

Abstract
Butyrylcholinesterase (BChE)-cocaine binding and the fundamental pathway for BChE-catalyzed hydrolysis of cocaine have been studied by molecular modelling, molecular dynamics (MD) simulations, and ab initio calculations. Modelling and simulations indicate that the structures of the prereactive BChE-substrate complexes for (−)-cocaine and (+)-cocaine are all similar to that of the corresponding prereactive BChE-butyrylcholine (BCh) complex. The overall binding of BChE with (−)-cocaine and (+)-cocaine is also similar to that proposed with butyrylthiocholine and succinyldithiocholine, i.e. (−)-cocaine/(+)-cocaine first slides down the substrate-binding gorge to bind to Trp-82 and stands vertically in the gorge between Asp-70 and Trp-82 (non-prereactive complex) and then rotates to a position in the catalytic site within a favorable distance for nucleophilic attack and hydrolysis by Ser-198 (prereactive complex). In the prereactive complex, cocaine lies horizontally at the bottom of the gorge. The fundamental catalytic hydrolysis pathway, consisting of acylation and deacylation stages similar to those for ester hydrolysis by other serine hydrolases, was proposed based on the simulated prereactive complex and confirmed theoretically by ab initio reaction coordinate calculations. Both the acylation and deacylation follow a double-proton-transfer mechanism. The calculated energetic results show that within the chemical reaction process the highest energy barrier and Gibbs free energy barrier are all associated with the first step of deacylation. The calculated ratio of the rate constant (kcat) for the catalytic hydrolysis to that (k0) for the spontaneous hydrolysis is ~ 9.0 × 107. The estimated kcat/k0 value of ~ 9.0 × 107 is in excellent agreement with the experimentally-derived kcat/k0 value of ~ 7.2 × 107 for (+)-cocaine, whereas it is ~ 2000 times larger than the experimentally-derived kcat/k0 value of ~ 4.4 × 104 for (−)-cocaine. All of the results suggest that the rate-determining step of the BChE-catalyzed hydrolysis of (+)-cocaine is the first step of deacylation, whereas for (−)-cocaine the change from the non-prereactive complex to the prereactive complex is rate determining and has a Gibbs free energy barrier higher than that for the first step of deacylation by ~ 4 kcal/mol. A further analysis of the structural changes from the non-prereactive complex to the prereactive complex reveals specific amino acid residues hindering the structural changes, providing initial clues for the rational design of BChE mutants with improved catalytic activity for (−)-cocaine.
Introduction
Cocaine abuse is a major medical and public health problem affecting over 40 million Americans since 1980.1 It is estimated that in the United States nearly 400,000 people use cocaine daily, and 5,000 new users are added each day.2 This widely abused drug, which reinforces self-administration in relation to the peak serum concentration of the drug, the rate of rise to the peak and the degree of change of the serum level, produces potent central nervous system and cardiovascular stimulation followed by depression.3 With overdose of the drug, respiratory depression, cardiac arrhythmia and acute hypertension are common effects. The disastrous medical and social consequences of cocaine addiction, such as violent crime, loss in individual productivity, illness and death, have made the development of an effective pharmacological treatment as a high priority.4 However, cocaine mediates its reinforcing and toxic effects by blocking neurotransmitter reuptake. Thus extensive efforts utilizing a classical pharmacodynamic approach that seeks small molecules to interfere with the cocaine-receptor interaction(s) have failed to identify an antagonist due to the difficulties inherent in blocking a blocker.1-4
As an alternative we embraced an alternative pharmacokinetic strategy that would act directly on the drug itself to alter its distribution and accelerate its clearance.4 Pharmacokinetic antagonism of cocaine could be implemented by administration of a molecule, such as an anti-cocaine antibody, which binds tightly to cocaine so as to prevent cocaine from crossing the blood-brain barrier.5 A more potent blocking action could also be implemented by administration of a more stoichiometric agent such as a catalytic antibody, an artificial enzyme, that not only binds but also degrades cocaine and thereby frees itself for further binding.1,6,7 To further the development of catalytic antibodies for hydrolysis of cocaine,1,6-20 we sought to understand the mechanism underlying hydrolysis by natural cocaine esterases. Also, natural esterases could themselves be useful therapeutic agents.
The primary pathway for metabolism of cocaine in primates is enzymatic hydrolysis, at the benzoyl ester or methyl ester group.4,21 Benzoyl ester hydrolysis yields ecgonine methyl ester (EME), whereas methyl ester hydrolysis yields benzoylecgonine (BE). The major cocaine-metabolizing enzymes in humans are butyrylcholinesterase (BChE) which catalyzes benzoyl ester hydrolysis and two liver carboxylesterases (denoted by hCE-1 and hCE-2) which catalyze hydrolysis at the methyl ester and the benzoyl ester, respectively. Hydrolysis accounts for about 95% of cocaine metabolism in humans and BChE is the principal cocaine hydrolase in human serum. The remaining 5% is deactivated through amine oxidation by the liver microsomal cytochrome P450 system, producing norcocaine.4,22 EME appears the least pharmacologically active of the cocaine metabolites and may even cause vasodilation, whereas both BE and norcocaine appear to cause vasoconstriction and lower the seizure threshold, similar to cocaine itself. Norcocaine is hepatotoxic and a local anesthetic.23 Thus, hydrolysis of cocaine at the benzoyl ester by BChE is the pathway most suitable for amplification.
BChE, designated in older literature as pseudo-cholinesterase or plasma cholinesterase to distinguish it from its close cousin acetylcholinesterase (AChE), is synthesized in the liver and widely distributed in the body, including plasma, brain, and lung.4,24 Studies in animals and humans demonstrate that enhancement of BChE activity by administration of exogenous enzyme substantially decreases cocaine half-life.25-28 For example, the addition of human BChE (extracted from donated blood) to human plasma containing cocaine (2 μg/ml) decreased the in vitro half-life of cocaine from 116 min at a BChE concentration of 3.02 μg/ml to 10 min at a BChE concentration of 37.6 μg/ml.25 In vivo studies in animals have also revealed significant enhancement of BChE activity on cocaine’s effects.26 Further, a single injection of enzyme may increase plasma BChE activity for several days.4
Clinical studies suggest that BChE has unique advantages. First, human BChE has a long history of clinic use, and no adverse effects have been noted with increased BChE plasma activity. Second, about 20 different naturally occurring mutants of human BChE have been identified,29 and there is no evidence that these mutants are antigenic. For these reasons, enhancement of cocaine metabolism by administration of BChE is considered a promising pharmacokinetic approach for treatment of cocaine abuse and dependence.4 However, the catalytic activity of this plasma enzyme is three orders-of-magnitude lower against the naturally occurring (−)-cocaine than that against the relatively biologically inactive (+)-cocaine enantiomer. (+)-cocaine can be cleared from plasma in seconds and prior to partitioning into the central nervous system (CNS), whereas (−)-cocaine has a plasma half-life of ~ 45 – 90 min, long enough for manifestation of the CNS effects which peak in minutes.30 Thus, positron emission tomography (PET) applied to mapping of the binding of (−)-cocaine and (+)-cocaine in baboon CNS showed marked uptake corresponding to (−)-cocaine at the striatum along with other areas of low uptake, whereas no CNS uptake corresponding to (+)-cocaine was observed.31 (+)-cocaine was hydrolyzed by BChE so rapidly that it never reached the CNS for PET visualization. Hence, BChE mutants with high activity against (−)-cocaine are highly desired for use in humans. One could anticipate a highly useful pharmacological treatment for cocaine if a BChE mutant were developed capable of hydrolyzing (−)-cocaine with the rate of (+)-cocaine hydrolysis by wild-type BChE.
