

Abstract
Ergot fungi in the genus Claviceps and several related fungal groups in the family Clavicipitaceae produce toxic ergot alkaloids. These fungi produce a variety of ergot alkaloids, including clavines as well as lysergic acid derivatives. Ergot alkaloids are also produced by the distantly related, opportunistic human pathogen Aspergillus fumigatus. However, this fungus produces festuclavine and fumigaclavines A, B, and C, which collectively differ from clavines of clavicipitaceous fungi in saturation of the last assembled of four rings in the ergoline ring structure. The two lineages are hypothesized to share early steps of the ergot alkaloid pathway before diverging at some point after the synthesis of the tricyclic intermediate chanoclavine-I. Disruption of easA, a gene predicted to encode a flavin-dependent oxidoreductase of the old yellow enzyme class, in A. fumigatus led to accumulation of chanoclavine-I and chanoclavine-I-aldehyde. Complementation of the A. fumigatus easA mutant with a wild-type allele from the same fungus restored the wild-type profile of ergot alkaloids. These data demonstrate that the product of A. fumigatus easA is required for incorporation of chanoclavine-I-aldehyde into more-complex ergot alkaloids, presumably by reducing the double bond conjugated to the aldehyde group, thus facilitating ring closure. Augmentation of the A. fumigatus easA mutant with a homologue of easA from Claviceps purpurea resulted in accumulation of ergot alkaloids typical of clavicipitaceous fungi (agroclavine, setoclavine, and its diastereoisomer isosetoclavine). These data indicate that functional differences in the easA-encoded old yellow enzymes of A. fumigatus and C. purpurea result in divergence of their respective ergot alkaloid pathways.
Different classes of ergot alkaloids are produced by members of two distinct fungal lineages. Clavicipitaceous species, which include Claviceps spp. and Neotyphodium spp., are in the order Hypocreales and typically synthesize lysergic acid derivatives (13, 16, 18). These alkaloids have a double bond in the last assembled of four rings (D ring) of the tetracyclic ergoline ring structure. Ergot alkaloids are also produced by the distantly related opportunistic human pathogen Aspergillus fumigatus, a member of the order Eurotiales (8, 14, 16, 18). Ergot alkaloids of A. fumigatus are of the clavine class and differ from the more complex profile of Claviceps purpurea and Neotyphodium spp. One important distinction between the ergot alkaloids produced by these different fungi is the saturation of the fourth ring of the ergoline structure in A. fumigatus (Fig. 1).
FIG. 1.

Structures and relationships of relevant ergot alkaloids. (A) Chanoclavine-I is oxidized to its aldehyde form before being incorporated into festuclavine (and downstream alkaloids) in A. fumigatus or agroclavine (and downstream alkaloids) in C. purpurea. (B) Conventional ring labeling and atom numbering referred to in the text.
Several genes involved in the ergot alkaloid pathways of A. fumigatus and clavicipitaceous fungi are found clustered together in the genome of each species (3, 4, 6, 18, 23). These distantly related fungi are hypothesized to share several early pathway steps, after which the pathways diverge to yield distinct sets of ergot alkaloids (3, 13, 16). The gene dmaW, which encodes dimethylallyltryptophan (DMAT) synthase, catalyzes the prenylation of tryptophan that initiates the ergot alkaloid pathway in clavicipitaceous fungi (22, 25) and functions similarly in A. fumigatus (3, 24). The region surrounding this gene in A. fumigatus contains homologues of genes also found in Neotyphodium lolii and C. purpurea ergot alkaloid gene clusters (3, 4, 6). One of the shared genes, easA, is predicted to encode a member of the old yellow enzyme (OYE) family of oxidoreductases. Old yellow enzymes are flavin-containing oxidoreductases initially found in the brewer's bottom yeast Saccharomyces carlsbergensis (26). Enzymes in this family use a reduced flavin cofactor and an active-site tyrosine residue to reduce the carbon-carbon double bond in an α/β-unsaturated aldehyde or ketone (7, 10). Subsequently, the enzymes require NADPH to restore the flavin cofactor to its reduced state. OYEs catalyze multiple reactions useful for both biotechnological and pharmaceutical applications; however, physiological roles and natural substrates for many of these enzymes presently are unknown (26). On the basis of the apparent need in the ergot alkaloid pathway of A. fumigatus for reduction of a carbon-carbon double bond in the intermediate chanoclavine-I-aldehyde, we hypothesized that the OYE-encoding gene easA is required for ergot alkaloid biosynthesis (3, 16). In this study, easA in A. fumigatus was disrupted and complemented to ascertain the role of its gene product in ergot alkaloid biosynthesis.
MATERIALS AND METHODS
Fungi and culture conditions.
