

Abstract
Pseudomonas aeruginosa PA4872 was identified by sequence analysis as a structurally and functionally novel member of the PEP mutase/isocitrate lyase superfamily and therefore targeted for investigation. Substrate screens ruled out overlap with known catalytic functions of superfamily members. The crystal structure of PA4872 in complex with oxalate (a stable analog of the shared family α-oxyanion carboxylate intermediate/transition state) and Mg2+ was determined at 1.9 Å resolution. As with other PEP mutase/isocitrate lyase superfamily members, the protein assembles into a dimer of dimers with each subunit adopting an α/β barrel fold and two subunits swapping their barrel's C-terminal α-helices. Mg2+ and oxalate bind in the same manner as observed with other superfamily members. The active site gating loop, known to play a catalytic role in the PEP mutase and lyase branches of the superfamily, adopts an open conformation. The Nε of His235, an invariant residue in the PA4872 sequence family, is oriented towards a C(2) oxygen of oxalate analogous to the C(3) of a pyruvyl moiety. Deuterium exchange into α-oxocarboxylate-containing compounds was confirmed by 1H-NMR spectroscopy. Having ruled out known activities, the involvement of a pyruvate enolate intermediate suggested a decarboxylase activity of an α-oxocarboxylate substrate. Enzymatic assays led to the discovery that PA4872 decarboxylates oxaloacetate (kcat = 7500 s−1 and Km = 2.2 mM) and 3-methyloxaloacetate (kcat = 250 s−1 and Km = 0.63 mM). Genome context of the fourteen sequence family members indicates that the enzyme is used by select group of Gram-negative bacteria to maintain cellular concentrations of bicarbonate and pyruvate; however the decarboxylation activity cannot be attributed to a pathway common to the various bacterial species.
The studies described in this paper were carried out to determine the biochemical function of the hypothetical protein PA4872 from Pseudomonas aeruginosa. PA4872 is a member of the PEP mutase/isocitrate lyase (PEP mutase/isocitrate lyase) superfamily (1). Enzymes of the PEP mutase/isocitrate lyase superfamily catalyze either P-C or C-C bond formation/cleavage. The P-C bond modifying enzymes that have been characterized to date include PEP mutase, carboxyPEP mutase and phosphonopyruvate hydrolase (reactions 1-3 of Figure 1). Enzymes known to act on C-C bonds include 5, 10-methylenetetrahydrofolate:3-methyl-2-oxobutanoate hydroxymethyl transferase (MOBH), isocitrate lyase, 2-methylisocitrate lyase, the plant petal death protein (a broad specificity 2-alkyl malate lyase having an additional oxaloacetate hydrolase activity) and fungal oxaloacetate hydrolase (reactions 4-7 of Figure 1). All of these enzymes catalyze the transformation of an α-oxocarboxylate substrate via an oxyanion intermediate and/or transition state (Figure 1) with the aid of a Mg2+ cofactor. Structure determinations have been carried out for each of these enzymes, with the exception of carboxyPEP mutase and oxaloacetate hydrolase, to define a conserved catalytic scaffold located at the C-terminal edge of the main-frame α/β-barrel (1-11). The catalytic scaffold provides a binding pocket for the α-oxocarboxylate substrate that includes residues engaged in hydrogen bond formation with the carboxylate substituent, and residues that bind the Mg2+ cofactor directly or via water molecules. The Mg2+ cofactor coordinates one oxygen atom of the substrate carboxylate group and the α-oxygen atom, thereby serving as an electron sink in the stabilization of the oxyanion intermediate and/or transition state. A stringently conserved arginine residue also functions to accommodate charge build up at the α-oxygen atom during catalytic turnover.
Figure 1.

The reactions catalyzed by the PEP mutase/isocitrate lyase superfamily. The reactions are depicted to occur stepwise with the formation of reaction intermediates (shown in brackets). The reactions might be concerted in which case the transition state will resemble the intermediate.
The divergence of function within the PEP mutase/isocitrate lyase superfamily is largely the result of changes in residue usage within the region of the catalytic scaffold. The long, flexible loop that connects the 4th β-strand to the 4th α-helix of the α/β-barrel plays an especially important role in defining chemical function. Moreover, this loop gates the active site; in the “open conformation”, ligands can enter or leave the active site whereas in the closed conformation, bulk solvent is excluded from the site and select loop residues participate in substrate binding and/or catalysis. With the exception of MOBH, the substrates for each of the enzymes identified to date are small enough to be accommodated by the active site in the loop-closed conformation. MOBH on the other hand, possesses a truncated loop, which presumably allows the large 5, 10-methylenetetrahydrofolate co-substrate to bind.
To discover novel catalytic functions within the PEP mutase/isocitrate lyase superfamily we have focused our attention on the residue usage and the length of the gating loop possessed by members having unknown function. An alignment of family sequences identified PA4872 as a representative of a unique clad containing eight members at the time of analysis (now 14 members) and having a novel sequence for the active site gating loop (1). We set forth to discover the reaction catalyzed by PA4872 by employing a functional genomic approach that combines structure determination, activity screening and genome context neighbor analysis. Here we present the results, which show that PA4872 is an oxaloacetate decarboxylase, used primarily by Pseudomonads. Inspection of genome contexts led us to speculate that the enzyme is employed by different bacterial species to maintain pyruvate and/or bicarbonate cellular levels in a variety of pathways.
MATERIALS AND METHODS
Preparation of recombinant wild-type PA4872
PA4872 was amplified from P. aeruginosa using Pfu Turbo DNA polymerase (Stratagene), genomic DNA, and oligonucleotide primers with restriction sites for NdeI and BamHI. The purified product was ligated to a NdeI- and BamHI-cut pET3c vector (Novagen). The resulting clone, PA4872-pET3c, was used to transform E. coli BL21 (DE3) cells (Novagen). The gene sequence was verified by DNA sequencing carried out at the Center for Genetics in Medicine, University of New Mexico School of Medicine. Transformed E. coli cells were grown to OD600nm ~ 0.8 at 25 °C in Luria broth (LB) media containing 50 μg/ml carbenicillin. Overexpression was induced with 0.4 mM isopropyl-D-thiogalactopyranoside (IPTG) at 25 °C. The cells were harvested by centrifugation at 6500 rpm (7,808g) for 15 min at 4 °C and suspended in 150 mL of lysis buffer (50 mM Na+Hepes (pH 7.5), 5 mM DTT, 1 mM EDTA, 1 mM benzamide hydrochloride, 0.05 g/L trypsin inhibitor, 1 mM 1,10-phenanthroline, and 0.1 mM phenylmethylsulphonylfluoride). Cells were lysed using a French press and then centrifuged for 60 min at 4 °C. The protein of the supernatant was fractionated by (NH4)2SO4 induced precipitation and harvested by centrifugation. The PA4872-containing fraction (40-60 % saturated (NH4)2SO4) was dissolved in buffer A (50 mM triethanolamine (pH 7.5), 5 mM MgCl2 and 0.5 mM DTT), and then dialyzed against 4 × 1 L buffer A at 4 °C before loading onto a 3.5 x 60 cm DEAE-cellulose column. The column was washed with 1 L buffer A and then eluted with a 1.4 L linear gradient of KCl (0 – 0.8 M) in buffer A. The chromatography was monitored by measuring the absorbance of the eluted fractions at 280 nm and by carrying out SDS-PAGE analysis. The desired fractions (eluted at ~ 0.4 M KCl) were combined, mixed with (NH4)2SO4 to 20% saturation, and loaded onto a 3 × 30 cm phenyl Sepharose column equilibrated with 20% saturated (NH4)2SO4 in buffer A. The column was washed at 4 °C with 500 mL of (NH4)2SO4 (20% saturation) in buffer A and then eluted with a 700 mL linear gradient of (NH4)2SO4 (20% - 0% saturation) in buffer A. The desired fractions (eluted at 0% of ammonium sulfate) were combined, mixed with (NH4)2SO4 to 25% saturation, and loaded onto a 3 × 30 cm butyl Sepharose column equilibrated with 25% saturated (NH4)2SO4 in buffer A. The column was washed at 4 °C with 500 mL of 25% saturated (NH4)2SO4 in buffer A and then eluted with a 800 mL linear gradient of (NH4)2SO4 (25% - 0% saturation) in buffer A. The desired fractions (eluted at ~12% saturated (NH4)2SO4) were combined and concentrated to 15 mL using an Amicon concentrator (Amicon), dialyzed against 4 × 1 L buffer A, and then concentrated to 35 mg/mL using MACROSEP 10K OMEGA (centrifugation at 6500 rpm). The purified protein (yield 4.4 mg/gm wet cells) was shown to be homogeneous by SDS-PAGE analysis and stored at − 80 °C. The molecular mass measured by TOF MS-ES mass spectrometry (University of New Mexico Mass Spectrometry Lab) is 31321 Da, identical to the protein's theoretical molecular mass.
Preparation of selenomethionine containing PA4872
For the preparation of selenomethionine (SeMet)-containing protein, E. coli BL21 (DE3) cells transformed with the PA4872-pET3c vector were grown in minimal media containing SeMet instead of methionine. The overexpression and purification protocols used are the same as those employed for the production of the wild type protein.
Preparation of PA4872 site directed mutants
Site-directed mutagenesis was carried out by using the Quikchange method and Turbopfu polymerase. The plasmid PA4872-pET3c was used as the template for the PCR amplification. The mutant proteins were purified using the same protocol as described for the wild-type recombinant enzyme. The homogeneity (>90%) of the purified mutant proteins was confirmed by SDS-PAGE analysis. Mutant protein yield: ~3 mg/gm of wet cells.
PA4872 crystallization and data collection
Wild-type PA4872 was crystallized in hanging drops by the vapor diffusion method at room temperature (approximately 20 °C). The drops contained equal volumes of reservoir solution and 10 mg/mL protein in 5 mM MgCl2, 5 mM oxalate and 10 mM Na+Hepes (pH 7.0). The reservoir solution contained 18% polyethylene glycol 3350, 20% glycerol and 0.1 M MES (pH 6.0). Crystals appeared within two days. SeMet-containing PA4872 was crystallized in sitting drops by the vapor diffusion method at room temperature. Aliquots of 10 mg/mL protein in 5 mM MgCl2, 5 mM phosphonopyruvate and 10 mM Na+Hepes (pH 7.0) were mixed with equal volumes of reservoir solution containing 12% polyethylene glycol 20,000 and 0.1 M MES (pH 6.0).