A detailed mechanistic understanding of the difference between the BChE activity for (+)-cocaine and for (−)-cocaine could lead to important insights for the rational design of esterases with a high activity for natural (−)-cocaine. The literature32-37 provides important background for such an endeavor. Harel et al.32 constructed a computer-based model of BChE from the solved X-ray crystal structure of Torpedo californica AChE. Other researchers have also reported similar BChE modeling33,34 or application of the BChE models.35-44 Masson, Legrand, Cynthia, Froment, Schopfer and Lockridge proposed a mechanism for human BChE with three discrete enzyme-substrate complexes to explain the observation that the Michaelis constant (Km) for butyrylthiocholine differs10-fold between wild-type human BChE and the Asp70Gly mutant, whereas the Km for succinyldithiocholine differs100-fold between wild-type human BChE and the Asp70Gly mutant.35 The first enzyme-substrate complex, ES1, was proposed to form when substrate binds to Asp-70. The second complex, ES2, was proposed to form when the substrate slides down the substrate-binding gorge to bind to Trp-82 and stays vertically in the gorge between Asp-70 and Trp-82. The third complex, ES3, forms when the substrate rotates to a horizontal position at the bottom of the gorge to come within a favorable distance for nucleophilic attack and hydrolysis by Ser-198. Additionally, the kinetic data reported for butyrylthiocholine suggested that activation of BChE by excess substrate takes place after binding of the second molecule on a peripheral anionic site (PAS) consisting of residues Asp-70 and Tyr-332, among others.35 Further, the dissociation constants (approximated by the inhibition constant Ki) of the human BChE-(−)-cocaine and BChE-(+)-cocaine complexes were determined by Berkman, Underiner and Cashman as 8.0 and 5.4 μM, respectively.36 The respective Km values determined recently are 14 and 10 μM; the respective kcat values are 3.9 and 7500 min−1.37 The similar Ki (and Km) values indicate that the significant difference in BChE activity between (−)-cocaine and (+)-cocaine is not determined by a difference in the binding efficiency, but rather by subsequent step(s), e.g. the change from ES2 to ES336 or other chemical steps.37
In order to understand the origin of this difference, we have examined BChE-cocaine binding and the fundamental reaction pathway for BChE-catalyzed hydrolysis of cocaine through molecular modelling, molecular dynamics (MD) simulations, and ab initio quantum mechanical (QM) calculations. First, the binding of BChE with (−)-cocaine and (+)-cocaine were modelled and simulated to provide the prereactive enzyme-substrate complexes. QM reaction coordinate calculations were then performed on an enzyme-substrate model system derived from the determined prereactive enzyme-substrate complex for (−)-cocaine to uncover the fundamental reaction pathway for hydrolysis by BChE.
Computational methods
Our computational studies began with a three-dimensional model of human BChE constructed by Harel et al.32 based on their X-ray crystal structure of Torpedo californica AchE. In their model, residues 4 – 534 of AChE are aligned with residues 2 – 532 of BChE. The reliability of this model and other similar ones33,34 is supported by their successful application to studies of BChE binding with a variety of substrates and inhibitors, including organophosphorus nerve agents.29,35,40,41,43,44 The overall features of BChE structure closely resemble those of AChE, but an interesting difference at the active site has been noted.34 The catalytic triad in the AChE structure is located near the bottom of a deep narrow cavity. In the modelled BChE structure, a similar ~ 20 Å deep cavity leads to the catalytic triad but it is ~ 5 Å wider such that a larger substrate, such as butyrylcholine (BCh) or cocaine, can bind in the active site. When Harel et al.’s three-dimensional model32 of human BChE was used in our computational studies, the missing residues 483 – 487 were added by comparing the BChE structure with that constructed by Ekholm et al..34 For each substrate, two kinds of possible enzyme-substrate complexes similar to ES2 and ES3 noted above were modelled and simulated by performing molecular dynamics (MD) simulations. We performed a 500 (or 400) ps MD simulation on each enzyme-substrate complex in a water bath by using the SANDER module of AMBER 4.145 with the Cornell et al. force field.46 The protein-solvent system was optimized prior to the simulation as follows. First, the protein was frozen and the solvent molecules with counterions were allowed to move during a 1000 step minimization and a 3 ps MD run. Second, the side chains were allowed to relax by several subsequent minimizations during which decreasing force constants were put on the backbone atoms. After full relaxation, the system was slowly heated to 250 K in 10 ps and then to 300 K in 40 ps.
The computational details for the MD simulations are much the same as we performed for phosphotriesterase.47,48 In particular, the simulations used a 2 fs time step and employed the particle-mesh Ewald (PME) method.49 The all-atom models were neutralized by adding two chloride counter ions and were immersed in rectangular boxes (with initial size 95.0 Å × 96.0 Å × 75.0 Å) filled with TIP3P water molecules.50 The simulations were performed with a periodic boundary condition in the NPT ensemble at 300 K with Berendsen temperature coupling51a and constant pressure (1 atm) with isotropic molecule-based scaling.51a The SHAKE algorithm,51b with a tolerance of 10−5, was applied to fix all bonds containing a hydrogen atom, and the non-bond pair list was updated every 10 steps. The restrained electrostatic potential (RESP) charges of cocaine (protonated) were determined at the Hartree-Fock (HF) level with the 6-31G* basis set following the standard RESP procedure.46
The QM methods used in our reaction coordinate calculations and subsequent energy evaluations include the HF, density functional theory (DFT) using Becke’s three-parameter hybrid exchange functional and the Lee-Yang-Parr correlation functional (B3LYP),52 and second-order Møller-Plesset (MP2) perturbation theory implemented in Gaussian98.53 The basis sets used include the 3-21G, 6-31G*, 6-31+G*, 6-31+G**, and 6-31++G**.54 Geometries of all transition states, reactants and intermediates considered in this study were first optimized at the HF/3-21G level, followed by energy calculations at the B3LYP/6-31+G*, B3LYP/6-31+G**, B3LYP/6-31++G**, and MP2/6-31+G* levels. To examine the reliability of the geometries optimized at the HF/3-21G level, the geometries of the transition state and the corresponding intermediate for the highest energy barrier step of reaction were further refined at the B3LYP/6-31G* level. Vibrational frequencies were evaluated at the optimized geometries to confirm all the first-order saddle points and local minima found on the potential energy surfaces, and to evaluate zero-point vibration energies (ZPVE) and thermal corrections to Gibbs free energies. Intrinsic Reaction Coordinate (IRC) calculations55 were also performed to verify the expected connections of the first-order saddle points with local minima found on the potential energy surfaces. We did not use diffuse basis functions for geometry optimisation because our previous calculations on the anionic systems indicate that the diffuse functions are not important for the geometry optimisations although they are critically important for the final energy calculations.56,57 All calculations were carried out on SGI Origin 200 multiprocessor computers.
Results and discussion
Similarity between structures of cocaine and butyrylcholine
Reaction coordinate calculations for a chemical reaction begin with a concept of the orientation of the reactants. Different starting structures for the enzyme-substrate complex can lead to completely different reactions. For such enzymatic reactions, we need to know structure of the prereactive enzyme-substrate complex, which may also be called the “near attack conformation (NAC)” defined by Bruice et al.58 and discussed more carefully by Shurki et al..59 Our initial insights into the enzyme-substrate binding came from a comparison of the optimized geometry of butyrylcholine (BCh) with those of (−)-cocaine and (+)-cocaine.
The geometries of BCh and (−)-cocaine optimized at the B3LYP/6-31+G* level are depicted in Figure 1. Note that cocaine mainly exists in its protonated form under physiological condition because its pKa is 8.6,60 and thus both BCh and cocaine have positively charged quaternary ammonium groups. In human BChE, Trp-82 is thought to be the key factor in the stabilization of positively charged substrates in the ES2 and ES3, although this interaction should be more properly classified as a cation-π interaction.35a While the positively charged quaternary ammonium is positioned to effectively bind with Trp-82 in the ES3 complex, the carbonyl carbon of the substrate must be positioned proximal to Ser-198 Oγ for nucleophilic attack. Thus the distance between the carbonyl carbon and the quaternary ammonium is critical and according to our optimized geometries depicted in Figure 1, this distance is 4.92 Å for the excellent substrate BCh.34 The optimized C to N distance for the substrate cocaine benzoyl ester (5.23 Å) is similar to that of BCh. The C to N distance for the cocaine methyl ester (2.95 Å) is remarkably shorter. This helps to explain why (−)-cocaine and (+)-cocaine bind with BChE in a way to hydrolyze at the benzoyl ester, instead of the methyl ester, whereas for the nonenzymatic hydrolysis of cocaine under physiological conditions (pH 7.4; 37°) the methyl ester hydrolyzes faster than the benzoyl.60
Figure 1.

Geometries of BCh and (−)-cocaine optimized at the B3LYP/6-31+G* level.
BChE-substrate complexes
Based on the structural similarity discussed above, the relative positions of the positively charged quaternary ammonium and the carbonyl group of the benzoyl ester moiety in the prereactive BChE-cocaine complexes could be similar to that in the corresponding BChE-BCh complex. The main structural difference between the BChE-(−)-cocaine and BChE-(+)-cocaine complexes exists only in the relative position of the methyl ester group (Scheme 1). Hence, the initial structures of both the BChE-(−)-cocaine and BChE-(+)-cocaine complexes used in our first step of molecular modelling and simulations were generated from the three-dimensional model of human BChE with substrate butyrylcholine (BCh) constructed by Harel et al.32 by replacement of substrate. (−)-cocaine and (+)-cocaine were positioned similarly to BCh: the carbonyl group of the benzoyl ester was superimposed on the carbonyl group of BCh, and the nitrogen at the positively charged tropane nucleus was superimposed on the nitrogen of the positively charged quaternary ammonium.
Scheme 1.

Structures of (−)-cocaine and (+)-cocaine.