Aspergillus fumigatus isolate FGSC A1141 (14) and its mutant derivatives were cultured on potato dextrose agar (PDA; 20 g dehydrated potato, 20 g glucose, and 15 g agar per liter). Liquid cultures used for preparation of protoplasts or DNA were grown at 37°C on an orbital shaker (150 rpm) overnight in a 250-ml Erlenmeyer flask containing 100 ml of malt extract medium (50 g malt extract, 50 g lactose, 5 g asparagine, and 0.4 g K2HPO4 per liter).
Disruption, complementation, and augmentation of easA.
A 595-bp fragment internal to the 1,131-bp coding sequence of A. fumigatus easA (GenBank accession number XM_751040), but lacking coding sequences on both ends, was amplified by PCR, primed with oligonucleotides oyeF (5′-ACCGAAGCAACAGACATCACC-3′) and oyeR (5′-ATATGCGACATCCAATCGCC-3′). The 50-μl PCR mixture contained 50 mM KCl, 10 mM Tris-HCl (pH 9.0), 0.1% (vol/vol) Triton X-100, 1.5 mM MgCl2, 200 μM each deoxyribonucleotide triphosphate (dNTP), 1 μM each primer, and 2.5 units of Taq DNA polymerase (Promega, Madison, WI), which was added once the thermocycler reached 95°C in the initial denaturing period. The reaction began with an initial denaturing step of 2 min 30 s at 95°C, followed by 35 cycles of 1 min at 94°C, 1 min at 55°C, and 1 min at 72°C, with a final extension step at 72°C for 7 min. A 4.5-kb disruption construct, pAFOYE1, was generated by ligating the PCR product into the T/A overlap vector pCR2.1 (Invitrogen, Carlsbad, CA). Prior to transformation into the fungus, pAFOYE1 was linearized by a restriction digest at the unique SmaI site within the easA region of the construct. Protoplast preparation and transformation were performed as described by Murray et al. (11), with modifications previously noted by Coyle and Panaccione (3). Protoplasts were separated from undigested hyphae by means of filtration through cheesecloth instead of step gradient centrifugation. Protoplasts in the filtrate were then pelleted by centrifugation (5 min at 3,400 × g) and washed twice with 0.7 M NaCl and once in STC (11) prior to a final resuspension in STC. Linearized disruption construct pAFOYE1 (Fig. 2 A) and a hygromycin resistance-encoding plasmid, pMOcosX (12), which had been linearized at a unique NotI site, were cotransformed into the protoplasts. Transformant colonies were purified to a homokaryotic state by culturing from single spores.
FIG. 2.

easA disruption strategy. (A) The disruption construct pAFOYE1 contains an incomplete, internal fragment of easA coding sequences (amplified from primers oyeF and oyeR) and was linearized at its sole SmaI site prior to transformation. The lower portion of this diagram shows tandem integration of three constructs in an easA disruptant (ΔeasA). The primer annealing sites are as follows: UF, universal (forward) primer sequences in the vector; 3S, primer flanking the 3′ border of the integration site; UR, primer derived from reverse primer sequences in the vector; 5S, primer flanking the 5′ border of the integration site. wt, wild type. (B) Primers 5S and 3S primed amplification of a 1.0-kb fragment only in the wild-type isolate. Primers UF and 3S primed amplification of the indicated 1.0-kb product from ΔeasA disruptant 8 (ΔeasA 8) and an easA complemented strain (easA ct). Primers UR and 5S primed amplification of the expected 0.8-kb band from strains with a disrupted easA allele (ΔeasA disruptant 8) and the ectopically complemented strain but not from the wild type. Relative mobilities of relevant fragments (sizes in kb) of BstEII-digested bacteriophage lambda are indicated. (C) Integration of transforming DNA at only the targeted site, as confirmed by Southern blot hybridization. DNA was digested with SalI and hybridized with a digoxigenin-dUTP-labeled easA probe. Relative mobilities of relevant fragments (sizes in kb) of BstEII-digested bacteriophage lambda are indicated. ΔeasA 9, ΔeasA disruptant 9.
Colonies growing on transformation plates were screened in three different PCR assays to identify strains in which the disruption construct had been inserted within the native easA gene. With the exception of different primers and annealing temperatures, the PCRs were conducted as described above. The 5′ border of the homologous recombination event was confirmed by PCR (annealing temperature at 57°C) primed with oligonucleotides UR (5′-AGCTATGACCATGATTACGCCA-3′), which anneals to vector sequences near the universal primer annealing site in pCR2.1, and 5S (5′-GGAAGATGTCATCTCCAACATAG-3′), which is complementary to easA sequences near the 5′ end of the gene and flanking the intended site of integration (Fig. 2A). The juxtaposition of sequences near the 3′ border of the integration event was verified by PCR (annealing temperature, 57°C) primed with oligonucleotides UF (5′-GCCAGTGAATTGTAATACGACTC-3′; complementary to vector sequences near the reverse primer annealing site of pCR2.1) and 3S (5′-AGCCTTCTCTTGATAGCGTGCT-3′), which anneals to easA sequences near the 3′ end of the gene and flanking the intended site of integration (Fig. 2A). PCR across the entire easA locus was conducted by priming with oligonucleotides 5S and 3S (both described above) with an annealing temperature of 55°C.