X-ray diffraction data for a SeMet-containing protein crystal were collected at IMCA-CAT 17-ID beam line at the Advanced Photon Source (Argonne National Laboratory, Argonne, IL). Prior to data collection, crystals were transferred to mother liquor containing 25% glycerol and flash-cooled in liquid propane cooled by liquid nitrogen. Multiple Anomalous Diffraction (MAD) data at 2.2Ǻ resolution were collected from a single crystal at three different wavelengths (Se absorption edge inflection, peak, and high energy remote wavelength).
Diffraction data for a wild-type PA4872 crystal were collected on the home facility using a Osmic Max-Flux monochromated X-rays generated by a Rigaku Micro Max 007 rotating anode equipped with a Rigaku AXIS IV++ detector. Data were processed with Crystal Clear/d*trek ((12), Rigaku/MSC, the Woodlands, TX). Data collection statistics are shown in Table 1.
TABLE 1.
Data collection statistics
| Sample | Wild-type | Se-Peak | Se-Inflection | Se-Remote |
|---|---|---|---|---|
| Wavelength (Å) | 1.0000 | 0.97944 | 0.97954 | 0.96485 |
| Space Group | C2 | C2 | ||
| Cell Dimensions (Å) | a=260.1 | a=258.7 | ||
| b= 83.8 | b= 83.9 | |||
| c=104.9 | c=104.9 | |||
| β=112.1 | β=111.9 | |||
| Resolution (Å) | 1.9 (1.97-1.9) | 2.2 (2.28-2.20) | 2.2 (2.28-2.20) | 2.2 (2.28-2.20) |
| No. observationsa | 522013 | 526067 | 525654 | 526511 |
| No. unique reflections | 159433 | 190525 | 190709 | 190732 |
| Completeness (%) | 97(77.8) | 91.8(94.7) | 91.8(94.6) | 91.6(94.3) |
| Rmerge b | 0.054(0.358) | 0.042 (0.230) | 0.042 (0.235) | 0.042 (0.242) |
| Mean I/σ(I) | 10.9(2.1) | 39.1 (7.8) | 21.3(4.1) | 15.2(2.7) |
| Redundancy | 3.3(2.1) | 2.8(2.7) | 2.8(2.7) | 2.8(2.7) |
Bijvot pairs are separated in the MAD experiment.
Rmerge = Σhkl [(Σj | Ij − < I > | ) / Σj | Ij | ]. Values in parentheses are for the highest resolution shell.
Structure determination and refinement
Selenium sites were identified using the computer program SHELXD (13). Initial phases were calculated using the program MLPHARE (14) in CCP4 (15). Phase improvement by density modification followed (16). The initial model was built automatically with the Solve/Resolve programs (17, 18). Further building of side chains and model modification were done manually on a Silicon Graphics workstation using the graphics program “O” (19). After adding the side chains to one molecule, the asymmetric unit model was generated by applying the non-crystallographic symmetry operators.
Refinement was carried out using the data collected from the wild-type PA4872 crystal, which diffracted to higher resolution (1.9 Ǻ) than the SeMet-containing protein crystal. CNS (20) was used initially. Simulated annealing at 2500K was followed by alternating positional and individual temperature factor refinement cycles. As the refinement advanced, REFMAC (15, 21) was used. Progress of the refinement was evaluated by the improvement in the quality of the sigma(a)-weighted 2Fobs - Fcalc and Fobs - Fcalc electron density maps (22), and the reduction of the conventional R-factor and Rfree values. Mg2+, oxalate and water molecules were added gradually as the model improved. They were assigned in the Fobs - Fcalc difference Fourier maps with a 3σ cutoff level for inclusion in the model. The refinement statistics are provided in Table 2.
TABLE 2.
Refinement statistics
| Resolution (Å) | 38.5 - 1.9 |
| No. reflections refined | 143540 |
| No. reflections not refined (10%) | 15883 |
| Rwork/Roverall/ Rfree a | 0.195/0.202/0.257 |
| No. Protein atoms | 12767 |
| No. Mg2+-oxalate | 6 |
| No. water molecules | 1728 |
| RMSD from ideal geometry | |
| Bonds (Å) | 0.019 |
| Angles (°) | 1.7 |
| Mean B value (Å)2 | 34 |
R = Σhkl | |Fo| − |Fc| | / Σhkl |Fo|, where Fo and Fc are the observed and calculated structure factors, respectively. Rwork corresponds to the reflections used in refinement, Rfree refers to randomly selected reflections omitted from the refinement, and Roverall corresponds to all reflections.
Modeling of the gating loop-closed conformation
A model of the active-site gating loop in a closed conformation was generated based on the Cα trace of the 2-methylisocitrate lyase loop. Side chains were added manually using the program “O”. Energy minimization of the modeled loop was carried out in CNS, keeping the remainder of the molecule fixed.
Structure analysis
Superposition of structures was carried out within the program ‘O’ (19). The stereochemical quality of the structure was assessed with the program PROCHECK (23). The programs PyMOL (24) and CCP4mg (25, 26) were used for structure depictions. The program Voidoo was used to calculate the active site cavity (27).
Activity Assays for Substrate Screening
The catalyzed conversion of phosphonopyruvate to PEP was tested using the pyruvate kinase (PK)/ADP and lactate dehydrogenase (LDH)/NADH coupling system to convert PEP to lactate with the coupled conversion of NADH to NAD+. The reaction solution (1 mL) contained 17 μM PA4872, 1 mM phosphonopyruvate, 5 mM MgCl2, 20 units/mL PK, 20 units/mL LDH, 2 mM ADP, and 0.2 mM NADH in 50 mM K+Hepes (pH 7.5 and 25°C). The absorbance of the solution was monitored at 340 nm (Δε= 6.2 mM−1 cm−1) over a 30 min time period.
The conversion of phosphonopyruvate to pyruvate was monitored using the LDH/NADH coupled assay. The reaction solution (1 mL) contained 17 μM PA4872, 1 mM phosphonopyruvate, 5 mM MgCl2, 20 units/mL LDH, and 0.2 mM NADH in 50 mM K+Hepes (pH 7.5 and 25°C). The absorbance of the solution was monitored at 340 nm (Δε= 6.2 mM−1 cm−1) over a 30 min time period.
The lyase activity with the reactants (R)-malic acid, (S)-malic acid, (2R,3S)-isocitric acid, threo-(2R,3S:2S,3R)-2-methylisocitrate, 3-butylmalate, 2R-citramalate, 2S-citramalate, 2R-ethyl-3S-methylmalate and (2R,3S)-dimethylmalate was measured at 25°C using the LDH/NADH coupled assay. The reaction solutions (1 mL) initially contained 1 mM substrate, 17 μM PA4872, 5 mM MgCl2, 20 units/mL LDH (except for 2R-ethyl-3S-methylmalate, for which 400 units/mL LDH was used), and 0.2 mM NADH in 50 mM K+Hepes (pH 7.5 and 25°C). The absorbance of the solution was monitored at 340 nm (Δε= 6.2 mM−1 cm−1) over a 30 min time period. Control reactions not including PA4872 were run in parallel.
The conversion of carboxyPEP to phosphinopyruvate was tested using the malate dehydrogenase/NADH coupled assay. The reaction solution (1 mL) contained 17 μM PA4872, 1 mM carboxyPEP, 5 mM MgCl2, 20 units/mL malate dehydrogenase, and 0.2 mM NADH in 50 mM K+Hepes (pH 7.5 and 25°C). The absorbance of the solution was monitored at 340 nm (Δε = 6.2 mM−1 cm−1) over a 30 min time period.
PA4872 catalyzed enolization of α-keto acids
The enolization rates of pyruvate, 2-keto-4-methyl pentanoate “α-ketoisocaproate”, 2-keto butanoate “α-ketobutyrate”, 2-keto pentanoate “α-ketovalerate”, 2-keto-3-methyl butanoate “α-ketoisovalerate” and 2-keto-3-methyl pentanoate “α-keto-β-methyl-valerate” catalyzed by PA4872 were determined by monitoring the exchange of the C(3) hydrogen atom(s) in buffered D2O using 1H-NMR techniques. The solutions contained 100 mM Tris-d11 (pH 7.5), 60 mM substrate, and 5 mM MgCl2 in a total volume of 0.5 mL. Assays were initiated by the addition of PA4872 (1.3 μM for α-ketovalerate, 25 μM for pyruvate and α-ketobutyrate, 74 μM for α-ketoisovalerate and α-keto-β-methyl-valerate). 1H-NMR spectra were recorded at 20 °C at 500 MHz with a Bruker Avance 500 NMR instrument spectrometer, using 10 mM 2-methyl-2-propan(ol-d) as the internal standard. The control reactions, which did not contain enzyme, were carried out in parallel. The rates of α-ketovalerate enolization catalyzed by the H235A (8 μM) and H235Q (10 μM) PA4872 mutants were measured using the same protocol as were the rates of pyruvate enolization catalyzed by phosphonopyruvate hydrolase (5 μM), petal death protein (4 μM) and PEP mutase (5 μM).
The observed rate constant for catalyzed proton exchange (kenol) was calculated from the 1H-NMR spectra using equation 1 where At/A0 is the ratio of the C(3) proton peak areas determined at time “t” and at t = 0, [S0] is the initial substrate concentration and [E] is the enzyme concentration.
| (1) |
13C-NMR analysis of PA4872 catalyzed decarboxylation of oxaloacetate
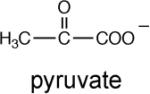
The 13C-NMR spectra were recorded at 25 °C by using a Bruker Avance 500 NMR spectrometer. Methanol served as an internal standard (49 ppm). Following a 30 min incubation period of a solution initially containing 31 μM PA4872, 600 mM oxaloacetate, 5 mM MgCl2 in deionized water at 25 °C (adjusted to pH 7.5 with aqueous NaOH), the PA4872 was removed by filtration using a centricon device. D2O was added to the filtrate to a final concentration of 10% (v/v). The 13C-NMR spectra of oxaloacetate, pyruvate, oxalate and bicarbonate in aqueous solution with 5 mM MgCl2 (pH 7.5) were measured at 25°C. Oxaloacetate signals appeared at 200.5 ppm (C(2)), 174.8 ppm (C(4)), 168.4 ppm (C(1)), 98.3 ppm (C(3)). The pyruvate signals appeared at 205.2 ppm (C(2)), 170.0 ppm (C(1)), 26.6 ppm (C(3)). The bicarbonate signal appeared at 161.2 ppm.
