

Abstract
Spore formation in Saccharomyces cerevisiae is driven by de novo assembly of new membranes termed prospore membranes. A vesicle-docking complex called the meiosis II outer plaque (MOP) forms on the cytoplasmic faces of the spindle-pole bodies at the onset of meiosis II and serves as the initiation site for membrane formation. In this study, a fluorescence-recovery assay was used to demonstrate that the dynamics of the MOP proteins change coincident with the coalescence of precursor vesicles into a membrane. Proteins within the MOP exchange freely with a soluble pool prior to membrane assembly, but after membranes are formed they remain stably within the MOP. By contrast, constitutive spindle-pole-body proteins display low exchange in both conditions. The MOP component Ady4p plays a role in maintaining the integrity of the MOP complex, but this role differs depending on whether the MOP is associated with docked vesicles or a fully formed membrane. These results suggest an architectural rearrangement of the MOP coincident with vesicle fusion.
Keywords: Spindle-pole body, Meiosis, Ady4p
Introduction
In the baker's yeast Saccharomyces cerevisiae, meiosis and sporulation are induced when diploid cells are cultured in the absence of nitrogen and the presence of a nonfermentable carbon source (Esposito and Klapholz, 1981). During spore formation, cell division occurs by capturing daughter nuclei within new membranes, termed the prospore membranes, which are discontinuous from the mother-cell plasma membrane (Moens and Rapport, 1971; Neiman, 1998). Formation of prospore membranes is initiated by the fusion of post-Golgi vesicles that are targeted to the cytoplasmic faces of the meiosis-II spindle-pole bodies by a developmentally controlled rearrangement of the secretory pathway (Neiman, 1998). As meiosis II progresses, chromosomes segregate into four lobes of the nucleus and each prospore membrane elongates towards the center of the spindle, engulfing the adjacent nuclear lobe as well as cytoplasm and organelles (Moens and Rapport, 1971; Nickas et al., 2003; Suda et al., 2007). At the completion of meiosis II and nuclear division, each of the four prospore membranes closes to form four immature spores (Neiman, 1998).
During meiosis II, each prospore membrane is attached to a spindle-pole body, which is embedded in the nuclear envelope and serves as the microtubule-organizing center of the cell, analogous to the centrosome in higher eukaryotes (Moens and Rapport, 1971; Rout and Kilmartin, 1990). Previous studies have shown that spindle-pole bodies are dynamic structures that contain components that can exchange or remain static depending on the stage of the cell cycle (Yoder et al., 2003). During mitosis and meiosis I, the cytoplasmic face, or outer plaque, of the spindle-pole body nucleates cytoplasmic microtubules. At the onset of meiosis II, the composition of the outer plaques, now called the meiosis II outer plaques (MOPs), change so that the cytoplasmic faces of the spindle-pole bodies serve as sites for nucleation of the prospore membrane (Knop and Strasser, 2000; Moens and Rapport, 1971).
The MOP is composed primarily of three meiosis-specific proteins: Spo21p, Spo74p and Mpc54p (Bajgier et al., 2001; Knop and Strasser, 2000; Nickas et al., 2003). Deletion of the gene encoding any one of these components leads to the absence of the MOP structure and a complete block of prospore-membrane formation, indicating that interaction of vesicles with this structure is necessary to promote vesicle fusion. Vesicles dock onto the surface of the MOP, and their subsequent fusion to form the prospore membrane is mediated by a soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex that includes Sso1p (Nakanishi et al., 2006; Neiman, 1998; Neiman et al., 2000). In sso1Δ cells, unfused vesicles accumulate on the MOP surface. Thus, the MOP functions upstream of the SNAREs to promote membrane fusion, similar to other vesicle-docking complexes (Rothman, 1994; Sogaard et al., 1994; Sollner et al., 1993; Wickner and Schekman, 2008).
Besides Mpc54p, Spo21p and Spo74p, the constitutive spindle-pole-body proteins Cnm67p and Nud1p are also found on the cytoplasmic side of the spindle-pole body during meiosis II (Adams and Kilmartin, 1999; Bullitt et al., 1997; Knop and Strasser, 2000). Cnm67p serves to link the MOP to the core of the spindle-pole body (Adams and Kilmartin, 1999; Bullitt et al., 1997; Schaerer et al., 2001). A fourth meiosis-specific component of the MOP, Ady4p, is distinct from Mpc54p, Spo21p and Spo74p in that it is not essential for MOP assembly and prospore-membrane formation. However, ady4Δ mutants display heterogeneous defects in MOP and prospore-membrane morphology that lead to failures in the packaging of individual nuclei and, often, to asci with fewer than four spores. These phenotypes have led to the proposal that Ayd4p functions to promote the stability of the MOP (Nickas et al., 2003).