The energy-minimized geometries of the BChE-(−)-cocaine and BChE-(+)-cocaine complexes are depicted in Figure 2. The interactions of the carbonyl group of the benzoyl ester with the hydroxyl oxygen (Oγ) of Ser-198 and with the oxyanion hole formed by the peptidic NH functions of Gly-116, Gly-117, and Ala-199, obtained from the MD simulations are summarized in Figure 3. RMSD in Figure 3 represents the root-mean-square deviation (for heavy atoms) of the enzyme structure from the original structure constructed by Harel et al..32 In the minimized structures of BChE binding with (−)-cocaine and (+)-cocaine, the internuclear distances between the carbonyl carbon of the benzoyl ester and Ser-198 Oγ are 3.19 and 3.18 Å, respectively. During the MD simulations from 100 ps to 500 ps, the time-average values of the distance between the carbonyl carbon and Ser-198 Oγ are ~ 3.51 and ~ 3.53 Å for (−)-cocaine and (+)-cocaine, respectively. These C to Oγ distances are all comparable to the distances between the carbonyl carbon and the hydroxide oxygen (2.99 − 3.56 Å)61 in our previously optimized geometries of the prereactive complexes of carboxylic acid esters with hydroxide.61-64 The distances between the carbonyl oxygen of the benzoyl ester and the NH hydrogen of Gly-116, Gly-117, and Ala-199 are 1.98, 2.59, and 1.92 Å, respectively, in the minimized structures of BChE with (−)-cocaine. The respective O to H distances in the minimized structures of BChE with (+)-cocaine are 1.82, 2.42, and 2.39 Å. The respective time-average values of the O to H distances are ~ 3.67, ~ 2.21, and ~ 2.46 Å for the MD simulation on BChE with (−)-cocaine, and ~ 3.80, ~ 2.39, and ~ 3.01 Å for the MD simulation with (+)-cocaine. In addition, the MD trajectories also reveal that in both the BChE-(−)-cocaine and BChE-(+)-cocaine complexes, the cocaine nitrogen atom stays at nearly the same position as the BCh nitrogen atom in the structure of BChE model constructed by Harel et al..32 These limited results suggest that both (−)-cocaine and (+)-cocaine may bind with human BChE so as to allow Ser-198 Oγ to approach the carbonyl carbon of the benzoyl ester.
Figure 2.

Minimized structures of prereactive BChE-(−)-cocaine and BChE-(+)-cocaine complexes. (a) Ribbon. (b) Residues forming the catalytic triad and residues surrounding substrate within 5 Å. (c) Residues forming the catalytic triad and the three-pronged oxyanion hole.
Figure 3.

The RMSD (A) of the BChE structure (heavy atoms) in the simulated prereactive BChE-cocaine complex from the original BChE structure constructed by Harel et al.. and important internuclear distances (C, E, F, and G) versus the simulation time. C represents the distance between the carbonyl carbon and Ser-198 Oγ; E, F and G represent the distances from the carbonyl oxygen to the peptidic NH hydrogen of Gly-116, Gly-117 and Ala-199, respectively.
Our modelled and simulated structures of the prereactive BChE-(−)-cocaine and BChE-(+)-cocaine complexes depicted in Figures 2 and 3 are similar to the ES3 structure proposed for BChE binding with other positively charged substrates, i.e. butyrylthiocholine and succinyldithiocholine;35 they are all positioned horizontally at the bottom of the substrate-binding gorge of BChE. To better understand BChE binding with cocaine, we also docked (−)-cocaine and (+)-cocaine to the BChE active site in a way similar to what described in literature37b in order to model the ES2 complex. Through energy minimization and MD simulations, we obtained the ES2-like BChE-(−)-cocaine and BChE-(+)-cocaine complexes in which (−)-cocaine and (+)-cocaine are positioned vertically in the substrate-binding gorge between Asp-70 and Trp-82. As illustrated in Figure 4, the MD trajectories for the ES2-like BChE-(−)-cocaine and BChE-(+)-cocaine complexes are also very stable. In addition, our simulated ES2-like BChE-(−)-cocaine and BChE-(+)-cocaine complexes are very close to the simulated Michaelis-Menten complexes reported by Sun et al..37b All these suggest that the binding of BChE with (−)-cocaine and (+)-cocaine is similar to those proposed with butyrylthiocholine and succinyldithiocholine.35 Both the non-prereactive (or ES2-like) complex and the prereactive (or ES3-like) complex could exist before going to the chemical reaction steps. Further, based on the results reported by Sun et al.,37b the Michaelis-Menten complex is likely the ES2-like complex for (−)-cocaine and (+)-cocaine, but it is not the prereactive complex in light of our new results. In other words, the Michaelis-Menten complex is different from the prereactive enzyme-substrate complex for BChE-catalyzed hydrolysis of (−)-cocaine and (+)-cocaine. The Michaelis-Menten complex must progress to the prereactive complex before the first chemical reaction step can occur.
Figure 4.
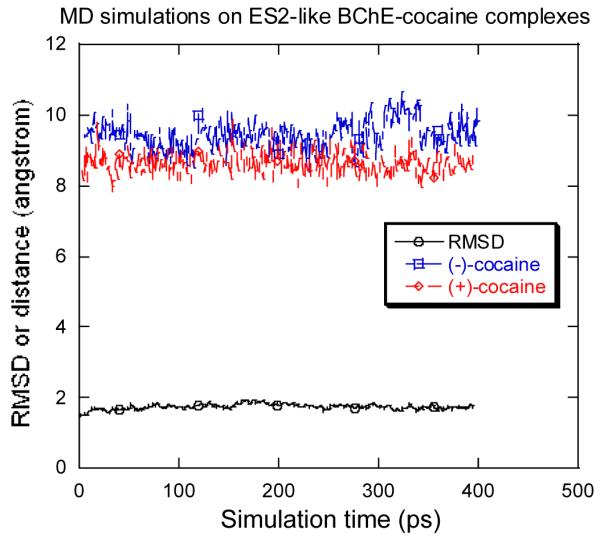
The RMSD of the BChE structure (heavy atoms) in the simulated ES2-like BChE-(+)-cocaine complex from the starting structure and the simulated distances between the benzoyl carbonyl carbon of cocaine and Ser-198 Oγ versus the simulation time.
To better compare the modelled ES2-like complex with the prereactive complex for (−)-cocaine and for (+)-cocaine, the protein backbone atoms in the ES2-like complex were superimposed with the corresponding atoms in the prereactive complex. It turned out that the overall protein structures in the ES2-like and prereactive complexes are very close to each other, while the orientations of the substrate are nearly vertical to each other. So, for both (−)-cocaine and (+)-cocaine, the substrate needs to rotate about 90° during the change from the ES2-like complex to the prereactive complex as depicted in Figure 5; more specifically, (−)-cocaine needs to rotate slightly more than (+)-cocaine. The energy barrier of the change from the ES2-like complex to the prereactive complex for (−)-cocaine is expected to differ from that for (+)-cocaine. This is because the relative positions of the C-2 methyl ester group of substrate are different and, therefore, some amino acid residues hindering the rotation of one substrate might not hinder the rotation of another. Specific residues possibly hindering substrate rotation will be discussed below.
Figure 5.


Relative positions of cocaine and some residues of BChE in the ES2-like and prereactive BChE-(−)-cocaine (a) and BChE-(+)-cocaine (b) complexes. The red and black refer to the ES2-like and prereactive complexes, respectively.
Possible reaction pathways
For both (−)-cocaine and (+)-cocaine, the relative positions of the nitrogen and the benzoyl carbonyl in the simulated prereactive BChE-substrate complex are essentially the same as those reported for BCh in BChE, we may expect that BChE-catalyzed hydrolysis of (−)-cocaine and (+)-cocaine follow a reaction pathway similar to that for BChE-catalyzed hydrolysis of BCh. A remarkable difference between (−)-cocaine and (+)-cocaine is associated with the relative positions of the C-2 methyl ester group. The C-2 methyl ester group of (−)-cocaine remains on the same side of the carbonyl of the benzoyl ester as the attacking hydroxyl oxygen (Ser-198 Oγ), whereas the C-2 methyl ester of (+)-cocaine remains on the opposite side. This difference could cause a difference in hydrogen bonding, electrostatic, and van der Waals interactions during the catalytic process, and result in a significant difference in free energies of activation. Nevertheless, the basic BChE mechanism for both enantiomers may resemble the common catalytic mechanism for ester hydrolysis in other serine hydrolases,65 including the thoroughly investigated AChE.66-69 Thus, based on our modelling and simulation of the prereactive complexes and the knowledge about ester hydrolysis in other serine hydrolases, a possible reaction pathway for BChE-catalyzed hydrolysis of cocaine can be hypothesized. Scheme 2 depicts (−)-cocaine and important groups from the catalytic triad (Ser-198, Glu-325, and His-438) and three-pronged oxyanion hole (Gly-116, Gly-117, and Ala-199). The proposed hydrolysis of cocaine consists of both the acylation and deacylation stages demonstrated for ester hydrolysis by other serine hydrolases.65 A significant difference might exist in the number of potential hydrogen bonds involving the carbonyl oxygen in the oxyanion hole. The three-pronged oxyanion hole formed by peptidic NH groups of Gly-116, Gly-117, and Ala-199 in BChE (or by peptidic NH groups of Gly-118, Gly-119, and Ala-201 in AChE) contrasts with the two-pronged oxyanion hole of many other serine hydrolases. Schematic representation of the pathway for (+)-cocaine hydrolysis should be similar to Scheme 2, differing only in the relative position of the C-2 methyl ester group in the acylation.