Oxaloacetate and 3-methyloxaloacetate decarboxylation assays
Direct Assay: the wild-type and mutant PA4872-catalyzed oxaloacetate decarboxylation reactions were monitored by using the continuous spectrophotometric assay for oxaloacetate consumption (decrease in solution absorbance at 255 nm; Δε = 1.1 mM−1) (28). Reactions (0.25 mL) were carried out in quartz cells of 0.1 cm path length at 25 °C. Reaction solutions initially contained oxaloacetate, 5 mM MgCl2 and 50 mM K+Hepes (pH 7.5). Coupled assay: the PA4872 catalyzed decarboxylation reactions of oxaloacetate or 3-methyloxaloacetate were monitored at 340 nm (Δε = 6.2 mM−1 cm−1) using a coupled assay for pyruvate or α-ketobutyrate formation. Reactions (1 mL volume) were carried out in quartz cells of 1 cm path length at 25 °C. Assay solutions contained oxaloacetate or 3-methyloxaloacetate, 10 U lactate dehydrogenase, 0.2 mM NADH, 5 mM MgCl2 and 50 mM K+Hepes (pH 7.5).
Steady-state kinetics analysis
The steady-state kinetic parameters (Km and kcat) were determined from the initial velocity data measured as a function of oxaloacetate concentration (varied from 0.5 to 5-fold Km). The initial velocity data were fitted to Equation 2 with KinetAsystI:
| (2) |
where [S] is the substrate concentration, V0 is the initial velocity, Vmax is the maximum velocity, and Km is the Michaelis-Menten constant. The kcat value was calculated from Vmax and the enzyme concentration using the equation kcat = Vmax/[E], where [E] is the PA4872 subunit molar concentration in the reaction.
pH rate profile determination
The steady-state rate constants kcat and kcat/Km were measured as function of reaction solution pH for PA4872-catalyzed oxaloacetate decarboxylation. The reactions were monitored using the direct assay (see above). Reaction solutions initially contained 5 mM MgCl2, various concentrations of oxaloacetate (0.5-5 Km) and PA4872 in the following buffers: (pH 5.0-5.5) 50 mM MES, (pH 5.5-8.0) 25 mM K+Hepes and 25 mM MES, (pH 8.0-9.0) 25 mM K+Hepes and 25 mM TAPS, (pH 9.0-9.5) 25 mM TAPS and 25 mM CAPSO.
1H-NMR analysis of PA4872 catalyzed 3-methyloxaloacetate decarboxylation
The reaction solution initially contained 5 mM MgCl2, 10 mM 3-methyloxaloacetate, 6 nM PA4872, and 1 mM sodium acetate (internal standard) in 50 mM KH2PO4 (pH 7.0, 20 °C). 1H-NMR spectra were recorded at 20°C with a Bruker Avance 500 NMR spectrometer. The decarboxylation reaction was monitored by measuring the disappearance of the C(3)CH3 1H-NMR signal. The two control reactions, one which lacked PA4872, and the other both PA4872 and MgCl2, were carried out in parallel.
The steady-state kinetic constant for catalyzed decarboxylation (kcat) was calculated from the 1H-NMR spectral data using equation 3, where At/A0 is the ratio of the C(3) proton peak areas determined at time “t” and at t = 0, [S0] is the initial substrate concentration and [E] is the enzyme concentration.
| (3) |
Inhibition Constant Determination
The competitive inhibition constant Ki was determined for 3,3-difluoroxaloacetate, pyruvate, oxalate, α-ketovalerate, acetopyruvate, and phosphonopyruvate vs. oxaloacetate by fitting initial velocity data (obtained using the direct assay described above) to equation 4 with KinetAsystI
| (4) |
Reaction solutions (0.25 mL) initially contained 0.8-8 mM oxaloacetate, 2 ×10−3 μM PA4872, 5 mM MgCl2 and 3,3-difluoroxaloacetate (0, 0.8, 1.6 mM), pyruvate (0, 4, 8, 16 mM), oxalate (0, 0.1, 0.2 mM), α-ketovalerate (0, 8, 16 mM), acetopyruvate (0, 2, 4 mM), or phosphonopyruvate (0, 4, 8 mM) in 50 mM K+Hepes (pH 7.5, 25 °C).
RESULTS AND DISCUSSION
A complete assignment of a biological function to an “unknown” member of an enzyme superfamily requires the identification of the type of chemical reaction that it catalyzes, the range of reactants that it can efficiently transform, and the metabolic context(s) of the reaction(s) in the organism. The active sites of the members of an enzyme superfamily have in common a conserved catalytic scaffold that positions core residues that function in stabilization of a common intermediate and/or transition state. Families within a superfamily diversify by acquiring active site residues that expand the range of chemical reactions. Accordingly, the core residues that stabilize the α-oxyanion intermediate shared by the PEP mutase/isocitrate lyase superfamily members are conserved in the PA4872 amino acid sequence. Key to the discovery of the structure of the intermediate stabilized by PA4872 is the determination of the spatial disposition of the diversity residues within the catalytic site. Thus, our first step in the PA4872 function assignment was the determination of the PA4872 X-crystal structure.
Overall Structure
The PA4872/Mg2+-oxalate crystal structure, refined at 1.9 Ǻ resolution, includes six molecules in the asymmetric unit. Four molecules denoted A-D form a tetramer (Figure 2A), the biological relevant assembly, and two molecules (E and F) form a dimer, which together with a crystallographic symmetry related dimer comprises another tetramer. The model includes residues 4-284 of molecule A except that no electron density is associated with the side chain of residue 128. Molecule B includes residues 4-128, and 130-285. Molecule C includes residues 4-255, and 259-284, with no electron density for the side chain of residue 128. Molecule D includes residues 4-126 and 129-284. Molecule E includes residues 4-285 with no electron density for the side chain of residue 273. Molecule F includes residues 4-284 with no electron density for the side chains of residues 128 and 259. The model includes 1728 water molecules. The tetramers obey 222 symmetry and pack into a dimer of dimers. The root mean-square deviation (rmsd) between Cα atoms of the molecules in the asymmetric unit is on averages 0.2 Å. Each molecule contains Mg2+ and an oxalate. All backbone torsion angle values are within the allowed regions of the Ramachandran plot, and the bond lengths and bond angles are well within the range observed in crystal structures of small molecules (Table 2).
Figure 2.

Overall fold of PA4872. (A) The tetramer structure highlighting each subunit in different color. Mg2+ is depicted as yellow sphere and oxalate as stick models with an atomic coloring scheme: Carbon - green, oxygen – red. (B) Superposition of the PA4872 (gold and salmon) and 2-methylisocitrate lyase (purple) monomers, illustrating the different paths of the C-terminal chains.
PA4872 adopts the α/β barrel fold characteristic of the PEP mutase/isocitrate lyase superfamily, in which the eighth α-helix of a subunit is swapped with that of an adjacent subunit. The gating loop adopts an open conformation and the active site, containing Mg2+ and oxalate (Figure 2B), is solvent accessible. PA4872 differs from other superfamily members in the C-terminus following the swapped α-helix. Whereas the C-terminus of all other superfamily members traverses the active site of the partner subunit, in PA4872 the polypeptide chain turns back after the swapping of the helix and traverses its own active site (Figure 2B).
Active-site
As with other α/β barrel enzymes (29), the active site is located at the C-terminal end of the barrel's β-strands. Mg2+ and oxalate bind in a highly polar cavity (Figure 3). Mg2+ coordinates the C(1)O and C(2)O oxygen atoms of oxalate, the Asp88 carboxylate and 3 water molecules. The 3 water ligands engage in hydrogen bond interactions with the side chain carboxylic groups of Asp61, Asp90, Glu117, and Asp88. The oxalate C(1)OO- carboxylate interacts with the Ser50 hydroxyl group (2.5 Å), and with an oxyanion hole formed by the backbone amides of Gly49 and Ser50 (3.5Å and 3.1Å, respectively). Gly49 and Ser50 are located at the N-terminus of a short α-helix (Figure 1), suggesting that the helix dipole also contributes to substrate binding. The oxalate C(2)OO- engages in an ionic interaction with the guanidinium group of Arg159 (3.1 Å). No restraints were imposed on the planarity of the oxalate during the refinement. The six molecules in the asymmetric unit exhibit on average a 19° departure from planarity (referring to the C(1)-C(2) torsion angle). This appears to be a significant distortion but should be taken with caution because the oxalate crystallographic temperature factors are relatively high at 42 Å2. The mode of oxalate interaction with the protein groups is the same as observed for the crystal structures of PEP mutase (7) and phosphonopyruvate hydrolase (5), and comparable to the mode of pyruvate interaction with protein groups observed for the crystal structure of 2-methylisocitrate lyase (1). The arginine, oxyanion hole and the Mg+2 binding residues comprise the core residues of the superfamily catalytic scaffold.
Figure 3.

Active site of PA4871. (A) The electron density associated with key active site residues. The sigma(a)-weighted 2Fo – Fc map is shown contoured at 2σ level. Atomic colors are used: Carbon – green, oxygen – red, nitrogen – blue, Mg2+ - yellow. Oxalate is labeled Oxl (B) stereoscopic representation of the active site. Atomic colors are: Carbon – gray, oxygen – red, nitrogen – blue, Mg2+ - green.
Two residues, located within the vicinity of the oxalate ligand, distinguish the PA4872 active site from those of other family members with known structure; Tyr212 positioned on the loop connecting β7 and α7, and His235 located on the loop connecting β8 and α8 (Figure 3). The His235 Nε is located within hydrogen bonding distance (2.5-3.0 Å) of the oxalate C(1)O (the same C(1)O that engages in hydrogen bond interaction with the Ser50 hydroxyl group; the other C(1)O is coordinated to Mg2+) and possibly within hydrogen bonding distance of the oxalate C(2)O (3.2 -3.8 Å) (the other C(2)O is coordinated to Mg2+). The H235 imidazole tautomer in which both the Nε and Nδ atoms are protonated is evidenced by the hydrogen bond network associated with Nδ. Specifically, the His235 Nδ functions as a hydrogen bond donor and Ser27 hydroxyl (2.5 – 3.0 Å) as a hydrogen bond acceptor (Figure 3). In turn, Ser27 hydroxyl serves as a hydrogen bond donor to an internal water molecule that donates its hydrogen atom to the backbone carbonyl groups of Val28 and Ile46, and accepts a hydrogen bond from the backbone amide group of Gly48 (not shown). The conclusion that His235 is charged is consistent with the pH of the crystals (6.0). Within the PA4872 sequence family (Figure 4), His235 is stringently conserved and position 27 is occupied by a serine or a threonine. The hydrogen bond network suggests that the His235-Ser27 pair plays an important structural role in maintaining the fold integrity and active site architecture. However, site directed mutagenesis studies described below show that His235 does not play a major mechanistic role despite its close proximity to the oxalate and by inference to the substrate.