Fluorescence recovery after photobleaching (FRAP) experiments have shown that the MOP is a stable structure with little exchange between incorporated and soluble subunits (Taxis et al., 2005). The high stability was proposed to be an intrinsic property of the assembled structure. This work demonstrates that, in fact, the composition of the MOP can change until constrained by the construction of a prospore-membrane cap on the MOP surface. Furthermore, we identify a role for the MOP component Ady4p in enhancing the stability of the MOP structure.
Results
The rate of exchange of MOP components depends on the presence of a prospore membrane
Formation of a properly assembled MOP is necessary for prospore-membrane formation (Bajgier et al., 2001; Knop and Strasser, 2000; Nickas et al., 2003). FRAP can be used to examine the stability of the MOP structure based on the rate and the degree of exchange of GFP-tagged MOP components (Taxis et al., 2005). Previous FRAP analysis of the MOP demonstrated that MOP components exchanged rapidly prior to the formation of an organized MOP structure, and this exchange was decreased once the MOP was formed and functioning (Taxis et al., 2005). This study did not address whether this stability is simply an intrinsic property of a completely constructed MOP or due to an exogenous factor such as the presence of a prospore membrane attached to the MOP. These two possibilities can be distinguished by looking at MOP-protein exchange in the absence of the prospore membrane. If the prospore membrane is required to stabilize the MOP, then MOP components should exchange when prospore-membrane formation is prevented.
One way to prevent prospore-membrane formation is to delete the gene encoding the SNARE protein Sso1p (Nakanishi et al., 2006). In the sso1Δ mutant, prospore-membrane precursor vesicles dock onto the surface of the MOP, but the fusion of these vesicles to create a larger membrane structure is blocked (Nakanishi et al., 2006). C-terminal GFP fusions to the MOP components were introduced into wild-type and sso1Δ cells. Each of the C-terminal GFP fusions is functional based on rescue of sporulation in the appropriate null mutant (Nickas et al., 2003) (supplementary material Table S1). FRAP was used to compare the exchange of each MOP protein in cells determined to be in mid-meiosis II based on the presence of four spindle-pole bodies (Fig. 1A). For all three MOP components, recovery of fluorescence was low in the presence of SSO1, consistent with earlier studies of Mpc54p (Fig. 1B-D) (Knop and Strasser, 2000; Taxis et al., 2005). By contrast, all of the MOP components displayed significant recovery of fluorescence in the absence of SSO1, reaching a plateau at about 25% of the initial fluorescence (Fig. 1B-D). This result indicates that, in the absence of a prospore membrane, MOP components are able to exchange with a soluble pool, and that the absence of exchange in wild-type cells depends upon the presence of an overlying prospore membrane.
Fig. 1.

The rate of exchange of MOP components depends on the presence of a growing prospore membrane. (A) Representative images from the fluorescence recovery of Spo21p-GFP. Time points are indicated. Pre-bleach images are shown in both the DIC and GFP channels. The bleached spindle-pole body is marked with an arrowhead. Scale bars: 1 μm. (B) Fluorescence recovery of Spo21p-GFP in wild-type and sso1Δ cells during meiosis II. The plots represent the average of six and ten experiments, respectively. Error bars represent the s.d. at each time point. (C) Fluorescence recovery of Spo74p-GFP in wild-type and sso1Δ cells during meiosis II. The plots represent the averages of 13 and 14 experiments, respectively. (D) Fluorescence recovery of Mpc54p-GFP in wild-type, sso1Δ and spo14Δ cells during meiosis II. The plots represent the averages of 12, 17 and 11 experiments, respectively.
