

Abstract
Objectives
We have investigated whether red cell infiltration of atheromatous lesions promotes the later stages of atherosclerosis.
Methods and results
We find that oxidation of ferro (FeII) hemoglobin in ruptured advanced lesions occurs generating ferri (FeIII) hemoglobin and via more extensive oxidation ferrylhemoglobin (FeIII/FeIV=O). The protein oxidation marker, dityrosine, accumulates in complicated lesions accompanied by the formation of crosslinked hemoglobin, a hallmark of ferrylhemoglobin. Exposure of normal red cells to lipids derived from atheromatous lesions causes hemolysis and oxidation of liberated hemoglobin. In the interactions between hemoglobin and atheroma lipids, hemoglobin and heme promote further lipid oxidation and subsequently endothelial reactions such as upregulation of heme oxygenase-1 and cytotoxicity to endothelium. Oxidative scission of heme leads to release of iron and a feed-forward process of iron-driven plaque lipid oxidation. The inhibition of heme release from globin by haptoglobin and sequestration of heme by hemopexin suppress hemoglobin-mediated oxidation of lipids of atheromatous lesions and attenuate endothelial cytotoxicity.
Conclusions
The interior of advanced atheromatous lesions is a pro-oxidant environment in which erythrocytes lyse, hemoglobin is oxidized to ferri- and ferrylhemoglobin, and released heme and iron promote further oxidation of lipids. These events amplify the endothelial cell cytotoxicity of plaque components.
Keywords: atherosclerosis, hemoglobin, lipid peroxidation, heme-oxygenase-1
Hematomas are formed either by fissures at the atherosclerotic lesion surface,1 or within the lesions as hemorrhages from neovasculature that sprouts from the vasa vasorum.2,3
Oxysterols and oxidation products of polyunsaturated fatty acids (PUFAs) are present in human atheromatous lesions.4,5 Atherosclerotic lesions are hazardous regions for nucleated cells, both endothelial cells and, quite probably, incoming macrophages.6 The major cytotoxic species may be oxidation products of lipids, particularly lipid hydroperoxides (LOOHs), aldehydes and carbonyls.6,7
In artificial systems, oxidation of PUFAs requires reactive transition metals such as iron and copper. Based on our earlier work,6,8,9 the metal in atheromatous lesions might be iron derived from heme. Non-protein bound heme is a particularly deleterious species inasmuch as it is hydrophobic and easily able to enter cell membranes.10
In previous studies we found that endothelial cells exposed to oxidized LDL upregulated both heme oxygenase-1 (HO-1) and ferritin,8,9 presumably as a defense mechanism.6,11-14 Up-regulation of HO-115 and ferritin H chain16 in endothelial cells has been reported in the early phase of progression of atherosclerotic lesions. Expression of HO-1 provides protection against atherosclerosis in several experimental models17,18 and HO-1 deficiency in humans has been associated with the appearance of vasculature fatty streaks and atheromatous plaques at age of six.19
We tested the hypothesis that heme-iron may accumulate in atherosclerotic lesions by intrusion and lysis of erythrocytes. Liberated hemoglobin is oxidized, and released heme-iron dependent oxidation of lipids is strongly favored, contributing to further plaque development.
Methods
Tissue samples
Specimens of human atherosclerotic lesions were obtained from aorta or its primary branches of beating-heart donors for organ transplantation, approved by the Scientific and Research Ethic Committee of Scientific Council of Health of the Hungarian Government. Tissue samples were washed with saline, dried, weighed, frozen in liquid nitrogen and stored at -70°C until assay. Samples showing no macroscopic evidence of atherosclerosis were designated as controls. Samples of lesions exhibiting thickened intima and large lipid cores with no sign of disruption represented atheromatous lesions. Disrupted plaques with hematomas were designated as complicated lesions. For histopathological examination tissues were fixed in 10% formalin and embedded in paraffin. 5μm sections were deparaffined with xylol for 8 minutes and rehydrated in a descending series of isopropyl-alcohol. H&E staining was performed (hematoxilin for 6 minutes, followed by wash in distilled water for 8 minutes, staining with eosin for 2 minutes, dehydrating and mounting on a coverslip). Stained slides were scanned with a Mirax Midi scanner (3D Histech, Budapest, Hungary) for digital documentation.
Lipids of vessel samples were extracted from tissue by chloroform-methanol (2:1 v/v).20 The organic phase was evaporated under N2 and the weight of the lipid extract was measured. The extract was redissolved in a small volume of chloroform and suspended in HBSS to produce a suspension of 2 mg lipid/mL solvent. Lipid aggregates were dispersed by the evaporation of chloroform coupled with vigorous vortexing.
Erythrocyte lysis by plaque material
Red cells were obtained from venous blood of a normal donor. The cells were incubated with extracts from vessels in a suspension containing 0.2 v/v% packed red cells, 1 mg/mL vessel lipid extracts in HBSS at 37°C. The amount of free hemoglobin was monitored spectrophotometrically for 72 hours13 after centrifuging.