Figure 4.

Structure based sequence alignment of PA4872 close relatives. Invariant active site residues that are conserved also in other PEP mutase/isocitrate lyase superfamily members are highlighted in blocked green. Other invariant residues are highlighted in blocked gray except for the active site His235, Tyr 212 (and the nearby glutamate of the 3-protein subgroup) and Ser27, which are highlighted in blocked red. Secondary structure motifs are shown above the sequences.
The Tyr212 hydroxyl group is positioned close to the oxalate ligand but not close enough for hydrogen bond interaction. Tyr212 is conserved among the majority of the PA4872 sequence family members (Figure 4).
Although the gating loop is ordered in the PA4872 crystal structure, it does not close over the active site. Other examples of an “active site-open loop conformation” have been observed in X-ray structures of superfamily members that are known to require loop closure for catalytic turnover (1, 5, 7, 30). Loop closure is facilitated by the substrates or substrate analogs that engage in favorable electrostatic interactions with one or more loop residues. An oxalate ligand, however, is an analog of pyruvate and does not possess the moiety that interacts with the gating loop in the structures of other PEP mutase/isocitrate lyase superfamily members. Thus, the observation that the gating loop does not close over the PA4872 active site in the present structure is not sufficient evidence to conclude that loop closure does not occur during catalytic turnover. On the other hand, a key determinant of loop closure is the interaction between a loop lysine and a catalytic site glutamate residue (exemplified by Lys120 and Glu114 in PEP mutase (7)). PA4872 is unique in that the loop lysine residue is absent. For the superfamily members that require a loop closed conformation for catalytic turnover, the Glu-Lys interaction serves to pin the gating loop at one side of the active site.
To explore the potential contribution that the gating loop residues might make to the catalytic site we modeled the loop in the active site-closed conformation, using input from the structures of other superfamily members as a template (model not shown). Although we do not know the accuracy of this model, it does provide an indication that the loop does not contribute residues that might function in binding substrate via electrostatic interaction or participate in proton transfer (i.e. general acid/base catalysis). On the other hand, the model suggests that in the closed conformation, a conserved aromatic loop residue, Phe125 (replaced only by Tyr in the sequence family), may restrict solvent access to the active site.
A second structural feature of PA4872 that is unique within the superfamily is the α-helical C-terminus. Among the α/β-barrel fold structures, the PEP mutase/isocitrate lyase superfamily is distinguished by the use of α/β-barrel 8th helix of one subunit to swap with that of the a neighboring subunit (7). The α-helical segment that follows the 8th helix traverses the active site of the neighboring subunit and assists in desolvation of the bound substrate. The unique conformation of the PA4872 C-terminus, in which the main chain reverses direction after the α8 helix swapping, creates a channel leading to the active site adjacent the C-terminus of β8 (Figure 5A). None of the other PEP mutase/isocitrate lyase superfamily enzyme structures exhibit such a channel (see for comparison, the analogous surface representation of 2-methylisocitrate lyase in Figure 5B, which shows no such channel). The channel exposes the PA4872 active site to solvent from the side of the oxalate/pyruvate C(1) carboxylate group. Closure of the gating loop, as observed for the loop-closed model, does not close this channel, and therefore, we can assume that exposure of this region to solvent occurs during catalytic turnover. Solvent exposure occurs with the binary product complex of MOBH bound with ketopantoate (11), however this would not occur with the ternary substrate complex in which the N5, N10-methylene tetrahydrofolate “sits on top” of the active site, thereby shielding it from solvent while acting as the hydroxymethyl donor to the ensuing α-ketoisovalerate anion intermediate (Figure 1). The solvent accessibility of the PA4872 active site may by required for catalytic turnover, which among the superfamily members is unique.
Figure 5.

Molecular surfaces in the vicinity of active sites. (A) The channel leading to the active site of PA4872 (indicated by an arrow). Mg2+-oxalate, His235 and Tyr212 are shown for reference. (B) The surface around the active site of 2-methylisocitrate lyase corresponding to the Mg2+-pyruvate where the gating loop is in the open conformation (1).
Assignment of PA4872 to the enolate-forming branch of the PEP mutase/isocitrate lyase superfamily
The reactions catalyzed by the PEP mutase/isocitrate lyase superfamily (Figure 1) fall into the two categories represented in Figure 6. The reactions catalyzed by PEP mutase, phosphonopyruvate hydrolase and MOBH proceed via a planar α-caboxy enolate intermediate (and/or transition state) whereas the reactions catalyzed by isocitrate lyase, 2-methylisocitrate lyase, petal death protein and oxaloacetate hydrolase proceed with the formation of a α-carboxy alkoxide and aci-carboxylate anion intermediate pair (and/or transition state). The conservation of the core residues known to stabilize the α-oxyanion intermediate, coupled with the absence of polar residues in the region of the active site that accommodates the aci-carboxylate anion intermediate formed by the C-C bond lyases (1-3, 6, 8-10, 31) suggested that PA4872 might belong to the α-caboxy enolate intermediate forming branch of the superfamily. Activity tests designed to evaluate the ability of the PA4872 catalytic scaffold to stabilize an enolate anion intermediate confirmed this hypothesis.
Figure 6.

The type of intermediates formed by the mutase/transferase and lyase branches of the superfamily.
Blanchard and coworkers carried out solvent deuterium exchange reactions monitored by 1H-NMR spectroscopy to demonstrate MOBH catalyzed enolization of α-ketoisovalerate (32). Although PA4872 lacks the MOBH general base (Glu181 in the E. coli enzyme (11)) that abstracts the C(3)H of the α-ketoisovalerate in the enolization partial reaction (Val188 occupies this position in PA4872), PA4872 does conserve the electropositive residues and the Mg2+ cofactor that function in stabilization of the α-carboxy enolate anion. Using the same 1H-NMR based approach, the rate constants for the enolization (kenol) of pyruvate catalyzed by phosphonopyruvate hydrolase and PEP mutase were measured. Like PA4872 these enzymes conserve the electropositive residues and the Mg2+ cofactor that stabilize the pyruvate enolate intermediate but lack an enzyme residue to function as a general base in the enolization of pyruvate. The kenol measured for phosphonopyruvate hydrolase is 12 min−1 and that for PEP mutase is 4 min−1. These rate constants are significantly smaller than the kcat values measured for the catalyzed reactions shown in Figure 1 (105 s−1 (5) and 34 s−1 (33), respectively) and thus, the turnover rates greatly exceed the enolization rates. In contrast, the reported kcat for the Mycobacterium tuberculosis MOBH is 47 min−1 and the rate constant for the enolization of α-ketoisovalerate (kenol) is 752 min−1 (32), which greatly exceeds the turnover rate. The difference in kinetic behavior is consistent with the fact that MOBH catalyzes the enolization of α-ketoisovalerate as the first partial reaction of the overall reaction (Figure 1) and that MOBH possesses a general base (Glu181) to assist in the catalysis of the α-ketoisovalerate enolization. Phosphonopyruvate hydrolase and PEP mutase, on the other hand, do not catalyze pyruvate enolization as a partial reaction of the overall reaction, nor do they possesses a general base that might facilitate pyruvate enolization. We assume that a water molecule functions in base catalysis of the pyruvate enolization. The rate enhancements observed with phosphonopyruvate hydrolase and PEP mutase are attributed to the delocalization of excess charge from the pyruvate enolate onto the Mg+2 cofactor and the electropositive active site residues.
The kenol determined for the petal death protein is 0.6 min−1 whereas isocitrate lyase and 2-methylisocitrate lyase do not catalyze exchange at a detectable level. These three enzymes do not form an enolate anion intermediate (Figure 1). Based on these results we conclude that the 2 branches of the superfamily are distinguished by the ability vs. inability to catalyze the enolization of α-ketoacids. In this context we interpret the kenol = 24 min−1 measured for PA4872 catalyzed pyruvate enolization (Table 3) as evidence that it is a member of the α-carboxy enolate anion intermediate forming branch of the superfamily.
TABLE 3.
The rate constant for enolization (kenol) determined from the rate of PA4872-catalyzed deuterium exchange from D2O solvent with the C(3)H of 2-ketoacids
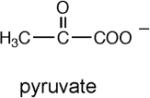


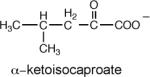
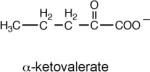
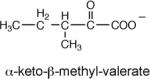
The rate constant for the PA4872 H235Q mutant.
The rate constant for the PA4872 H235A mutant.
To gain information about the structure of the α-carboxy enolate anion intermediate formed during PA4872 catalytic turnover of its physiological substrate, the kenol for a variety of C(3) substituted pyruvate compounds were measured (Table 3). Whereas only α-ketoisovalerate is substrate for the overall reaction catalyzed by MOBH (32), this enzyme catalyzes the enolization of pyruvate and C(3) substituted pyruvate with the following efficiencies: for the native substrate α-ketoisovalerate kenol = 752 min−1; for α-keto-β-methyl-valerate kenol = 482 min−1; for α-ketobutyrate kenol = 354 min−1; for α-ketovalerate kenol = 229 min−1; for pyruvate kenol = 129 min−1; and for α-ketoisocaproate kenol = 19 min−1. In contrast, PA4872 is only significantly active with the linear α-ketoacids for which the order of reactivity is α-ketovalerate > α-ketobutyrate > pyruvate (Table 3). This result suggested that the native substrate is a small linear α-ketoacid.