One alternative explanation for increased exchange of GFP-tagged MOP components observed in the sso1Δ mutants would be if the deletion of SSO1 resulted in higher expression of the MOP proteins and created a larger pool of soluble protein available for exchange. To test this possibility, western blotting was employed to compare the expression levels of the MOP components in wild-type and sso1Δ cells. For all three MOP components, there were no significant differences in expression between wild-type and sso1Δ cells (Fig. 2A,B). Quantification of the bands validated this result, because the expression of each MOP component in sso1Δ mutants was within 10% of the expression of the same MOP component in wild-type cells. A significant fraction of the Spo21p-GFP protein seemed to be degraded in the extracts, although the expression levels of full-length Spo21p-GFP were similar in wild-type and sso1Δ cells (Fig. 2A). No significant degradation of Mpc54p-GFP and Spo74p-GFP was seen (Fig. 2A,B). This result is consistent with the idea that the increase in exchange observed in the sso1Δ mutant is due to the absence of the prospore membrane.
Fig. 2.

GFP-tagged proteins were expressed at the same levels in wild-type and sso1Δ cells. (A,B) Immunoblots of protein extracts from wild-type and sso1Δ cells expressing Spo21p-GFP (A), Mpc54p-GFP (A) or Spo74p-GFP (B). Extracts were probed with anti-GFP or anti-porin antibodies.
To ensure that the changes in Mpc54p-GFP dynamics seen by FRAP were caused by the lack of prospore membranes in the sso1Δ mutant and not as a result of the absence of SSO1 per se, an alternative approach to preventing prospore-membrane formation was employed. Cells lacking the phospholipase-D enzyme encoded by SPO14 also accumulate unfused vesicles on the MOP surface, similar to sso1Δ cells (Nakanishi et al., 2006; Riedel et al., 2005). The FRAP values for Mpc54p-GFP in the spo14Δ mutant were similar to those seen in the sso1Δ strain (Fig. 1D). This result demonstrates that the stability of the MOP observed in wild-type cells is due specifically to the lack of a prospore membrane.
The behavior of constitutive spindle-pole-body components is independent of the prospore membrane
The core of the spindle-pole body is a central plaque composed of a crystal lattice of Spc42p that is embedded in the nuclear envelope (Adams and Kilmartin, 1999; Jaspersen and Winey, 2004; Muller et al., 2005). Cnm67p is a component of both the vegetative and meiotic outer plaques and connects the central plaque to the MOP (Bajgier et al., 2001; Schaerer et al., 2001). To determine whether the absence of the prospore membrane has a destabilizing effect on these more-interior spindle-pole-body components, FRAP measurements were taken of Spc42p-GFP and Cnm67p-GFP in sporulating wild-type and sso1Δ cells. Cells expressing the C-terminal GFP fusions to Spc42p-GFP and Cnm67p-GFP had no growth or sporulation defects (supplementary material Fig. S1 and Table S2). Exchange of Spc42p-GFP and Cnm67p-GFP remained low in both wild-type and sso1Δ cells (Fig. 3A,B). These observations demonstrate that the central plaque and the interior layer of the outer plaque are stable whether or not a prospore membrane is present. Thus, the dependence on the prospore membrane for stability is unique to the MOP.
Fig. 3.

The prospore membrane uniquely stabilizes the MOP structure. (A) Fluorescence recovery of Spc42p-GFP in wild-type and sso1Δ cells during meiosis II. The plots represent the average of ten and eight experiments, respectively. Fluorescence recovery of Mpc54-GFP wild-type cells from Fig. 1D is also shown for comparison. (B) Fluorescence recovery of Cnm67p-GFP in wild-type and sso1Δ cells during meiosis II. The plots represent the average of 11 experiments each. Fluorescence recovery of Mpc54p-GFP wild-type cells from Fig. 1D is also shown for comparison.
The prospore membrane is not a barrier to the exchange of cytoplasmic proteins
How does the prospore membrane reduce the exchange of MOP components? One possibility is that the membrane acts sterically to inhibit exchange. This could result either from the membrane separating the MOP from a pool of exchangeable proteins in the mother-cell cytoplasm, or from physical restrictions conferred by the attachment of the membrane to the surface of the MOP, which prevents the photobleached proteins from leaving the MOP. To test whether the prospore membrane blocks the exchange of contents between the presumptive cytoplasms of the spore and the mother cell, FRAP analysis was used to examine the exchange of cytosolic proteins localized within the growing prospore membrane.