Glutathione/glutathione peroxidase treatment
Equimolar amounts of glutathione (GSH) added to oxidized LDL or plaque lipids of known hydroperoxide content. Glutathione peroxidase (Px) was applied according to the manufacturer's instructions. Reduction of lipid hydroperoxides took 1 hour for LDL and 16 hours for plaque lipids at 37 °C.
Measurement of dityrosine in hemoglobin
Hemoglobin from minced complicated lesions was extracted by vortexing with saline. After centrifugation to remove insoluble material the supernatant was precipitated and hydrolyzed. The residue was injected into an HPLC (Merck, Darmstadt, Germany) using a reverse-phase C18 column (4.6 × 150mm, 5 mm particle size) and the eluate was monitored by fluorescence detection (ex=280 nm, em=410). The mobile phase consisted of 20% methanol and 0.2% trifluoroacetic acid in water. Control dityrosine samples were prepared as described earlier.21 Results are expressed as area under the curve (AUC).
Detection of crosslinked hemoglobin by Western blot
150 nmol of hemoglobin from complicated lesions was applied to 12.5% SDS-PAGE gel. Hemoglobin polymerization was detected using a chicken anti-human polyclonal hemoglobin antibody (ab17542, Abcam, Cambridge, UK).
Endothelial cell cytotoxicity assay
Endothelial cell monolayers were exposed for 16 hours to lipid suspensions (2 mg/mL in HBSS) that were either untreated or treated with heme (5 μmol/L), heme and antioxidants (10 μmol/L butylated hydroxytoluene; BHT or 40 μmol/L α-tocopherol; α-toc); or an iron chelator, 100 μmol/L desferroxamine (DFO) or the heme-binding protein, hemopexin (Hpx, 5 μmol/L). In additional experiments, the lipid suspension (2 mg/ml in HBSS) was treated with 10 μmol/L ferro- or ferrihemoglobin in the presence or absence of haptoglobin (Hpg, 20 μmol/L) or hemopexin (40 μmol/L) for 16 hours. Cell viability was determined by MTT-reduction (3-[4,5-dimethylthiazol-2yl]-2,5-diphenyl-tetrazolium-bromide).
Statistical analysis
Statistical analysis was performed by ANOVA test followed by post hoc, Tukey's test for multiple comparisons. Significance is indicated on figures by one (p < 0.05) or two (p < 0.01) asterisks. Results are expressed as mean ± standard deviation of at least three independent experiments.
Results
Hematomas occur in atheromatous lesions and plaque material contains lipid oxidation products including lipid hydroperoxide22 which can mediate not only the oxidation of hemoglobin but might also lyse intact red cells.23 We therefore tested whether oxidized LDL and lipids from atheroma have hemolytic activity. Lipids of atheromatous (Fig 1A, 1b and 2b panels) and ruptured complicated lesions (Fig 1A, 1c and 2c panels) as well as oxidized LDL caused significant lysis of red cells within 24 hours (Fig 1B, black bars). Moreover, the cell-free hemoglobin underwent oxidation (Fig 1C). Preincubation of lipid extract derived from atheroma, complicated lesion or oxidized LDL with glutathione/glutathione peroxidase (which specifically reduced lipid hydroperoxide to alcohol by 35%, 38% and 90%, respectively) significantly lowered the lytic effect (Fig 1B, empty bars). Oxidation of liberated hemoglobin was also reduced (Fig 1C, empty bars). These observations support the notion that red cells infiltrating an atherosclerotic lesion will undergo hemolysis and release free hemoglobin that is subsequently oxidized.
Figure 1. Hemolysis and hemoglobin oxidation are provoked by exposure to atheromatous lipid.
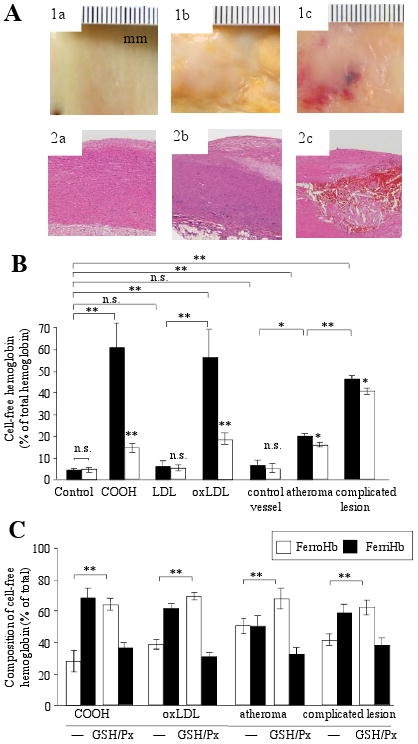
(A) Intact, normal arterial intima (control,1a), atheromatous plaque showing irregular yellowish discoloration due to fibrofatty material (1b) and complicated plaque with a prominent hemorrhagic lesion (1c) representing a continuous spectrum of atheromatous changes of the aorta. Histopathology demonstrates no change in control (2a), extracellular intimal fat accumulation and cholesterol crystal formation in atheroma (2b) and red blood cell extravasation (haemorrhage) into the fatty plaque (2c). (B) Cumene hydroperoxide (COOH; 50 μmol/L), LDL (250 μg/mL) and 1 mg/mL lipid from control (n=7), atheromatous (n=11) and complicated lesions (n=7) were added to red blood cell suspensions. Amounts of liberated hemoglobin were measured at 24 hours, with (empty bars) and without (black bars) glutathione/glutathione peroxidase pretreatment (GSH/Px). (C) Composition of liberated hemoglobin is expressed in percent of total free hemoglobin.