PA4872 chemical function determination
The PA4872 active site provides space in a hydrophobic pocket lined by Met208, Val210 and Val232, for a small C(3) substituent such as a methyl or ethyl group. Adjacent to the Tyr212, there is also space for a second C(3) substituent oriented towards the active site exit at the C-terminus edge of the α/β barrel, albeit with slight adjustment of the Tyr212 side chain conformation. This suggested the possibility that PA4872 catalyzes a β-elimination reaction in an α-ketoacid substrate. The obvious candidate for such a reaction is oxaloacetate. The PA4872 active site, modeled with oxaloacetate is shown in Figure 7 along with models built with 3-methyloxaloacetate and 3-ethyloxaloacetate as the ligands. All three models conserve the electrostatic interactions with the core residues that stabilize the pyruvate enolate while the carboxylate group is confined to a position above the plane of the developing pyruvate enolate intermediate, in accord with the accepted geometry for decarboxylation in a β-ketoacid (34).
Figure 7.

The active site cavity (shown as a cage) with docked potential substrates shown as stick models with the atomic colors green – carbon, red – oxygen. The Mg2+ is depicted as a magenta sphere.
The addition of PA4872 to a buffered solution of oxaloacetate and Mg2+ was accompanied by rapid bubble formation (viz. CO2). 13C-NMR analysis of the solution confirmed that the oxaloacetate had been quantitatively converted to pyruvate and bicarbonate. The steady state rate constants for this reaction were measured using two different assay methods. The first monitors the disappearance of oxaloacetate (absorbance decrease at 255 nm) and the second monitors the formation of pyruvate via coupled reaction with lactate dehydrogenase and NADH (absorbance decrease at 340 nm). The results are presented in Table 4. Owing to the fact that oxaloacetate is a slow substrate for lactate dehydrogenase (hence a background rate must be subtracted) the direct assay is likely to be the more accurate measurement. The kcat = 7500 s−1 is 2-3 orders of magnitude larger than the kcat values of the following superfamily enzymes in reaction with their physiological substrates: PEP mutase (kcat = 34 s−1 for the enzyme from Mytilus edulis (33)), phosphonopyruvate hydrolase (kcat = 105 s−1 for the enzyme from Variovorax sp. Pal2 (5)), oxaloacetate hydrolase (kcat = 10 s−1 for the enzyme from Botrytis cinerea (31)), isocitrate lyase (kcat = 100 s−1 for the enzyme from E. coli (1)), 2-methylisocitrate lyase (kcat = 19 s−1 for the enzyme from E. coli (1)), and petal death protein (kcat = 8 s−1 for the best substrate, 2R-ethyl-3S-methylmalate (35)). The kcat is 4 orders of magnitude larger than that of the MOBH (kcat= 0.8 s−1).
TABLE 4.
Steady-state kinetic constants for PA4872 catalyzed decarboxylation of oxaloacetate and 3-methyloxaloacetate
| Substrate | Assay | kcat (s−1) | Km (mM) | kcat/Km (M−1s−1) |
|---|---|---|---|---|
| oxaloacetate | direct | 7500 ± 200 | 2.2 ± 0.1 | 3 × 106 |
| oxaloacetate | coupled | 7200 ± 400 | 1.3 ± 0.2 | 6 × 106 |
| 3-methyl-oxaloacetate | coupled | 250 ± 10 | 0.63 ± 0.08 | 4 × 105 |
| 3-methyl-oxaloacetate a | 1H-NMR | 800 | -------- | -------- |
The reaction mixture in this assay initially contained 10 mM 3-methyloxaloacetate, 5 mM MgCl2, 6 nM PA4872, 50 mM KH2PO4 (pH 7.0, 20 °C), and 1 mM sodium acetate (as the internal standard). The 1H-NMR of 3-methyloxaloacetate measured under the reaction conditions defines the relative ratio of enol:ketone:gem diol as 1.7: 0.8: 0.8. The rate of decarboxylation of 3-methyloxaloacetate in buffer is 0.052 mM/min, the rate in MgCl2-containing buffer is 0.078 mM/min whereas the rate measured in the presence of the 6 nM PA4872 is 0.38 mM/min.
Knowledge of the kcat for PA4872 catalysis of its physiological reaction allows us to contrast the efficiency by which the pyruvate enolate is formed by decarboxylation of oxaloacetate to the efficiency at which the enzyme catalyzes the enolization of bound pyruvate; at kcat = 7500 s−1 vs. kenol = 0.4 s−1, the trend is the same as PEP mutase and phosphonopyruvate hydrolase, and the reverse of MOBH. Although the His235 Nε is located near the C(3) of the modeled oxaloacetate it does not make a significant contribution to catalysis of α-ketoacid enolization. Specifically, kenol values measured for H235A and H235Q PA4872 with the best enolization substrate (α-ketovalerate) are only 3-fold smaller than that of wild-type PA4872 (Table 3).
The models of PA4872 bound with 3-methyl- and 3-ethyloxaloacetate (Figure 7) suggested that these compounds might also be good substrates for PA4872 decarboxylation. 3-methyloxaloacetate is a metabolite formed from L-threo-3-methylaspartate within the C-5 branched dibasic acid metabolic pathway (http://www.genome.ad.jp/kegg/pathway.html; (36)) and thus a potential physiological substrate for PA4872. 3-Methyloxaloacetate was prepared by chemical synthesis (see Supporting Information) and the rate of PA4872 decarboxylation was monitored by 1H-NMR to define a turnover rate of 790 s−1 at 10 mM 3-methyloxaloacetate (Table 4). The steady-state kinetic constants were measured using the LDH/NADH coupled assay for formation of α-ketobutyrate defining kcat = 250 ± 10 s−1, Km = 0.63 ± 0.08 mM and kcat/Km = 4 × 105 M−1 s−1. Although the kcat/Km value is an order of magnitude smaller than that measured for oxaloacetate, it is still large enough to support the possible physiological relevance of this reaction. Whereas, the C(3)CH3 substituent increased the rate of the enolization of the α-ketoacid 2-fold (α-ketovalerate kenol = 56 min−1 vs. pyruvate kenol = 24 min−1) it decreased the rate of decarboxylation in the corresponding diacid 3-methyloxaloacetate relative to oxaloacetate by ~20-fold. Thus, the PA4872 catalyzed decarboxylation reaction is hindered by the C(3)CH3 substituent and based on this observation we hypothesize that the PA4872 evolved to function in the decarboxylation of the main stream metabolite oxaloacetate rather than the decarboxylation of the more obscure metabolite 3-methyloxaloactate.
PA4872 catalytic proficiency
The rate of oxaloacetate decarboxylation in aqueous solution was investigated earlier using 3-methyloxaloacetate in place oxaloacetate so that the reaction can be monitored by 1H-NMR. The rate measured at pH 4.6 and 31°C was 7.8 ×10−5 s−1 (37). The rate measured at pH 7 and 25°C defines k = 1.2 × 10−4 s−1 in the absence of Mg+2 and k = 1.8 × 10−4 s−1 in the presence of 5 mM (Table 4). Thus, the approximate value for kcatalyzed/ksolution is 4 ×107.
The steady state kinetic constants measured for decarboxylation of oxaloacetate catalyzed by PEP mutase are kcat =0.269 ± 0.004 s−1 and Km = 0.22 ± 0.01 mM (kcat/Km = 1.2 × 103 M−1 s−1). The values measured for phosphonopyruvate hydrolase are kcat = 1.04 ± 0.02 s−1 and Km = 1.6 ± 0.1 mM (kcat/Km = 6.5 × 102 M−1 s−1). Our “control enzyme” 2-methylisocitrate lyase did not catalyze oxaloacetate decarboxylation at a detectable rate (kcat < 1 × 10−3 s−1). Thus, the ability of the members of the pyruvate enolate intermediate forming branch of the PEP mutase/isocitrate lyase superfamily to stabilize the pyruvate enolate anion (Figure 6) is sufficient to confer a weak decarboxylase activity on them. The proficiency of the PA4872 (kcatalyzed/ksolution ~ 108) is substantially greater than that of PEP mutase (kcatalyzed/ksolution ~ 103) and phosphonopyruvate hydrolase (kcatalyzed/ksolution ~ 104). Hence, PA4872 diverged in function through optimization of the active site environment to facilitate decarboxylation.
PA4872 Substrate Recognition
The binding constants for substrate and intermediate analogs were determined by measuring the competitive (vs. oxaloacetate) inhibition constant (Ki) for these compounds. The pyruvate enolate analog used in the X-ray structure determination, oxalate, was found to have a Ki = 43 μM (Table 5). This value is comparable to the Ki = 25 μM measured for PEP mutase (38). In contrast, phosphonopyruvate, which binds very tightly to PEP mutase (Km = 1 μM and Ki for sulfopyruvate = 40 μM) (33, 38), binds very poorly to PA4872 (Ki = 3 mM). Thus, the two enzymes share tight binding to the pyruvate enolate analog but diverge in substrate recognition. Oxalate is also the product of oxaloacetate hydrolase and petal death protein catalyzed oxaloacetate cleavage. The Ki values measured for these two enzymes are 19 μM (31) and 4 μM (35), respectively.
TABLE 5.
The competitive inhibition constant determined for PA4872 catalyzed oxaloacetate decarboxylation
| Inhibitor | Ki (mM) | Inhibitor | Ki (mM) |
|---|---|---|---|
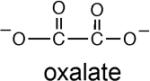
|
0.043 ± 0.002 |
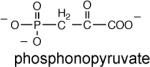
|
3.0 ± 2 |
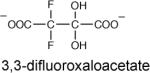
|
0.45 ± 0.03 |
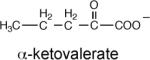
|
6.7 ± 0.3 |
| 3.2 ± 0.3a | |||
|
1.09 ± 0.08 |
|
7.2 ± 0.4 |
| 0.56 ± 0.04a |
The inhibition constant was measured for the PA4872 Y212F mutant rather than the wild-type PA4872.
3,3-difluoroxaloacetate, which differs from oxaloacetate by replacement of the C(3) hydrogens with fluorine atoms, is not a substrate for PA4872 decarboxylation. Nor is it a substrate for oxaloacetate hydrolase catalyzed hydrolytic cleavage (31). Whereas oxaloacetate exists predominantly in the C(2) keto form (35), the 3,3-difluoroxaloacetate exists predominantly in the C(2) gem diol (hydrate) form (31). Oxaloacetate hydrolase catalyzes C(2)-C(3) bond cleavage in the oxaloacetate gem diol. The tight binding of 3,3-difluoroxaloacetate (Ki = 68 nM, (31)) suggests that the enzyme active site is specialized in recruiting or generating the gem diol form of oxaloacetate. It is not clear why the fluorine atoms at C(3) impair the C(2)-C(3) cleavage step. PA4872 binds 3,3-difluoroxaloacetate 7500-fold less tightly than does oxaloacetate hydrolase at Ki = 450 μM and is unable to decarboxylate this substrate analog. The comparatively higher stability of the 3,3-difluoroxaloacetate to decarboxylation in solution and in the presence of PA4872 may be due to the predominance of the gem diol and/or another inherent property of the keto form.