Integration of pAFOYE1 into the native easA gene was also analyzed by Southern hybridization according to the method described previously (3). Genomic DNA was digested by SalI prior to Southern blot analysis. An easA-specific probe was generated by amplifying wild-type A. fumigatus genomic DNA in a reaction primed with oyeF and oyeR (described above) according to the PCR conditions described above, but with substitution of 1× digoxigenin (DIG) DNA labeling mix (Roche, Indianapolis, IN) for unlabeled dNTPs.
A strain in which the native easA gene was disrupted (a ΔeasA mutant termed ΔeasA disruptant 8) was complemented by ectopic insertion of a functional copy of A. fumigatus easA into the mutant genome. A 3.3-kb fragment containing the entire coding sequence along with approximately 1.2 kb of 5′-flanking sequences and 1.0 kb of 3′-flanking sequences was amplified by PCR primed with WeasAF (5′-AGAATCGAGGTGAGCGTGTAGAATGC-3′) and WeasAR (5′-AAGCTGGAACACTACTGATCGAGTGC-3′). The PCR conditions were as described above, except the annealing temperature was 58°C and the extension time at 72°C was set at 3 min 30 s.
In a parallel experiment, the ΔeasA mutant was augmented with the easA homologue from C. purpurea strain ATCC 20102 (termed the ΔeasA easACp mutant). Amplification of this gene was primed with CpoyeF (5′-TGAGTTCGAACGATGATCTGTAGCG-3′) and CpoyeR (5′-TGAGTTCGAAAGAGTCTTCTACGCC-3′), with the PCR conditions as described above, except for an annealing temperature of 54°C and an extension time of 2 min 30 s. The resulting 1,985-bp PCR product contained the 1,143-bp coding sequence of C. purpurea easA plus 531 bp of 5′-flanking sequences and 311 bp of 3′-flanking sequences.
In both complementation and augmentation experiments, the PCR product was cotransformed into an easA-disrupted mutant of A. fumigatus along with a phleomycin resistance plasmid, pBC-phleo (Fungal Genetics Stock Center, University of Missouri—Kansas City, Kansas City, MO), linearized at a unique NotI site. Transformants were selected on complete regeneration medium (15) containing 100 μg/ml phleomycin and incubated at 37°C. Phleomycin-resistant transformants were cultured from individual germinated conidia in order to generate homokaryotic cultures. To test for the presence of the original disruption construct, PCRs were conducted as described above, primed with 5S and UR (for the 5′ flank of the integration) and UF and 3S (for the 3′ flank). An additional PCR screen was performed with either primers WeasAF and WeasAR (described above) or primers CpoyeF and CpoyeR (also described above) to test for the presence of the introduced wild-type A. fumigatus or C. purpurea allele, respectively, in the complemented or augmented transformants.
Analysis of ergot alkaloids.
To analyze ergot alkaloids qualitatively, small cubes (containing approximately 8 mm by 8 mm of colony surface area) were removed from sporulating colonies that had been grown on PDA at 37°C for 5 or more days. Samples were submerged in 0.4 ml of 80% methanol, vortexed briefly, rotated end-over-end (44 rpm) for 2 h, and centrifuged at 14,500 rpm (10 min) to pellet fungus and agar. Supernatants were analyzed by high-performance liquid chromatography (HPLC) over a C18 column (Prodigy 5-μm ODS3 [150 mm by 4.6 mm]; Phenomenex, Torrance, CA) and subjected to a multilinear binary gradient from 5% (vol/vol) acetonitrile plus 95% (vol/vol) aqueous 50 mM ammonium acetate to 75% acetonitrile plus 25% aqueous 50 mM ammonium acetate at a flow rate 1 ml/min, as described previously (14, 17). Ergot alkaloids were detected in two serially arranged fluorescence detectors set at excitation and emission wavelengths of 272 nm/372 nm (for ergot alkaloids without a double bond conjugated to the indole ring system) and 310 nm/410 nm (for setoclavine and isosetoclavine, which fluoresce more intensely under these conditions due to the 9,10 double bond that they possess).
The identities of peaks corresponding to festuclavine and fumigaclavines A, B, and C were previously established by mass spectral analyses of native and deesterified fractions (14). These peak identities were also confirmed by their elimination in extracts from a dmaW-disrupted strain in which the first gene of the ergot alkaloid pathway was inactivated (3). Chanoclavine-I, agroclavine, and the diastereoisomeric pair comprising setoclavine and isosetoclavine were identified by coelution with standards and by electrospray ionization liquid chromatography-mass spectrometry (ESI LC-MS). Chanoclavine-I standard was provided by B. A. Tapper (AgResearch, Palmerston North, New Zealand), agroclavine standard was purchased from Sigma (St. Louis, MO), and setoclavine/isosetoclavine standard was prepared by oxidizing agroclavine with horseradish peroxidase and H2O2 or with peroxidase-rich plant extract (in this case endophyte-free perennial ryegrass extract), as previously described (17, 19, 20, 21). Analytes that coeluted with standards were collected, and their masses were measured by ESI LC-MS to confirm their identities. Chanoclavine-I-aldehyde was identified based on ESI LC-MS analyses and was further characterized by 1H nuclear magnetic resonance (NMR) analyses (2) in which spectra that matched those previously published for chanoclavine-I-aldehyde were provided (5).
