

Abstract
The purpose of this study was to determine the role of the pro-inflammatory cytokine IL-6 in cold-induced hypertension (CIH). Four groups of male Sprague Dawley rats were used (6 rats/group). After blood pressure (BP) was stabilized, 3 groups received intravenous delivery of AAV carrying IL-6shRNA, AAV carrying scrambled shRNA (ScrshRNA), and phosphate buffered solution (PBS), respectively, prior exposure to a cold environment (5°C). The last group received PBS and was kept at room temperature (25°C, warm) as a control. AAV delivery of IL-6shRNA significantly attenuated cold-induced elevation of systolic BP and kept it at the control level for up to 7 weeks (length of the study). Chronic exposure to cold up-regulated IL-6 expression in aorta, heart and kidneys and increased macrophage and T-cell infiltration in kidneys, suggesting that cold exposure increases inflammation. IL-6shRNA delivery abolished the cold-induced up-regulation of IL-6, indicating effective silence of IL-6. Interestingly, RNAi knockdown of IL-6 prevented cold-induced inflammation as evidenced by a complete inhibition of TNF-α expression and leukocyte infiltration by IL-6shRNA. RNAi knockdown of IL-6 significantly decreased the cold-induced increase in vascular superoxide production. It is noted that IL-6shRNA abolished the cold-induced increase in collagen deposition in the heart, suggesting that inflammation is involved in cold-induced cardiac remodeling. Cold exposure caused glomerular collapses which could be prevented by knockdown of IL-6, suggesting an important role of inflammation in cold-induced renal damage. Conclusions: Cold exposure increased IL-6 expression and inflammation which play a critical role in the pathogenesis of CIH and cardiac and renal damage.
Keywords: cold exposure, inflammation, interleukin-6, short-hairpin siRNA, RNAi, adeno-associated virus
Introduction
It is well documented that cold temperatures have adverse effects on the human cardiovascular system.1 The prevalence of hypertension and related cardiovascular disease is higher in people who live in colder climates. The mortality and morbidity of cardiovascular diseases peak during the winter months in the United States.2-4 In addition, cold temperatures exacerbate hypertension in hypertensive patients.5-7 Therefore it is important to fully understand the mechanism mediating cold-induced elevation of blood pressure (BP).
Cold-induced hypertension (CIH) represents an excellent model for studying environmentally induced hypertension. CIH is a “naturally occurring” form of experimental hypertension that requires no genetic manipulation, surgical intervention or excessive drug or hormone administration.1, 8-10 Previous studies have provided a central role for the sympathetic nervous system (SNS) in initiating CIH via activation of the renin-angiotensin system (RAS).1, 11-12 Activation of the RAS results in increased levels of angiotensin II (AngII) and aldosterone. Besides its potent vasoactive effects, AngII has been demonstrated to influence many inflammatory processes.13-14 Excessive aldosterone levels have also been shown to increase inflammation.15
Located in the short arm of chromosome 7 (7p21) in humans, IL-6 is a pleotropic cytokine with multiple biological roles in many different types of cell.16 The functions of IL-6 include induction of both local and systemic inflammatory responses, regulation of immune reaction and hematopoiesis.17 In addition, IL-6 also induces proliferation and differentiation of T cells and terminal differentiation of autoantibody-producing B cells.17 An overproduction of IL-6 thus exacerbates the immune reaction. Also, circulating IL-6 has been linked to central obesity, hypertension and insulin resistance.16 In human males, the pro-inflammatory cytokine interleukin (IL)-6 has been shown to be associated with elevated BP.18 Several reports have shown increased plasma levels of IL-6 in hypertensive patients.19-21 In addition, infusion of IL-6 into pregnant female rats induces pre-eclampsia (pregnancy-induced hypertension).22 Furthermore, angiotensin receptor blockers (ARBs) have been reported to reduce inflammatory mediator levels in hypertensive patients, specifically IL-6 and tumor necrosis factor-α (TNF-α) levels.15
Although the association of IL-6 and hypertension has been reported, the cause and effect relationship is not clear. The purpose of this study was to examine the role of IL-6 in the development of hypertension using the CIH model. We hypothesized that in vivo knockdown of IL-6 by RNAi silencing would decrease inflammatory infiltrates and attenuate cold-induced elevation of BP.
Methods
For a full description of the Materials and Methods, please see http://hyper.ahajournals.org”.
AAV.IL-6 Generation
The procedure for constructing the recombinant adeno-associated virus (AAV)-2 carrying the rat IL-6shRNA sequence was described in detail in http://hyper.ahajournals.org.
Additional AAV generation
AAV carrying Scrambled shRNA (AAV.ScrshRNA) was constructed and used as a control construct. Scrambled shRNA has been confirmed by BD Biosciences (Palo Alto, CA. USA) not to match any known gene sequence.
Packaging of recombinant plasmids of AAV with IL-6-shRNA
AAV.IL-6 and AAV.ScrshRNA were packaged with pHelper and pAAV-Helper to produce recombinant viruses as described in our recent studies.23-24 For the virus packaging procedure, please see http://hyper.ahajournals.org.
Animal Study protocols
This study was carried out according to the guidelines of the National Institutes of Health on the Care and Use of Laboratory Animals. This project was approved by the Institutional Animal Care and Use Committee (IACUC). Four groups of male Sprague–Dawley rats (145–180g, 6 rats/group) were allowed to acclimate for a week. After acclimation, resting systolic BP was measured twice weekly at room temperature from the tail of each unanesthetized rat using by tail-cuff method with slight warming (28°C) but not heating of the tail using a CODA 6 BP Monitoring System (Kent Scientific, Torrington, CT USA). The volume-based tail-cuff measurements of BP have been validated by using a telemetry system25. After two stable BP readings were obtained, the four groups of rats received a single injection via tail vein of AAV.IL-6shRNA, AAV.ScrshRNA, PBS and PBS, respectively. The AAV complexes were delivered at 1.2 × 108 PFU/rat (0.5 ml). After injection, three groups (AAV.IL-6shRNA, AAV.ScrshRNA and one PBS) were moved into to a climate-controlled walk-in chamber (5 ± 2°C), whereas the remaining PBS group was kept in an identical chamber maintained at RT (25 ± 2°C, warm). BP and body weight (BW) were measured at least once a week until week 8 post-injection, when all animals were euthanized. For detailed procedures, please see http://hyper.ahajournals.org.