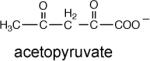
The Ki values of α-ketovalerate (6.7 mM) and pyruvate (7.2 mM) were measured to determine whether the C(3) ethyl substituent enhances binding affinity, which it does not (Table 5). Lastly, we synthesized acetopyruvate (see Supporting Information) to test as an oxaloacetate analog. The Ki = 1 mM indicates a ~7-fold tighter binding than observed for pyruvate.
PA4872 Catalytic Mechanism
Shown in Figure 8 are the key interactions of oxaloacetate modeled in the PA4872 active site. These are analogous to the set of interactions of the phosphonopyruvate substrate in the active sites of PEP mutase and phosphonopyruvate hydrolase, which share with PA4827 the capacity to catalyze oxaloacetate decarboxylation (albeit at a much reduced rate). The modest oxaloacetate decarboxylase activity of PEP mutase and phosphonopyruvate hydrolase is evidence that these interactions are applicable to oxaloacetate binding and decarboxylation and that the difference in catalytic efficiency originates in differences in the active site environments.
Figure 8.

Proposed catalytic mechanism of PA4872 and comparison with the catalytic machinery of the malic enzyme oxaloacetate decarboxylase.
Not shown in Figure 8 are the two polar residues of the PA4872 active site with the greatest potential to facilitate the decarboxylation are His235 and Tyr212 (Figure 3), conserved residues in the PA4872 sequence family (Figure 4) that are absent in PEP mutase and phosphonopyruvate hydrolase. At first glance one might envision hydrogen bond interaction between the Tyr212 hydroxyl group and the oxaloacetate C(3)COO- and a role for the His235 in general acid catalysis. These features, in particular the positioning of His235 with respect to oxalate, inspired us to test whether PA4872 stabilizes a pyruvate enolate intermediate, which ultimately led to the discovery of the decarboxylation activity. Further analysis, however did not support either catalytic role because replacements of these two residues did not impact enzyme efficiency significantly (Table 6). Perhaps noteworthy is the 25-fold reduction in the Km value of the Y212F mutant, which suggests that the Tyr212 hydroxyl group impairs substrate binding yet at the same time it facilitates catalysis (the kcat of Y212F PA4872 is reduced by 23-fold). This conjecture is supported by the observation that the Ki values measured for acetopyruvate (aceto group in place of the oxaloacetate C(3) carboxylate group) and α-ketovalarate (ethyl group in place of the oxaloacetate C(3) carboxylate group) are 2-fold smaller than those measured for the wild-type enzyme (Table 5).
TABLE 6.
Steady-state kinetic constants for mutant PA4872 catalyzed decarboxylation of oxaloacetate.a
| kcat (s−1) | Km (μM) | kcat/Km (M−1s−1) | |
|---|---|---|---|
| WT | 7500 ± 200 | 2200 ± 100 | 3.4 × 106 |
| Y212F | 323 ± 6 | 87 ± 4 | 3.7 × 106 |
| H235A | 510 ± 7 | 1020 ± 50 | 5.0 × 105 |
| H235Q | 2580 ± 90 | 2500 ± 200 | 1.0 × 106 |
0.25 mL reaction mixture contained 50 mM K+Hepes (pH 7.5, 25 °C), 5 mM MgCl2, 0.5-10 Km of oxaloacetate. Reactions were monitored at 255 nm.
Relevant to the catalytic mechanism, the pH rate profile data (Figure 9) shows that there is no change in the kcat value over the pH 5-10 range and there is not sufficient variation in the kcat/Km to assign to the ionization of an essential residue. This rules out that a protonated His235 serves as the general acid in the decarboxylation reaction. Otherwise we would anticipate a drop in catalytic efficiency above pH 6.5. Moreover, the kcat and kcat/Km values determined for H235A and H235A catalysis of oxaloacetate decarboxylation are less than an order of magnitude smaller than the kcat and kcat/Km value measured for wild-type PA4872. The stringent conservation of the histidine residue among the PA4872 sequence family members is attributed to its structural role (described in a previous section) rather than to a catalytic role. Because Tyr212 and His235 do not appear to play a direct role in the catalytic mechanism, they are not included in Figure 8.
Figure 9.

pH profile of PA4872. The buffers used in various pH ranges are listed in the Methods section.
The origin of the greater rate enhancement observed with PA4872 vs. the PEP mutase and phosphonopyruvate hydrolase is currently unknown but may arise from two unique structural features. The first is a sterically restricted hydrophobic region of the active site which is able to confine the C(3)COO- in an orientation optimal for the dissociation of CO2 (Figure 7). If the gating loop closes, the desolvation of the C(3)COO- into a nonpolar region of the active site is also expected to contribute to ground state destabilization that is relieved as the charge is transferred to the C(3) upon CO2 dissociation. The second important feature of the PA4872 active site is the solvent access to C(3) provided by the channel (Figure 6), which is unique to PA4872. A water molecule that approaches C(3) from that direction may serve as the acid that protonates the pyruvate enolate.
There are several other enzyme superfamilies from which an oxaloacetate decarboxylase has emerged. Pyruvate kinase possesses an intrinsic oxaloacetate decarboxylase activity owing to its ability to stabilize a pyruvate enolate intermediate, but oxaloacetate is not considered its physiological substrate (39, 40). The malic enzyme catalyzes oxaloacetate decarboxylation as the second partial reaction in the conversion of malate to CO2 plus pyruvate with the reduction of NAD to NADH (41). CitM within the malic enzyme family has evolved to function in a specialized citrate degradation pathway in a small group of Gram- positive bacteria (42). The CitM cannot convert malate to oxaloacetate but it does decarboxylate oxaloacetate at an efficiency of kcat = 20 s−1 and Km = 0.5 mM using the same catalytic residues of the malic enzyme. The catalytic residues of CitM and the malic enzyme (43) are depicted in Figure 8 along with two possible pathways for the catalyzed decarboxylation. As with PA4872, a Mg+2 cofactor is used along with several electropositive active residues to bind the α-keto acid moiety. There are however distinct differences in catalytic mechanism. Firstly, the carboxylate is oriented for the dissociation by binding to an asparagine side chain and an internal water molecule. Secondly, a tyrosine residue is positioned close to the C(3) so that it might function as the acid catalyst (pathway a). Alternatively, the Lys residue that engages in hydrogen bond formation with the substrate C(2)=O may transfer its proton as the reaction proceeds (pathway b). Despite its seemingly more sophisticated catalytic site, the catalytic efficiency of CitM is significantly lower than that of PA4872.
A third known “dedicated” oxaloacetate decarboxylase is the oxaloacetate decarboxylase Na+ pump present in a wide range of bacterial species. This membrane protein system catalyzes the transfer of the C(3) carboxyl group of oxaloacetate to a biotin prosthetic group and the decarboxylation of the carboxybiotin, coupled to Na+ translocation. The recent crystal structure of the enzyme shows that the Zn2+-dependent oxaloacetate decarboxylase domain adopts an α/β barrel fold, but otherwise the catalytic mechanism is entirely different than that of PA4872 (44); a catalytic lysine residue serves as the acceptor of the CO2 from oxaloacetate and transfers it to biotin, which induces the sodium translocation.
A fourth dedicated oxaloacetate decarboxylase from Pseudomonas species has been characterized. This large (~600 amino acids) 3-domain decarboxylase is found in a wide range of Gram-negative bacteria, including the same species of bacteria that produce the PA4872 family members. Although the structure of this enzyme has not been determined, the stereochemistry of the protonation with inversion at C(3) has been reported (45) as well as the results from an in depth examination of transition state structure, metal cofactor activation and pH effects (46).
PA4872-like species distribution and biological function
PA4872 orthologs are found in all deposited Pseudomonad genomes (genus of γ-proteobacteria) and in only a few other Gram-negative bacteria. The most distinguishing sequence features in this group of enzymes is the invariant His235-Ser27 (or Thr) pair, the lack of the conserved lysine that regulates the closure of the gating active site loop, and the very different sequence of the gating loop compared with other enzymes in the PEP mutase/isocitrate lyase superfamily (Figure 4). Two of the Pseudomonad species each have a strain that contain two copies of the gene (paralogs), well separated within the genome. One paralog shares high sequence identity with PA4872 (>70%) while the other shares 50-60% identity. The identity level of both paralogs within a genome is sufficiently high to indicate the same chemical function although their substrate specificity may vary. Within the Pseudomonad group the sequence identity between PA4872 and the closest homologues is 70-99 %. Outside of the Pseudomonad group it is in the 70-80% range for Chromohalobacter salexigens, Reinekea sp. MED297 and Marinomonas sp. MWYL1, but drops to the 50-52% for Rhodopseudomonas palustris strains (genus of α-proteobacteria), 44-47% for Bradyrhizobium strains (α-division) and down to 40% for the Burkholderia xenovorans (β- division). Importantly, B. xenovorans is the only species of the many Burkholderia species whose genome sequences have been determined that contains a PA4872 ortholog. This was confirmed by a BLAST search of the Burkholderiaceae group using the B. xenovorans as query that showed only lyase sequences (clearly identified by the active site gating loop sequence) at 30-36% identity.
The second PA4872-like copy is found in Pseudomonas fluorescens PfO-1 and in Pseudomonas putida W619 but in no other reported species. This paralog is expected to have identical substrate specificity in both organisms because the sequence identity between the two is 84%.
Based on the species distribution it is evident that PA4872 performs a specialized metabolic function in a select group of Gram-negative bacteria restricted to all Pseudomonads of defined genome sequence (γ-proteobacteria), a few among the many α-proteobacteria, and a single, environmental, β-proteobacteria. Because oxaloacetate and pyruvate are key intermediary metabolites that link numerous pathways and because carbonate is used as a precursor for biotin mediated caboxylation reactions, the oxaloacetate decarboxylase could have many potential metabolic uses. Indeed, the genome context analysis described below lead us to the conclusion that the Pseudomonads, which exhibit great metabolic diversity, may utilize the oxaloacetate in a variety of processes that require the supply of pyruvate or bicarbonate.
