case letters. The RNA template contains partial repeats of the telomere repeat known as the alignment region (lighter gray box). The larger dark grey box contains the templating region which is utilized to ensure correct base-pairing and correct translocation and re-alignment for the next round of template synthesis. Critical bases involved in the triple helix structure are highlighted in bold. Thermodynamics and mutational analysis suggest that nucleotides 171-172AA form a Hoogsteen base-pair with 97-98UG, which are helically-bound by conventional Watson-Crick base-pairing to 116-117CA. Nucleotides 100-102UUU then form a Hoogsteen base-pair with 174-176AAA, which are helically-bound by conventional Watson-Crick base-pairing to 113-115UUU. Nucleotides 99U and 173A can then pair up between these two triple helices to bridge the physical gap. The Watson-Crick base-pairing is denoted by gray lines while the dashed black lines represent the Hoogsteen basepairing. The de novo mutations described in this paper are in italics and are highlighted with *. B. While the two novel de novo mutations are not directly involved in the loop formed during the proposed molecular switch, the complementary nucleotides G110 and C112 are critical to the correct formation of the new TERC RNA structure.
Another hypothesis is that functional differences within the P3 stem region could correlate with the ability of the resulting mutant TERC to either dimerize with other TERC molecules or telomerase-associated proteins. In vitro research shows that human telomerase appears to be highly processive such that a single telomerase enzyme usually continues to extend a single telomere rather than the enzyme being released at the end of each round of repeat extension.22 It has been hypothesized that the conserved TERC pseudoknot structure is required for template recognition and for defining the template boundaries for telomere synthesis.23,24 TERC control over processivity could occur through base pair interactions between the telomeric products and TERC during the process of translocation25,26 or through protein- TERC-protein interactions.27,28 This difference in clinical presentation and telomerase restoration in vitro could be due to fact that G178 and C180 are localized to the internalized strand of the triple helix, in comparison to nucleotides G107, C108, C116, and A117 which rest on an external strand of the triple helix (Figure 6). Further studies are required to fully elucidate TERC pseudoknot function and the resulting telomerase complex activity in the presence of WT and/or TERC mutations. The identification of template and pseudoknot TERC mutations (53-87del and 110-113delGACT) enhances the observation that disruption of telomerase activity and resulting haplo-insufficiency can cause a variety of clinical presentations. Single-point substitutions, deletions, insertions and complete substitutions have been previously made in ciliate, yeast and human TERC templates. In some cases template mutations appear to alter the in vitro and in vivo nature of the telomerase enzyme.29 The 53-87del TERC mutation identified in family D removes a significant proportion of the template and pseudoknot domain in affected members. Even with such a dramatic loss of TERC structure, affected members of this family initially presented with pulmonary fibrosis (grandmother and father of index case) rather than the classical muco-cutaneous features of DC. The finding of pulmonary disease as the primary presentation of disease in two members of this family suggests that screening for TERC mutation should be conducted in other patients presenting with cryptogenic/idiopathic pulmonary fibrosis.3
A surprising result from this subset of patients was the discovery of a recurring pseudoknot deletion, 110-113delGACT, in family C. Comparisons between a previously reported family and family C in this study show that this 4bp deletion in TERC can induce a variety of clinical phenotypes, ranging from AA to MDS or AML. These findings suggest that it might be useful to study the TERC gene, not only in patients presenting with AA or MDS, but also in patients presenting with acute leukemia, especially since 10-30% of AA patients develop AML and/or MDS.30 This is the second report of a conclusively pathogenic recurring TERC mutation. The C116T mutation has also been reported in two apparently unrelated families in which the index cases presented with thrombocytopenia or were diagnosed as having acquired AA.14,31 The only other recurring TERC mutations that have been described to date are G58A and G228A, which have been found to be polymorphic, and C-99G, the functional significance of which is not entirely clear.16,31-33 This highlights the diversity of clinical phenotypes resulting from TERC mutations. This also raises the semantic question of whether mutations in TERC result in DC, whose clinical phenotype can vary between cryptic presentations similar to AA through to more severe disease presentations, or whether TERC mutations result in a variety of BM failure syndromes? The patients reported here do not present with the classical triad of muco-cutaneous features and therefore would not be classified as having DC based on clinical criteria. It is possible that these additional clinical features will present over time, but this is not always the case.
The G2C change identified in family E had no observable effect on telomerase activity in vitro (Figure 5). It is located in the 5′ region of TERC, which shows low sequence conservation among vertebrates. Prior to this report, five other TERC mutations had been described that appeared to have no effect on TRAP activity. Two of these (G58A and G228A) are polymorphic, but three others (28-34del, A37G and G450A) have been observed only once in patients with AA, DC (in trans to a deleterious 216-229del mutation) and severe AA. Although it may well be that these TERC mutations are rare non-pathogenic variants, it is still possible that they could be disease risk factors that might be important particularly when associated with other uncharacterized genetic mutations not detected by the in vitro TRAP assays described here. These data highlight the importance of including functional analysis of TERC mutations identified through mutation screening in patient groups.
