

Abstract

A natural product chemistry-based approach was employed to discover small molecule inhibitors of the important tumor-selective molecular target hypoxia-inducible factor-1 (HIF-1). Bioassay-guided isolation of an active lipid extract of a Saipan collection of the marine sponge Lendenfeldia sp. afforded the terpene-derived furanolipid furospongolide as the primary inhibitor of hypoxia-induced HIF-1 activation (IC50 2.9 μM, T47D breast tumor cells). The active component of the extract also contained one new cytotoxic scalarane sesterterpene and two previously reported scalaranes. Furospongolide blocked the induction of the downstream HIF-1 target secreted vascular endothelial growth factor (VEGF) and was shown to suppress HIF-1 activation by inhibiting the hypoxic induction of HIF-1α protein. Mechanistic studies indicate that furospongolide inhibits HIF-1 activity primarily by suppressing tumor cell respiration via the blockade of NADH-ubiquinone oxidoreductase (complex I)-mediated mitochondrial electron transfer.
Tumor hypoxia (low oxygenation) arises when rapidly proliferating tumor cells demand more oxygen than the tumor vasculature can supply. Clinical studies have indicated that tumor hypoxia is an important prognostic factor for the malignancy of cancers found in many tissues (e.g breast, brain, etc.).1 Hypoxic tumors are more resistant to radiation and chemotherapeutic drugs than their normoxic counterparts.1–2 Experimental approaches to overcome tumor hypoxia include improving tumor oxygenation via enhanced delivery2 and developing hypoxic radiosensitizers and cytotoxins.3 Currently, there is no approved single method for specifically treating hypoxic tumor masses.1
The transcription factor hypoxia-inducible factor-1 (HIF-1) has emerged as an important molecular target for anticancer drug discovery. As a heterodimer of the bHLH-PAS proteins HIF-1α and HIF-1β/ARNT, HIF-1 activates the expression of genes that promote cellular adaptation and survival under hypoxic conditions.1,4 The HIF-1α protein is rapidly degraded under normoxic conditions and stabilized under hypoxic conditions, while HIF-1β protein is constitutively expressed.5 Chemicals such as iron chelators (e.g. 1,10-phenanthroline, desferroxamine, etc.) and transition metals can each activate HIF-1 by blocking the Fe(II)-dependent degradation and inactivation of HIF-1α protein. Upon induction and activation, HIF-1 binds to the hypoxia response element (HRE) present in the promoters of target genes and activates transcription. Clinical studies revealed that the oxygen regulated HIF-1α subunit is overexpressed in common human cancers and their metastases, and is associated with poor prognosis and advanced stage cancers.6–9 In animal models, HIF-1 inhibition retards tumor growth and improves treatment outcome when combined with chemotherapeutic agents or radiation.10–15 Numerous research efforts are underway to discover small molecule HIF-1 inhibitors for cancer treatment.4
Using a human breast tumor T47D cell-based reporter assay, we have evaluated over 15,000 natural product-rich extracts from marine organisms and plants for HIF-1 inhibitory activity. This screening effort has yielded an array of structurally diverse natural product-derived HIF-1 inhibitors such as manassantin B (1),16 7-hydroxyneolamellarin A (2),17 laurenditerpenol (3),18 and tetrahydroisoquinoline alkaloids klugine (4) and emetine (5).19 This report describes the identification and characterization of furospongolide (6) from a marine sponge Lendenfeldia sp. as a structurally unique inhibitor of HIF-1 activation. In addition, one new cytotoxic scalarane sesterterpene and two previously reported scalaranes were isolated from the active fractions and characterized for their HIF-1 inhibitory activity and tumor cell line cytotoxicity.

Results and Discussion
In search of novel natural product-derived HIF-1 inhibitors, over 10,000 lipid extracts of marine organisms obtained from the National Cancer Institute Open Repository were evaluated in a human breast tumor T47D cell-reporter assay for HIF-1 inhibitory activity.16 An active extract from the marine sponge Lendenfeldia sp. (5 μg mL-1) inhibited hypoxia (1% O2)-induced HIF-1 activation by 91%. This extract also inhibited iron chelator 1,10-phenanthroline (10 μM)-induced HIF-1 activation by 78% in the T47D cell-based reporter assay. Bioassay-guided isolation yielded compounds 6 – 9. The spectroscopic data for compounds 6, 8, and 9 match those reported in the literature for the furanolipid furospongolide (6), and two scalarane-type sesterterpenes 24-methyl-12,24,25-trioxoscalar-16-en-22-oate (8), and 22-hydroxy-24-methyl-12,24-dioxoscalar-16-en-25-al (9).20,21 Compound 7 appeared to be a new scalarane sesterterpene.