The genome contexts of the PA4872 and its close relatives were examined to obtain clues about the biological function, underscoring the variety of contexts in which they are found. Only in the R. palustris strains and the B. japonicum UDSA110 are the oxaloacetate decarboxylase encoding genes organized in an operon. In R. palustris strains, the flanking genes (separated by 1-6 nucleotides) are homologs of acyl phosphatase (hydrolysis of acylphosphate metabolites to the corresponding carboxylic acid and orthophosphate) and the α subunit of acyl-CoA carboxylase (phosphorylation of carbonate with ATP to form carboxyphosphate, followed by the transfer of CO2 from the carboxyphosphate to biotin). We speculate that in these two bacterial species, the bicarbonate produced by the oxaloacetate decarboxylase is fixed in the cell by reaction with the biotin carboxylase subunit of the acyl-CoA carboxylase. The connection with the acyl phosphatase is not clear. However we note that in other bacteria (for example Nitrobacter hamburgensis X14) that do not contain the oxaloacetate decarboxylase encoding gene, the genes encoding the acyl phosphatase and the biotin carboxylase are co-transcribed.
The biotin carboxylase, oxaloacetate decarboxylase and acyl phosphatase homologues are also adjacent to one another in the genome of B. japonicum UDSA110. However, only the oxaloacetate decarboxylase and acyl phosphatase genes are clearly co-transcribed (3 nucleotide separation). The biotin carboxylase gene is separated from the oxaloacetate decarboxylase by 149 nucleotides. Perhaps coincidental but nonetheless intriguing, the citrate lyase gene cluster is located in the neighborhood of the biotin carboxylase, oxaloacetate decarboxylase and acyl phosphatase genes within the R. palustris BisB18 genome. The citrate lyase converts citrate to oxaloacetate.
The genome contexts of other PA4872 sequence family members are not as informative; the genes are not located on operons and the neighboring genes tend to be different. Yet, it is intriguing that Pseudomonas stutzeri A1501, and Pseudomonas mendocina ymp, and the two annotated P. aeruginosa genomes, PAO1 and PA7, position the oxaloacetate decarboxylase gene in the same neighborhood as the urease operon. Urease, which plays an important role in nitrogen acquisition and recycling requires CO2 for the synthesis of its nickel metallo center (47). Next to the urease operon is an amino acid transporter. It is possible that the close arrangement of the amino acid transporter, urease and oxaloacetate decarboxylase genes is not coincidental.
The P. fluorescens Pf-5 oxaloacetate decarboxylase gene is included among the genes encoding acetylornithine aminotransferase (of the urea cycle) and the enzymes of the nicotinamide metabolic pathway. P. fluorescens PfO-1 has two PA4872 homologs; the closest to PA4872 in sequence (78%) is clustered with arginine N-succinyltransferase, N-formylglutamate amidohydrolase, glutamine synthetase and an amino acid transporter. The respective gene clusters are suggestive of amino acid transport and recycling. The pyruvate generated by the oxaloacetate decarboxylase may serve as a NH3 acceptor in the synthesis of alanine. The genomes of the Pseudomonas syringae strains include the gene encoding oxaloacetate decarboxylase as neighbor to the gene encoding isopropyl malate synthase (leucine biosynthesis) and the genes encoding an amidase and an aminotransferase.
A common denominator in each of the gene contexts described above is the metabolism of amino acids, in which pyruvate might participate. The oxaloacetate decarboxylase gene of P. fluorescens PfO-1 (the second homolog having 55% identity with PA4872) along with that of the Burholderia xenovans and P. putida F1 are clustered with genes that form a carboxylate transporter and degradation pathway. The genome context of the oxaloacetate decarboxylase gene of Pseudomonas entomophila has no relationship to any of the genes listed above, and its neighboring genes share no common function with one another except that there are several genes related to antibiotics resistance.
Based on essential gene analysis in P. aeruginosa PAO1 using transposon insertion technology, PA4872 is not required for survival (48). Taken together, the currently available information suggests that the oxaloacetate decarboxylase gene was acquired by all Pseudomonads and by a few other organisms as an auxiliary enzyme that helps maintain the supply of two key metabolites for a variety of cellular processes.
Conclusion
The determination of the biological function of a novel gene product presents a challenge in the era of genome sequencing. The rapidly developing discipline of genomic enzymology relies on sequence analysis and/or structure determination to assign the gene product to an enzyme superfamily, which is achieved by recognizing a conserved overall molecular fold that supports a common catalytic scaffold. The discovery of the biochemical function of the new enzyme falls into two classes. Either the chemistry catalyzed is known but the substrate specificity needs to be defined, or in the more difficult situation, both the chemical reaction and substrate range are unknown. In the letter cases, which PA4872 belongs to, the recognizable catalytic scaffold is joined by residues that expand the core chemistry to facilitate the novel chemical pathway.
In studying PA4872, we identified the core chemistry of the PEP mutase/isocitrate lyase superfamily that supports Mg2+/Mn2-dependent C-C or P-C bond cleavage/formation of α-keto acid substrates. The initial activity screening that followed the assignment of enzyme superfamily ruled out the known chemistry but did not lead to the discovery of the new chemistry. The genome context may provide clues about the protein function, but this did not apply in the initial examination of PA4872 genome context. Thus, we relied solely on structure determination and chemical intuition to develop a hypothesis of catalytic function that could then be tested by in vitro activity screening. The structure provided a clue that PA4872 may belong to the branch of the superfamily that forms an α-carboxy enolate anion intermediate/transition state, and we verified this assignment by demonstrating catalysis of the enolization in α-keto acids. Using a model of PA8472(Mg+2)(α-keto acid) complex and our understanding of the mechanisms of catalysis in the other PEP mutase/isocitrate lyase superfamily members, we developed the hypothesis that PA8472 catalyzes the decarboxylation of oxaloacetate and/or a 3-alkyloxaloacetate metabolite. Once the chemistry was identified, a focused activity screen together with further scrutiny of genome context helped define oxaloacetate as the most probable physiological substrate. One would expect that a similar discovery process will apply to other proteins of unknown function.
Supplementary Material
Footnotes
Grant sponsors: NSF MCB9813271 (O.H.) and NIH RO1GM28688 (D.D.-M.). Use of the Argonne National Laboratory Structural Biology Center beamlines at the Advanced Photon Source, was supported by the U. S. Department of Energy, Office of Energy Research, under Contract No. W-31-109-ENG-38.
The coordinates have been deposited in the Protein Data Bank (entry codes 3B8I)
Abbreviations used are: MOBH, 5, 10-methylenetetrahydrofolate: 3-methyl-2-oxobutanoate hydroxymethyl transferase; PEP, phosphoenolpyruvate; LDH, lactate dehydrogenase; MES, 2-(N-Morpholino)ethanesulfonic acid hydrate 4-Morpholineethanesulfonic acid; HEPES, 4-(2-Hydroxyethyl)piperazine-1-ethanesulfonic acid; TAPS, N-Tris(hydroxymethyl)methyl-3-aminopropanesulfonic; CAPSO, 3-(Cyclohexylamino)-2-hydroxy-1-propanesulfonic acid.
Supporting Information Available: preparation of 3-methyloxaloacetate; preparation of acetopyruvate; 1H NMR 3-methyloxaloacetate; 13C NMR 3-methyloxaloacetate; 1H NMR acetopyruvate; 3C NMR acetopyruvate. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Liu S, Lu Z, Han Y, Melamud E, Dunaway-Mariano D, Herzberg O. Crystal structures of 2-methylisocitrate lyase in complex with product and with isocitrate inhibitor provide insight into lyase substrate specificity, catalysis and evolution. Biochemistry. 2005;44:2949–2962. doi: 10.1021/bi0479712. [DOI] [PubMed] [Google Scholar]
- 2.Britton K, Langridge S, Baker PJ, Weeradechapon K, Sedelnikova SE, De Lucas JR, Rice DW, Turner G. The crystal structure and active site location of isocitrate lyase from the fungus Aspergillus nidulans. Structure. 2000;8:349–362. doi: 10.1016/s0969-2126(00)00117-9. [DOI] [PubMed] [Google Scholar]
- 3.Britton KL, Abeysinghe IS, Baker PJ, Barynin V, Diehl P, Langridge SJ, McFadden BA, Sedelnikova SE, Stillman TJ, Weeradechapon K, Rice DW. The structure and domain organization of Escherichia coli isocitrate lyase. Acta Crystallogr D Biol Crystallogr. 2001;57:1209–1218. doi: 10.1107/s0907444901008642. [DOI] [PubMed] [Google Scholar]
- 4.Chaudhuri BN, Sawaya MR, Kim CY, Waldo GS, Park MS, Terwilliger TC, Yeates TO. The crystal structure of the first enzyme in the pantothenate biosynthetic pathway, ketopantoate hydroxymethyltransferase, from M tuberculosis. Structure. 2003;11:753–764. doi: 10.1016/s0969-2126(03)00106-0. [DOI] [PubMed] [Google Scholar]
- 5.Chen CC, Han Y, Niu W, Kulakova AN, Howard A, Quinn JP, Dunaway-Mariano D, Herzberg O. Structure and kinetics of phosphonopyruvate hydrolase from Variovorax sp. Pal2: new insight into the divergence of catalysis within the PEP mutase/isocitrate lyase superfamily. Biochemistry. 2006;45:11491–11504. doi: 10.1021/bi061208l. [DOI] [PubMed] [Google Scholar]