Western blot analysis of IL-6 and TNF-α protein expression in tissue
IL-6 and TNF-α protein expressions in kidneys and arteries were measured using Western blot as described in our previous studies.23-24 For details, please see http://hyper.ahajournals.org.
Immunohistochemical (IHC) analysis of IL-6 expression and macrophage and T cell infiltration
IL-6 expression and macrophage and T cell infiltration were analyzed using IL-6 antibody and CD-3 and CD-68 markers, respectively. For details, please see http://hyper.ahajournals.org.
Measurement of in situ vascular superoxide production
The in situ superoxide production was measured in aortas using the oxidation-sensitive dye dihydroethidium (DHE). For details, please see http://hyper.ahajournals.org.
Statistical analysis
The data for BP and BW were analyzed by a repeated measures one-way analysis of variance (ANOVA). The protein expression and ROS levels were analyzed by one-way ANOVA. Tukey’s multiple comparison tests were used to assess the significance of differences between means. Significance was set at a 95% confidence limit.
Results
IL-6shRNA delivery attenuated the cold-induced BP increase
IL-6shRNA significantly decreased cold-induced elevation of systolic BP compared to the PBS Cold and ScrshRNA Cold groups (Fig. 1). RNAi knockdown of IL-6 maintained BPs at the level of the PBS Warm group (control). It is noted that one single dose of AAV.IL-6shRNA controlled hypertension for at least 7 weeks (length of the study) (Fig. 1). Body weight was not significantly different between groups both before and after the single injection of viral complexes (Supplemental Figure, S2), suggesting that AAV-2 had no adverse effects on body weight gains.
Figure 1. IL-6shRNA delivery attenuated cold-induced elevation of blood pressure.
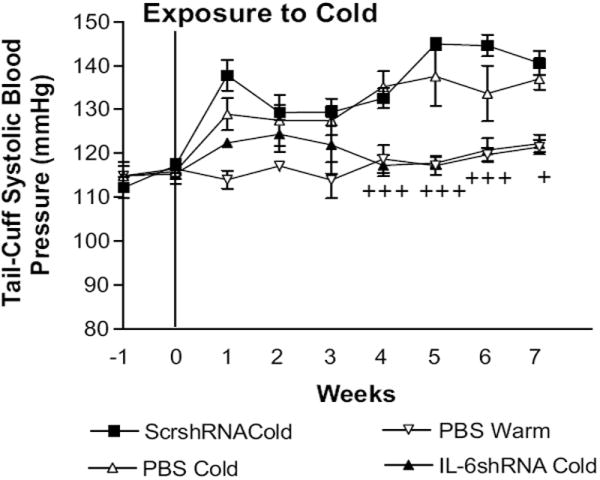
Intravenous injections of AAV.ScrshRNA, AAV.IL-6shRNA and PBS were administered prior exposure to cold. Systolic Blood pressure (BP) was measured weekly. Data=means±SE. +p<0.05, +++p<0.001 vs the ScrshRNA Cold. n=6.
IL-6shRNA delivery decreased cold-induced increases in IL-6 expression in the kidney
The PBS Cold group showed a significant increase in IL-6 protein expression compared to the PBS Warm group (Fig. 2A&B), suggesting that cold exposure may increase inflammation. IL-6shRNA significantly decreased the cold-induced increase in IL-6 expression vs the ScrshRNA Cold group and the PBS Cold group (Fig. 2A&B). The immunohistochemical (IHC) analysis revealed that cold exposure increased IL-6 expression (brown staining) around the glomeruli and tubules in the ScrshRNA Cold group and the PBS Cold group vs the PBS Warm group (Fig. 2C&D). IL-6 expression was decreased significantly in the IL-6shRNA Cold group (Fig. 2C&D). These results indicate that IL-6 gene was effectively silenced by IL-6shRNA.
Figure 2. IL-6shRNA delivery decreased IL-6 expression in the kidney.
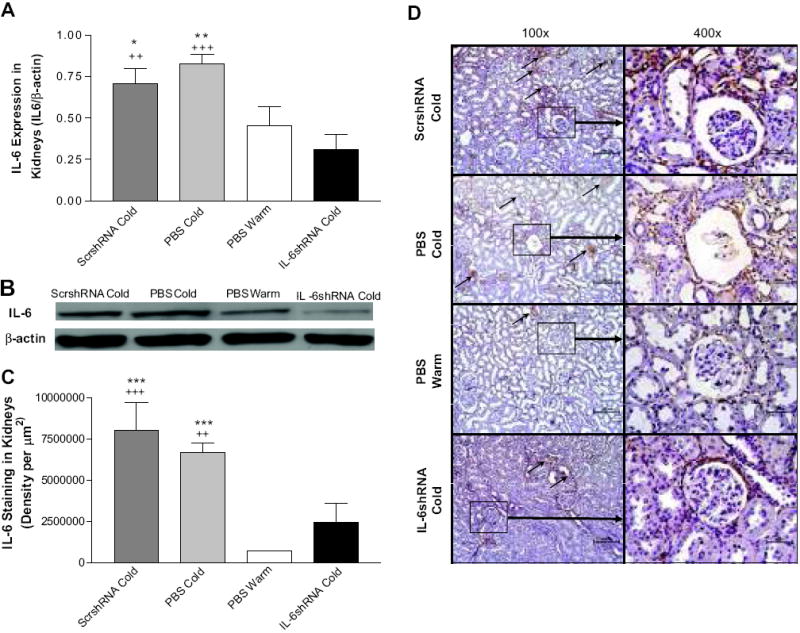
Western blot analysis of IL-6 protein expression in kidney homogenates (A,B). Immunohistochemical analysis of IL-6 expression in the kidney (C,D). Arrows indicate IL-6 expression (brown staining). *p<0.05, **p<0.01, ***p<0.001 vs the PBS Warm group; ++p<0.01, +++p<0.001 vs the IL-6shRNA Cold group. Data=means+SE. n=6.