Sec7p is a Golgi-associated protein that localizes to the cytoplasmic region captured within the growing prospore membrane during meiosis II (Reinke et al., 2004; Suda et al., 2007). Wild-type cells were co-transformed with plasmids expressing Sec7p-GFP and plasmids expressing the prospore-membrane marker RFP-Spo20p51-91 [RFP fused to the lipid-binding domain of Spo20p (Nakanishi et al., 2004)]. RFP-labeled prospore membranes that contained Sec7p-GFP signals were identified and the area within the prospore membrane was bleached (Fig. 4A). Sec7p-GFP fluorescence recovered rapidly (Fig. 4A,B), demonstrating that Sec7p-GFP is able to exchange between the two cytoplasmic regions of the cell. Similar results have been reported monitoring a different cytoplasmic GFP fusion using a fluorescence loss in photobleaching (FLIP) assay (Diamond et al., 2009). Taken together, these results indicate that the prospore membrane does not prevent the exchange of proteins between the mother cytoplasm and the cytoplasm that is captured within the growing prospore membrane.
Fig. 4.

The prospore membrane is not a diffusion barrier. (A) Representative images from fluorescence recovery of Sec7p-GFP in wild-type cells. RFP-Spo20p51-91 labels the prospore membrane. Time points are indicated. Pre-bleach images are shown as both merged DIC/RFP/GFP and merged RFP/GFP. The region that was bleached is marked with a white ellipse in the pre-bleach RFP/GFP image. Fluorescence recovery is marked with an arrowhead. Scale bars: 1 μm. (B) Fluorescence recovery of Sec7-GFP in wild-type cells during meiosis II. The plot represents the average of five experiments. Error bars represent the s.d. at each time point. (C) Representative images from the fluorescence recovery of Ady4p-GFP in an sso1Δ cell. Time points are indicated. Pre-bleach images are shown in both the DIC and GFP channels. The bleached spindle-pole body is marked with an arrowhead. Scale bars: 1 μm. (D) Fluorescence recovery of Ady4p-GFP in wild-type and sso1Δ cells during meiosis II. The plots represent the average of thirteen and ten experiments, respectively. Fluorescence recovery of Mpc54p-GFP wild-type and sso1Δ cells from Fig. 1D are also shown for comparison.
The prospore membrane does not sterically prevent the exchange of MOP components
Although cytoplasmic proteins are free to exchange, the prospore membrane might specifically obstruct the exchange of MOP components and thereby block recovery from photobleaching. Ady4p is a fourth protein of the MOP, although it is a non-essential component because loss of ADY4 does not block MOP assembly (Nickas et al., 2003). Ady4p-GFP was introduced on a plasmid into both wild-type and sso1Δ cells. Ady4p-GFP was functional because it rescued the sporulation defect of ady4Δ cells (supplementary material Table S3). In sharp contrast to the other MOP components, Ady4p-GFP displayed rapid recovery of fluorescence at the spindle-pole body in both wild-type and sso1Δ cells (Fig. 4C,D). The ability of Ady4p-GFP to exchange even in the presence of a prospore membrane indicates that the prospore membrane does not create an impermeable barrier to the exchange of all MOP components. Together, the Sec7p-GFP and Ady4p-GFP FRAP results argue against models in which the prospore membrane impedes fluorescence recovery by acting as a passive diffusion barrier.
Increased MOP stability correlates with membrane assembly
The experiments described demonstrate a requirement for prospore-membrane formation to stabilize the MOP, but do not address the issue of whether formation of a prospore-membrane cap is sufficient for stabilization, or whether more extensive elongation of the prospore membrane is necessary. To look more closely at the timing of the change in stability of the MOP, the exchange of components was examined in staged wild-type cells. As seen with a fluorescent marker, the prospore membrane expands in a series of distinct morphological stages (Diamond et al., 2009). An initial dot of fluorescence associated with the MOP resolves into a small horseshoe shape and then subsequently expands into larger structures (Fig. 5A). The transition from dot to horseshoe is thought to mark the change from clustered vesicles to an assembled membrane cap.
Fig. 5.

Vesicle fusion into a membrane structure reduces the exchange of MOP components. (A) Stages of prospore-membrane formation. Images are from wild-type cells coexpressing Mpc54p-GFP and RFP-Spo20p51-91. Arrowheads indicate prospore membranes. Scale bars: 1 μm. (B) Fluorescence recovery of Mpc54p-GFP from cells with RFP dots and from cells with RFP during meiosis II. The plots represent the average of nine and eight experiments, respectively. Error bars represent the s.d. at each time point. Fluorescence recovery of Mpc54p-GFP wild-type and sso1Δ cells from Fig. 1D are also shown for comparison.