- 6.Grimm C, Evers A, Brock M, Maerker C, Klebe G, Buckel W, Reuter K. Crystal structure of 2-methylisocitrate lyase (PrpB) from Escherichia coli and modelling of its ligand bound active centre. J Mol Biol. 2003;328:609–621. doi: 10.1016/s0022-2836(03)00358-9. [DOI] [PubMed] [Google Scholar]
- 7.Huang K, Li Z, Jia Y, Dunaway-Mariano D, Herzberg O. Helix swapping between two alpha/beta barrels: crystal structure of phosphoenolpyruvate mutase with bound Mg(2+)-oxalate. Structure. 1999;7:539–548. doi: 10.1016/s0969-2126(99)80070-7. [DOI] [PubMed] [Google Scholar]
- 8.Sharma V, Sharma S, Hoener zu Bentrup K, McKinney JD, Russell DG, Jacobs WR, Jr., Sacchettini JC. Structure of isocitrate lyase, a persistence factor of Mycobacterium tuberculosis. Nat Struct Biol. 2000;7:663–668. doi: 10.1038/77964. [DOI] [PubMed] [Google Scholar]
- 9.Simanshu DK, Satheshkumar PS, Savithri HS, Murthy MR. Crystal structure of Salmonella typhimurium 2-methylisocitrate lyase (PrpB) and its complex with pyruvate and Mg(2+) Biochem Biophys Res Commun. 2003;311:193–201. doi: 10.1016/j.bbrc.2003.09.193. [DOI] [PubMed] [Google Scholar]
- 10.Teplyakov A, Liu S, Lu Z, Howard A, Dunaway-Mariano D, Herzberg O. Crystal structure of the petal death protein from carnation flower. Biochemistry. 2005;44:16377–16384. doi: 10.1021/bi051779y. [DOI] [PubMed] [Google Scholar]
- 11.von Delft F, Inoue T, Saldanha SA, Ottenhof HH, Schmitzberger F, Birch LM, Dhanaraj V, Witty M, Smith AG, Blundell TL, Abell C. Structure of E. coli ketopantoate hydroxymethyl transferase complexed with ketopantoate and Mg2+, solved by locating 160 selenomethionine sites. Structure. 2003;11:985–996. doi: 10.1016/s0969-2126(03)00158-8. [DOI] [PubMed] [Google Scholar]
- 12.Pflugrath JW. The finer things in X-ray diffraction data collection. Acta Crystallogr D Biol Crystallogr. 1999;55:1718–1725. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- 13.Schneider TR, Sheldrick GM. Substructure solution with SHELXD. Acta Crystallogr D Biol Crystallogr. 2002;58:1772–1779. doi: 10.1107/s0907444902011678. [DOI] [PubMed] [Google Scholar]
- 14.Otwinowski Z. Proc. Daresbury Study Weekend. SERC Daresbury Laboratory; Warrington, England: 1991. “Maximum Likelihood Refinement of Heavy Atom Parameters” In Isomorphous Replacement and Anomalous Scattering; pp. 80–85. [Google Scholar]
- 15.CCP4 The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 16.Cowtan K. DM: an automated procedure for phase improvement. Joint CCP4 and ESF-EACBM Newsletter on Protein Crystallography. 1994:34–38. [Google Scholar]
- 17.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr D Biol Crystallogr. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Terwilliger TC. Automated main-chain model building by template matching and iterative fragment extension. Acta Crystallogr D Biol Crystallogr. 2003;59:38–44. doi: 10.1107/S0907444902018036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones TA, Zou JY, Cowan SW, Kjeldgaard GJ. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 20.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 21.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 22.Read RJ. Improved Fourier coefficients for maps using phases from partial structures with errors. Acta Crystallographica section A. 1986;42:140–149. [Google Scholar]
- 23.Laskowski RA, MacArthur MW, Moss DS, Thornton J. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;26:283–291. [Google Scholar]
- 24.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; Palo Alto, CA, USA: 2002. [Google Scholar]
- 25.Potterton E, McNicholas S, Krissinel E, Cowtan K, Noble M. The CCP4 molecular-graphics project. Acta Crystallogr D Biol Crystallogr. 2002;58:1955–1957. doi: 10.1107/s0907444902015391. [DOI] [PubMed] [Google Scholar]
- 26.Potterton L, McNicholas S, Krissinel E, Gruber J, Cowtan K, Emsley P, Murshudov GN, Cohen S, Perrakis A, Noble M. Developments in the CCP4 molecular-graphics project. Acta Crystallogr D Biol Crystallogr. 2004;60:2288–2294. doi: 10.1107/S0907444904023716. [DOI] [PubMed] [Google Scholar]
- 27.Kleywegt GJ, Jones TA. Detection, delineation, measurement and display of cavities in macromolecular structures. Acta Crystallogr D Biol Crystallogr. 1994;50:178–185. doi: 10.1107/S0907444993011333. [DOI] [PubMed] [Google Scholar]
- 28.Lenz H, Wunderwald P, Eggerer H. Partial purification and some properties of oxalacetase from Aspergillus niger. European journal of biochemistry / FEBS. 1976;65:225–236. doi: 10.1111/j.1432-1033.1976.tb10409.x. [DOI] [PubMed] [Google Scholar]
- 29.Brändén C-I. Relation between structure and function of α/β-proteins. Q. Rev. Biophys. 1980;13:317–338. doi: 10.1017/s0033583500001712. [DOI] [PubMed] [Google Scholar]
- 30.Liu S, Lu Z, Han Y, Jia Y, Howard A, Dunaway-Mariano D, Herzberg O. Conformational flexibility of PEP mutase. Biochemistry. 2004;43:4447–4453. doi: 10.1021/bi036255h. [DOI] [PubMed] [Google Scholar]
- 31.Han Y, Joosten HJ, Niu W, Zhao Z, Mariano PS, McCalman M, van Kan J, Schaap PJ, Dunaway-Mariano D. Oxaloacetate hydrolase, the C-C bond lyase of oxalate secreting fungi. J Biol Chem. 2007;282:9581–9590. doi: 10.1074/jbc.M608961200. [DOI] [PubMed] [Google Scholar]
- 32.Sugantino M, Zheng R, Yu M, Blanchard JS. Mycobacterium tuberculosis ketopantoate hydroxymethyltransferase: tetrahydrofolate-independent hydroxymethyltransferase and enolization reactions with alpha-keto acids. Biochemistry. 2003;42:191–199. doi: 10.1021/bi020516q. [DOI] [PubMed] [Google Scholar]
- 33.Kim A, Kim J, Martin BM, Dunaway-Mariano D. Isolation and characterization of the carbon-phosphorus bond-forming enzyme phosphoenolpyruvate mutase from the mollusk Mytilus edulis. J Biol Chem. 1998;273:4443–4448. doi: 10.1074/jbc.273.8.4443. [DOI] [PubMed] [Google Scholar]
- 34.O'Leary MH. Catalytic Strategies in Enzymatic Carboxylation and Decarboxylation. In: Sigman DS, editor. Mechanism of catalysis. Academic Press, Inc.; 1992. p. 239. [Google Scholar]
- 35.Lu Z, Feng X, Song L, Han Y, Kim A, Herzberg O, Woodson WR, Martin BM, Mariano PS, Dunaway-Mariano D. Diversity of function in the isocitrate lyase enzyme superfamily: the Dianthus caryophyllus petal death protein cleaves alpha-keto and alpha-hydroxycarboxylic acids. Biochemistry. 2005;44:16365–16376. doi: 10.1021/bi051776l. [DOI] [PubMed] [Google Scholar]
- 36.Kanehisa M, Goto S, Hattori M, Aoki-Kinoshita KF, Itoh M, Kawashima S, Katayama T, Araki M, Hirakawa M. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 2006;34:D354–357. doi: 10.1093/nar/gkj102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kubala G, Martell AE. Nuclear Magnetic Resonance Investigation of the Spontaneous Decarboxylation of 2-Oxalopropionic Acid. 2. Species in Solution. J. Am. Chem. Soc. 1981;103:7609–7615. [Google Scholar]
- 38.Liu S, Lu Z, Jia Y, Dunaway-Mariano D, Herzberg O. Dissociative phosphoryl transfer in PEP mutase catalysis: structure of the enzyme/sulfopyruvate complex and kinetic properties of mutants. Biochemistry. 2002;41:10270–10276. doi: 10.1021/bi026024v. [DOI] [PubMed] [Google Scholar]
- 39.Creighton DJ, Rose IA. Studies on the mechanism and stereochemical properties of the oxalacetate decarboxylase activity of pyruvate kinase. J Biol Chem. 1976;251:61–68. [PubMed] [Google Scholar]
- 40.Jursinic SB, Robinson JL. The active site of rabbit muscle pyruvate kinase. Evidence for a site common to the oxalacetate decarboxylase and pyruvate kinase reactions. Biochimica et biophysica acta. 1978;523:358–367. doi: 10.1016/0005-2744(78)90038-4. [DOI] [PubMed] [Google Scholar]
- 41.Park SH, Harris BG, Cook PF. pH dependence of kinetic parameters for oxalacetate decarboxylation and pyruvate reduction reactions catalyzed by malic enzyme. Biochemistry. 1986;25:3752–3759. doi: 10.1021/bi00361a004. [DOI] [PubMed] [Google Scholar]
- 42.Sender PD, Martin MG, Peiru S, Magni C. Characterization of an oxaloacetate decarboxylase that belongs to the malic enzyme family. FEBS letters. 2004;570:217–222. doi: 10.1016/j.febslet.2004.06.038. [DOI] [PubMed] [Google Scholar]
- 43.Chang GG, Tong L. Structure and function of malic enzymes, a new class of oxidative decarboxylases. Biochemistry. 2003;42:12721–12733. doi: 10.1021/bi035251+. [DOI] [PubMed] [Google Scholar]
- 44.Studer R, Dahinden P, Wang WW, Auchli Y, Li XD, Dimroth P. Crystal structure of the carboxyltransferase domain of the oxaloacetate decarboxylase Na+ pump from Vibrio cholerae. J Mol Biol. 2007;367:547–557. doi: 10.1016/j.jmb.2006.12.035. [DOI] [PubMed] [Google Scholar]
- 45.Piccirilli JA, Rozzell JD, Benner SA. The stereochemistry of oxaloacetate decarboxylase: A stereochemical imperative? J. Amer Chem Soc. 1987;109:8084–8085. [Google Scholar]
- 46.Waldrop GL, Braxton BF, Urbauer JL, Cleland WW, Kiick DM. Secondary 18O and primary 13C isotope effects as a probe of transition-state structure for enzymatic decarboxylation of oxalacetate. Biochemistry. 1994;33:5262–5267. doi: 10.1021/bi00183a032. [DOI] [PubMed] [Google Scholar]
- 47.Park IS, Hausinger RP. Science. Vol. 267. New York, N.Y: 1995. Requirement of carbon dioxide for in vitro assembly of the urease nickel metallocenter; pp. 1156–1158. [DOI] [PubMed] [Google Scholar]
- 48.Jacobs MA, Alwood A, Thaipisuttikul I, Spencer D, Haugen E, Ernst S, Will O, Kaul R, Raymond C, Levy R, Chun-Rong L, Guenthner D, Bovee D, Olson MV, Manoil C. Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A. 2003;100:14339–14344. doi: 10.1073/pnas.2036282100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.