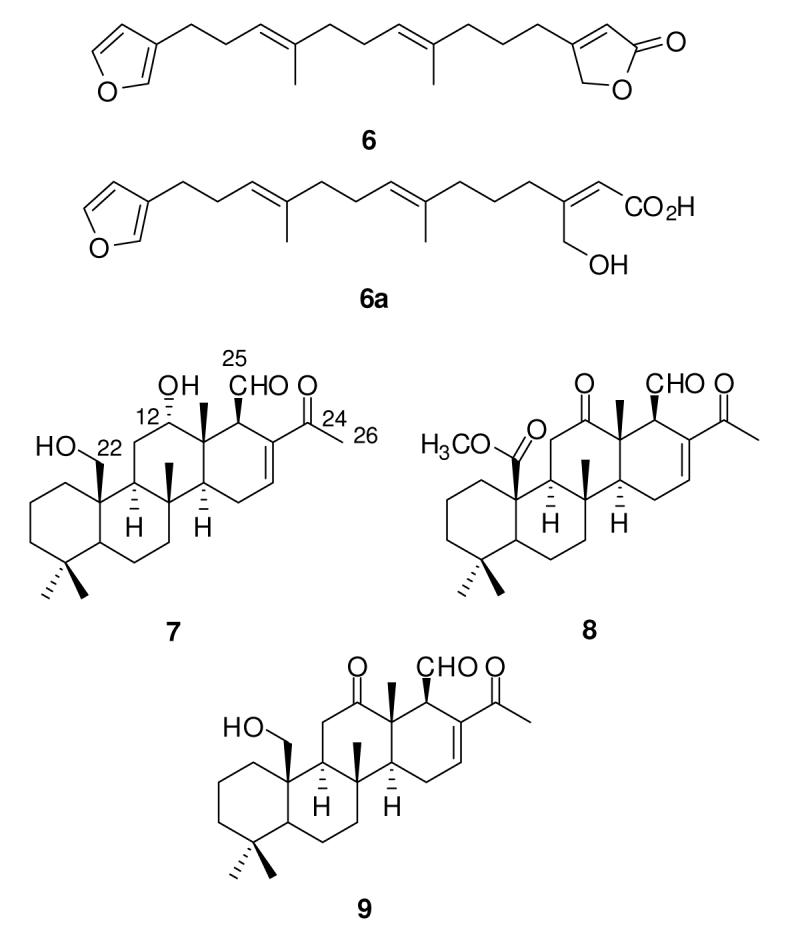
Compound 7 was isolated as yellow oil. The HRESIMS, 1H and 13C NMR data shown in Table 1 support the molecular formula C26 H40O4. The 1H NMR spectrum indicated the presence of one aldehyde proton at β 9.75 (H-25), one low field olefinic proton at β 7.03 (H-16), oxygenated methylene protons at β 3.77, 3.95 (2H-22), and five singlet methyl proton resonances. The 13C NMR and DEPT data revealed 26 carbon resonances, including two ketone carbons at β 204.4 (C-25), β 199.1 (C-24), two olefinic carbons at β 138.6 (C-17), β 143.0 (C-16), one oxygenated methine carbon at β 81.8 (C-12), one oxygenated methylene carbon at β 62.1 (C-22), and sp3 methylene, methine and methyl groups. These 1H and 13C NMR data indicated that 7 possessed a structural skeleton similar to the scalaranes 8 and 9. Comparison of the 1H and 13C NMR spectra of 7 with those of 9 revealed that the C-12 ketone 13C resonance at β 214.3 observed in the known compound 9 was replaced by a methylene carbon resonance at β 81.8 in the new compound 7. Similarly, the 1H NMR spectrum of 7 displayed an additional oxygenated proton resonance at β 3.46 (H-12), relative to 9. Therefore, the only difference between compounds 7 and 9 is that the C-12 ketone in 9 was reduced to a secondary alcohol in 7. This proposed structure of 7 is also supported by 1H-1H COSY and HMBC correlations. The relative configuration of 7 was determined by comparing the chemical shifts of 7 with those of the previously reported scalaranes.20,21 Further, observed NOESY correlations between H-12 and CH3-21 and CH3-23 indicated that the C-12 hydroxyl group was orientation “α” to the C-ring.
Table 1.
13C and 1H NMR Dataa for 7
| No. | δ c | δH (J in Hz) |
|---|---|---|
| 1 | 34.3 CH2 | 2.27 (2H, m) |
| 2 | 17.7 CH2 | 1.44 (2H, m) |
| 3 | 41.6 CH2 | 1.41 (2H, m) |
| 4 | 32.8 C | --- |
| 5 | 56.7 CH | 0.83 (1H, m) |
| 6 | 18.3 CH2 | 1.34 (2H, m) |
| 7 | 41.9 CH2 | 1.76 (2H, m) |
| 8 | 37.3 C | --- |
| 9 | 58.5 CH | 0.87 (1H, m) |
| 10 | 42.0 C | --- |
| 11 | 29.8 CH2 | 1.86 (2H, m) |
| 12 | 81.8 CH | 3.46 (1H, m) |
| 13 | 43.9 C | --- |
| 14 | 52.9 CH | 1.08 (1H, m) |
| 15 | 23.2 CH2 | 2.22 (2H, m) |
| 16 | 143.0 CH | 7.03 (1H, s) |
| 17 | 138.6 C | --- |
| 18 | 61.3 CH | 3.22 (1H, s) |
| 19 | 33.7 CH3 | 0.80 (3H, s) |
| 20 | 21.7 CH3 | 0.71 (3H, s) |
| 21 | 16.4 CH3 | 1.05 (3H, s) |
| 22 | 62.1 CH2 | 3.77 (1H, d,11.4) 3.95 (1H, d, 11.4) |
| 23 | 9.6 CH3 | 0.76 (3H, s) |
| 24 | 199.1 C | --- |
| 25 | 204.4 CH | 9.75 (1H, s) |
| 26 | 25.0 CH3 | 2.24 (3H, s) |
CDCl3; 150 MHz for 13C; 600 MHz for 1H.
A concentration-response study was performed to determine the effects of 6 – 9 on HIF-1 activation in T47D and PC-3 (human prostate tumor) cell-based reporter assays (Figure 1 and Table 2). Compound 7 inhibited both hypoxia and 1,10-phenanthroline induced HIF-1 activation in both cell lines with comparable IC50 values (hypoxia-induced HIF-1: 0.4 μM in T47D and 0.5 μM in PC-3; 1,10-phenanthroline-induced HIF-1: 0.2 μM in T47D and 0.8 μM in PC-3). The observation that 7 also suppressed luciferase expression from a control construct pGL3-Control (Promega) suggested that 7 may have a narrow window between HIF-1 inhibitory activity and cytotoxicity (Figure 1B; Table 2). The only structural difference between compounds 7 and 9 is that 7 is hydroxylated at C-12, while in 9 it was oxidized to a ketone. Compound 9 exerted similar HIF-1 inhibitory activity and a similar selectivity profile as those observed for 7 except that 9 inhibited luciferase expression from the pGL3-Control to a lesser extent (Figure 1D). The structurally related compound 8 inhibited HIF-1 activation in both cell lines with reduced potency (5 to 7-fold reduction in T47D and 11 to 15-fold reduction in PC-3 cells, in comparison to 7, Figure 1C). At the range of concentrations examined, 6 exhibited more pronounced HIF-1 inhibitory activity in T47D cells, relative to the activity observed in PC-3 cells (IC50 values in T47D: 2.9 μM for hypoxia-induced and 10.9 μM for 1,10-phenanthroline-induced HIF-1; Figure 1A; Table 2).
Figure 1. Concentration-response effects of 6 – 9 on HIF-1 activity in T47D (left panel of AD) and PC-3 (right panel of A – D) cell-based reporter assays.

Data shown are averages from one representative experiment performed in triplicate and the bars represent standard error. The label “pHRE_Hyp” indicates that the cells were transfected with the pHRE-TK-Luc reporter to monitor the ability of HIF-1 to direct the expression of luciferase under the control of hypoxia-response element (HRE) and the transfected cells were exposed to hypoxic conditions for 16 h. The label “pGL3Control_Hyp” indicates that the cells were transfected with a control plasmid pGL3-Control (Promega) that expresses luciferase under the control of the constitutively active CMV promoter and exposed to hypoxic conditions for 16 h. The label “pHRE_1,10-phen” indicates that cells transfected with the pHRE-TK-Luc reporter were treated with a hypoxia mimetic 1,10-phenanthroline (10 μM) for 16 h.