Extracts for ESI LC-MS analyses were prepared from cultures inoculated on the surface of liquid potato dextrose medium (20 g dehydrated potato and 20 g glucose per liter; 50 ml in a 250-ml Erlenmeyer flask) and grown at 37°C with no agitation for 1 to 2 weeks. The resulting mat of conidiating culture was removed from the liquid medium and blotted dry on filter paper (nonsporulating side down). Ergot alkaloids were extracted from the dried culture by agitation in 4 ml of 100% methanol. The methanol and dislodged conidia and hyphal fragments were transferred to microcentrifuge tubes, vortexed, and rotated end-over-end for a 2-h period. Then, the sample was centrifuged (10 min at 14,000 × g) to pellet fungal material. Methanol extracts and individual peaks collected from HPLC analysis (performed as described above) were concentrated under a vacuum. LC-MS samples were separated on an Acquity Ultra Performance BEH C18 column with a 1.7-μm particle size and 2.1- by 100-mm dimensions and were eluted with a linear gradient (0.5 ml/min) of 20 parts acetonitrile plus 80 parts 0.1% formic acid in water, initially, to 80 parts acetonitrile plus 20 parts 0.1% formic acid in water at 5 min. Masses of analytes were detected by a Micromass LCT Premier time-of-flight mass spectrometer (TOF MS; Waters, Milford, MA) with an electrospray ionization source set in positive mode.
To quantify ergot alkaloids from five cultures each of the wild type, an easA disruptant (ΔeasA disruptant 8), and the ΔeasA easACp augmented strain, cultures were grown on PDA for 5 days at 37°C. Samples containing 50.3 mm2 of colony surface area were excised from cultures with the broader end of a disposable 1-ml pipette tip (catalog number 02-681-172; Fisher Scientific, Pittsburgh, PA), submerged in 1 ml 80% methanol, agitated by vortexing, and rotated end-over-end at 44 rpm for 2 h. Aliquots were removed for HPLC analyses conducted as described above. All samples from each of these strains were analyzed quantitatively for chanoclavine-I, for chanoclavine-I-aldehyde, for festuclavine, for fumigaclavines A, B, and C, for agroclavine, and for setoclavine/isosetoclavine; samples in which a particular ergot alkaloid was not found were reported as below the detection limit. Quantities of setoclavine and isosetoclavine (fluorescing maximally at 310 nm/410 nm) were based on comparison of peak areas relative to those of the external standards of ergotamine (Sigma, St. Louis, MO). Quantities of all other measured ergot alkaloids (fluorescing more intensely at 272 nm/372 nm) were based on comparison of peak areas relative to those of the external standards of agroclavine, as previously described (14).
RESULTS
Disruption and complementation of easA.
Integration of the disruption construct pAFOYE1 into the native easA gene of A. fumigatus via homologous recombination occurred in 2 out of the 64 hygromycin-resistant transformants. Insertion of pAFOYE1 within easA was verified by PCR, with screens of the 5′ and 3′ borders of the integration site resulting in the predicted 811-bp and 992-bp fragments, respectively (Fig. 2B). In the mutants, no 1-kb amplification product was detected when PCR was primed with oligonucleotides with binding sites separated from each other by 1 kb in the wild type due to separation of the primer annealing sites resulting from the insertion of multiple disruption constructs within the easA locus (Fig. 2B). Southern blot analysis showed integration of multiple copies of pAFOYE1 within easA by the shift in mobility of the easA-containing fragment (Fig. 2C).
The easA disruption mutants did not produce festuclavine or fumigaclavines A, B, and C (Table 1 and Fig. 3), which are four ergot alkaloids typically detectable in A. fumigatus (13, 14). However, an important intermediate in the ergot alkaloid pathway, chanoclavine-I, which eluted from the HPLC column at 32.0 min, was abundant in the mutants' chemical profiles (Table 1 and Fig. 3). The analyte coeluted with chanoclavine-I standard in the fluorescence HPLC and LC-MS systems and showed an m/z of 257.11 when analyzed by ESI LC-MS, consistent with the [M+H]+ of chanoclavine-I. In addition to the increased accumulation of chanoclavine-I, a novel peak with a retention time of 37.7 min was observed in HPLC traces of easA disruptants. In ESI LC-MS analyses this analyte had an m/z of 255.15, which matches the [M+H]+ of chanoclavine-I-aldehyde. Further analysis of this analyte by 1H NMR (2) provided spectra that matched those previously published for chanoclavine-I-aldehyde (5). Lesser quantities of peaks that coeluted with agroclavine, setoclavine, and isosetoclavine were also observed in mutant extracts (Fig. 3 and Table 1).