Figure 2. Lipid hydroperoxides of atheroma lipids convert ferrohemoglobin to ferrihemoglobin.

(A) 200 μg/mL native LDL and 1 mg/mL lipid suspension from atheromatous lesions (n=16) were incubated with 10 μmol/L ferro-or ferrihemoglobin and lipid hydroperoxide content was measured. (B) LDL samples of different lipid hydroperoxide content were incubated with 10 μmol/L ferrohemoglobin for 90 minutes. Ferrihemoglobin ratios with and without (empty and black bars) glutathione/glutathione peroxidase pretreatment (GSH/Px) are expressed as percent of total hemoglobin. (C) Lipids (1 mg/mL) from atheromatous lesion were incubated with 10 μmol/L of ferrohemoglobin. Changes in ferro (阳)-and ferrihemoglobin (■) levels were monitored over 24 hours. (D) Effect of glutathione/glutathione peroxidase pretreatment on the reaction of 1 mg/mL lipid from atheromatous lesion with 10 μmol/L of ferrohemoglobin at 24 hours.
We earlier reported that both ferrihemoglobin and heme can mediate oxidative modification of LDL.9 In contrast, ferrohemoglobin – in which the heme group is bound tightly – cannot initiate oxidation of LDL.24 Therefore, we incubated lipids from atheromatous lesions with ferro- and ferrihemoglobin. Surprisingly, both ferro- and ferrihemoglobin induced the oxidative modification of atheromatous lesion lipids, as indicated by lipid hydroperoxide formation (Fig 2A). Since oxidatively modified LDL was shown to oxidize ferrohemoglobin,25 and the degree of oxidation strongly depended on the concentration of LDL-associated lipid hydroperoxides (Fig 2B), we assessed whether lipid hydroperoxides in the lipid extracts of atheromatous lesion might also oxidize ferrohemoglobin. Indeed, lipids derived from atheromatous lesions promoted the oxidation of ferrohemoglobin to ferrihemoglobin (Fig 2C). Treatment of oxidized LDL (Fig 2B), or atheroma lipids (Fig 2D) with glutathione/glutathione peroxidase lowered the lipid hydroperoxide content as well as the oxidation of hemoglobin.
As is true of intact hemoglobin, lipid extracts from atheromatous lesions exposed to heme also underwent lipid peroxidation as reflected by the accumulation of thiobarbituric acid-reactive substances (TBARs) and lipid hydroperoxides (supplemental Fig I). The results suggest that hemoglobin-derived heme can promote oxidation within atheromatous lesions and that such oxidation requires oxidant species present in atheromatous plaque but not in normal vasculature.
Heme/iron-mediated oxidative modification of LDL can cause endothelial cytotoxicity8,24 and – at sublethal doses – the expression of stress-response genes.9,11-14 Therefore, we tested whether the same effect was observed when endothelial cells were exposed to lipids isolated from atheromatous lesions pre-exposed to heme or not (Fig 3A). As shown in Fig 3B, lipids from atherosclerotic lesions were cytotoxic to endothelium, an effect strikingly enhanced when lipids were pre-oxidized by exposure to heme. Equal amounts of lipid isolated from control blood vessels were not cytotoxic. At sublethal doses, atheroma lipid - whether pre-treated with heme or not - induced the expression of the stress-responsive gene HO-1, at both mRNA (Fig 3C) and protein levels (Fig 3D). In contrast, lipids from control blood vessels failed to affect the expression of HO-1 in endothelium. Pre-treatment of heme-oxidized lipids with glutathione/glutathione peroxidase reduced the lipid hydroperoxide content (113±30 vs. 74±22 nmol LOOH/mg extract, p<0.01) and inhibited the endothelial cytotoxicity by 25% (p<0.05). Furthermore, the induction of HO-1 was decreased (153 ± 16 versus 105 ± 3 pmol bilirubin formed per milligram of cell protein per 60 minutes, p<0.01) in endothelial cells.
Figure 3. Heme with atheroma lipids augments lipid peroxidation and subsequent endothelial cell reactions.

Lipids from atheromatous lesions (n=9) or controls (n=9) were treated with heme. LOOH content (A), specific cytotoxicity (B), HO-1 mRNA expression (C) and and HO-1 enzyme activity (D) were measured.
In an attempt to explain the different effect of heme on lipid extracts of atheromatous lesions versus control blood vessels, we measured the amounts of lipid peroxidation products in control and atheromatous samples. Levels of conjugated dienes, lipid hydroperoxides and TBARs were significantly higher in atheromatous lesions compared to controls (supplemental Table I). Elevated cholesterol, oxy-cholesterol, lyso-phospholipid and decreased phosphatidylserine were found in atheromatous lipids compared to controls (supplemental Table II). Moreover, lipids extracted from atheromatous lesions contained 1.9 times more monounsaturated fatty acids than control extracts. Significantly lower amounts of PUFA were found likely reflecting enhanced oxidation of PUFA within the lesions.