Scheme 2.

Schematic representation of the single-proton-transfer mechanism for BChE-catalyzed hydrolysis of (−)-cocaine at the benzoyl ester.
As depicted in Scheme 2, the acylation is initialized by Ser-198 Oγ attack at the carbonyl carbon of the cocaine benzoyl ester to form the first tetrahedral intermediate (INT1) through the first transition state (TS1). During the formation of INT1, the C–O bond between the carbonyl carbon and Ser-198 Oγ gradually forms, while the proton at Ser-198 Oγ gradually transfers to the imidazole N atom of His-438 which acts as a general base. The second step of the acylation is the decomposition of INT1 to the metabolite ecgonine methyl ester and acyl-BChE (INT2a) through the second transition state (TS2). During the change from INT1 to INT2a, the proton gradually transfers to the benzoyl ester oxygen, while the C–O bond between the carbonyl carbon and the ester oxygen gradually breaks. Also, during the first step of acylation, the carbonyl oxygen may potentially form up to three hydrogen bonds with the NH groups of Gly-116, Gly-117, and Ala-199. Note that these potential hydrogen bonds do not necessarily exist in the prereactive enzyme-substrate complex (ES) and in other structures, even though they are, for convenience, indicated throughout the reaction process in Scheme 2.
As seen in Figure 3, in the simulated prereactive enzyme-substrate complex only one or two of the three NH groups weakly hydrogen-bond to the carbonyl oxygen of (+)-cocaine or (−)-cocaine during the simulations. No hydrogen bonding was noted between the carbonyl oxygen and the NH group of Gly-116. These potential hydrogen bonds are expected to increase in strength from ES to TS1 and to INT1 due to the expected increase of net negative charge on the carbonyl oxygen. By the same logic, these potential hydrogen bonds are expected to progressively weaken from INT1 to TS2 and to INT2. The deacylation is initialized by water (oxygen) attack at the carbonyl carbon with participation of His-438 as a general base, and is the reverse of acylation with respect to bond breaking/formation and potential hydrogen bonds.
A common feature of the four reaction steps indicated in Scheme 2 is that a C–O bond formation/breaking process always incorporates a single proton transfer along the hydrogen bond between the His-438 nitrogen and the oxygen of the C–O bond, whereas there is no proton transfer between His-438 and Glu-325. The pathway shown in Scheme 2 is thus a single-proton-transfer mechanism. A double-proton-transfer mechanism, depicted in Scheme 3, would involve an additional, concurrent proton transfer between His-438 and Glu-325, as discussed by Hu, Brinck, and Hult for ester hydrolysis catalyzed by a serine hydrolase.65
Scheme 3.

Schematic representation of the double-proton-transfer mechanism for BChE-catalyzed hydrolysis of (−)-cocaine at the benzoyl ester.
Hu, Brinck and Hult performed QM calculations on a simplified model system for hydrolysis of methyl formate catalyzed by a serine hydrolase.65 Their enzyme model includes the two-pronged oxyanion hole represented by two water molecules, and the catalytic triad represented by formate anion, imidazole and methanol. They examined both a single-proton-transfer mechanism and double-proton-transfer mechanism for the first step of acylation, and the later mechanism was found to be energetically unfavorable by ~ 2 kcal/mol.65 Questions for further study include whether the conclusion for methyl formate hydrolysis catalyzed by a serine hydrolase involving the two-pronged oxyanion hole is correct also for hydrolysis of (−)-cocaine and (+)-cocaine catalyzed by BChE involving the three-pronged oxyanion hole. Only by performing extensive reaction coordinate calculations, can we know whether the two mechanisms co-exist in all the reaction steps of BChE-catalyzed hydrolysis of (−)-cocaine and (+)-cocaine, and which is favorable if the two mechanisms co-exist. If the single-proton-transfer mechanism is favorable for some reaction steps and the double-proton-transfer mechanism is favorable for other reaction steps, then the favorable reaction pathway should be a proper combination of the steps shown in Schemes 2 and 3. The combination might be very simple, if one mechanism is favorable for the whole acylation process and the other is favorable for the whole deacylation process. If one mechanism is favorable for the first step of acylation (or deacylation) and the other is favorable for the second step of acylation (or deacylation), then an additional reaction step, i.e. an individual proton transfer along the hydrogen bond between His-438 and Glu-325, may exist between the two major steps. Thus, it is also possible that the favorable acylation (or deacylation) pathway includes three reaction steps.
Reaction coordinates and energy profiles
To examine the above mechanistic hypotheses, we performed detailed reaction coordinate calculations on the fundamental reaction pathways for BChE-catalyzed hydrolysis of (−)-cocaine with a BChE active site model. The general approach of using fully quantum mechanical methods for active site residues has previously been used by other researchers.70 The BChE active site model used includes only the six amino acids indicated in Schemes 2 and 3 with the following simplifications: Ser-198 is represented by methanol, His-438 is represented by imidazole, Glu-325 is represented by acetate (CH3COO−), and Gly-116, Gly-117, and Ala-199 are all represented by ammonia molecules. This BChE active site model consists of 34 atoms. So, a total of 78 atoms were included in the ab initio calculations on the model of (−)-cocaine hydrolysis. Reaction coordinate calculations on this model system are expected to provide a qualitative picture for the formation and breaking of covalent bonds at the reaction center and to estimate the intrinsic energy barriers and Gibbs free energy barriers for the enzymatic reaction.
We examined both the single-proton-transfer mechanism and double-proton-transfer mechanism by testing different initial geometries in the transition state geometry optimisations. However, the calculations always pointed to the same transition states expected from the hypothetical double-proton-transfer mechanism, consisting of four individual steps (ES → TS1→ INT1 → TS2 → INT2 → TS3 → INT3 → TS4 → EB). Depicted in Figure 6 are the geometries of the transition states (TS1, TS2, TS3, and TS4), intermediates (INT1, INT2, and INT3), and prereactive BChE-(−)-cocaine complex model (ES) optimized at the HF/3-21G level. The calculated energy barriers (ΔEa) and Gibbs free energy barriers (ΔGa) are summarized in Table 1. Our reaction coordinate calculations argue against a single-proton-transfer mechanism and support a double-proton-transfer mechanism for BChE-catalyzed hydrolysis of cocaine.
Figure 6.


The model geometries of the prereactive enzyme-substrate (ES) complex, transition states, and intermediates for the acylation (a) and deacylation (b) stages of the BChE-catalyzed hydrolysis of (−)-cocaine optimized at the HF/3-21G level.
Table 1.
Energy barriers (ΔEa) and Gibbs free energy barriers (ΔGa), in kcal/mol, calculated for BChE-catalyzed hydrolysis of (−)-cocaine at 298 K and 1 atm.a
| Method | Δ Ea | Δ Ga | ||||||
|---|---|---|---|---|---|---|---|---|
| Step 1 | Step 2 | Step 3 | Step 4 | Step 1 | Step 2 | Step 3 | Step 4 | |
| MP2/6-31+G* | 4.0 | 3.1 | 16.6 (17.0) |
6.5 | 5.6 | 3.6 | 19.0 (18.2) |
6.6 |
| B3LYP/6-31+G* | 6.2 | 16.9 (18.5) |
7.8 | 19.2 (19.8) |
||||
| B3LYP/6-31+G** | 5.5 | 16.2 (17.5) |
7.1 | 18.5 (18.7) |
||||
| B3LYP/6-31++G** | 5.6 | 16.2 (17.5) |
7.2 | 18.5 (18.7) |
||||
The values in parentheses were calculated using the geometries optimized at the B3LYP/6-31G* level. The other values were calculated using the geometries optimized at the HF/3-21G level.