IL-6shRNA delivery attenuated cold-induced macrophage and T-cell infiltration in the kidney
The IHC analysis showed that CD-68 staining and the number of CD-68 positive cells were increased significantly in kidneys in PBS Cold and ScrshRNA Cold groups (Fig. 3A,B&C), indicating that cold exposure increased macrophage infiltration. Macrophages infiltrated in the renal tubules and glomeruli (Fig. 3C). IL-6shRNA abolished the cold-induced increase in macrophage infiltration (Fig 3A,B&C). Cold exposure also increased T cell (CD-3 positive cell) infiltration in kidneys which could be abolished by IL-6shRNA (Fig. 3D,E&F). T cells infiltrated in the renal tubules (Fig. 3F). These results suggest that that silence of IL-6 prevented cold-induced inflammation.
Figure 3. IL-6shRNA delivery attenuated macrophage and T-cell infiltration in the kidney.
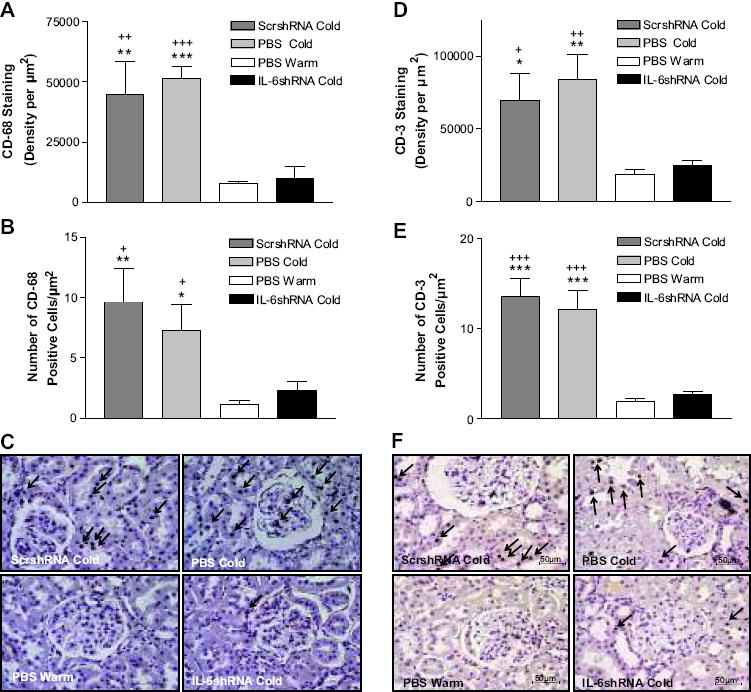
Semi-quantitative analysis of macrophage infiltration in the kidney as measured by the density of CD-68 expression (A) and the average count of CD-68-positive cells (B). The IHC analysis of macrophage infiltration in the kidney as measured by CD-68 staining (C). Semi-quantitative analysis of T-cell infiltration in the kidney as measured by the density of CD-3 expression (D) and the average count of CD-3-positive cells (E). The IHC analysis of T-cell infiltration in the kidney was measured by CD-3 staining (F). Arrows indicate positive staining of CD-68 or CD-3. Photos were taken at 200x. Scale bars indicate 100 μm. *p<0.05, **p<0.01, ***p<0.001 vs The PBS Warm group; +p<0.05, ++p<0.01, +++p<0.001 vs the IL-6shRNA Cold group. Data=means+SE. n=6.
IL-6shRNA delivery attenuated cold-induced kidney damage
Figure 4 showed glomerular collapses in the ScrshRNA Cold and PBS Cold groups, indicating that chronic exposure to cold caused structural damage in kidneys. RNAi knockdown of IL-6 abolished cold-induced glomerular collapses (Fig. 4A&B).
Figure 4. IL-6shRNA delivery prevents cold-induced kidney damage.
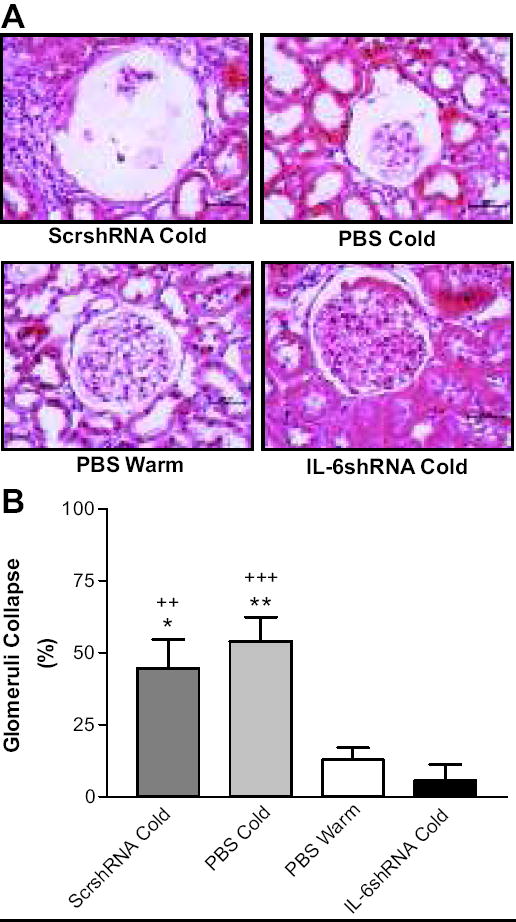
A, H&E staining in the kidney showing collapsed glomeruli. B, Quantitative analysis of glomerular collapse. Photos were taken at 400x. Scale bars represent 50μm. *p<0.05, **p<0.01 <0.001 vs The PBS Warm group; ++p<0.01, +++p<0.001 vs the IL-6shRNA Cold group. Data=means+SE. n=6.