The chemical changes exerted by heme on lipids isolated from atheromatous lesions were attenuated by antioxidants such as BHT, α-tocopherol, the iron chelator deferoxamine, and the heme binding protein, hemopexin26 (supplemental Fig II A). Because haptoglobin stabilizes the binding of heme to globin and inhibits heme release from hemoglobin,27 we exposed lipid derived from atheromatous lesions to hemoglobin in the presence of haptoglobin. Hemoglobin-mediated oxidative modification of lipid extracted from atheromatous lesions was inhibited by haptoglobin (Fig 4A). Moreover, the heme-binding protein, hemopexin, also suppressed the oxidation of lipid by ferro- and ferrihemoglobin, indicating the necessity for heme release from ferrihemoglobin for this oxidative process. Inhibition of lipid oxidation by either haptoglobin or hemopexin reduced the cytotoxicity (Fig 4B) and HO-1 induction caused by sublethal amounts of pretreated atheromatous lesion lipids (Fig 4C and D).
Figure 4. Oxidation of atheroma lipids by hemoglobin is inhibited by haptoglobin or hemopexin.
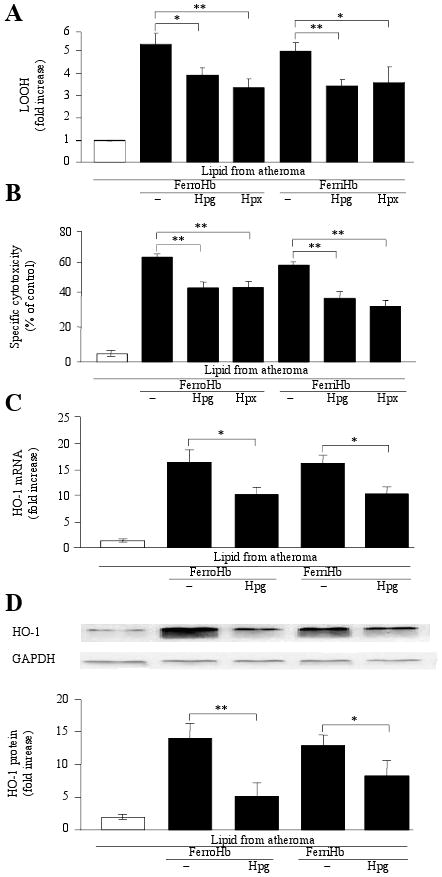
Atheroma lipids (n=16) were incubated with ferro- or ferrihemoglobin in the presence or absence of haptoglobin (n=5) or hemopexin (n=3) for 16 hours. LOOH content (A), specific cytotoxicity (B), HO-1 mRNA (C) and protein expression (D) were measured.
Since ruptured complicated lesions contain hemoglobin, and DFO prevented lipid oxidation, we next measured levels of iron and lipid peroxidation products in extracts of these lesions. Concentrations of iron, conjugated dienes and lipid hydroperoxides were elevated by about 2-fold in ruptured complicated lesions, as compared to atheromatous lesions (0.433 ± 0.075 vs. 0.185 ± 0.096 nmol Fe/mg tissue; 0.047 ± 0.019 vs. 0.021 ± 0.003 A234 conjugated dienes/mg tissue and 0.465 ± 0.110 vs. 0.248 ± 0.106 nmol LOOH/mg tissue, respectively) and complicated lesions contained 5.6 times more TBARs than atheromatous lesions (0.028 ± 0.012 vs. 0.005 ± 0.001 nmol/mg tissue). Importantly, hemoglobin derived from ruptured complicated lesions was mainly oxidized (Fig 5A). Moreover, the amounts of a protein oxidation marker, dityrosine,21 were elevated in complicated lesions (Fig 5B) whereas atheromatous lesions did not contain detectable dityrosine. Supporting the assumption that hemoglobin was oxidized by lipid hydroperoxides, we found that dityrosine also forms in hemoglobin exposed to oxidized LDL and oxidized atheroma lipids, whereas hemoglobin oxidized by K3Fe(CN)6 did not contain this marker (not shown). Accumulation of dityrosine was accompanied by the formation of crosslinked hemoglobin (Fig 5C) a hallmark of precedent formation of ferrylhemoglobin.
Figure 5. Hemoglobin derived from complicated lesions is oxidized.