Table 2.
IC50 Values (μM) of 6 – 9 on Luciferase Expression from the HIF-1-Regulated pHRE-TK-Luc and Control (pGL3-control) Constructs (one experiment in triplicate).
| Hypoxia (1% O2) | 1,10-phen (10 μM) | |||||
|---|---|---|---|---|---|---|
| T47D | PC-3 | T47D | PC-3 | |||
| pHRE3-TK- luc (HIF) |
pGL3- control |
pHRE3-TK- luc (HIF) |
pHRE3-TK- luc (HIF) |
pGL3- control |
pHRE3-TK- luc (HIF) |
|
| 6 | 2.9 | >30 | >30 | 10.9 | >30 | >30 |
| 7 | 0.4 | 2.6 | 0.5 | 0.2 | 1.6 | 0.8 |
| 8 | 2.7 | > 30 | 7.7 | 1.0 | 23.0 | 9.0 |
| 9 | 0.5 | > 10 | 0.9 | 0.2 | 2.6 | 1.0 |
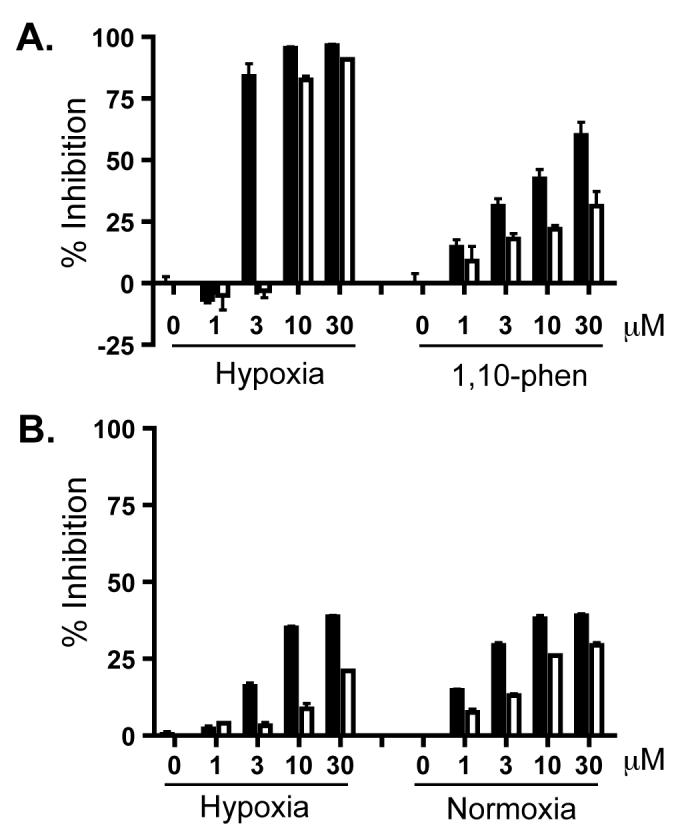
The scalarane class of sesterterpenes have been reported to be cytotoxic.22 To differentiate between cytotoxicity and the HIF-1 inhibitory activity observed for 6 – 9, a concentration-response study was conducted in a panel of human tumor cell lines (Figure 2 and Table 3). Exponentially grown cells were exposed to test compounds at the indicated concentrations under normoxic (95% air) and hypoxic (1% O2) conditions for 48 h, and cell viability/proliferation was determined by the neutral red method. For compounds 7 – 9, there is a narrow window between the HIF-1 inhibitory activity and the suppression of cell proliferation/viability in T47D cells (3 to 4-fold difference in IC50 values; Tables 2 and 3), while the HIF-1 inhibitory activity correlates with the decrease in cell proliferation/viability in PC-3 cells (Right panels, Figures 1B-1D and 2B-2D). Compound 6 exhibited more than 10-fold selectivity towards HIF-1 inhibition relative to its suppression of hypoxic T47D cell proliferation/viability (for example: 97% inhibition of hypoxia-induced HIF-1 and 26% inhibition of hypoxic cell proliferation/viability at 30 μM, Figures 1A and 2A). Among the cell lines examined (MCF-7, MDA-MB-231, T47D, and PC-3), the estrogen-independent metastatic MDA-MB-231 cells appear to be the most sensitive to the growth inhibitory/cytotoxic effects of the scalarane sesterterpenes 7 – 9 (Figure 2B-2D; Table 3).
Figure 2. Concentration-response effects of 6 – 9 on tumor cell proliferation/viability.

Exponentially grown cells plated into 96-well plates were exposed to compounds at the concentrations indicated for 48 h under normoxic (5% CO2/95% Air, “Norm”) or hypoxic (1% O2/5% CO2/94% N2, “Hyp”) conditions, as appropriate. Cell viability was determined by the neutral red method except the MDA-MB-231 and PC-3 cell panel for compound 6. Data shown are averages from one representative experiment with each data point performed in triplicate and the bars represent standard error.
Table 3.
IC50 Values (μM) of 6 – 9 on Tumor Cell Proliferation/Viability under Normoxic (Norm, 95% air) and Hypoxic (Hyp, 1% O2) Conditions Following 48 h Exposure(one experiment in triplicate).
| MCF-7 | MDA-MB-231 | T47D | PC-3 | |||||
|---|---|---|---|---|---|---|---|---|
| Norm | Hyp | Norm | Hyp | Norm | Hyp | Norm | Hyp | |
| 6 | 29 | >30 | >30 | >30 | 14 | >30 | >30 | >30 |
| 7 | 0.8 | 0.7 | 0.2 | 0.2 | 1.0 | 1.6 | 0.8 | 0.9 |
| 8 | 6.1 | 6.8 | 1.9 | 2.0 | 5.5 | 10.8 | 6.4 | 10.2 |
| 9 | 0.6 | 0.6 | 0.3 | 0.3 | 1.2 | 1.8 | 0.8 | 1.1 |
Among the four Lendenfeldia sp. metabolites isolated, compound 6 was the only relatively non-cytotoxic inhibitor of HIF-1 activation. The effect of 6 on the induction of HIF-1 downstream target gene VEGF was examined and the results are shown in Figure 3A. Compound 6 decreased hypoxic induction of secreted VEGF proteins in a concentration-dependent manner. At the same concentration (30 μM), 6 did not affect the increase in secreted VEGF protein level caused by treatment with the hypoxia mimetic 1,10-phenanthroline. This observation mirrors that observed in the T47D cell-based reporter assay for HIF-1 activity - compound 6 preferentially inhibits HIF-1 activation induced by hypoxia, relative to its affect on 1,10-phenanthroline-induced HIF-1 activation (Figure 1A). In general, the availability and activity of the oxygen-regulated HIF-1α subunit determines the activity of HIF-1. Under normoxic conditions, HIF-1α protein is rapidly degraded and HIF-1 is inactive. Under hypoxic conditions or exposure to chemical hypoxia (i.e., 1,10-phenanthroline), HIF-1α protein accumulates in the nucleus, forms a heterodimer with the HIF-1β subunit, and regulates transcription of HIF-1 target genes. The effect of 6 on the induction of HIF-1α protein was examined in T47D cells by Western blot and the results are shown in Figure 3B. Compound 6 blocked the induction of HIF-1α protein by hypoxia at the concentration that inhibited HIF-1 activation as well as the induction of downstream target VEGF. As anticipated, compound 6 only weakly inhibited the induction of HIF-1α protein by 1,10-phenanthroline. Thus, 6 selectively inhibits hypoxic activation of HIF-1 by blocking the induction of HIF-1α protein.