Our previous computational studies61-64,71,72 on the reaction pathways for various ester hydrolyses demonstrated that electron correlation effects are important only for final energy evaluations, but are not important for the geometry optimisations. The geometry optimisations at a lower HF level (using a smaller basis set) followed by single-point energy calculations at the MP2/6-31+G* level are adequate for predicting energy barriers in excellent agreement with the corresponding experimental data. The calculations at higher levels, e.g. replacing MP2 with QCISD(T) or using larger basis set, do not change the calculated energy barriers substantially.61,72 In the present study, we further tested this issue. Our results calculated at the MP2/6-31+G*//HF/3-21G level show that for both the acylation and deacylation stages, the energy barrier and free energy barrier predicted for the first step (i.e. the first or third step of the entire chemical reaction process) is always higher than that for the corresponding second step (i.e. the second or fourth step of the entire chemical reaction process). Hence, we performed the B3LYP energy calculations using the 6-31+G* and larger basis sets on these two critical reaction steps. The barriers calculated at the B3LYP/6-31+G*//B3LYP/6-31G* level are close to the corresponding values at the MP2/6-31+G*//HF/3-21G level, although the change for the first step (~ 2.2 kcal/mol) is significant. Increasing the basis set from 6-31+G* to 6-31++G**, the changes of the calculated barriers are smaller than 0.7 kcal/mol.
Since the third reaction step, i.e. the first step of deacylation, is associated with the highest energy barrier and the highest Gibbs free energy barrier, we further optimized the geometries of TS3 and INT2 at the B3LYP/6-31G* level to evaluate the barrier for this highest-barrier reaction step using the B3LYP/6-31G* geometries. We found that the barriers calculated using the B3LYP/6-31G* geometries are close to those from the HF/3-21G geometries, particularly for the Gibbs free energy barriers. These comparisons indicate that our energy barriers and Gibbs free energy barriers predicted at the MP2/6-31+G*//B3LYP/6-31G* and MP2/6-31+G*//HF/3-21G levels are reliable.
We note that our conclusion about the rate-determining step for BChE-catalyzed hydrolysis of (−)-cocaine is different from that for other similar model serine protease enzymatic systems with amide substrate HC(O)NH273 or CH3C(O)NHCH3.74 The formation of the tetrahedral intermediate during the deacylation is rate-determining for the cocaine hydrolysis, whereas the formation of the tetrahedral intermediate during the acylation is rate-determining for the amide hydrolysis. This difference might be due to the remarkable difference between substrate cocaine and substrate amide, particularly for the identity of the leaving group of the hydrolysis. For acylation stage of the cocaine hydrolysis a C–O bond will be broken and the leaving group includes a tropanecarboxylic acid methyl ester, whereas for acylation stage of the amind hydrolysis a C–N bond will be broken and the leaving group is NH2 or NHCH3. For both the cocaine and amide hydrolyses, the deacylation stage is always associated with breaking a C–O bond. Our previous reaction pathway calculations on base-catalyzed hydrolysis of various esters61-64,72 indicate that the calculated energy barriers are very sensitive to the identity of the leaving group of the ester. The tropanecarboxylic acid methyl ester existing in the leaving group significantly stabilizes the transition state for the formation of the tetrahedral intermediate so that the energy barrier calculated for the cocaine hydrolysis (7.6 kcal/mol) is ~ 4 kcal/mol lower than that for the CH3C(O)OCH3 hydrolysis (11.4 kcal/mol) calculated at the same level of theory.63a The energy barrier of 11.4 kcal/mol calculated for the CH3C(O)OCH3 hydrolysis is in excellent agreement with the available experimental activation energy 12.2 kcal/mol75 or 10.45 kcal/mol.76
Because the third reaction step is predicted to have the highest barrier, this reaction step is expected to be rate determining if the effects of the remaining protein environment on the calculated barriers can be neglected. Further, because the third reaction steps for (−)-cocaine and for (+)-cocaine are identical, their hydrolysis rates in BChE would be expected to be the same if this step of the chemical reaction process were really rate determining for the entire catalytic process. In fact, (−)-cocaine hydrolysis in BChE is about 1000 to 2000 times slower than (+)-cocaine, suggesting that some other factors, such as the change from the ES2-like complex to the prereactive ES complex, are important for (−)-cocaine or for both (−)-cocaine and for (+)-cocaine. To better understand the mechanism, below we compare the predicted highest barriers in Table 1 with those predicted for the alkaline hydrolysis of cocaine predicted in our previous computational studies and with available experimental rate constants.
BChE-catalyzed hydrolysis vs spontaneous hydrolysis
The highest energy barrier, ~ 17 kcal/mol, predicted at the MP2/6-31+G*//B3LYP/6-31G* level for the BChE-catalyzed hydrolysis of (−)-cocaine is ~ 9 kcal/mol higher than the energy barrier of ~ 7.6 cal/mol predicted for hydroxide ion-catalyzed hydrolysis of cocaine at the benzoyl ester in aqueous solution.63,71 For the hydroxide ion-catalyzed hydrolysis of cocaine in aqueous solution, we reported two competing reaction pathways for the second step, one is associated with a direct proton transfer and the other is associated with a water-assisted proton transfer.63 The energy barrier for the second step corresponding to the direct proton transfer is higher than that (7.6 cal/mol) for the first step, whereas the barrier for the second step corresponding to the water-assisted proton transfer is lower than that for the first step. So, the first step is rate-determining for the hydroxide ion-catalyzed hydrolysis.63 Nevertheless, the entropy contributions to the Gibbs free energy barriers for the hydroxide ion-catalyzed hydrolysis are considerably different from those for the BChE-catalyzed hydrolysis. The energy barriers, ~ 17.0 kcal/mol vs ~ 7.6 kcal/mol, refer to the barriers at the ideal temperature of 0 K. Further accounting for the thermal motion at 298 K and 1 atm (including the entropy contributions), the Gibbs free energy barrier, denoted by ΔGa0, predicted for the hydroxide ion-catalyzed hydrolysis of cocaine at the benzoyl ester should be ~ 19.5 kcal/mol,63b about 1 kcal/mol higher than the Gibbs free energy barrier of ~ 18.2 kcal/mol predicted for the BChE-catalyzed hydrolysis.
The catalytic rate constant, kcat defined by (d[S]/dt)cat = kcat[ES], for the BChE-catalyzed hydrolysis is determined by the predicted Gibbs free energy barrier, ΔGa = ~ 18.2 kcal/mol, through
| (1) |
in which ν is the frequency factor. ν = kBT/h, where kB is Boltzmann’s constant and h is Planck’s constant. [S] and [ES] represent the concentrations of the substrate and the enzyme-substrate complex, respectively. It has been known that the spontaneous, nonenzymatic hydrolysis of cocaine in aqueous solution is dominated by the hydrolysis catalyzed by hydroxide ion. The rate constant of the “spontaneous” hydrolysis, k0, is dependent on the concentration of hydroxide ion, [HO−], and the second order reaction rate constant, k2:
| (2) |
| (3) |
Thus we have
| (4) |
Equations (1) and (4) give the ratio of kcat to k0, i.e.
| (5) |
When ΔGa = 18.2 kcal/mol, ΔGa0 = 19.5 kcal/mol, [HO−] = 10−7 M (i.e. pH = 7), and T = 298 K, Eq.(5) provides kcat/k0 = ~ 9.0 × 107.
The predicted kcat/k0 value of ~ 9.0 × 107 can be compared with available experimental rate constants. The experimental k0 value for the cocaine benzoyl ester hydrolysis in aqueous solution was reported as 0.00536 h−1 = 8.93 × 10−5 min−1.60 The kcat values for the (−)-cocaine and (+)-cocaine hydrolyses in BChE were recently determined to be 3.9 ± 0.8 and 6423 ± 24 min−1, respectively.37b Thus, the kcat/k0 values derived from experimental rate constants for (−)-cocaine and (+)-cocaine are ~ 4.4 × 104 and ~ 7.2 × 107, respectively. Our predicted kcat/k0 value of ~ 9 × 107 is ~ 2000 times larger than the experimentally-derived kcat/k0 value of ~ 4.4 × 104 for (−)-cocaine, but is in excellent agreement with the experimentally-derived kcat/k0 value of ~ 7.2 × 107 for (+)-cocaine. The excellent agreement for (+)-cocaine implies that the rate-determining step of the BChE-catalyzed hydrolysis of (+)-cocaine is likely the third step of the chemical reaction process, i.e. the first step of the deacylation stage, and that the overall effect of the remaining protein environment on the third reaction step can be ignored.
The large difference of ~ 2000 times between the theoretical and experimental kcat/k0 values for (−)-cocaine implies that another step in the acylation or the change from the ES2-like complex to the prereactive BChE-(−)-cocaine complex has a Gibbs free energy barrier higher than the third step (~ 18 kcal/mol) by ~ 4 kcal/mol. So the remaining protein environment considerably affects a process other than the deacylation. Furthermore, if a chemical reaction step in the acylation, rather than the transformation from ES2-like complex to the prereactive BChE-(−)-cocaine complex, is rate determining, the enzymatic reaction rate is expected to be pH-dependent. A very recent experimental study37b revealed that the rate of the BChE-catalyzed hydrolysis of (−)-cocaine is not significantly affected by the pH of the reaction solution, whereas the rate of the BChE-catalyzed hydrolysis of (+)-cocaine is clearly pH-dependent. So, it is likely that the rate-determining step is the change from the ES2-like complex to the prereactive complex for BChE-catalyzed hydrolysis of (−)-cocaine.