Both a prospore-membrane marker (RFP-Spo20p51-91) and GFP-tagged Mpc54p were transformed into wild-type cells. FRAP was performed on MOPs associated with RFP-labeled dots (operationally defined as having a diameter of 0.4 to 0.65 μm) or horseshoes (0.65-1.1 μm at their longest point) (Fig. 5A). Mpc54p-GFP fluorescence recovered very slowly in MOPs associated with horseshoe-shaped prospore membranes, indicating that the presence of even a small prospore membrane was sufficient to stabilize the MOP (Fig. 5B). By contrast, Mpc54p-GFP fluorescence recovered significantly in MOPs associated with dots, comparable to the level of recovery seen in sso1Δ cells (Fig. 5B). The range of recovery was somewhat broader in wild-type cells with dots than in sso1Δ cells. Presumably this is because the dot-staged wild-type cells represent a mixture of MOPs associated with unfused, newly fused or fusing vesicles. Together, these data indicate that the change in MOP stability corresponds with initial formation of the prospore membrane.
Ady4p reduces the rate of exchange of Spo21p and Spo74p in the absence of a prospore membrane
Ady4p is unique among the MOP components in that it is not essential for MOP assembly (Nickas et al., 2003) and that it exchanges rapidly out of the MOP complex (Fig. 4C,D). In earlier work, we predicted that Ady4p might be an auxiliary stabilizer of the MOP (Nickas et al., 2003). To further define the role of Ady4p, FRAP measurements were taken for GFP-tagged MOP components both when ADY4 was overexpressed and in the absence of ADY4. If Ady4p acts as a stabilizing factor, then overexpression of ADY4 might rescue the high exchange of MOP components observed in sso1Δ mutants. To identify the cells containing the ADY4-overexpression plasmid, the prospore-membrane marker Dtr1p-RFP was coexpressed from the same plasmid (pADY4-DTR1-RFP). ADY4 in this construct was functional because it rescued the sporulation defect of ady4Δ cells (supplementary material Table S3). Genes encoding individual GFP-tagged MOP components and pADY4-DTR1-RFP were co-transformed into sso1Δ cells and the fluorescence recovery of GFP at the spindle-pole body was measured in RFP-labeled cells as the cells progressed through meiosis II. Overexpression of ADY4 reduced the rate and the degree of fluorescence recovery of both Spo21p-GFP and Spo74p-GFP, although not to wild-type levels (Fig. 6A,B). By contrast, Mpc54p-GFP exchange was not altered (Fig. 6C).
Fig. 6.

Overexpression of ADY4 decreases the rate of exchange of Spo21p and Spo74p in the absence of a prospore membrane. (A) Fluorescence recovery of Spo21p-GFP in sso1Δ cells overexpressing ADY4. Cells were analyzed during meiosis II. The green Spo21p-GFP plot represents the average of ten experiments. Fluorescence recovery of Spo21p-GFP in wild-type and sso1Δ cells from Fig. 1B is also shown for comparison. Error bars represent the s.d. at each time point. (B) Fluorescence recovery of Spo74p-GFP in sso1Δ cells overexpressing ADY4. The green Spo74p-GFP plot represents the average of ten experiments. Fluorescence recovery of Spo74p-GFP wild-type and sso1Δ cells from Fig. 1C are also shown for comparison. (C) Fluorescence recovery of Mpc54p-GFP in sso1Δ cells overexpressing ADY4. The green Mpc54p-GFP plot represents the average of eight experiments. Fluorescence recovery of Mpc54p-GFP wild-type and sso1Δ cells from Fig. 1D are also shown for comparison.