IL-6shRNA delivery abolished the cold-induced increase in IL-6 and TNF-α expression in aorta
Western blot analysis showed significant increases in IL-6 and TNF-α protein expression in aortas in the ScshRNA Cold and PBS Cold groups compared to the PBS Warm group (Fig. 5A-D), indicating that cold exposure may increase inflammation in the aorta. IL-6shRNA delivery abolished the cold-induced increases in IL-6 and TNF-α in aortas (Fig. 5A-D). This result suggests that IL-6 may mediate the cold-induced increase in vascular TNF-α.
Figure 5. IL-6shRNA delivery decreased IL-6 and TNF-α expression in aorta.
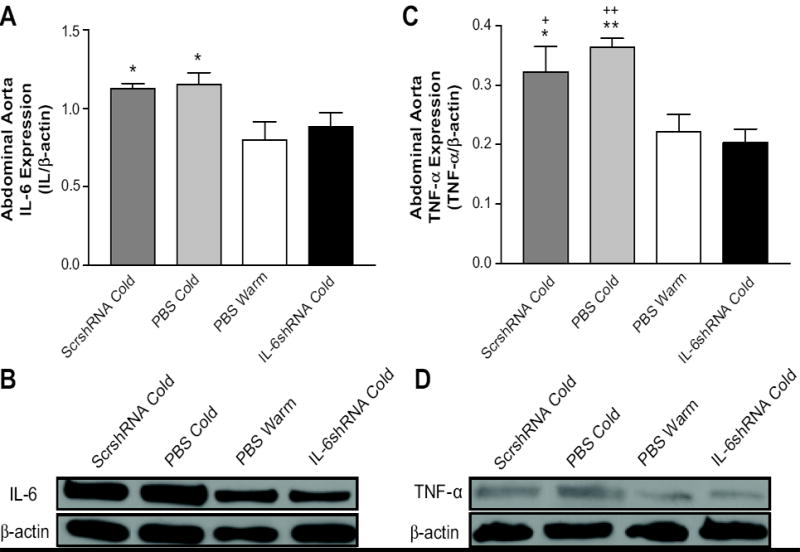
Quantitative analysis of IL-6 protein expression in abdominal aorta (A) and the representative Western blot bands of IL-6 (B). Quantitative analysis of TNF-α protein expression in abdominal aorta (C) and the representative Western blot bands of TNF-α (D). *p<0.05, **p<0.01 vs PBS Warm group; +P<0.05, ++P<0.01 vs the IL-6shRNA Cold group. Data=means+SE. n=6.
IL-6shRNA delivery abolished the cold-induced increase in vascular superoxide production
In situ vascular superoxide production was evaluated using DHE staining. Cold exposure significantly increased superoxide production in aortas (Fig. 6A&B). RNAi silencing of IL-6 abolished the cold-induced increase in vascular superoxide production.
Figure 6. IL-6shRNA delivery decreased in situ superoxide production in the abdominal aorta.
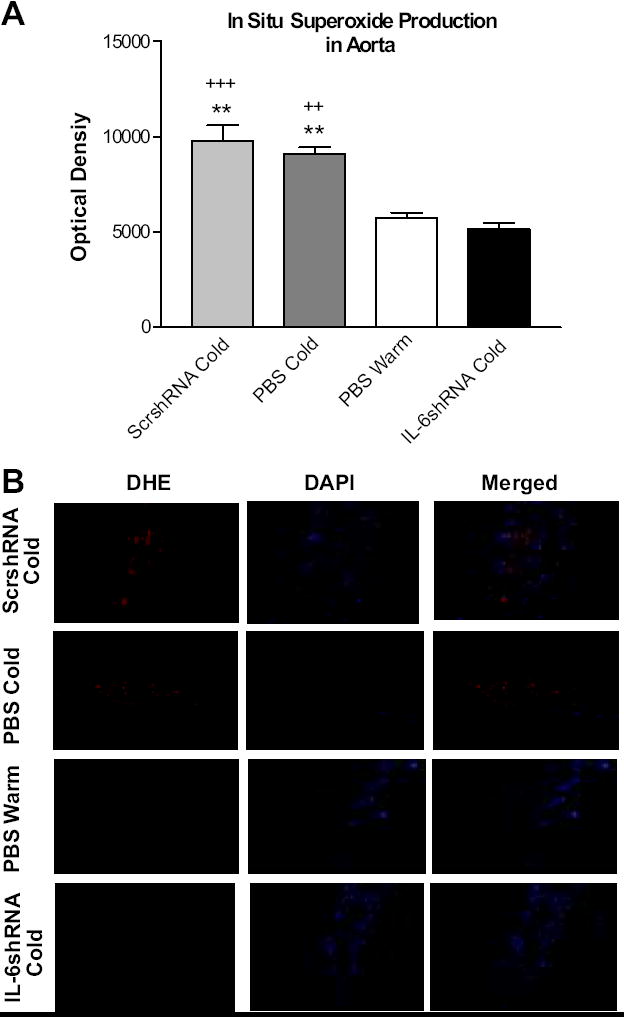
Quantification of superoxide staining in aortas (A). Sections of aortas were incubated with DHE and red fluorescence was viewed using TRITC. DAPI was used to view nuclear staining. Aortic segments showing DHE staining (red), DAPI staining (blue), and a merge of DHE and DAPI respectively (B). Sections were viewed at 200x. **p<0.01 vs the PBS Warm group; ++P<0.01, +++P<0.001 vs the IL-6shRNA Cold group. Data=means+SE. n=6.
IL-6shRNA delivery abolished the cold-induced increases in IL-6 expression in the heart and prevented collagen deposition around the coronary arteries
IHC analysis showed a significant increase in cardiac IL-6 expression in the PBS Cold and ScrshRNA Cold groups (Fig. 7A&B), suggesting that cold exposure increased inflammation in the heart. IL-6shRNA significantly decreased IL-6 expression compared to the PBS Cold and ScrshRNA Cold groups (Figure 7A), confirming effective silencing of IL-6 gene. A significant decrease in collagen deposition (blue staining) around the coronary arteries of the IL-6shRNA Cold group was observed compared to the PBS Cold and ScrshRNA Cold groups (Figure 7C&D), suggesting that RNAi silencing of IL-6 prevented heart remodeling. IL-6shRNA decreased the cold-induced increase in heart weight but did not reduce it to the control level (S1), suggesting partial attenuation of cold-induced cardiac hypertrophy.