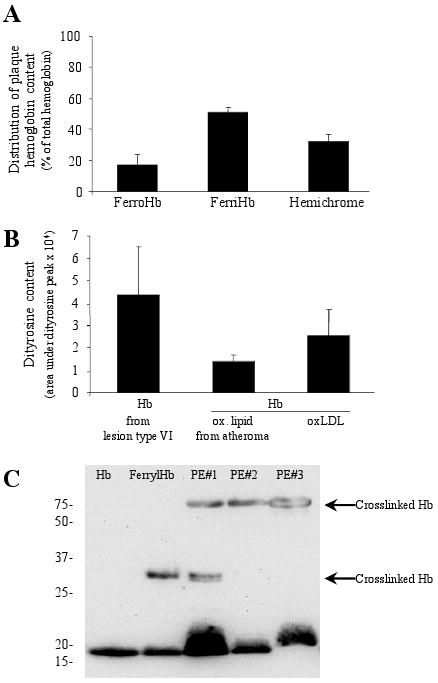
(A) Hemoglobin composition was determined. (B) Dityrosine content of oxidized hemoglobin was measured by HPLC. Oxidation of hemoglobin was induced by heme pre-treated lipid from atheromatous lesion (n=3) or oxidized LDL. (C) Hemoglobin polymers were detected in H2O2-treated hemoglobin and in hemoglobin obtained from complicated lesions (PE I, II, III).
Discussion
Atheromatous lesions are prone to disruption leading to hematoma or hemorrhage. Erythrocytes can also enter developing atherosclerotic lesions through the porous neovasculature in the vasa vasorum underlying atherosclerotic plaque.1-3 The results reported here indicate that, once exposed to oxidized plaque material, erythrocytes are lysed, the liberated hemoglobin is oxidized and heme dissociates from the resultant ferrihemoglobin. The free heme can be oxidatively cleaved by oxidized plaque components, through reaction with lipid hydroperoxides. This engenders the release of iron, which can promote further the oxidation of plaque lipids through redox cycling reactions. The result of these chemical reactions is the formation of deleterious oxidized ‘gruel’ which, among other things, leads to endothelial oxidative stress and ultimately to cytotoxicity.
These oxidation reactions involve initial interactions between the excess lipid in the vessel wall (mainly derived from LDL) and heme and heme-derived iron. Previously we have shown that heme can enter the lipid moiety of LDL and induce iron-dependent lipid peroxidation.8 Here we demonstrate that lipids isolated from human atheromatous lesions – which are already in an oxidized state – can be further oxidized in the presence of heme, whereas this effect is not observed using lipids isolated from normal vasculature. Heme treatment of the atheroma and its extracted lipid increased the amount of conjugated dienes, LOOHs and TBARs. Kinetics of the reaction between heme and atheroma lipid in the presence or absence of antioxidants and an iron chelator were similar to that reactions between LDL and heme suggesting that the mechanisms of the two reactions are similar. The heme-binding protein, hemopexin, which likely prevents heme:lipid interactions and blocks the oxidative scission of heme,26 significantly inhibited the oxidative reactions. This serum protein, present at remarkably high concentrations in plasma (≈1g/L), binds heme with extraordinary avidity (Kd less than 1 pmol/L) and promotes its clearance.
Importantly, atheroma lipid, whether exposed to heme or not, is cytotoxic, while lipids isolated from control vessels do not exert toxicity towards endothelial cells. We have found that atheroma lipids when oxidized by heme are highly cytotoxic to human endothelial cells, and hemopexin reduced this cytotoxicity. The inhibition provided by DFO supports the idea that as heme-mediated oxidation of plaque material proceeds, there is a concomitant increase in free iron from the heme molecule.
Since hemoglobin, when oxidized, releases its heme28 we asked whether ferrihemoglobin might also modify the lipids of artery walls. After a co-incubation of ferrihemoglobin and atheroma lipid, peroxidation products were formed to an extent similar to that of heme treatment. Both the hemoglobin-binding protein, haptoglobin,27 and the heme-binding protein, hemopexin, inhibited such oxidative modification of lipids indicating the importance of heme loss and scission in hemoglobin-provoked oxidation of lipids derived from atheromatous lesions. Ferrohemoglobin also exerted the same effect on plaque extracts. Thus, it appears that these extracts oxidize ferrohemoglobin to ferrihemoglobin, thereby leading to heme instability and heme-mediated initiation of lipid peroxidation.
Heme oxygenase-1, a key antioxidant enzyme that exerts cytoprotective effects in endothelial cells8,11-14 also plays an important role in preventing the development of atherosclerosis. Elevated amounts of HO-1 were found in macrophages and medial smooth muscle cells of human atherosclerotic lesions.15 The central importance of HO-1 in atherosclerosis is highlighted by the case of a HO-1 deficient boy who suffered from severe atherosclerosis.19 Earlier we found that HO-1 induction occurs when endothelial cells are treated with sublethal amounts of LDL oxidized by ferrihemoglobin-derived heme.24 Now we demonstrate that heme and hemoglobin-treated atheroma lipids also induce HO-1 in endothelial cells exposed in sublethal doses. The degree of HO-1 induction is partially a function of LOOH levels of the lipid as inhibition of lipid oxidation moderates its HO-1 induceability. Supporting this, 13-HPODE, a hydroperoxide derivative of linoleic acid acts as a transcriptional factor for HO-1 via a regulatory element in the promoter region of the human HO-1 gene.29
Supporting the relevance of our model of plaque development, we examined complicated lesions containing hematomas. Lipids of these plaques were highly oxidized as reflected by increased amounts of conjugated dienes, LOOHs and TBARs. Earlier chemical investigations of gruel from advanced lesions revealed that it contains ceroid-like insoluble material composed mainly of hydoxyapatite, iron and calcium. These materials are cytotoxic to macrophages and this was ameliorated by chelating iron and calcium.6 Gruel from advanced plaque also contains large amounts of organic soluble carbonyls and aldehydes that are also cytotoxic.