Figure 3. Compound 6 inhibits hypoxic induction of HIF-1 target VEGF protein (A) and blocks the induction of nuclear HIF-1 α protein accumulation (B). Compound 6 inhibits oxygen consumption in T47D cells (C) by disrupting complex I (E) but not complex II, III, and IV (D).
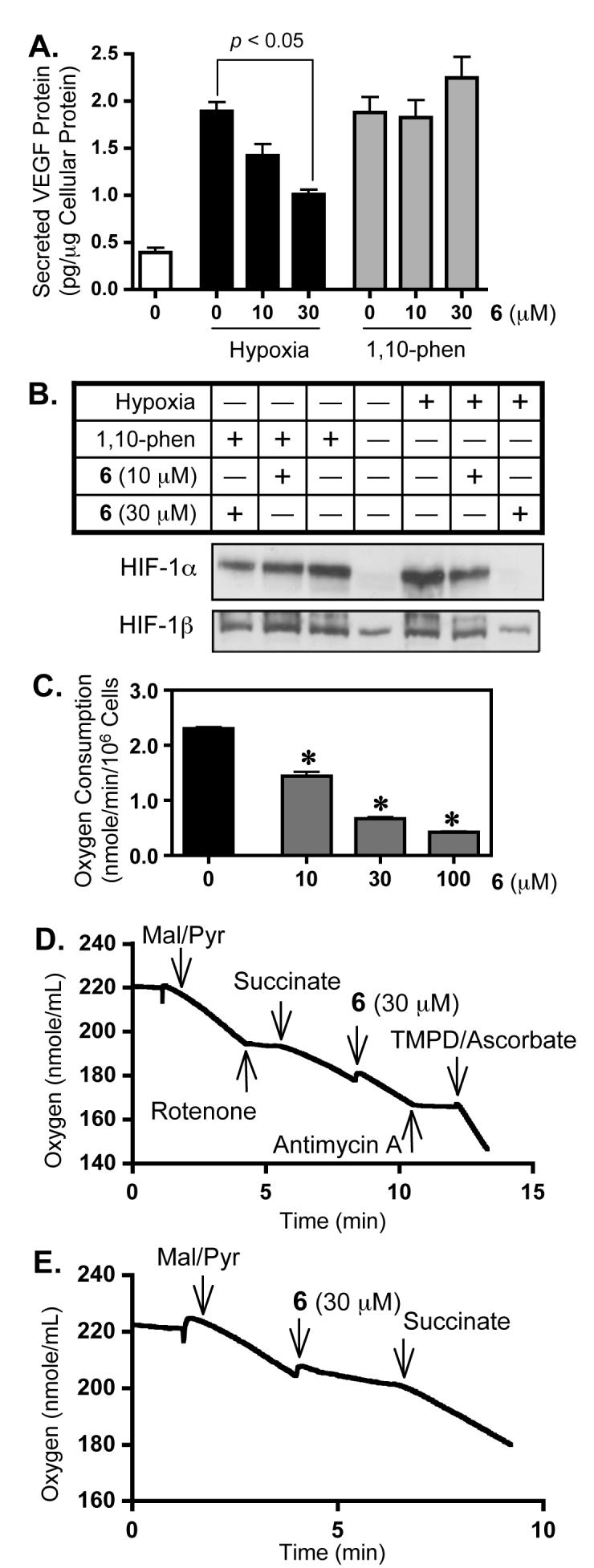
Data shown in (A) are averages from one representative experiment performed in T47D cells (n = 3 for each data point) and the bars represent standard error. Data were analyzed by one-way ANOVA, followed by Bonferroni post test using GraphPad Prism 4 software. The p value is provided in the figure when the inhibition is statistically significant. Data shown in (B) are Western blot results from a study evaluating the effects of 6 on the induction of nuclear HIF-1α protein and the nuclear accumulation of the constitutively expressed HIF-1β protein in T47D cells. The label “Hypoxia” correlates to a 4 h hypoxic exposure (1% O2/5% CO2/94% N2, 37°C), while “1,10-phen” represents a 4 h treatment with 1,10-phenanthroline at 10 μM. The concentration-dependent effects of 6 on oxygen consumption were examined in intact T47D cells with glucose as a substrate (C). Data shown are averages from one representative experiment performed in triplicate and an asterisk (*) indicates p < 0.05 when compared to the control by one-way ANOVA, followed by Bonferroni post test using GraphPad Prism 4 software. The effects of 6 on respiration of digitonin permeabilized T47D cells in the presence of various combinations of inhibitors and substrates were examined and representative data are shown (D and E). Abbreviations: Mal = malate; Pyr = pyruvate; TMPD = N,N,N’,N’-tetramethyl-p-phenylenediamine.
Mitochondria electron transport chain (ETC) inhibitors constitute one group of recently recognized small molecule HIF-1 inhibitors.23 Several opposing theories have emerged to explain the role of mitochondria in HIF-1 regulation.24 Many now believe that, under hypoxic conditions, reactive oxygen species (ROS) produced by the Qo site of mitochondrial complex III serve as “signal molecules” that 1) oxidize the catalytic iron in the Fe(II)-dependent HIF-prolyl hydroxylases that are responsible for initiating the ubiquitin-mediated proteasomal degradation of HIF-1α protein; and 2) may similarly block the Fe(II)-dependent asparaginyl hydroxylases that have been shown to interfere with the transcriptional activation of HIF-1.25 In this respect, inhibitors of the mitochondrial ETC (e.g. complex I inhibitors) could prevent the production of essential ROS signaling molecules required to stabilize HIF-1α under hypoxic conditions. We investigated the effects of 6 on mitochondrial respiration using the same T47D breast tumor cells that were used to examine HIF-1 activation and the data are shown in Figure 3C. Compound 6 significantly inhibited T47D oxygen consumption at concentrations as low as 10 μM (38% inhibition) in a concentration-dependent manner.