Insights into rational design of BChE mutants
Generally speaking, for rational design of a more active mutant of an enzyme for a given substrate, one needs to find a mutation that can accelerate the rate-determining step of the entire catalytic reaction while the other steps are not slowed down by the mutation. The enzyme-substrate binding and fundamental reaction pathways discussed above provide a rational base for the design of more active BChE mutants for (−)-cocaine. Now that the change from the ES2-like complex to the prereactive complex is likely the rate-determining step of the BChE-catalyzed hydrolysis of (−)-cocaine, useful BChE mutants should be designed to specifically accelerate the change from the ES2-like complex to the prereactive BChE-(−)-cocaine complex. Plausible mutations would focus on the residues surrounding the C-2 methyl ester group during the (−)-cocaine rotation process. The amino acid residues surrounding the C-2 methyl ester group of (−)-cocaine include Try-82, Leu-125, and Tyr-128 in the simulated ES2-like BChE-(−)-cocaine complex, and Gln-223, Ser-224, and His-438 in the simulated prereactive BChE-(−)-cocaine complex. Furthermore, during the rotation of (−)-cocaine from the ES2-like complex to the prereactive complex, other residues, such as Tyr-332, could also participate.
The residues that most likely hinder the rotation of (−)-cocaine from the ES2-like complex to the prereactive complex include Tyr-332, Trp-82, and Tyr-128. The relative positions of the large side chains of these three residues were significantly changed from the ES2-like complex to the prereactive complex for (−)-cocaine (Figure 5a), whereas significant changes of the relative positions of these three residues were not observed for (+)-cocaine (Figure 5b). We propose that the side chains of these three residues be replaced with smaller ones. A problem could come with the mutation on Trp-82, because the side chain of Trp-82 was considered to be important for formation of the ES2 complex for other positively charged substrates, i.e. butyrylthiocholine and succinyldithiocholine.35 If Trp-82 is also important for formation of the ES2-like BChE-(−)-cocaine complex, with a BChE mutant including such as Try82Gly or Try82Ala mutation, the formation of the ES2-like complex will be impassed even as the change from the ES2-like complex to prereactive complex is facilitated. It would be interesting to test BChE mutants that reduce the bulk sizes of these two (i.e. Tyr-332 and Tyr-128) or three (i.e. Tyr-332, Trp-82, and Tyr-128) residues are reduced, e.g. by replacing each with Gly or Ala. It should also be interesting to test the BChE mutants including some of these mutations and an additional mutation on Ala-328 because the Ala328Tyr mutant is 4-fold more efficient than the wild type.37a Other residues that could also hinder the rotation of (−)-cocaine include Tyr-440. In the simulated ES2-like BChE-(−)-cocaine complex, the Tyr-440 hydroxyl group binds the carbonyl oxygen of (−)-cocaine benzoyl ester. A similar interaction was not observed in the simulated ES2-like BChE-(+)-cocaine complex. As a result, the benzoyl ester group was positioned closer to Tyr-440 and farther from the expected position of the prereactive complex for (−)-cocaine than for (+)-cocaine. Note that the benzoyl ester group must move away from Tyr-440 during the change from the ES2-like complex to the prereactive complex. Thus, the binding between the Tyr-440 hydroxyl group and the carbonyl oxygen of (−)-cocaine benzoyl ester group could stabilize ES2-like complex, but it could slow down the rotation of (−)-cocaine to the desired prereactive complex. A mutation, such as Tyr440Phe, could destabilize the ES2-like complex, but could speed up the necessary rotation of (−)-cocaine towards the position in the prereactive complex.
A recently reported study37b on the Michaelis-Menten complex (i.e. the ES2-like complex discussed here) suggested that the rotation of (−)-cocaine could be hindered by interactions of its phenyl ring with Phe-329 and Trp-430. Our results for both the ES2-like and prereactive complexes indicate that these interactions might, in fact, not be critical because the relative positions of Phe-329 and Trp-430 did not significantly change when the BChE-(−)-cocaine binding changed from the ES2-like complex to the prereactive complex.
Conclusion
We have carried out a variety of molecular modelling, molecular dynamics (MD) simulations, and ab initio calculations to explore butyrylcholinesterase (BChE)-cocaine binding and the fundamental reaction pathway for BChE-catalyzed hydrolysis of cocaine. The molecular modelling and MD simulations indicate that the structures of the prereactive BChE-substrate complexes for (−)-cocaine and (+)-cocaine are similar to that of the corresponding prereactive BChE-butyrylcholine (BCh) complex. The overall binding of BChE with (−)-cocaine and (+)-cocaine is also similar to that proposed for butyrylthiocholine and succinyldithiocholine, i.e. the substrate first advances down the substrate-binding gorge to bind to Trp-82 and remains vertically in the gorge between Asp-70 and Trp-82 in the non-prereactive complex. The substrate then rotates to a horizontal position at the bottom of the gorge at a favorable distance for nucleophilic attack and hydrolysis by Ser-198 in the prereactive complex. Thus, for both (−)-cocaine and (+)-cocaine, the substrate must rotate about 90° during the transformation from non-prereactive complex to prereactive complex. The main structural difference between the BChE-(−)-cocaine complexes and the corresponding BChE-(+)-cocaine complexes is in the relative position of the cocaine methyl ester group.
Based on the modelled and simulated prereactive BChE-(−)-cocaine and BChE-(+)-cocaine complexes, we have proposed possible reaction pathways for BChE-catalyzed hydrolysis of cocaine. The proposed fundamental reaction pathway, either a single-proton-transfer or a double-proton-transfer mechanism, includes both acylation and deacylation steps similar to ester hydrolysis by other serine hydrolases. The pathway was examined by ab initio reaction coordinate calculations on a catalytic reaction model consisting of substrate cocaine and functional groups from the catalytic triad (Ser-198, Glu-325, and His-438) and three-pronged oxyanion hole (Gly-116, Gly-117, and Ala-199) of BChE. The geometries of all states involved in the deacylation are independent of whether the hydrolysed cocaine is (−)-cocaine or (+)-cocaine. The ab initio reaction coordinate calculations strongly support the pathway involving the double-proton-transfer, and further show that the highest energy barrier/Gibbs free energy barrier is associated with the first step of deacylation.
Based on the Gibbs free energy barrier calculated for the first step of deacylation, the estimated ratio of the rate constant (kcat) for the catalytic hydrolysis to that (k0) for the spontaneous hydrolysis is ~ 9.0 × 107. This value is in excellent agreement with the experimentally-derived kcat/k0 value of ~ 7.2 × 107 for (+)-cocaine. However, it is ~ 2000 times larger than the experimentally-derived value for (−)-cocaine (~ 4.4 × 104). The excellent agreement for (+)-cocaine reveals that the rate-determining step of the BChE-catalyzed hydrolysis of (+)-cocaine is likely the first step of deacylation, and that the overall effect of the remaining protein environment on this critical step can be ignored. In contrast, for BChE-(−)-cocaine the transformation from the ES2-like complex to the prereactive complex is apparently rate determining and has a Gibbs free energy barrier higher than that for the first step of deacylation by ~ 4 kcal/mol. Specific amino acid residues hindering the structural changes from the non-prereactive complex to the prereactive complex provide a useful target for the rational design of BChE mutants with improved catalytic activity for (−)-cocaine.
Supplementary Material
Footnotes
Supporting Information Available. Cartesian coordinates of the optimized model geometries (ES, TS1, INT1, TS2, INT2, TS3, INT3, TS4, and EB) together with the calculated energies (16 pages).