TABLE 1.
Ergot alkaloid accumulation in strains of Aspergillus fumigatus with modified easA compared to the wild-type level
| Strain | Ergot alkaloid concn (nmol/cm2 culture surface area) (mean ± SE) |
|||||||
|---|---|---|---|---|---|---|---|---|
| Chanoclavine | Chanoclavine aldehyde | Festuclavine | Fumigaclavine B | Fumigaclavine A | Fumigaclavine C | Agroclavine | Setoclavine-isosetoclavine | |
| Wild type | TRa | <DLb | 2,778 ± 173 | 31 ± 9 | 359 ± 16 | 1,519 ± 89 | <DL | <DL |
| ΔeasA disruptant 8 | 299 ± 6 | 52 ± 6 | <DL | <DL | <DL | <DL | 24 ± 5 | 25 ± 3 |
| ΔeasA easACp mutant | TR | TR | <DL | <DL | <DL | <DL | 1,058 ± 77 | 1,267 ± 155 |
TR, trace: the peak was detected in some but not all samples of this strain (mean, <3 nmol/cm2).
<DL, the value was below the detection limit of 0.6 nmol/cm2; this alkaloid could not be detected in any sample of this strain.
FIG. 3.

Qualitative fluorescence HPLC analyses of wild-type A. fumigatus (wt), an easA disrupted strain (ΔeasA disruptant 8 [ΔeasA 8]), and ΔeasA disruptant 8 augmented with a functional copy of C. purpurea easA (ΔeasA easACp mutant). B, fumigaclavine B; Ch, chanoclavine-I; Ald, chanoclavine-I-aldehyde; S/I, setoclavine or isosetoclavine; F, festuclavine; A, fumigaclavine A; Ag, agroclavine; and C, fumigaclavine C. The relative sequence of elution of the diastereoisomers setoclavine and isosetoclavine has not been determined in our chromatographic system; thus, the two peaks resulting from oxidation of agroclavine and having spectral properties of setoclavine and isosetoclavine are both labeled with S/I in the figure. Excitation and emission wavelengths are provided in the upper left corner of each chromatogram.
Cotransformation of a copy of A. fumigatus easA into an easA mutant restored a normal ergot alkaloid profile (not shown). PCR analysis of the complemented strain showed that the wild-type allele had been inserted ectopically in the genome, since the original disruption construct pAFOYE1 was still present in the native easA locus (Fig. 2B).
Augmentation of a ΔeasA mutant of A. fumigatus with an easA homologue from C. purpurea.
Ectopic insertion of the easA homologue from C. purpurea into an A. fumigatus easA disruptant resulted in a distinct modification of the ergot alkaloid profile (Fig. 3 and Table 1). Chanoclavine-I and chanoclavine-I-aldehyde, typically present in the ΔeasA mutant background, were greatly reduced in concentration in transformants augmented with C. purpurea easA. Peaks corresponding to the ergot alkaloids that typically accumulate in A. fumigatus (festuclavine and fumigaclavines A, B, and C) were not detectable in the augmented strain (Fig. 3 and Table 1). Instead, a relatively large peak that coeluted with the agroclavine standard was evident when eluates were monitored with excitation and emission wavelengths of 272 nm and 372 nm, respectively (Fig. 3). This analyte was isolated and produced an ion with a 239.12 m/z in ESI LC-MS analyses, consistent with the [M+H]+ for agroclavine. Two additional peaks were evident in extracts of the ΔeasA easACp mutant strains. These peaks fluoresced more strongly when monitored at 310 nm (excitation) and 410 nm (emission) than they did when monitored at 272 nm (excitation) and 372 nm (emission), typical of ergot alkaloids with a double bond conjugated to the indole ring. The analytes coeluted with the standard for the diastereoisomeric pair setoclavine/isosetoclavine prepared by oxidation of agroclavine (Fig. 3). Both peaks (39.5 min and 42.3 min) were isolated and yielded ions with m/z values of 255.13 and 255.14, respectively, in ESI LC-MS analyses. These values are consistent with the [M+H]+ values for setoclavine and isosetoclavine. PCR analyses of a C. purpurea easA-augmented strain showed that it was positive for the presence of the original easA disruption construct pAFOYE1 and for the introduced allele of easA originating from C. purpurea (Fig. 4).
FIG. 4.