Further studies were performed to discern where within the ETC compound 6 acts to inhibit electron transport and the results are shown in Figures 3D and 3E. A mixture of malate and pyruvate was added to permeabilized T47D cells to initiate mitochondrial respiration at complex I (NADH-ubiquinone oxidoreductase). This respiration was inhibited by the complex I inhibitor rotenone (Figure 3D). Succinate, a substrate for complex II, overcomes the inhibition exerted by complex I inhibitors such as rotenone. The observation that 6 did not affect mitochondrial respiration in the presence of succinate indicated that 6 does not inhibit complex II, III, or IV. In order to ensure that the ETC remained functional, the complex III inhibitor antimycin A was added to inhibit respiration and complex IV substrates [N,N,N’,N’-tetramethyl-p-phenylenediamine (TMPD)/ascorbate] were added to re-initiate respiration (Figure 3D). As in the case of the mitochondrial complex I inhibitor rotenone, addition of the complex II substrate succinate reestablishes mitochondrial respiration and electron transport following inhibition with 6 (Figure 3E). Taken together, the data presented in Figures 3 D and 3E indicate that 6 selectively inhibits mitochondrial complex I-mediated respiration.
To test the hypothesis that the furan ring cleaved form of 6 may be responsible for the HIF-1 inhibitory activity, compound 6 was hydrolyzed to yield the lactone-opened form. Following the chemical reaction and purification efforts, a mixture of 6 and the lactone-hydrolyzed form (6a) was obtained at the ratio of 1:1. This mixture was subjected to the T47D cell-based reporter assay for HIF-1 activity and the results are shown in Figure 4A. A similar hypoxia-selective HIF-1 inhibitory activity as that observed for 6 was obtained with this mixture, although the 6/6a mixture was less potent than 6. In the cell proliferation/viability study conducted with T47D, MDA-MB-231, and PC-3 cells, the mixture exhibited a similar profile to that observed with 6. Neither 6 nor the 6/6a mixture affected MDA-MB-231 or PC-3 cell proliferation/viability under normoxic or hypoxic conditions (data not shown). In T47D cells, the mixture has less effect on cell proliferation/viability than produced by a purified sample of 6 at the same concentration (Figure 4B). Therefore, the hydrolyzed form of 6 does not appear to be the active form that contributes to the HIF-1 inhibitory activity of 6. The apparently inactive hydrolysis product 6a was not further purified.
Figure 4. Comparison between 6 and a mixture of 6 and the hydrolyzed form 6a on HIF-1 inhibition (A) and suppression of cell proliferation/viability in T47D cells (B).
Data shown in (A) are averages from one representative T47D cell-based reporter assay performed in triplicate and the bars indicate standard error. The solid bars indicate pure 6 at each specified concentration and the open bars represent a mixture of 6 and 6a at equivalent concentrations. The label “Hypoxia” correlates to a 16 h hypoxic exposure (1% O2/5% CO2/94% N2, 37°C), while “1,10-phen” represents a 16 h treatment with 1,10-phenanthroline at 10 μM. Results from the 48 h cell proliferation/viability study are shown in (B). Data representation is similar to those specified in (A) except that the incubation time was 48 h and the label “Normoxia” refers to 95% Air/5% CO2.
While 6 represents only a moderate potency inhibitor, it is the first marine-derived furanolipid found to inhibit hypoxia-induced HIF-1 activation and its relatively simple structure provides an attractive target for structural modification and possible pharmacological optimization.
Experimental Section
General Experimental Procedures
Optical rotations were obtained on an AP IV/589-546 digital polarimeter. The IR spectrum was obtained using a Bruker Tensor 27 genesis Series FTIR. UV spectra were recorded on a Varian 50 Bio spectrophotometer. The NMR spectra were recorded in CDCl3 on Bruker AMX-NMR spectrometers operating at either 400 MHz or 600 MHz for 1H and either 100 MHz or 150 MHz for 13C, respectively. The NMR spectra were recorded running gradients and residual solvent peaks (β 7.27 for 1H) and (β 77.0 for 13C) were used as internal references. The HRESIMS spectra were measured using a Bruker Daltonic micro TOF with electrospray ionization. Silica gel (200-400 mesh) was used for column chromatography. The TLCs were run on Merck Si60F254 or Si60RP18F254 plates and visualized under UV at 254 nm and by heating after spraying with a 1% anisaldehyde solution in acetic acid:H2SO4 (20:1). All the chemicals were purchased from Sigma, unless specified.
Sponge Material
The sponge extract was obtained from the National Cancer Institute’s Open Repository Program. Lendenfeldia sp. was collected at 8 m depth on February 18, 1993 (collection C019633) from Saipan, Commonwealth of the Northern Mariana Islands. The sample was identified by Dr. Michele Kelly (National Institute of Water and Atmospheric Research Limited, Auckland, New Zealand). It was frozen at -20°C and ground in a meat grinder. A voucher specimen was placed on file with the Department of Invertebrate Zoology, National Museum of Natural History, Smithsonian Institution, Washington, D.C.
Extraction and Isolation
Ground sponge material was extracted with H2O. The residual sample was lyophilized and extracted with CH2Cl2:MeOH (1:1), residual solvents were removed under vacuum, and the crude extract stored at -20°C in the NCI repository at the Frederick Cancer Research and Development Center (Frederick, Maryland). The crude extract (5.8 g) (inhibited HIF-1 activation by 97% at 5 μg mL-1 in T47D cells) was separated into two fractions by Sephadex LH-20 chromatography (CH2Cl2 in MeOH [1:1]). The active second fraction (inhibited HIF-1 activation in T47D cells by 99% at 5 μg mL-1) was further separated into five subfractions by C18 VLC column (eluted with step gradients of 50% to 100% MeOH in H2O). The second fraction (inhibited HIF-1 activation in T47D cells by 76% at 1 μg mL-1, eluted with 70% MeOH in H2O), was further separated by Si gel CC (eluted with petroleum ether:acetone 9:1) to produce 6 (100 mg, 1.7% yield, ACS Registry No. 76343-80-1) and four other subfractions. The third subfraction was further purified by repeated Si gel CC [1) 85% EtOAc in hexanes; 2) 60% EtOAc in hexanes] and HPLC (Luna® 5μ, ODS-3 100 Å, 250 × 10.0 mm, isocratic 50% CH3CN in H2O, 4.0 mL min-1) to produce 7 (46 mg, 0.79% yield, tR 16 min). A slightly less polar fraction that eluted from the second Si gel CC (60% EtOAc in hexanes) was separated by HPLC (Prodigy® 5μ, ODS-3 100 Å, 250 × 21.2 mm, isocratic 65% CH3CN in H2O, 10.0 mL min-1), to produce 8 (10 mg, 0.17% yield, tR 18 min, ACS Registry No. 81575-81-7), and 9 (18 mg, 0.31% yield, tR 22 min, ACS Registry No. 75587-67-6).