In conclusion, this study highlights the importance of functional characterization of identified TERC mutations in evaluating their pathogenic significance. The study also shows that constitutional TERC mutations can present with clinical features in the first generation and that the mutations are associated with highly variable clinical phenotypes. These overlap with classical DC and AA but may also include leukemia and pulmonary fibrosis as the first clinical presentation.
Acknowledgments
We thank our colleagues Nahla Abbas, Natalie Killeen, David Stevens and Kate Sullivan for their technical assistance as well as David Ballard and Denise Syndercombe-Court for micro-satellite analysis. We would also like to thank all the patients and their clinicians for supporting the Dyskeratosis Congenita Registry (DCR). Work in our laboratory is supported by the Wellcome Trust and the MRC.
Funding: the work submitted within this article was funded by the Wellcome Trust and MRC.
Footnotes
Conflict of Interest
The authors have no affiliations within the last five years with any companies, trade associations, unions or other groups that would have a direct financial interest in the subject matter or materials discussed in this manuscript.
References
- 1.Heiss NS, Knight SW, Vulliamy TJ, Klauck SM, Wiemann S, Mason PJ, et al. X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nat Genet. 1998;19:32–8. doi: 10.1038/ng0598-32. [DOI] [PubMed] [Google Scholar]
- 2.Vulliamy T, Marrone A, Goldman F, Dearlove A, Bessler M, Mason PJ, et al. The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita. Nature. 2001;413:432–5. doi: 10.1038/35096585. [DOI] [PubMed] [Google Scholar]
- 3.Armanios M, Chen JL, Chang YP, Brodsky RA, Hawkins A, Griffin CA, et al. Haploinsufficiency of telomerase reverse transcriptase leads to anticipation in autosomal dominant dyskeratosis congenita. Proc Natl Acad Sci USA. 2005;102:15960–4. doi: 10.1073/pnas.0508124102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen JL, Greider CW. Telomerase RNA structure and function: implications for dyskeratosis congenita. Trends Biochem Sci. 2004;29:183–92. doi: 10.1016/j.tibs.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 5.Marrone A, Dokal I. Dyskeratosis congenita: a disorder of telomerase deficiency and its relationship to other diseases. Expert Rev Derm. 2006;1:463–79. [Google Scholar]
- 6.Fu D, Collins K. Distinct biogenesis pathways for human telomerase RNA and H/ACA small nucleolar RNAs. Mol Cell. 2003;11:1361–72. doi: 10.1016/s1097-2765(03)00196-5. [DOI] [PubMed] [Google Scholar]
- 7.Marrone A, Stevens D, Vulliamy T, Dokal I, Mason PJ. Heterozygous telomerase RNA mutations found in dyskeratosis congenita and aplastic anemia reduce telomerase activity via haploinsufficiency. Blood. 2004;104:3936–42. doi: 10.1182/blood-2004-05-1829. [DOI] [PubMed] [Google Scholar]
- 8.Ly H, Calado RT, Allard P, Baerlocher GM, Lansdorp PM, Young NS, et al. Functional characterization of telomerase RNA variants found in patients with hematologic disorders. Blood. 2005;105:2332–9. doi: 10.1182/blood-2004-09-3659. [DOI] [PubMed] [Google Scholar]
- 9.Vulliamy TJ, Marrone A, Knight SW, Walne A, Mason PJ, Dokal I. Mutations in dyskeratosis congenita: their impact on telomere length and the diversity of clinical presentation. Blood. 2006;107:2680–5. doi: 10.1182/blood-2005-07-2622. [DOI] [PubMed] [Google Scholar]
- 10.Vulliamy T, Marrone A, Szydlo R, Walne A, Mason PJ, Dokal I. Disease anticipation is associated with progressive telomere shortening in families with dyskeratosis congenita due to mutations in TERC. Nat Genet. 2004;36:447–9. doi: 10.1038/ng1346. [DOI] [PubMed] [Google Scholar]
- 11.Marrone A, Walne A, Dokal I. Dyskeratosis congenita: telomerase, telomeres and anticipation. Curr Opin Genet Dev. 2005;15:249–57. doi: 10.1016/j.gde.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 12.Notaro R, Cimmino A, Tabarini D, Rotoli B, Luzzatto L. In vivo telomere dynamics of human hematopoietic stem cells. Proc Natl Acad Sci USA. 1997;94:13782–5. doi: 10.1073/pnas.94.25.13782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brummendorf TH, Maciejewski JP, Mak J, Young NS, Lansdorp PM. Telomere length in leukocyte subpopulations of patients with aplastic anemia. Blood. 2001;97:895–900. doi: 10.1182/blood.v97.4.895. [DOI] [PubMed] [Google Scholar]