Hemoglobin derived from hematomas in complicated lesions was mainly present in the oxidized form (ferrihemoglobin). Under inflammatory conditions, ferrihemoglobin is formed in erythrocytes exposed to activated polymorphonuclear cells.30 Cell-free hemoglobin is readily oxidized to ferrihemoglobin by oxidants such as H2O2 produced by activated polymorphonuclear cells or monocyte/macrophages. Hemoglobin can also be oxidized by reacting with lipid hydroperoxides.25 In addition to ferrihemoglobin these reactions generate ferrylhemoglobin,31 an unstable oxidized form of hemoglobin detected in humans under physiologic32 and pathophysiologic conditions.33 This highly unstable oxidized form of hemoglobin (ferryl state, FeIII/IV=O) rapidly returns to the ferric (FeIII) state through protein electron transfer in which the α–chain tyrosine 42 acts as a redox center, cycling between the tyrosine and the tyrosyl radical while delivering electrons to ferryl heme.10,34,35 Tyrosyl radicals can react with each other to generate dityrosine leading to inter- and intramolecular cross-linking and formation of hemoglobin multimers. We recently found that, contrary to other forms of oxidized hemoglobin, ferrylhemoglobin acts as a potent pro-inflammatory agonist in endothelial cells, leading to the up-regulation of adhesion molecules that support the recruitment of macrophages into the vessel wall.36 Detection of dityrosine and crosslinked hemoglobin in complicated plaques suggests that lipid hydroperoxide-mediated oxidation of hemoglobin occurs within the lesion. Further evidence for this mechanism is the finding that oxidation of hemoglobin with either LDL or atheroma lipid with elevated lipid hydroperoxide levels also led to the formation of dityrosine residues in the globin moiety of hemoglobin.
Overall, our results support the concept that erythrocytes invading atheromatous lesions are lysed by lipid oxidation products within the lesions. Ferrohemoglobin released by this event is converted to ferryl- and ferrihemoglobin by the same oxidized materials. This, in turn, destabilizes the heme group promoting its release from the globin and the free hydrophobic heme group readily enters atheroma lipid. Oxidative scission of the heme group leads to iron release and a feed-forward process of further plaque lipid oxidation. The inhibition of heme release from hemoglobin by haptoglobin and sequestration of heme by hemopexin suppress hemoglobin-mediated oxidation of lipids of atheromatous lesions and attenuate subsequent endothelial cell damage (Fig 6). These events likely amplify the oxidation of plaque components which are cytotoxic for endothelial cells and, quite likely, infiltrating phagocytic cells which would otherwise help resolve these lesions.
Figure 6. A model for red cell-mediated progression of atherogenesis.

1) Red blood cells infiltrate the atheromatous lesion; 2) Erythrocyte lysis and liberation of ferrohemoglobin by lipids of atheroma; 3) Oxidation of ferrohemoglobin to ferrylhemoglobin (a) and to ferrihemoglobin (b); 4) Heme is released from ferrihemoglobin; 5) Heme uptake by atheroma lipid; 6) Amplification of lipid oxidation in atheroma; 7) Damage and activation of endothelium induced by reactive lipid metabolites of atheroma; and 8) Induction of heme oxygenase-1 and ferritin by atheroma lipid and heme.
Supplementary Material
Acknowledgments
We thank Erika Barna for technical assistance.
Funding Sources: This work was supported by Hungarian Government grants OTKA-K61546, OTKA- K75883, ETT-337/2006, ETT-147/2009, RET-06/2004, MTA-DE-11003; JWE is supported by the Commonwealth of Kentucky Research Challenge Trust Fund and by NIH DK073586; VJ is supported by the European Commission's 7th Framework, PEOPLE-2007-2-1-IEF “GasMalaria”; MPS is supported by POCTI/BIA-BCM/56829/2004, POCTI/SAU-MNO/56066/2004 and POCTI/SAU/56066/2007 grants from Fundação para a Ciência e a Tecnologia, Portugal, XENOME (LSHB-CT-2006037377), GEMI fund (Linde Health care), and European Commission's sixth Framework Program; AS is supported by the University of Missouri at Kansas City Research Incentive Funds. GMV is supported by NIH NHLBI R01 HL67367 and P01HL055552.