Hydrolysis of furospongolide (6)
Compound 6 (4.5 mg, 0.014 mmol) was dissolved in NaOH solution (1.5 N, 1.5 mL) and stirred for 24 h at room temperature. The lipid portion of the reaction mixture was removed by partitioning with Et2O (3 × 1 mL). The remaining aqueous portion was extracted by partitioning with sec-BuOH (2 × 2 mL). The sec-BuOH fraction was washed 3 times with H2O and evaporated to dryness under vacuum to yield 6/6a (3.32 mg, a 1:1 mixture of 6 and 6a).
6a from 6/6a mixture
White powder; 1H NMR (CD3OD, 400 MHz) δ 7.36 (1H, br s, H-1), 7.24 (1H, br s, H-4), 6.29 (1H, br s, H-2), 5.77 (1H, br s, H-17), 5.17 (1H, t, J = 7.6 Hz, H-11), 5.12 (1H, t, J = 7.6 Hz, H-7), 4.22 (1H, s, H-19), 2.44 (2H, t, J = 7.6 Hz, H-5), 2.42 (2H, t, J = 6.0 Hz, H-15), 2.25 (2H, br q, J = 7.2 Hz, H-6), 2.08 (6H, m, H-9, 10, 13), 1.71 (2H, br q, J = 7.6 Hz, H-14), 1.59 (6H, s, H-20, 21); 13C NMR (CD3OD, 400 MHz) 154.0 (C, C-16), 143.9 (CH, C-1), 140.2 (CH, C-4), 136.7 or 136.6 (C, C-8), 135.8 (CH, C-12), 126.6 or 126.4 (CH, C-11), 126.3 (C, C-3), 126.0 (CH, C-17), 125.4 (CH, C-7), 112.2 (CH, C-2), 63.5 (CH2, C-19), 40.9 or 40.8 (CH2, C-13), 40.5 or 40.2 (CH2, C-9), 29.7(CH2, C-14), 28.9(CH2, C-6), 27.5 (CH2, C-10), 26.1 (CH2, C-5, 15), 16.0 or 16.2 (CH3, C-20, 21), C-18 not observed; ESIMS, m/z 345 [M-H]- .
22-Hydroxy-24-methyl-12α-hydroxy-24-oxoscalar-16-en-25-al (7)
Yellow oil; [α]D25 +30.42 (c 0.23, CH2Cl2); UV (CH2Cl2) λmax (log ε) 210 (3.82), 235 (3.82)nm; IR (film) vmax cm-1 3423, 2929, 2850, 1706, 1660, 1457, 1388, 1374, 1255, 1053, 1029, 1003, 912, 733; 1H and 13C NMR, see Table 1; HRESIMS m/z 439.2813 [M+Na]+ (calcd for C26H40O4Na, 439.2824).
Cell-Based Reporter Assay
Human breast tumor T47D and prostate tumor PC-3 cells (ATCC) were maintained in DMEM/F12 media with glutamine (Mediatech) supplemented with 10% fetal calf serum (FCS, v/v, Hyclone), and 50 U mL-1 penicillin G sodium and 50μg mL-1 streptomycin (Biowhittaker) in a humidified environment under 5% CO2/95% Air at 37°C. Transient transfection, compound treatment, exposure to hypoxic conditions (1% O2) or a hypoxia mimetic (10 μM 1,10-phenanthroline), and determination of luciferase activity were performed as described previously (18). Data were presented as % inhibition of the induced control, calculated using the following formula: % inhibition = (1 - light outputtreated/ light outputinduced) × 100.
ELISA Assay for VEGF Protein
Exponentially grown T47D cells were plated at the density of 356,000 cells per well (12-well Corning plate) and incubated at 37°C overnight. Compounds diluted in serum-free DMEM/F12 medium supplemented with penicillin and streptomycin were added in equal volumes. After a 30 min incubation under 5% CO2/95% Air, the treatment continued for another 16 h under hypoxic conditions (1% O2/5% CO2/94% N2) or in the presence of 1,10-phenanthroline (10 μM). The cells were lysed in a volume of 250 μL M-PER mammalian protein extraction reagent (Pierce) per well of a 12-well plate (Corning) and the protein concentration determined using the Micro BCA Protein Assay (Pierce). Levels of secreted VEGF protein in the conditioned media and cellular VEGF protein in the cell lysates were measured by ELISA as previously described.16 The quantity of VEGF protein was normalized to the amount of total protein in the cell lysate.
Cell Proliferation/Viability Assay
Human breast tumor MCF-7, MDA-MB-231, T47D, and human prostate tumor PC-3 cells (ATCC) were maintained as described in the cell-based reporter assay section. For the cell proliferation/viability study, exponentially grown cells were plated at a density of 30,000 cells per well of a 96-well plate in a volume of 100 μL DMEM/F12 media supplemented with 10% FCS and antibiotics. After incubating overnight, compounds diluted in serum-free DMEM/F12 with antibiotics were added to the cells in a volume of 100 μL per well of a 96-well plate. After an initial 30 min exposure, the incubation continued for another 48 h under normoxic (5% CO2/95% Air) or hypoxic (1% O2/5% CO2/94% N2) conditions as appropriate. Cell proliferation/viability was determined using either the neutral red method16 or sulforhodamine B method26 as indicated in the figure legend. For the sulforhodamine B method, 100 μL per well of a 20% TCA solution (v/v, in 1x PBS, pH 7.4) was added to the cells to replace 100 μL conditioned media at the end of incubation. Following incubation at 4°C for 1 h, the supernatant was removed, the fixed cells washed 4 times with tap water, and air dried. A 0.4% sulforhodamine B solution (v/v, in 1% acetic acid) was added in a volume of 100 μL per well and incubated under room temperature for 10 min. The stained cells were washed 4 times with 1% acetic acid and air dried. A Trizma® base solution (10 mM, pH 7.4) was added in a volume of 200 μL per well to extract sulforhodamine B from the stained cells by shaking at room temperature for 5 min. Light absorbance at 490 nm was measured on a BioTek Synergy plate reader, with background at 690 nm subtracted. A formula similar to that used in the cell-based reporter assay section was used to calculate percentage inhibition.