Figure 7. IL-6shRNA delivery reduced IL-6 expression in the heart and prevented cardiac collagen deposition.
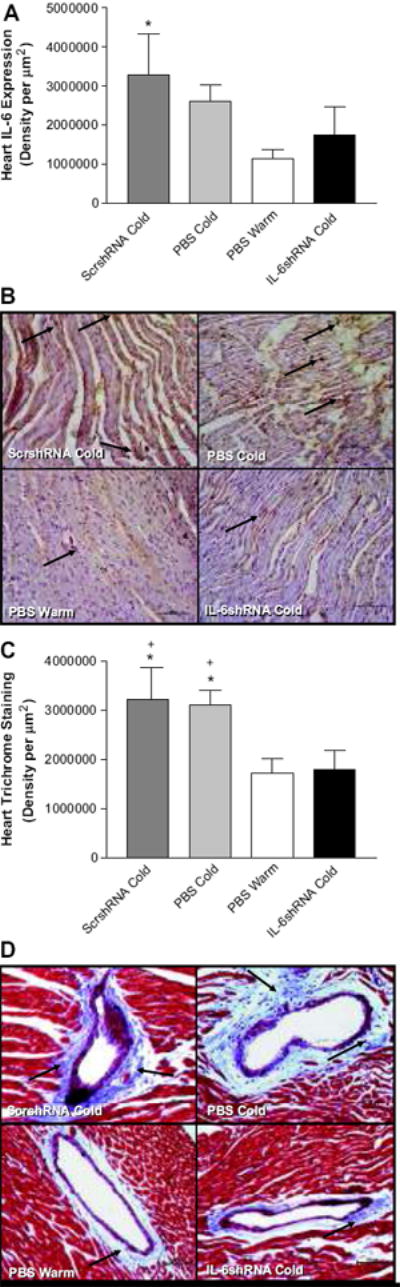
Semi-quantitative analysis of IL-6 expression in the heart (A) and the IHC analysis of cardiac IL-6 expression (B). Semi-quantitative analysis of Tri-chrome staining in the heart (C) and the representative photos showing collagen deposition (indicated by arrows) around coronary arteries (D). Photos were taken at 200x. Scale bars=100μm. *p<0.05 vs the PBS Warm group; +p<0.01 vs the IL-6shRNA Cold group. Data=means+SE. n=6.
Discussion
The present data demonstrated, for the first time, that chronic exposure to cold caused inflammation in kidneys, hearts, and blood vessels as evidenced by increased levels of IL-6 and TNF-α expression and leukocyte infiltration. Interestingly, RNAi knockdown of IL-6 attenuated cold-induced inflammation and elevation of BP, suggesting that that inflammation plays a vital role in the pathogenesis of CIH and that IL-6 is a key mediator in this process. The finding is significant because of its potential to provide novel preventive and therapeutic strategies for cold-related cardiovascular and renal dysfunction, which is important for people who live in cold regions and in winter. The prolonged attenuation of CIH is likely to due to the long-term expression vector, AAV. AAV is an effective and non-pathogenic vector that has been used to deliver therapeutic genes to the cardiovascular system and kidneys for long-term control of hypertension.23-24, 26 AAV could express for months after gene delivery.24, 27
Our previous studies have established that over-activation of the renin-angiotensin system (RAS) is responsible for the development of CIH. In addition to its vasoconstrictor properties, AngII has been demonstrated to exert pro-inflammatory effects in the vasculature by inducing production of integrins, adhesion molecules, cytokines and growth factors through activation of the transcription factor NFκB.13, 28 NFκB in turn, regulates at the transcriptional level, a variety of pro-inflammatory genes.29 AngII has also been shown to directly contribute to the up-regulation of IL-6 and has the ability to influence various stages of the inflammatory process.14, 30-33 It will be interesting to test if inflammation mediates the role of the RAS in CIH.
The molecular mechanisms involved in the pathogenesis of hypertension are not fully understood. Inflammation has been shown to increase the generation of reactive oxygen species (ROS) by activating NADPH oxidases.34 IL-6 is a strong activator of NADPH oxidases.35-36 Normal superoxide production via NADPH oxidases is necessary for biological processes such as cell signaling, post-translational modification and host defense.34 An overproduction of ROS such as superoxide, however, can lead to oxidative damage in the vasculature resulting in endothelial dysfunction, vascular senescence and remodeling, and hypertension.34, 37 The present study clearly showed that knockdown of IL-6 led to a decrease in cold-induced vascular superoxide production, which may contribute to its anti-hypertensive effect.