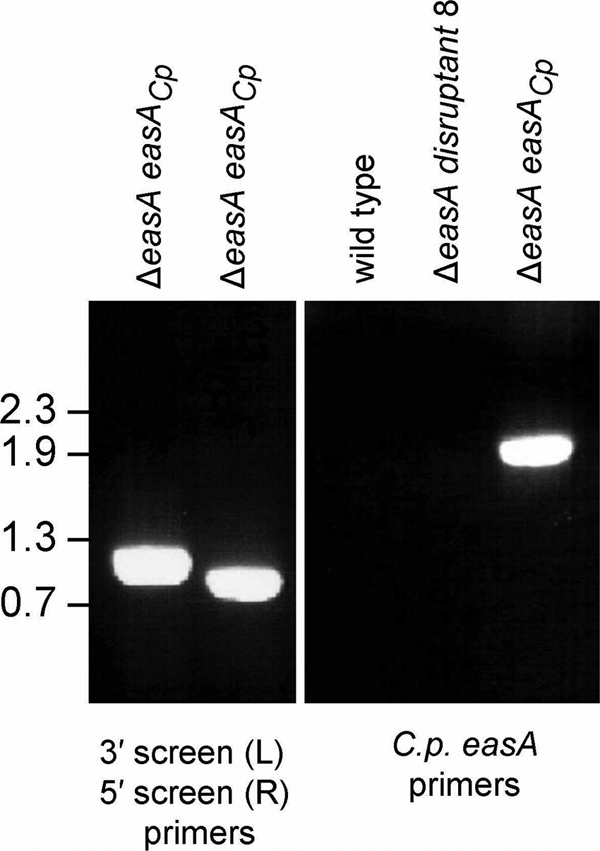
PCR characterization of A. fumigatus ΔeasA disruptant 8 augmented with C. purpurea easA (ΔeasA easACp mutant). In the left panel, the presence of the A. fumigatus easA disruption construct in the mutant background was detected by PCR with primers flanking the 3′ end of the integration site (3S and UR; left lane) and primers flanking the 5′ end of the integration site (UF and 5S; right lane). Primer annealing positions are shown in Fig. 2. In the right panel, the presence of an introduced copy of C. purpurea easA in the augmented strain but not in the wild type or the ΔeasA disruptant 8 recipient was shown by PCR with the primer set comprising CpoyeF and CpoyeR. The relative mobilities of relevant fragments of BstEII-digested bacteriophage lambda DNA (sizes in kb) are indicated to the left of the photographs.
DISCUSSION
Disruption and complementation analyses of easA demonstrated that this gene is required for ergot alkaloid biosynthesis in A. fumigatus. Disruption of easA in A. fumigatus halted the pathway at chanoclavine-I-aldehyde and prevented the biosynthesis of festuclavine and downstream fumigaclavines B, A, and C. In addition to chanoclavine-I-aldehyde, ΔeasA mutants also accumulated chanoclavine-I, the immediate precursor to chanoclavine-I-aldehyde and the first accumulating intermediate typically observed in ergot alkaloid producers. A spontaneous mutant of C. purpurea with the same chemical profile has been characterized chemically (9) but to our knowledge has not been characterized genetically. Complementation of an A. fumigatus ΔeasA mutant with the wild-type allele of easA from A. fumigatus restored the pathway to festuclavine and downstream alkaloids. In contrast, augmentation of the same ΔeasA mutant with the easA allele obtained from C. purpurea resulted in the accumulation of agroclavine and its oxidation products setoclavine/isosetoclavine. These data indicate that chanoclavine-I-aldehyde is the last shared intermediate in the ergot pathways of these two fungal lineages and that functional differences in the products of the easA alleles from these different fungi determine whether agroclavine (and its derivatives) or festuclavine (and its derivatives) are produced.
The enzyme encoded by easA (here referred to as EasA) has sequence similarity to members of the old yellow enzyme class of oxidoreductases (3). Enzymes in this class reduce double bonds conjugated to aldehydes or ketones (26). Reduction of the double bond conjugated to the aldehyde group of chanoclavine-I-aldehyde would allow for free rotation of that aldehyde functional group to bring it into close proximity to the secondary amine, facilitating ring closure via Schiff base formation. This mechanism for closure of the D-ring has been proposed previously (5, 16, 18). More details of the enzymology and reaction product of EasA in vitro have been provided by Cheng et al. (2).
The chemical profile of the easA disruptants contained small quantities of agroclavine and its diastereoisomeric oxidation products setoclavine/isosetoclavine. We hypothesize that chanoclavine-I-aldehyde, which accumulates due to the disruption of easA, tautomerizes to its enol form, thus allowing free rotation around carbon 8, bringing carbon 17 (the aldehyde or enol carbon) into close proximity to the secondary amine at position 6. Tautomerization back to the favored aldehyde form would facilitate ring closure via formation of a Schiff base (Fig. 5). In this noncatalyzed ring closure, the 8,9 double bond would not have been reduced (due to the lack of an intact easA gene). Thus, upon reduction of the resulting cyclic iminium ion, agroclavine as opposed to festuclavine would be the product. Although it has not been characterized, an enzyme capable of reducing the cyclic iminium ion must be a standard component of ergot alkaloid producers. Our data, which showed the production of festuclavine or agroclavine, depending upon the status of easA, suggest that the A. fumigatus iminium reductase is able to accept as a substrate iminium ions with single or double bonds between carbons 8 and 9.