Nuclear Extract Preparation and Western Blot for HIF-1α and HIF-1β Proteins
Exponentially grown T47D cells were plated at the density of 10 × 106 cells per 100 mm diameter plate (Corning) in a volume of 10 mL DMEM/F12 medium supplemented with 10% FCS and antibiotics as described in the cell-based reporter assay section. After 16 h, compound was added at the desired concentration. Following a short incubation (30 min at 37°C), the cells were exposed to hypoxic conditions (1%) O2/94% N2/5% CO2) or a hypoxia mimetic (10 μM 1,10-phenanthroline) for another 4 h. Nuclear extract samples were prepared using NE-PER nuclear and cytoplasmic extraction kit (PIERCE) and the protein concentration determined with a Micro BCA assay kit (PIERCE). Sixty micrograms of each nuclear extract protein sample were loaded onto an 8% SDS-PAGE gel and separated by electrophoresis. The proteins were transferred onto a nitrocellulose membrane (BioRad) by standard methods using a semi-dry blotter (Bio-Rad). The membrane was blocked in TTBS (20 mM Tris, PH 6.8, 150 mM NaCl, 0.05% Tween-20) with 5% non-fat milk (Bio-Rad) at 4°C overnight. The blot was incubated with a monoclonal anti-HIF-1α antibody (BD Biosciences) at 1:500 dilution in TTBS with 1% BSA at room temperature for 1 h. For HIF-1β Western blot, a monoclonal anti-HIF-1β antibody (BD Biosciences) was used at 1:1000 dilution. The secondary antibody (ECL™-HRP linked sheep anti-mouse IgG, Amersham) was used at 1:30,000 in TTBS with 1% BSA at room temperature for 40 min. The blot was developed with the SuperSignal West Femto Maximum Sensitivity Substrate (PIERCE) and exposed to a CL-XPosure film (PIERCE).
Mitochondrial Respiration Assay
A method used to monitor respiration in isolated mitochondria27 was modified to measure the level of oxygen consumption in T47D cells and to investigate the specific target within the mitochondrial electron transport chain affected by active compounds. To determine the effect of the isolated compounds on cell respiration, 5 × 106 T47D cells (equilibrated to 30°C) were added to the chamber of an Oxytherm Clarke-type electrode System (Hansatech) containing 1 mL DMEM/F12 (Dulbecco’s Modified Eagle’s Medium/Ham’s Nutrient Mixture F12, JRH) medium free of serum and antibiotics. Glucose (17.5 mM) in the DMEM/F12 media served as the metabolic substrate. Once baseline respiration had been established (for a 15 min interval), compounds dissolved in isopropyl alcohol were injected into the chamber at 3 min intervals using a 100 μL syringe (Hamilton). For mechanistic studies, the plasma membrane was selectively permeabilized with digitonin (30 μM) so that the substrates available to the mitochondria could be manipulated. This required the use of a mitochondrial buffer containing 20 mM HEPES (pH 7.3), 120 mM KCl, 2 mM KH2PO4, 2 mM MgCl2, 1 mM EGTA [ethylene glycol bis(2-aminoethyl ether)-N,N,N’,N’-tetraacetic acid], and 0.3% BSA (fat-free) was used in place of the DMEM/F12 medium. The buffer was supplemented with the following substrates that provide electrons to different sites within the mitochondrial respiratory chain: 5 mM sodium pyruvate and 5 mM sodium malate (complex I), 5 mM sodium succinate dibasic hexahydrate (complex II), and 5 mM L-ascorbic acid + 0.2 mM TMPD (N,N,N’,N’-tetramethyl-p-phenylenediamine) (complex IV). Known complex I and III inhibitors - rotenone and antimycin A, respectively, were added from EtOH stock solutions to final concentrations of 1 μM where indicated.
Statistical Analysis
Data were compared using one-way ANOVA and Bonfferoni post hoc analyses (GraphPad Prism 4). Differences were considered significant when p < 0.05.
Supplementary Material
Acknowledgment
The authors thank the Natural Products Branch Repository Program at the National Cancer Institute for providing marine extracts from the NCI Open Repository used in these studies, D.J. Newman and E.C. Brown (NCI — Frederick, MD) for assistance with sample logistics and collection information, and S.L. McKnight (University of Texas Southwestern Medical Center at Dallas) for providing the pTK-HRE3-luc construct. This research was supported by the National Institutes of Health-NCI CA98787 (DGN/YDZ) and NOAA NURP/NIUST NA16RU1496. This investigation was conducted in a facility constructed with support from Research Facilities Improvement Grant C06 RR-14503-01 from the National Institutes of Health.