Patients with cardiovascular disease have increased protein expression and plasma concentration of many inflammatory markers including selectins (P, E and L-selectins), intracellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule (VCAM-1).38 Human hypertension is associated with increased levels of TNF-α, IL-6 as well as ICAM-1, VCAM-1 and selectins.39 However, the role of IL-6 in inflammation in the context of hypertension is largely unknown. The present study revealed that IL-6 is likely a key mediator of inflammation because knockdown of IL-6 prevented the cold-induced increases in TNF-α and leukocyte infiltration The molecular mechanism leading to increased levels of macrophages and T-cells in cold exposed animals needs to be further investigated but increased levels of IL-6 could up-regulate ICAM-1, VCAM-1 and the selectins through NFκB activation leading to the recruitment of inflammatory infiltrates. These inflammatory factors are directly responsible for the recruitment of leukocytes including macrophages and lymphocytes to the vasculature and have been demonstrated to be up-regulated in several animal models of hypertension.38, 40-43
In the present study, systolic BP was monitored using tail-cuff method. The tail-cuff procedure is a common method used by our laboratory12, 23 and others44-45 to delineate CIH. It has been confirmed by the intra-arterial cannulation that the noninvasive tail-cuff method is effective and reliable in monitoring BP responses to cold.8-9, 46
It is noted that chronic exposure to cold for 8 weeks is sufficient to cause glomerular collapse, a sign of renal structural damage. RNAi knockdown of IL-6 effectively prevented cold-induced glomerular damage, suggesting that IL-6 may mediate inflammatory responses that lead to renal vascular damage and ultimately glomerular collapse. Previous studies from our laboratory have demonstrated that chronic cold exposure also leads to cardiac hypertrophy.8, 12, 47 The mechanism of cold-induced cardiac hypertrophy (CICH) is unknown, but is independent of high BP because prevention of CIH does not attenuate the development of CICH.1, 12 The present data revealed that inflammation may play a role in the pathogenesis of CICH because knockdown of IL-6 attenuated the cold-induced increase in heart weight. Interestingly, cold exposure also resulted in formation of fibrosis around the coronary artery, which may ultimately impair the coronary circulation. It is notable that cold-induced formation of fibrosis was abolished by inhibition of IL-6, suggesting an important role of inflammation in this pathological process.
TNF-α is a primary pro-inflammatory cytokine that has been shown to increase the production of cytokines including IL-6 and to initiate inflammatory cascades.48-49 Both TNF-α and IL-6 blockade therapies can reduce inflammation and improve prognosis in patients with arthritis, a chronic inflammatory disease.50 Although TNF-α blockers have been shown to decrease IL-6 levels, this is the first study demonstrating that knockdown of IL-6 prevented cold-induced up-regulation of TNF-α. Further investigation into the molecular mechanism mediating the regulation of TNF-α by IL-6 would be of particular interest.
Perspectives
The current study yields two major findings in the pathogenesis of CIH. First, chronic exposure to cold caused inflammation in the cardiovascular system and kidneys as evidenced by up-regulation of pro-inflammatory cytokines (IL-6 and TNF-α) and an increase in leukocyte infiltration. Second, RNAi knockdown of IL-6 abolished cold-induced inflammation and attenuated cold-induced elevation of BP and organ damage. These results suggest that inflammation plays a critical role in the development of CIH and that IL-6 is a critical mediator in this pathological process. This study also reveals that AAV.RNAi knockdown of IL-6 may serve as a new approach for effective control of hypertension.
Supplementary Material
Acknowledgments
Sources of Funding This work was supported by National Institute of Health grant R01 NHLBI-077490.
Footnotes
Disclosures None.
References
- 1.Sun Z. Cardiovascular responses to cold exposure. Front Biosci (Elite Ed) 2010;2:495–503. doi: 10.2741/e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baker-Blocker A. Winter weather and cardiovascular mortality in Minneapolis-St. Paul. Am J Pub Health. 1982;72:261–265. doi: 10.2105/ajph.72.3.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seretakis D, Lagiou P, Lipworth L, Signorello LB, Rothman KJ, Trichopoulos D. Changing seasonality of mortality from coronary heart disease. JAMA. 1997;278:1012–1014. [PubMed] [Google Scholar]
- 4.Sheth T, Nair C, Muller J, Yusuf S. Increased winter mortality from acute myocardial infarction and stroke: the effect of age. J Am Coll Cardiol. 1999;33:1916–1919. doi: 10.1016/s0735-1097(99)00137-0. [DOI] [PubMed] [Google Scholar]
- 5.Verdon F, Boudry JF, Chuat M, Studer JP, Truong CB, Jacot E. Seasonal variations in arterial pressure in hypertensive patients. Schweizerische medizinische Wochenschrift. 1993;123:2363–2369. [PubMed] [Google Scholar]
- 6.Minami J, Kawano Y, Ishimitsu T, Yoshimi H, Takishita S. Seasonal variations in office, home and 24 h ambulatory blood pressure in patients with essential hypertension. J hypertens. 1996;14:1421–1425. doi: 10.1097/00004872-199612000-00006. [DOI] [PubMed] [Google Scholar]
- 7.Hata T, Ogihara T, Maruyama A, Mikami H, Nakamaru M, Naka T, Kumahara Y, Nugent CA. The seasonal variation of blood pressure in patients with essential hypertension. Clin exp hypertens. 1982;4:341–354. doi: 10.3109/10641968209060747. [DOI] [PubMed] [Google Scholar]