- 14.Fogarty PF, Yamaguchi H, Wiestner A, Baerlocher GM, Sloand E, Zeng WS, et al. Late presentation of dyskeratosis congenita as apparently acquired aplastic anaemia due to mutations in telomerase RNA. Lancet. 2003;362:1628–30. doi: 10.1016/S0140-6736(03)14797-6. [DOI] [PubMed] [Google Scholar]
- 15.Yamaguchi H, Baerlocher GM, Lansdorp PM, Chanock SJ, Nunez O, Sloand E, et al. Mutations of the human telomerase RNA gene (TERC) in aplastic anemia and myelodysplastic syndrome. Blood. 2003;102:916–8. doi: 10.1182/blood-2003-01-0335. [DOI] [PubMed] [Google Scholar]
- 16.Field JJ, Mason PJ, An P, Kasai Y, McLellan M, Jaeger S, et al. Low frequency of telomerase RNA mutations among children with aplastic anemia or myelodysplastic syndrome. J Pediatr Hematol Oncol. 2006;28:450–3. doi: 10.1097/01.mph.0000212952.58597.84. [DOI] [PubMed] [Google Scholar]
- 17.Vulliamy T, Marrone A, Dokal I, Mason PJ. Association between aplastic anaemia and mutations in telomerase RNA. Lancet. 2002;359:2168–70. doi: 10.1016/S0140-6736(02)09087-6. [DOI] [PubMed] [Google Scholar]
- 18.Ly H, Blackburn EH, Parslow TG. Comprehensive structure-function analysis of the core domain of human telomerase RNA. Mol Cell Biol. 2003;23:6849–56. doi: 10.1128/MCB.23.19.6849-6856.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen JL, Blasco MA, Greider CW. Secondary structure of vertebrate telomerase RNA. Cell. 2000;100:503–14. doi: 10.1016/s0092-8674(00)80687-x. [DOI] [PubMed] [Google Scholar]
- 20.Theimer CA, Blois CA, Feigon J. Structure of the human telomerase RNA pseudoknot reveals conserved tertiary interactions essential for function. Mol Cell. 2005;17:671–82. doi: 10.1016/j.molcel.2005.01.017. [DOI] [PubMed] [Google Scholar]
- 21.Chen JL, Greider CW. Functional analysis of the pseudoknot structure in human telomerase RNA. Proc Natl Acad Sci USA. 2005;102:8080–5. doi: 10.1073/pnas.0502259102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maine IP, Chen SF, Windle B. Effect of dGTP concentration on human and CHO telomerase. Biochemistry. 1999;38:15325–32. doi: 10.1021/bi991596+. [DOI] [PubMed] [Google Scholar]
- 23.Miller MC, Collins K. Telomerase recognizes its template by using an adjacent RNA motif. Proc Natl Acad Sci USA. 2002;99:6585–90. doi: 10.1073/pnas.102024699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen JL, Greider CW. Template boundary definition in mammalian telomerase. Genes Dev. 2003;17:2747–52. doi: 10.1101/gad.1140303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gavory G, Farrow M, Balasubramanian S. Minimum length requirement of the alignment domain of human telomerase RNA to sustain catalytic activity in vitro. Nucleic Acids Res. 2002;30:4470–80. doi: 10.1093/nar/gkf575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen JL, Greider CW. Determinants in mammalian telomerase RNA that mediate enzyme processivity and cross-species incompatibility. EMBO J. 2003;22:304–14. doi: 10.1093/emboj/cdg024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lai CK, Miller MC, Collins K. Roles for RNA in telomerase nucleotide and repeat addition processivity. Mol Cell. 2003;11:1673–83. doi: 10.1016/s1097-2765(03)00232-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moriarty TJ, Marie-Egyptienne DT, Autexier C. Functional organization of repeat addition processivity and DNA synthesis determinants in the human telomerase multimer. Mol Cell Biol. 2004;24:3720–33. doi: 10.1128/MCB.24.9.3720-3733.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Drosopoulos WC, Direnzo R, Prasad VR. Human telomerase RNA template sequence is a determinant of telomere repeat extension rate. J Biol Chem. 2005;280:32801–10. doi: 10.1074/jbc.M506319200. [DOI] [PubMed] [Google Scholar]
- 30.Socie G, Rosenfeld S, Frickhofen N, Gluckman E, Tichelli A. Late clonal diseases of treated aplastic anemia. Semin Hematol. 2000;37:91–101. [PubMed] [Google Scholar]
- 31.Ortmann CA, Niemeyer CM, Wawer A, Ebell W, Baumann I, Kratz CP. TERC mutations in children with refractory cytopenia. Haematologica. 2006;91:707–8. [PubMed] [Google Scholar]
- 32.Wilson DB, Ivanovich J, Whelan A, Goodfellow PJ, Bessler M. Human telomerase RNA mutations and bone marrow failure. Lancet. 2003;361:1993–4. doi: 10.1016/S0140-6736(03)13575-1. [DOI] [PubMed] [Google Scholar]
- 33.Keith WN, Vulliamy T, Zhao J, Ar C, Erzik C, Bilsland A, et al. A mutation in a functional Sp1 binding site of the telomerase RNA gene (hTERC) promoter in a patient with paroxysmal nocturnal haemoglobinuria. BMC Blood Disord. 2004;4:3. doi: 10.1186/1471-2326-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]