References
- 1.Landry DW, Yang GX-Q. J. Addict. Diseases. 1997;16:1. doi: 10.1300/J069v16n03_01. [DOI] [PubMed] [Google Scholar]
- 2.Singh S. Chem. Rev. 2000;100:925. doi: 10.1021/cr9700538. [DOI] [PubMed] [Google Scholar]
- 3.Sparenborg S, Vocci F, Zukin S. Drug Alcohol Depend. 1997;48:149. doi: 10.1016/s0376-8716(97)00072-0. [DOI] [PubMed] [Google Scholar]
- 4.Gorelick DA. Drug Alcohol Depend. 1997;48:159. doi: 10.1016/s0376-8716(97)00119-1. [DOI] [PubMed] [Google Scholar]
- 5.Fox BS. Drug Alcohol Depend. 1997;100:153. doi: 10.1016/s0376-8716(97)00121-x. [DOI] [PubMed] [Google Scholar]
- 6.Landry DW, Zhao K, Yang GX-Q, Glickman M, Georgiadis TM. Science. 1993;259:1899. doi: 10.1126/science.8456315. [DOI] [PubMed] [Google Scholar]
- 7.Yang G, Chun J, Arakawa-Uramoto H, Wang X, Gawinowicz MA, Zhao K, Landry DW. J. Am. Chem. Soc. 1996;118:5881. [Google Scholar]
- 8.Mets B, Winger G, Cabrera C, Seo S, Jamdar S, Yang G, Zhao K, Briscoe RJ, Almonte R, Woods JH, Landry DW. Proc. Natl. Acad. Sci. USA. 1998;95:10176. doi: 10.1073/pnas.95.17.10176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Almonte RE, Deng S-X, De Prada P, Stojanovic MN, Landry DW. Phosphorus. Sulfur. 1999;146:239. [Google Scholar]
- 10.Baird TJ, Deng S-X, Landry DW, Winger G, Woods JH. J. Pharmacol. Exp. Ther. 2000;295:1127. [PubMed] [Google Scholar]
- 11.De Prada P, Winger G, Landry DW. Ann. NY Acad. Sci. 2000;909:159. doi: 10.1111/j.1749-6632.2000.tb06681.x. [DOI] [PubMed] [Google Scholar]
- 12.Cashman JR, Berkman CE, Underiner GE. J. Pharmacol. Exp. Ther. 2000;293:952. [PubMed] [Google Scholar]
- 13.Kuhar MJ, Carroll FI, Bharat N, Landry DW. Synapse. 2001;41:176. doi: 10.1002/syn.1072. [DOI] [PubMed] [Google Scholar]
- 14.Briscoe RJ, Jeanville PM, Cabrera C, Baird TJ, Woods JH, Landry DW. Int. Immunopharmacol. 2001;1:1189. doi: 10.1016/s1567-5769(01)00054-6. [DOI] [PubMed] [Google Scholar]
- 15.Matsushita M, Hoffman TZ, Ashley JA, Zhou B, Wirsching P, Janda KD. Bioorg. Med. Chem. Lett. 2001;11:87. doi: 10.1016/s0960-894x(00)00659-4. [DOI] [PubMed] [Google Scholar]
- 16.Deng S-X, Bharat N, Fischman MC, Landry DW. Proc. Natl, Acad, Sci. USA. 2002;99:3412. doi: 10.1073/pnas.042700599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Homayoun P, Mandal T, Landry DW, Childress V, Zhou M, Komiskey H. Faseb. J. 2002;16:A586. [Google Scholar]
- 18.Isomura S, Hoffman TZ, Wirsching P, Janda KD. J. Am. Chem. Soc. 2002;124:3661. doi: 10.1021/ja012376y. [DOI] [PubMed] [Google Scholar]
- 19.Wang J, Han YQ, Liang SZ, Wilkinson MF. Biochem. Bioph. Res. Co. 2002;291:605. doi: 10.1006/bbrc.2002.6484. [DOI] [PubMed] [Google Scholar]
- 20.Tellier C. Transfus. Clin. Biol. 2002;9:1. doi: 10.1016/s1246-7820(01)00208-7. [DOI] [PubMed] [Google Scholar]
- 21.Kamendulis LM, Brzezinski MR, Pindel EV, Bosron WF, Dean RA. J. Pharmacol. Exp. Ther. 1996;279:713. [PubMed] [Google Scholar]
- 22.Poet TS, McQueen CA, Halpert JR. Drug Metab. Dispos. 1996;24:74. [PubMed] [Google Scholar]
- 23.Pan W-J, Hedaya MA. J. Pharm. Sci. 1999;88:468. doi: 10.1021/js980283h. [DOI] [PubMed] [Google Scholar]
- 24.Sukbuntherng J, Martin DK, Pak Y, Mayersohn M. J. Pharm. Sci. 1996;85:567. doi: 10.1021/js960026h. [DOI] [PubMed] [Google Scholar]
- 25.Browne SP, Slaughter EA, Couch RA, Rudnic EM, McLean AM. Biopharmaceutics & Drug Disposition. 1998;19:309. doi: 10.1002/(sici)1099-081x(199807)19:5<309::aid-bdd108>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 26 (a).Carmona GN, Baum I, Schindler CW, Goldberg SR, Jufer R. Life Sci. 1996;59:939. doi: 10.1016/0024-3205(96)00392-x. [DOI] [PubMed] [Google Scholar]; (b) Lynch TJ, Mattes CE, Singh A, Bradley RM, Brady RO, Dretchen KL. Toxicol. Appl. Pharmacol. 1997;145:363. doi: 10.1006/taap.1997.8187. [DOI] [PubMed] [Google Scholar]
- 27.Mattes CE, Lynch TJ, Singh A, Bradley RM, Kellaris PA, Brady RO, Dretchen KL. Toxicol. Appl. Pharmacol. 1997;145:372. doi: 10.1006/taap.1997.8188. [DOI] [PubMed] [Google Scholar]
- 28.Mattes CE, Belendiuk GW, Lynch TJ, Brady RO, Dretchen KL. Addict. Biol. 1998;3:171. doi: 10.1080/13556219872236. [DOI] [PubMed] [Google Scholar]
- 29.Lockridge O, Blong RM, Masson P, Froment M-T, Millard CB, Broomfield CA. Biochemistry. 1997;36:786. doi: 10.1021/bi961412g. [DOI] [PubMed] [Google Scholar]
- 30.Gately SJ. Biochem. Pharmacol. 1991;41:1249. doi: 10.1016/0006-2952(91)90665-r. [DOI] [PubMed] [Google Scholar]
- 31.Gately SJ, MacGregor RR, Fowler JS, Wolf AP, Dewey SL, Schlyer DJ. J. Neurochem. 1990;54:720. doi: 10.1111/j.1471-4159.1990.tb01933.x. [DOI] [PubMed] [Google Scholar]
- 32.Harel M, Sussman JL, Krejci E, Bon S, Chanal P, Massoulie J, Silman I. Proc. Natl. Acad. Sci. USA. 1992;89:10827. doi: 10.1073/pnas.89.22.10827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Millard CB, Broomfield CA. Biochem. Biophys. Res. Commun. 1992;189:1280. doi: 10.1016/0006-291x(92)90212-4. [DOI] [PubMed] [Google Scholar]
- 34.Ekholm M, Konschin H. J. Mol. Struct. (Theochem) 1999;467:161. [Google Scholar]
- 35 (a).Masson P, Legrand P, Bartels CF, Froment M-T, Schopfer LM, Lockridge O. Biochemistry. 1997;36:2266. doi: 10.1021/bi962484a. [DOI] [PubMed] [Google Scholar]; (b) Masson P, Xie W, Froment M-T, Levitsky V, Fortier P-L, Albaret C, Lockridge O. Biochim. Biophys. Acta. 1999;1433:281. doi: 10.1016/s0167-4838(99)00115-6. [DOI] [PubMed] [Google Scholar]
- 36.Berkman CE, Underiner GE, Cashman JR. Biochem. Pharmcol. 1997;54:1261. doi: 10.1016/s0006-2952(97)00403-6. [DOI] [PubMed] [Google Scholar]
- 37 (a).Xie W, Altamirano CV, Bartels CF, Speirs RJ, Cashman JR, Lockridge O. Mol. Pharmacol. 1999;55:83. doi: 10.1124/mol.55.1.83. [DOI] [PubMed] [Google Scholar]; (b) Sun H, Yazal JE, Lockridge O, Schopfer LM, Brimijoin S, Pang Y-P. J. Biol. Chem. 2001;276:9330. doi: 10.1074/jbc.M006676200. [DOI] [PubMed] [Google Scholar]
- 38.Millard CB, Lockridge O, Broomfield CA. Biochemistry. 1995;34:15925. doi: 10.1021/bi00049a007. [DOI] [PubMed] [Google Scholar]
- 39.Millard CB, Lockridge O, Broomfield CA. Biochemistry. 1998;37:237. doi: 10.1021/bi972057c. [DOI] [PubMed] [Google Scholar]
- 40.Saxena A, Redman AMG, Jiang X, Lockridge O, Doctor BP. Biochemistry. 1997;36:14642. doi: 10.1021/bi971425+. [DOI] [PubMed] [Google Scholar]
- 41.Saxena A, Redman AMG, Jiang X, Lockridge O, Doctor BP. Chemico-Biological Interactions. 1999;119:61. doi: 10.1016/s0009-2797(99)00014-9. [DOI] [PubMed] [Google Scholar]
- 42.Broomfield CA, Lockridge O, Millard CB. Chemico-Biological Interactions. 1999;119:413. doi: 10.1016/s0009-2797(99)00053-8. [DOI] [PubMed] [Google Scholar]
- 43.Masson P, Fortier P-L, Albaret C, Clery C, Guerra P, Lockridge O. Chemico-Biological Interactions. 1999;119:17. doi: 10.1016/s0009-2797(99)00010-1. [DOI] [PubMed] [Google Scholar]