- 8.Fregly MJ, Kikta DC, Threatte RM, Torres JL, Barney CC. Development of hypertension in rats during chronic exposure to cold. J Appl Physiol. 1989;66:741–749. doi: 10.1152/jappl.1989.66.2.741. [DOI] [PubMed] [Google Scholar]
- 9.Sun Z, Cade R, Katovich MJ, Fregly MJ. Body fluid distribution in rats with cold-induced hypertension. Physiol Behav. 1999;65:879–884. doi: 10.1016/s0031-9384(98)00250-9. [DOI] [PubMed] [Google Scholar]
- 10.Sun Z, Cade JR, Fregly MJ. Cold-induced hypertension. A model of mineralocorticoid-induced hypertension. Ann N Y Acad Sci. 1997;813:682–688. doi: 10.1111/j.1749-6632.1997.tb51767.x. [DOI] [PubMed] [Google Scholar]
- 11.Sun Z, Bello-Roufai M, Wang X. RNAi inhibition of mineralocorticoid receptors prevents the development of cold-induced hypertension. Am J Physiol Heart Circ Physiol. 2008;294:H1880–1887. doi: 10.1152/ajpheart.01319.2007. [DOI] [PubMed] [Google Scholar]
- 12.Sun Z, Cade R, Zhang Z, Alouidor J, Van H. Angiotensinogen gene knockout delays and attenuates cold-induced hypertension. Hypertension. 2003;41:322–327. doi: 10.1161/01.hyp.0000050964.96018.fa. [DOI] [PubMed] [Google Scholar]
- 13.Marchesi C, Paradis P, Schiffrin EL. Role of the renin-angiotensin system in vascular inflammation. Trends Pharmacol Sci. 2008;29:367–374. doi: 10.1016/j.tips.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 14.Moriyama T, Fujibayashi M, Fujiwara Y, Kaneko T, Xia C, Imai E, Kamada T, Ando A, Ueda N. Angiotensin II stimulates interleukin-6 release from cultured mouse mesangial cells. J Am Soc Nephrol. 1995;6:95–101. doi: 10.1681/ASN.V6195. [DOI] [PubMed] [Google Scholar]
- 15.Androulakis ES, Tousoulis D, Papageorgiou N, Tsioufis C, Kallikazaros I, Stefanadis C. Essential hypertension: is there a role for inflammatory mechanisms? Cardiol Rev. 2009;17:216–221. doi: 10.1097/CRD.0b013e3181b18e03. [DOI] [PubMed] [Google Scholar]
- 16.Di Bona D, Vasto S, Capurso C, Christiansen L, Deiana L, Franceschi C, Hurme M, Mocchegiani E, Rea M, Lio D, Candore G, Caruso C. Effect of interleukin-6 polymorphisms on human longevity: a systematic review and meta-analysis. Ageing Res Rev. 2009;8:36–42. doi: 10.1016/j.arr.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 17.Mima T, Nishimoto N. Clinical value of blocking IL-6 receptor. Curr Opin Rheumatol. 2009;21:224–230. doi: 10.1097/BOR.0b013e3283295fec. [DOI] [PubMed] [Google Scholar]
- 18.Boos CJ, Lip GY. Is hypertension an inflammatory process? Curr Pharm Des. 2006;12:1623–1635. doi: 10.2174/138161206776843313. [DOI] [PubMed] [Google Scholar]
- 19.Vazquez-Oliva G, Fernandez-Real JM, Zamora A, Vilaseca M, Badimon L. Lowering of blood pressure leads to decreased circulating interleukin-6 in hypertensive subjects. J Hum Hypertens. 2005;19:457–462. doi: 10.1038/sj.jhh.1001845. [DOI] [PubMed] [Google Scholar]
- 20.Manabe S, Okura T, Watanabe S, Fukuoka T, Higaki J. Effects of angiotensin II receptor blockade with valsartan on pro-inflammatory cytokines in patients with essential hypertension. J Cardiovasc Pharmacol. 2005;46:735–739. doi: 10.1097/01.fjc.0000185783.00391.60. [DOI] [PubMed] [Google Scholar]
- 21.Fliser D, Buchholz K, Haller H. Antiinflammatory effects of angiotensin II subtype 1 receptor blockade in hypertensive patients with microinflammation. Circulation. 2004;110:1103–1107. doi: 10.1161/01.CIR.0000140265.21608.8E. [DOI] [PubMed] [Google Scholar]
- 22.Gadonski G, LaMarca BB, Sullivan E, Bennett W, Chandler D, Granger JP. Hypertension produced by reductions in uterine perfusion in the pregnant rat: role of interleukin 6. Hypertension. 2006;48:711–716. doi: 10.1161/01.HYP.0000238442.33463.94. [DOI] [PubMed] [Google Scholar]
- 23.Wang X, Skelley L, Cade R, Sun Z. AAV delivery of mineralocorticoid receptor shRNA prevents progression of cold-induced hypertension and attenuates renal damage. Gene Ther. 2006;13:1097–1103. doi: 10.1038/sj.gt.3302768. [DOI] [PubMed] [Google Scholar]
- 24.Wang Y, Sun Z. Klotho gene delivery prevents the progression of spontaneous hypertension and renal damage. Hypertension. 2009;54:810–817. doi: 10.1161/HYPERTENSIONAHA.109.134320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feng M, Whitesall S, Zhang Y, Beibel M, D’Alecy L, DiPetrillo K. Validation of volume-pressure recording tail-cuff blood pressure measurements. Am J Hypertens. 2008;21:1288–1291. doi: 10.1038/ajh.2008.301. [DOI] [PubMed] [Google Scholar]
- 26.Phillips MI, Mohuczy-Dominiak D, Coffey M, Galli SM, Kimura B, Wu P, Zelles T. Prolonged reduction of high blood pressure with an in vivo, nonpathogenic, adeno-associated viral vector delivery of AT1-R mRNA antisense. Hypertension. 1997;29(1 Pt 2):374–380. doi: 10.1161/01.hyp.29.1.374. [DOI] [PubMed] [Google Scholar]
- 27.Kimura B, Mohuczy D, Tang X, Phillips MI. Attenuation of hypertension and heart hypertrophy by adeno-associated virus delivering angiotensinogen antisense. Hypertension. 2001;37(2 Part 2):376–380. doi: 10.1161/01.hyp.37.2.376. [DOI] [PubMed] [Google Scholar]
- 28.Touyz RM. Molecular and cellular mechanisms in vascular injury in hypertension: role of angiotensin II. Curr Opin Nephrol Hypertens. 2005;14:125–131. doi: 10.1097/00041552-200503000-00007. [DOI] [PubMed] [Google Scholar]
- 29.Savoia C, Schiffrin EL. Inflammation in hypertension. Curr Opin Nephrol Hypertens. 2006;15:152–158. doi: 10.1097/01.mnh.0000203189.57513.76. [DOI] [PubMed] [Google Scholar]