FIG. 5.

Noncatalyzed closure of ergoline D ring via keto-enol tautomerism. Heavier arrows indicate the direction favored by equilibrium. The shift in double bonds in the enol tautomer allows rotation around the alpha carbon, positioning functional groups for Schiff base formation, resulting in ring closure as an iminium ion. An additional reductase is required to reduce the iminium ion to agroclavine (18).
Agroclavine is oxidized to the diastereoisomeric pair comprising setoclavine and isosetoclavine (Fig. 1) by horseradish peroxidase (20), and the same oxidation products result from feeding agroclavine to many bacteria, fungi, or plants. The oxidation of exogenously supplied agroclavine to yield setoclavine/isosetoclavine has previously been demonstrated to occur in A. fumigatus (1). In fact, Béliveau and Ramstad (1) reported that 68 of 71 fungal cultures and 18 of 19 bacterial cultures that accumulated exogenously supplied agroclavine oxidized it into setoclavine/isosetoclavine. Many plants and plant extracts catalyze a similar oxidation (17, 19, 21). Thus, it is reasonable that much of the agroclavine left to accumulate in the mutant strains in our present study would be oxidized to setoclavine/isosetoclavine.
Augmentation of the A. fumigatus easA knockout with C. purpurea easA resulted in significant increases in agroclavine, setoclavine, and isosetoclavine (Table 1). The DNA fragment introduced contains fragments of no other C. purpurea genes. The product of C. purpurea easA presumably must temporarily reduce the 8,9 double bond of chanoclavine-I-aldehyde to allow rotation of the aldehyde group for ring closure but must then catalyze reoxidation of the 8,9 double bond. In this way, the C. purpurea easA protein is proposed to act more as an isomerase than as a reductase.
Directed mutational analyses of the A. fumigatus and C. purpurea alleles and expression of the mutant alleles in the ΔeasA background or in vitro may provide insight into the functional differences that result in the production of festuclavine (and its derivatives) versus agroclavine (and its derivatives). Such analyses are currently under way in our laboratories.
Acknowledgments
This project was supported by a National Research Initiative Competitive Grant (number 2008-35318-04549) from the USDA National Institute of Food and Agriculture.
This article is published with the approval of the Director of the West Virginia Agricultural and Forestry Experiment Station as scientific article number 3070.
We thank Christopher Schardl (University of Kentucky) for helpful discussions on potential mechanisms of EasA.
Footnotes
Published ahead of print on 30 April 2010.
REFERENCES
- 1.Béliveau, J., and E. Ramstad. 1967. 8-Hydroxylation of agroclavine and elymoclavine by fungi. Lloydia 29:234-238. [Google Scholar]
- 2.Cheng, J. Z., C. M. Coyle, D. G. Panaccione, and S. E. O'Connor. 2010. A role for old yellow enzyme in ergot alkaloid biosynthesis. J. Am. Chem. Soc. 132:1776-1777. [DOI] [PubMed] [Google Scholar]
- 3.Coyle, C. M., and D. G. Panaccione. 2005. An ergot alkaloid biosynthesis gene and clustered hypothetical genes from Aspergillus fumigatus. Appl. Environ. Microbiol. 71:3112-3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fleetwood, D. J., B. Scott, G. A. Lane, A. Tanaka, and R. D. Johnson. 2007. A complex ergovaline gene cluster in Epichloe endophytes of grasses. Appl. Environ. Microbiol. 73:2571-2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Floss, H. G., M. Tcheng-Lin, C. Chang, B. Naidoo, G. E. Blair, C. I. Abou-Chaar, and J. M. Cassady. 1974. Biosynthesis of ergot alkaloids. Studies on the mechanism of conversion of chanoclavine-I into tetracyclic ergolines. J. Am. Chem. Soc. 96:1898-1909. [DOI] [PubMed] [Google Scholar]
- 6.Haarmann, T., C. Machado, Y. Lübbe, T. Correia, C. L. Schardl, D. G. Panaccione, and P. Tudzynski. 2005. The ergot alkaloid gene cluster in Claviceps purpurea: extension of the cluster sequence and intra species evolution. Phytochemistry 66:1312-1320. [DOI] [PubMed] [Google Scholar]
- 7.Kohli, R. M., and V. Massey. 1998. The oxidative half-reaction of old yellow enzyme. The role of tyrosine 196. J. Biol. Chem. 273:32763-32770. [DOI] [PubMed] [Google Scholar]
- 8.Kozlovsky, A. G. 1999. Producers of ergot alkaloids out of Claviceps genus, p. 479-499. In V. Kren and L. Cvak (ed.), Ergot: the genus Claviceps. Harwood Academic Publishers, Amsterdam, Netherlands.