References
- (1).Tatum JL, Kelloff GJ, Gillies RJ, Gillies RJ, Arbeit JM, Brown JM, Chao KS, Chapman JD, Eckelman WC, Fyles AW, Giaccia AJ, Hill RP, Koch CJ, Krishna MC, Krohn KA, Lewis JS, Mason RP, Melillo G, Padhani AR, Powis G, Rajendran JG, Reba R, Robinson SP, Semenza GL, Swartz HM, Vaupel P, Yang D, Croft B, Hoffman J, Liu G, Stone H, Sullivan D. Int. J. Radiat. Biol. 2006;82:699–757. doi: 10.1080/09553000601002324. [DOI] [PubMed] [Google Scholar]
- (2).Dewhirst MW, Navia IC, Brizel DM, Willett C, Secomb TW. Clin. Cancer Res. 2007;13:375–377. doi: 10.1158/1078-0432.CCR-06-2629. [DOI] [PubMed] [Google Scholar]
- (3).Nagasawa H, Uto Y, Kirk KL, Hori H. Biol. Pharm. Bull. 2006;29:2335–2342. doi: 10.1248/bpb.29.2335. [DOI] [PubMed] [Google Scholar]
- (4) a).Semenza GL. Drug Discov. Today. 2007;12:853–859. doi: 10.1016/j.drudis.2007.08.006. [DOI] [PubMed] [Google Scholar]; b) Nagle DG, Zhou Y-D. Curr. Drug Targets. 2006;7:355–369. doi: 10.2174/138945006776054979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Wang GL, Jiang BH, Rue EA, Semenza GL. Proc. Natl. Acad. Sci. U.S.A. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Zhong H, de Marzo AM, Laughner E, Lim M, Hilton DA, Zagzag D, Buechler P, Isaacs WB, Semenza GL, Simons JW. Cancer Res. 1999;59:5830–5835. [PubMed] [Google Scholar]
- (7).Bos R, Zhong H, Hanrahan CF, Mommers EC, Semenza GL, Pinedo HM, Abeloff MD, Simons JW, van Diest PJ, van der Wall E. J. Natl. Cancer Inst. 2001;93:309–314. doi: 10.1093/jnci/93.4.309. [DOI] [PubMed] [Google Scholar]
- (8).Birner P, Schindl M, Obermair A, Plank C, Breitenecker G, Oberhuber G. Cancer Res. 2000;60:4693–4696. [PubMed] [Google Scholar]
- (9).Aebersold DM, Burri P, Beer KT, Laissue J, Djonov V, Greiner RH, Semenza GL. Cancer Res. 2001;61:2911–2916. [PubMed] [Google Scholar]
- (10).Maxwell PH, Dachs GU, Gleadle JM, Nicholls LG, Harris AL, Stratford IJ, Hankinson O, Pugh CW, Ratcliffe PJ. Proc. Natl. Acad. Sci. U.S.A. 1997;94:8104–8109. doi: 10.1073/pnas.94.15.8104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Ryan HE, Lo J, Johnson RS. EMBO J. 1998;17:3005–3015. doi: 10.1093/emboj/17.11.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Kung AL, Wang S, Klco JM, Kaelin WG, Livingston DM. Nat. Med. 2000;6:1335–1340. doi: 10.1038/82146. [DOI] [PubMed] [Google Scholar]
- (13).Kung AL, Zabludoff SD, France DS, Freedman SJ, Tanner EA, Vieira A, Cornell-Kennon S, Lee J, Wang B, Wang J, Memmert K, Naegeli HU, Petersen F, Eck MJ, Bair KW, Wood AW, Livingston DM. Cancer Cell. 2004;6:33–43. doi: 10.1016/j.ccr.2004.06.009. [DOI] [PubMed] [Google Scholar]
- (14).Unruh A, Ressel A, Mohamed HG, Johnson RS, Nadrowitz R, Richter E, Katschinski DM, Wenger RH. Oncogene. 2003;22:3213–20. doi: 10.1038/sj.onc.1206385. [DOI] [PubMed] [Google Scholar]
- (15).Moeller BJ, Dreher MR, Rabbani ZN, Schroeder T, Cao Y, Li CY, Dewhirst MW. Cancer Cell. 2005;8:99–110. doi: 10.1016/j.ccr.2005.06.016. [DOI] [PubMed] [Google Scholar]
- (16) a).Hodges TW, Hossain CF, Kim Y-P, Zhou Y-D, Nagle DG. J. Nat. Prod. 2004;67:767–771. doi: 10.1021/np030514m. [DOI] [PubMed] [Google Scholar]; b) Hossain CF, Kim Y-P, Baerson SR, Zhang L, Bruick RK, Mohammed KA, Agarwal AK, Nagle DG, Zhou Y-D. Biochem. Biophys. Res. Commun. 2005;333:1026–1033. doi: 10.1016/j.bbrc.2005.05.191. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Hanessian S, Reddy GJ, Chahal N. Org. Lett. 2006;8:5477–5480. doi: 10.1021/ol0621710. [DOI] [PubMed] [Google Scholar]
- (17).Liu Rui, Liu Yang, Zhou Y-D, Nagle DG. J. Nat. Prod. 2007;70:1741–1745. doi: 10.1021/np070206e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18) a).Mohammed KA, Hossain CF, Zhang L, Bruick RK, Zhou Y-D, Nagle DG. J. Nat. Prod. 2004;67:2002–2007. doi: 10.1021/np049753f. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chittiboyina AG, Gundluru MK, Carvalho P, Liu Y, Zhou Y-D, Nagle DG, Avery MA. J. Med. Chem. 2007;50:6299–6302. doi: 10.1021/jm7011062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Zhou Y-D, Kim Y-P, Mohammed KA, Jones DK, Muhammad I, Dunbar DC, Nagle DG. J. Nat. Prod. 2005;68:947–950. doi: 10.1021/np050029m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Kazlauskas R, Murphy PT, Wells RJ, Daly JJ. Aust. J. Chem. 1980;33:1783–1797. [Google Scholar]
- (21).Kazlauskas R, Murphy PT, Wells RJ. Aust. J. Chem. 1982;35:51–59. [Google Scholar]
- (22).Dai J, Liu Y, Zhou YD, Nagle DG. J. Nat. Prod. 2007;70:1824–1826. doi: 10.1021/np070337f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23) a).Baby SM, Roy A, Lahiri S. Histochem. Cell Biol. 2005;124:69–76. doi: 10.1007/s00418-005-0028-6. [DOI] [PubMed] [Google Scholar]; b) Semenza GL. Biochem. J. 2007;405:1–9. doi: 10.1042/BJ20070389. [DOI] [PubMed] [Google Scholar]
- (24) a).Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Proc. Natl. Acad. Sci. U.S.A. 1998;95:11715–11720. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Vaux EC, Metzen E, Yeates KM, Ratcliffe PJ. Blood. 2001;98:296–302. doi: 10.1182/blood.v98.2.296. [DOI] [PubMed] [Google Scholar]; c) Bell EL, Klimova TA, Chandel NS. Antioxid. Redox Signal. 2008;10:1–6. doi: 10.1089/ars.2007.1655. [DOI] [PubMed] [Google Scholar]
- (25) a).Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, Simon MC, Hammerling U, Schumacker PT. Cell Metab. 2005;1:401–408. doi: 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]; b) Simon MC. Adv. Exp. Med. Biol. 2006;588:165–170. doi: 10.1007/978-0-387-34817-9_15. [DOI] [PubMed] [Google Scholar]; c) Pan Y, Mansfield KD, Bertozzi CC, Rudenko V, Chan DA, Giaccia AJ, Simon MC. Mol. Cell Biol. 2007;27:912–925. doi: 10.1128/MCB.01223-06. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Bell EL, Klimova TA, Eisenbart J, Moraes CT, Murphy MP, Budinger GRS, Chandel NS. J. Cell Biol. 2007;177:1029–1036. doi: 10.1083/jcb.200609074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR. J. Natl. Cancer Inst. 1990;82:1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- (27).Pierre J, Buckingham JA, Roebuck SJ, Brand MD. J. Biol. Chem. 2002;277:44784–44790. doi: 10.1074/jbc.M207217200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.