Footnotes
Disclosures: None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Stary HC, Chandler AB, Dinsmore RE, Fuster V, Glagov S, Insull W, Jr, Rosenfeld ME, Schwartz CJ, Wagner WD, Wissler RW. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. Circulation. 1995;92:1355–1374. doi: 10.1161/01.cir.92.5.1355. [DOI] [PubMed] [Google Scholar]
- 2.Paterson JC. Vascularization and hemorrhage of the intima of arteriosclerotic coronary arteries. Arch Pathol. 1936;22:313–324. [Google Scholar]
- 3.Barger AC, Beeuwkes R, III, Lainey LL, Silverman KJ. Hypothesis: vasa vasorum and neovascularization of human coronary arteries-a possible role in the pathophysiology of atherosclerosis. N Engl J Med. 1984;310:175–177. doi: 10.1056/NEJM198401193100307. [DOI] [PubMed] [Google Scholar]
- 4.Fogelman AM, Shechter I, Seager J, Hokom M, Child FS, Edwards PA. Malondialdehyde alteration of low density lipoproteins leads to cholesteryl ester accumulation in human monocyte-macrophages. Proc Natl Acad Sci USA. 1980;77:2214–2218. doi: 10.1073/pnas.77.4.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sevanian A, McLeod LL. Cholesterol autoxidation in phospholipid bilayers. Lipids. 1987;22:627–631. doi: 10.1007/BF02533940. [DOI] [PubMed] [Google Scholar]
- 6.Li W, Ostblom M, Xu LH, Hellsten A, Leanderson P, Liedberg B, Brunk UT, Eaton JW, Yuan XM. Cytocidal effects of atheromatous plaque components: the death zone revisited. FASEB J. 2006;20:2281–90. doi: 10.1096/fj.06-6114com. [DOI] [PubMed] [Google Scholar]
- 7.Therond P, Abella A, Laurent D, Couturier M, Chalas J, Legrand A, Lindenbaum A. In vitro study of the cytotoxicity of isolated oxidized lipid low density lipoprotein fractions in human endothelial cells: relationship with the glutathione status and cell morphology. Free Radic Biol Med. 2000;28:585–596. doi: 10.1016/s0891-5849(99)00265-8. [DOI] [PubMed] [Google Scholar]
- 8.Balla G, Jacob HS, Eaton JW, Belcher JD, Vercellotti GM. Hemin: a possible physiological mediator of low density lipoprotein oxidation and endothelial cell injury. Arterioscler Thromb. 1991;11:1700–1711. doi: 10.1161/01.atv.11.6.1700. [DOI] [PubMed] [Google Scholar]
- 9.Balla J, Vercellotti GM, Jeney V, Yachie A, Varga Z, Jacob HS, Eaton JW, Balla G. Heme, heme oxygenase, and ferritin: how the vascular endothelium survives (and dies) in an iron-rich environment. Antioxid Redox Signal. 2007;9:2119–2137. doi: 10.1089/ars.2007.1787. [DOI] [PubMed] [Google Scholar]
- 10.Miller YI, Shaklai N. Oxidative crosslinking of LDL protein induced by hemin: involvement of tyrosines. Biochem Mol Biol Int. 1994;34:1121–1129. [PubMed] [Google Scholar]
- 11.Abraham NG, Lavrovsky Y, Schwartzman ML, Stoltz RA, Levere RD, Gerritsen ME, Shibahara S, Kappas A. Transfection of the human heme oxygenase gene into rabbit coronary microvessel endothelial cells: protective effect against heme and hemoglobin toxicity. Proc Natl Acad Sci USA. 1995;92:6798–6802. doi: 10.1073/pnas.92.15.6798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Balla G, Jacob HS, Balla J, Rosenberg M, Nath K, Apple F, Eaton JW, Vercellotti GM. Ferritin: a cytoprotective antioxidant strategem of endothelium. J Biol Chem. 1992;267:18148–18153. [PubMed] [Google Scholar]
- 13.Balla J, Jacob HS, Balla G, Nath K, Eaton JW, Vercellotti GM. Endothelial-cell heme uptake from heme proteins: induction of sensitization and desensitization to oxidant damage. Proc Natl Acad Sci USA. 1993;90:9285–9289. doi: 10.1073/pnas.90.20.9285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soares MP, Bach FH. Heme oxygenase-1: from biology to therapeutic potential. Trends Mol Med. 2009;15:50–58. doi: 10.1016/j.molmed.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 15.Wang LJ, Lee TS, Lee FY, Pai RC, Chau LY. Expression of heme oxygenase-1 in atherosclerotic lesions. Am J Pathol. 1998;152:711–720. [PMC free article] [PubMed] [Google Scholar]
- 16.Juckett MB, Balla J, Balla G, Jessurun J, Jacob HS, Vercelotti GM. Ferritin protects endothelial cells from oxidized low density lipoprotein in vitro. Am J Pathol. 1995;147:782–789. [PMC free article] [PubMed] [Google Scholar]
- 17.Ishikawa K, Sugawara D, Goto J, Watanabe Y, Kawamura K, Shiomi M, Itabe H, Maruyama Y. Heme oxygenase-1 inhibits atherogenesis in Watanabe Heritable Hyperlipidemic rabbits. Circulation. 2001;104:1831–1836. doi: 10.1161/hc3901.095897. [DOI] [PubMed] [Google Scholar]