- 30.Funakoshi Y, Ichiki T, Takeda K, Tokuno T, Iino N, Takeshita A. Critical role of cAMP-response element-binding protein for angiotensin II-induced hypertrophy of vascular smooth muscle cells. The Journal of biological chemistry. 2002;277:18710–18717. doi: 10.1074/jbc.M110430200. [DOI] [PubMed] [Google Scholar]
- 31.Kandalam U, Clark MA. Angiotensin II activates JAK2/STAT3 pathway and induces interleukin-6 production in cultured rat brainstem astrocytes. Regulatory peptides. 2009 doi: 10.1016/j.regpep.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 32.Wu J, Yang X, Zhang YF, Zhou SF, Zhang R, Dong XQ, Fan JJ, Liu M, Yu XQ. Angiotensin II upregulates Toll-like receptor 4 and enhances lipopolysaccharide-induced CD40 expression in rat peritoneal mesothelial cells. Inflamm Res. 2009;58:473–482. doi: 10.1007/s00011-009-0012-z. [DOI] [PubMed] [Google Scholar]
- 33.Ruef J, Browatzki M, Pfeiffer CA, Schmidt J, Kranzhofer R. Angiotensin II promotes the inflammatory response to CD40 ligation via TRAF-2. Vascular medicine (London, England) 2007;12:23–27. doi: 10.1177/1358863X07076766. [DOI] [PubMed] [Google Scholar]
- 34.Sedeek M, Hebert RL, Kennedy CR, Burns KD, Touyz RM. Molecular mechanisms of hypertension: role of Nox family NADPH oxidases. Curr Opin Nephrol Hypertens. 2009;18:122–127. doi: 10.1097/MNH.0b013e32832923c3. [DOI] [PubMed] [Google Scholar]
- 35.Behrens MM, Ali SS, Dugan LL. Interleukin-6 mediates the increase in NADPH-oxidase in the ketamine model of schizophrenia. J Neurosci. 2008;28:13957–13966. doi: 10.1523/JNEUROSCI.4457-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Volk T, Hensel M, Schuster H, Kox WJ. Secretion of MCP-1 and IL-6 by cytokine stimulated production of reactive oxygen species in endothelial cells. Mol Cell Biochem. 2000;206:105–112. doi: 10.1023/a:1007059616914. [DOI] [PubMed] [Google Scholar]
- 37.Sedeek M, Gilbert JS, LaMarca BB, Sholook M, Chandler DL, Wang Y, Granger JP. Role of reactive oxygen species in hypertension produced by reduced uterine perfusion in pregnant rats. American journal of hypertension. 2008;21:1152–1156. doi: 10.1038/ajh.2008.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Savoia C, Schiffrin EL. Vascular inflammation in hypertension and diabetes: molecular mechanisms and therapeutic interventions. Clin Sci (Lond) 2007;112:375–384. doi: 10.1042/CS20060247. [DOI] [PubMed] [Google Scholar]
- 39.Preston RA, Ledford M, Materson BJ, Baltodano NM, Memon A, Alonso A. Effects of severe, uncontrolled hypertension on endothelial activation: soluble vascular cell adhesion molecule-1, soluble intercellular adhesion molecule-1 and von Willebrand factor. Journal of hypertension. 2002;20:871–877. doi: 10.1097/00004872-200205000-00021. [DOI] [PubMed] [Google Scholar]
- 40.Haverslag R, Pasterkamp G, Hoefer IE. Targeting adhesion molecules in cardiovascular disorders. Cardiovascular & hematological disorders drug targets. 2008;8:252–260. doi: 10.2174/187152908786786188. [DOI] [PubMed] [Google Scholar]
- 41.Muller WA. Mechanisms of transendothelial migration of leukocytes. Circulation research. 2009;105:223–230. doi: 10.1161/CIRCRESAHA.109.200717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.De Ciuceis C, Amiri F, Brassard P, Endemann DH, Touyz RM, Schiffrin EL. Reduced vascular remodeling, endothelial dysfunction, and oxidative stress in resistance arteries of angiotensin II-infused macrophage colony-stimulating factor-deficient mice: evidence for a role in inflammation in angiotensin-induced vascular injury. Arteriosclerosis, thrombosis, and vascular biology. 2005;25:2106–2113. doi: 10.1161/01.ATV.0000181743.28028.57. [DOI] [PubMed] [Google Scholar]
- 43.Ando H, Zhou J, Macova M, Imboden H, Saavedra JM. Angiotensin II AT1 receptor blockade reverses pathological hypertrophy and inflammation in brain microvessels of spontaneously hypertensive rats. Stroke; a journal of cerebral circulation. 2004;35:1726–1731. doi: 10.1161/01.STR.0000129788.26346.18. [DOI] [PubMed] [Google Scholar]
- 44.Peng JF, Kimura B, Fregly MJ, Phillips MI. Reduction of cold-induced hypertension by antisense oligodeoxynucleotides to angiotensinogen mRNA and AT1-receptor mRNA in brain and blood. Hypertension. 1998;31:1317–1323. doi: 10.1161/01.hyp.31.6.1317. [DOI] [PubMed] [Google Scholar]
- 45.Zhu Z, Zhu S, Zhu J, van der Giet M, Tepel M. Endothelial dysfunction in cold-induced hypertensive rats. Am J Hypertens. 2002;15(2 Pt 1):176–180. doi: 10.1016/s0895-7061(01)02268-3. [DOI] [PubMed] [Google Scholar]
- 46.Wang X, Sun Z. RNAi silencing of brain klotho potentiates cold-induced elevation of blood pressure via the endothelin pathway. Physiol Genomics. 2010 doi: 10.1152/physiolgenomics.00192.2009. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sun Z, Fregly MJ, Cade JR. Effect of renal denervation on elevation of blood pressure in cold-exposed rats. Can J Physiol Pharmacol. 1995;73:72–78. doi: 10.1139/y95-010. [DOI] [PubMed] [Google Scholar]
- 48.Desmet C, Warzee B, Gosset P, Melotte D, Rongvaux A, Gillet L, Fievez L, Seumois G, Vanderplasschen A, Staels B, Lekeux P, Bureau F. Pro-inflammatory properties for thiazolidinediones. Biochem Pharmacol. 2005;69:255–265. doi: 10.1016/j.bcp.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 49.Navarro-Gonzalez JF, Mora-Fernandez C. The role of inflammatory cytokines in diabetic nephropathy. J Am Soc Nephrol. 2008;19:433–442. doi: 10.1681/ASN.2007091048. [DOI] [PubMed] [Google Scholar]
- 50.Brennan FM, McInnes IB. Evidence that cytokines play a role in rheumatoid arthritis. J Clin Invest. 2008;118:3537–3545. doi: 10.1172/JCI36389. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.