- 9.Maier, W., D. Erge, J. Schmidt, and D. Gröger. 1980. A blocked mutant of Claviceps purpurea accumulating chanoclavine-I-aldehyde. Experientia 36:1353-1354. [Google Scholar]
- 10.Meah, Y., and V. Massey. 2000. Old yellow enzyme: stepwise reduction of nitro-olefins and catalysis of aci-nitro tautomerization. Proc. Natl. Acad. Sci. U. S. A. 97:10733-10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murray, F. R., G. C. M. Latch, and D. B. Scott. 1992. Surrogate transformation of perennial ryegrass, Lolium perenne, using genetically modified Acremonium endophyte. Mol. Gen. Genet. 233:1-9. [DOI] [PubMed] [Google Scholar]
- 12.Orbach, M. J. 1994. A cosmid with a HyR marker for fungal library construction and screening. Gene 150:159-162. [DOI] [PubMed] [Google Scholar]
- 13.Panaccione, D. G. 2005. Origins and significance of ergot alkaloid diversity in fungi. FEMS Microbiol. Lett. 251:9-17. [DOI] [PubMed] [Google Scholar]
- 14.Panaccione, D. G., and C. M. Coyle. 2005. Abundant respirable ergot alkaloids from the common airborne fungus Aspergillus fumigatus. Appl. Environ. Microbiol. 71:3106-3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Panaccione, D. G., R. D. Johnson, J. Wang, C. A. Young, P. Damrongkool, B. Scott, and C. L. Schardl. 2001. Elimination of ergovaline from a grass-Neotyphodium endophyte symbiosis by genetic modification of the endophyte. Proc. Natl. Acad. Sci. U. S. A. 98:12820-12825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Panaccione, D. G., C. Schardl, and C. M. Coyle. 2006. Pathways to diverse ergot alkaloid profiles in fungi, p. 23-52. In J. T. Romeo (ed.), Recent advances in phytochemistry, vol. 40. Elsevier, Amsterdam, Netherlands. [Google Scholar]
- 17.Panaccione, D. G., B. A. Tapper, G. A. Lane, E. Davies, and K. Fraser. 2003. Biochemical outcome of blocking the ergot alkaloid pathway of a grass endophyte. J. Agric. Food Chem. 51:6429-6437. [DOI] [PubMed] [Google Scholar]
- 18.Schardl, C., D. G. Panaccione, and P. Tudzynski. 2006. Ergot alkaloids—biology and molecular biology, p. 45-86. In G. A. Cordell (ed.), The alkaloids: chemistry and biology, vol. 63. Academic Press, San Diego, CA. [DOI] [PubMed] [Google Scholar]
- 19.Scigelova, M., T. Macek, A. Minghetti, M. Mackova, P. Sedmera, V. Prikrylova, and V. Kren. 1995. Biotransformation of ergot alkaloids by plant cell cultures with high peroxidase activity. Biotechnol. Lett. 17:1213-1218. [Google Scholar]
- 20.Taylor, E. H., and H. R. Shough. 1967. Enzymology of ergot alkaloid biosynthesis. II. The oxidation of agroclavine by horseradish peroxidase. Lloydia 30:197-201. [PubMed] [Google Scholar]
- 21.Taylor, E. H., K. J. Goldner, S. F. Pong, and H. R. Shough. 1966. Conversion of Δ8,9 ergolines to Δ9,10-8-hydroxyergolines in plant homogenates. Lloydia 29:239-244. [Google Scholar]
- 22.Tsai, H.-F., H. Wang, J. C. Gebler, C. D. Poulter, and C. L. Schardl. 1995. The Claviceps purpurea gene encoding dimethylallyltryptophan synthase, the committed step for ergot alkaloid biosynthesis. Biochem. Biophys. Res. Commun. 216:119-125. [DOI] [PubMed] [Google Scholar]
- 23.Tudzynski, P., K. Holter, T. Correia, C. Arntz, N. Grammel, and U. Keller. 1999. Evidence for an ergot alkaloid gene cluster in Claviceps purpurea. Mol. Gen. Genet. 261:133-141. [DOI] [PubMed] [Google Scholar]
- 24.Unsöld, I. A., and S.-M. Li. 2005. Overproduction, purification and characterization of FgaPT2, a dimethylallyltryptophan synthase from Aspergillus fumigatus. Microbiology 151:1499-1505. [DOI] [PubMed] [Google Scholar]
- 25.Wang, J., C. Machado, D. G. Panaccione, H.-F. Tsai, and C. L. Schardl. 2004. The determinant step in ergot alkaloid biosynthesis by an endophyte of perennial ryegrass. Fungal Genet. Biol. 41:189-198. [DOI] [PubMed] [Google Scholar]
- 26.Williams, R. E., and N. C. Bruce. 2002. ‘New uses for an old enzyme’—the old yellow enzyme family of flavoenzymes. Microbiology 148:1607-1614. [DOI] [PubMed] [Google Scholar]