- 18.Juan SH, Lee TS, Tseng KW, Liou JY, Shyue SK, Wu KK, Chau LY. Adenovirus-mediated heme oxygenase-1 gene transfer inhibits the development of atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2001;104:1519–1525. doi: 10.1161/hc3801.095663. [DOI] [PubMed] [Google Scholar]
- 19.Yachie A, Niida Y, Wada T, Igarashi N, Kaneda H, Toma T, Ohta K, Kasahara Y, Koizumi S. Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1deficiency. J Clin Invest. 1999;103:129–135. doi: 10.1172/JCI4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Biophys. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 21.Giulivi C, Davies KJA. Mechanism of the formation and proteolytic release of H2O2-induced dityrosine and tyrosine oxidation products in hemoglobin and red blood cells. J Biol Chem. 2001;276:24129–24136. doi: 10.1074/jbc.M010697200. [DOI] [PubMed] [Google Scholar]
- 22.Carpenter KL, Taylor SE, Ballantine JA, Fussel B, Halliwell B, Mitchinson MJ. Lipids and oxidised lipids in human atheroma and normal aorta. Biochim Biophys Acta. 1993;1167:121–130. doi: 10.1016/0005-2760(93)90151-x. [DOI] [PubMed] [Google Scholar]
- 23.van den Berg JJ, Op den Kamp JA, Lubin BH, Roelofsen B, Kuypers FA. Kinetics and site specificity of hydroperoxide-induced oxidative damage in red blood cells. Free Radic Biol Med. 1992;12:487–98. doi: 10.1016/0891-5849(92)90102-m. [DOI] [PubMed] [Google Scholar]
- 24.Jeney V, Balla J, Yachie A, Varga Z, Vercellotti GM, Eaton JW, Balla G. Pro-oxidant and cytotoxic effect of circulating heme. Blood. 2002;100:879–887. doi: 10.1182/blood.v100.3.879. [DOI] [PubMed] [Google Scholar]
- 25.Nagy E, Jeney V, Yachie A, Szabó RP, Wagner O, Vercellotti GM, Eaton JW, Balla G, Balla J. Oxidation of hemoglobin by lipid hydroperoxide associated with low-density lipoprotein (LDL) and increased cytotoxic effect by LDL oxidation in heme oxygenase-1 (HO-1) deficiency. Cell Mol Biol (Noisy-le-grand) 2005;51:377–85. [PubMed] [Google Scholar]
- 26.Gutteridge JM, Smith A. Antioxidant protection by haemopexin of haem-stimulated lipid peroxidation. Biochem J. 1988;256:861–865. doi: 10.1042/bj2560861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gutteridge JM. The antioxidant activity of haptoglobin towards haemoglobin-stimulated lipid peroxidation. Biochim Biophys Acta. 1987;917:319–223. doi: 10.1016/0005-2760(87)90125-1. [DOI] [PubMed] [Google Scholar]
- 28.Bunn HF, Jandl JH. Exchange of heme among hemoglobins and between hemoglobin and albumin. J Biol Chem. 1968;243:465–475. [PubMed] [Google Scholar]
- 29.Hill-Kapturczak N, Voakes C, Garcia J, Visner G, Nick HS, Agarwal A. A cis-acting region regulates oxidized lipid-mediated induction of the human heme oxygenase-1 gene in endothelial cells. Arterioscler Thromb Vasc Biol. 2003;23:1416–1422. doi: 10.1161/01.ATV.0000081656.76378.A7. [DOI] [PubMed] [Google Scholar]
- 30.Weiss SJ. Neutrophil-mediated methemoglobin formation in the erythrocyte. The role of superoxide and hydrogen peroxide. J Biol Chem. 1982;257:2947–53. [PubMed] [Google Scholar]
- 31.Giulivi C, Davies KJ. Hydrogen peroxide-mediated ferrylhemoglobin generation in vitro and in red blood cells. Methods Enzymol. 1994;231:490–496. doi: 10.1016/0076-6879(94)31032-7. [DOI] [PubMed] [Google Scholar]
- 32.Svistunenko DA, Patel RP, Voloshchenko SV, Wilson MT. The globin-based free radical of ferryl hemoglobin is detected in normal human blood. J Biol Chem. 1997;272:7114–7121. doi: 10.1074/jbc.272.11.7114. [DOI] [PubMed] [Google Scholar]
- 33.Vollaard NB, Reeder BJ, Shearman JP, Menu P, Wilson MT, Cooper CE. A new sensitive assay reveals that hemoglobin is oxidatively modified in vivo. Free Radic Biol Med. 2005;39:1216–1228. doi: 10.1016/j.freeradbiomed.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 34.Reeder BJ, Grey M, Silaghi-Dumitrescu RL, Svistunenko DA, Bülow L, Cooper CE, Wilson MT. Tyrosine Residues as Redox Cofactors in Human Hemoglobin: Implications for engineering nontoxic blood substitutes. J Biol Chem. 2008;283:30780–30787. doi: 10.1074/jbc.M804709200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maples KR, Kennedy CH, Jordan SJ, Mason RP. In vivo thiyl free radical formation from hemoglobin following administration of hydroperoxides. Arch Biochem Biophys. 1990;277:402–409. doi: 10.1016/0003-9861(90)90596-q. [DOI] [PubMed] [Google Scholar]
- 36.Silva G, Jeney V, Chora A, Larsen R, Balla J, Soares MP. Oxidized hemoglobin is an endogenous proinflammatory agonist that targets vascular endothelial cells. J Biol Chem. 2009;284:29582–29595. doi: 10.1074/jbc.M109.045344. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.