- 44.Kovarik Z, Radic Z, Grgas B, Skrinjaric-Spoljar M, Reiner E, Simeon-Rudolf V. Biochim. Biophys. Acta. 1999;1433:261. doi: 10.1016/s0167-4838(99)00124-7. [DOI] [PubMed] [Google Scholar]
- 45.Pearlman DA, Case DA, Caldwell JW, Ross WS, Cheatham TE, III, Ferguson DM, Seibel GL, Singh UC, Weiner PK, Kollman PA. AMBER 4.1. University of California; San Francisco: 1995. [Google Scholar]
- 46.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Jr., Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. J. Am. Chem. Soc. 1995;117:5179. [Google Scholar]
- 47.Zhan C-G, de Souza O. Norberto, Rittenhouse R, Ornstein RL. J. Am. Chem. Soc. 1999;121:7279. [Google Scholar]
- 48 (a).Koca J, Zhan C-G, Rittenhouse R, Ornstein RL. J. Am. Chem. Soc. 2001;123:817. doi: 10.1021/ja000439r. [DOI] [PubMed] [Google Scholar]; (b) Koca J, Zhan C-G, Rittenhouse R, Ornstein RL. J. Comp. Chem. Soc. doi: 10.1021/ja000439r. in press. [DOI] [PubMed] [Google Scholar]
- 49.Essmann U, Perera L, Berkowitz ML, Darden TA, Lee H, Pedersen LG. J. Chem. Phys. 1995;98:10089. [Google Scholar]
- 50.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. J. Chem. Phys. 1983;79:926. [Google Scholar]
- 51 (a).Berendsen HC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. J. Comp. Phys. 1984;81:3684. [Google Scholar]; (b) Ryckaert JP, Ciccotti G, Berendsen HC. J. Comp. Phys. 1977;23:327. [Google Scholar]
- 52 (a).Becke AD. J. Chem. Phys. 1993;98:5648. [Google Scholar]; (b) Lee C, Yang W, Parr RG. Phys. Rev. B. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]; (c) Stephens PJ, Devlin FJ, Chabalowski CF, Frisch MJ. J. Phys. Chem. 1994;98:11623. [Google Scholar]
- 53.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Zakrzewski VG, Montgomery JA, Stratmann RE, Burant JC, Dapprich S, Millam JM, Daniels AD, Kudin KN, Strain MC, Farkas O, Tomasi J, Barone V, Cossi M, Cammi R, Mennucci B, Pomelli C, Adamo C, Clifford S, Ochterski J, Petersson GA, Ayala PY, Cui Q, Morokuma K, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Cioslowski J, Ortiz JV, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Gomperts R, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Gonzalez C, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Andres JL, Gonzalez AC, Head-Gordon M, Replogle ES, Pople JA. Gaussian 98. Gaussian, Inc.; Pittsburgh PA: 1998. Revision A.6. [Google Scholar]
- 54.Hehre WJ, Radom L, Schleyer P. v. R., Pople JA. Ab Initio Molecular Orbital Theory. John Wiley & Sons; New York: 1987. [Google Scholar]
- 55 (a).Gonzalez C, Schlegel HB. J. Chem. Phys. 1989;90:2154. [Google Scholar]; (b) Gonzalez C, Schlegel HB. J. Phys. Chem. 1990;94:5523. [Google Scholar]
- 56 (a).Zhan C-G, Iwata S. J. Chem. Phys. 1996;104:9058. [Google Scholar]; (b) Zhan C-G, Iwata S. J. Phys. Chem. A. 1997;101:591. [Google Scholar]; (c) Zhan C-G, Iwata S. J. Chem. Phys. 1997;107:7323. [Google Scholar]
- 57.Zhan C-G, Dixon DA, Sabri MI, Kim M-S, Spencer PS. J. Am. Chem. Soc. 2002;124:2744. doi: 10.1021/ja0113394. [DOI] [PubMed] [Google Scholar]
- 58.Bruice TC, Lightstone FC. Acc. Chem. Res. 1999;32:127. [Google Scholar]
- 59.Shurki A, Štrajbl M, Villà J, Warshel A. J. Am. Chem. Soc. 2002;124:4097. doi: 10.1021/ja012230z. [DOI] [PubMed] [Google Scholar]
- 60.Li P, Zhao K, Deng S, Landry DW. Helvetica Chim. Acta. 1999;82:85. [Google Scholar]
- 61.Zhan C-G, Landry DW, Ornstein RL. J. Am. Chem. Soc. 2000;122:1522. [Google Scholar]
- 62.Zhan C-G, Landry DW, Ornstein RL. J. Am. Chem. Soc. 2000;122:2621. [Google Scholar]
- 63 (a).Zhan C-G, Landry DW. J. Phys. Chem. A. 2001;105:1296. (b) The energy barrier, 7.6 kcal/mol for the rate-determining step, given in ref.63a was determined by using the calculated total energies plus zero-point vibration energy corrections. Determination of the Gibbs free energy barrier requires the thermal corrections to the Gibbs free energies for all relevant species (cocaine, hydroxide ion, and the first transition state), instead of the zero-point vibration energy corrections. The required thermal corrections to the Gibbs free energies were not presented in ref.63a and we use these results in this paper for the first time. The value after the thermal correction to Gibbs free energy in the Gaussian98 output actually includes the zero-point vibration energy and all thermal corrections (through corrections to both enthalpy and entropy) to Gibbs free energy. Hence, the Gibbs free energy in the gas phase was evaluated to be the calculated total energy plus the value after the thermal correction to Gibbs free energy. The free energy in solution was determined to be the gas phase free energy plus the solvent shift obtained from the SVPE or FPCM solvation calculation (see refs.63a and 77 for the solvation method used). So, we obtained the Gibbs free energy barrier of 19.5 kcal/mol for the rate-determining step of the hydroxide ion-catalyzed hydrolysis of cocaine at the benzoyl ester.
- 64.Zhan C-G, Landry DW, Ornstein RL. J. Phys. Chem. A. 2000;104:7672. [Google Scholar]
- 65.Hu C-H, Brinck T, Hult K. Int. J. Quantum Chem. 1998;69:89. [Google Scholar]
- 66.Wlodek ST, Clark TW, Scott L, McCammon JA. J. Am. Chem. Soc. 1997;119:9513. [Google Scholar]
- 67.Wlodek ST, Antosiewicz J, Briggs JM. J. Am. Chem. Soc. 1997;119:8159. [Google Scholar]
- 68.Zhou H-X, Wlodek ST, McCammon JA. Proc. Natl. Acad. Sci. USA. 1998;95:9280. doi: 10.1073/pnas.95.16.9280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Malany S, Sawai M, Sikorski RS, Seravalli J, Quinn DM, Radic Z, Taylor P, Kronman C, Velan B, Shafferman A. J. Am. Chem. Soc. 2000;122:2981. [Google Scholar]
- 70 (a).Tantillo DJ, Chen J, Houk KN. Curr. Op. Chem. Biol. 1998;2:743. doi: 10.1016/s1367-5931(98)80112-9. [DOI] [PubMed] [Google Scholar]; (b) Tantillo DJ, Houk KN. In: Stimulating Concepts in Chemistry. Vogtle F, Stoddart JF, Shibasaki M, editors. Wiley-VCH; Weinheim, Germany: 1998. p. 79. [Google Scholar]
- 71.Deng S-X, Zhan C-G, Shields GC, Landry DW. (unpublished results)
- 72.Zheng F, Zhan C-G, Ornstein RL. J. Chem. Soc. Perkin Trans. 2001;2:2355. [Google Scholar]
- 73.Štrajbl M, Florián J, Warshel A. J. Am. Chem. Soc. 2000;122:5354. [Google Scholar]
- 74.Daggett V, Schröder S, Kollman P. J. Am. Chem. Soc. 1991;113:8926. [Google Scholar]
- 75.Rylander PN, Tarbell DS. J. Am. Chem. Soc. 1950;72:3021. [Google Scholar]
- 76.Fairclough RA, Hinshelwood CN. J. Chem. Soc. 1937:538. [Google Scholar]
- 77 (a).Zhan C-G, Bentley J, Chipman DM. J. Chem. Phys. 1998;108:177. [Google Scholar]; (b) Zhan C-G, Chipman DM. J. Chem. Phys. 1998;109:10543. [Google Scholar]; (c) Zhan C-G, Chipman DM. J. Chem. Phys. 1999;110:1611. [Google Scholar]; (d) Zhan C-G, Landry DW, Ornstein RL. J. Phys. Chem. A. 2000;104:7672. [Google Scholar]; (e) Zhan C-G, Dixon DA. J. Phys. Chem. A. 2001;105:11534. [Google Scholar]; (f) Zhan C-G, Dixon DA. J. Phys. Chem. A. 2002 in press. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.