Ady4p reduces the exchange of Mpc54p during prospore-membrane growth
The effect of ADY4 overexpression suggests that Ady4p might be important in stabilizing Spo21p and Spo74p at the MOP. If true, then the absence of ADY4 should increase the rate of exchange of these proteins. In fact, the rate of exchange for Spo21p, Spo74p and Mpc54p in an sso1Δ ady4Δ background was comparable to the sso1Δ single mutant (Fig. 7A), demonstrating that the loss of ADY4 had no additive effect on fluorescence recovery. However, it is possible that the high rate of exchange seen in an sso1Δ mutant might mask any modest increase caused by the deletion of ADY4; therefore, the FRAP of GFP-tagged MOP components was analyzed in ady4Δ SSO1 cells. An ady4Δ strain displays heterogeneous MOP and prospore-membrane morphologies within a single cell, including some MOPs that lack prospore membranes (Nickas et al., 2003). To ensure that the MOPs examined in the ady4Δ background were associated with prospore membranes, and were therefore functional, RFP-Spo20p51-91 was expressed in these cells and only MOPs with associated RFP-labeled prospore membranes were photobleached (Fig. 7B). In the sso1Δ background, deletion of ADY4 had no effect on the exchange of Spo21p-GFP or Spo74p-GFP (Fig. 7C). Both proteins display the same limited recovery seen in wild-type cells. By contrast, modest fluorescence recovery of Mpc54p-GFP was observed in the ady4Δ strain. As analyzed by Student's t-test, the difference between the fluorescence recovery in wild-type and ady4Δ cells is significant to a confidence level of P<0.01 (Fig. 7B,D). These findings suggest that Ady4p assists in retaining Mpc54p in MOPs that are associated with prospore membranes.
Fig. 7.

Ady4p retains Mpc54p in the MOP during prospore-membrane growth. (A) Fluorescence recovery at 5 minutes after photobleaching for Spo21p-GFP, Mpc54p-GFP, and Spo74p-GFP. The graph compares fluorescence recovery in sso1Δ ady4Δ double mutants and sso1Δ single mutants. Cells were analyzed during meiosis II. The sso1Δ ady4Δ bars represent the average of nine, seven and nine experiments, respectively from left to right. Fluorescence recovery values of Spo21p-GFP, Mpc54p-GFP and Spo74p-GFP in sso1Δ cells are from Fig. 1B-D. Error bars represent s.d. (B) Representative images from the fluorescence recovery of Mpc54p-GFP in ady4Δ cells. RFP-Spo20p51-91 labels the prospore membrane. Time points are indicated. Pre-bleach images are shown in merged DIC/RFP/GFP channels and the GFP channel alone. The bleached spindle-pole body is marked with an arrowhead. Scale bars: 1 μm. (C) Fluorescence recovery of Spo21p-GFP and Spo74p-GFP in ady4Δ cells. Cells were analyzed during meiosis II. The green Spo21p-GFP plot represents the average of 14 experiments. The dark blue Spo74p-GFP plot represents the average of nine experiments. Fluorescence recovery of Spo21p-GFP in wild-type and sso1Δ cells from Fig. 1B are also shown for comparison. (D) Fluorescence recovery of Mpc54p-GFP in ady4Δ cells. The plot represents the average of 13 experiments. Fluorescence recovery of Mpc54p-GFP in wild-type and sso1Δ cells from Fig. 1D are also shown for comparison.
The physical connection between the MOP and the growing prospore membrane is unstable in ady4Δ mutants
To examine the effect of Ady4p on MOP structural integrity, three-dimensional (3D) electron microscope (EM) tomography was used to generate images of the MOP in both wild-type and ady4Δ cells. Consistent with earlier thin-section EM studies (Nickas et al., 2003), heterogeneous MOP defects were seen in the ady4Δ cells, including some spindle-pole bodies that had largely lost connection to the prospore membrane and others with fragmented MOPs. Importantly, even in those MOPs that seemed to be more similar to wild type, defects were seen (supplementary material Movies 1-6). In wild-type cells, the prospore membrane was always found in close apposition to the surface of the MOP (Fig. 8A,C,E). By contrast, in ady4Δ cells, MOPs frequently harbored regions in which this connection between the MOP and the prospore membrane was lost and the prospore membrane seemed to bubble away from the MOP surface (Fig. 8B,D,F). Additionally, the structure of the MOP seemed disorganized opposite these regions at which the prospore membrane was disconnected from the MOP surface (Fig. 8B,D,F). These disrupted areas were often quite small. In the example in Fig. 8F, the area of disorganization is ~60 nm in diameter and might be too small to be seen by thin-section EM. It is unclear whether the loss of connection leads to MOP disassembly or vice versa, but it is clear that Ady4p plays an integral role in maintaining the connection between the MOP and the prospore membrane.
Fig. 8.
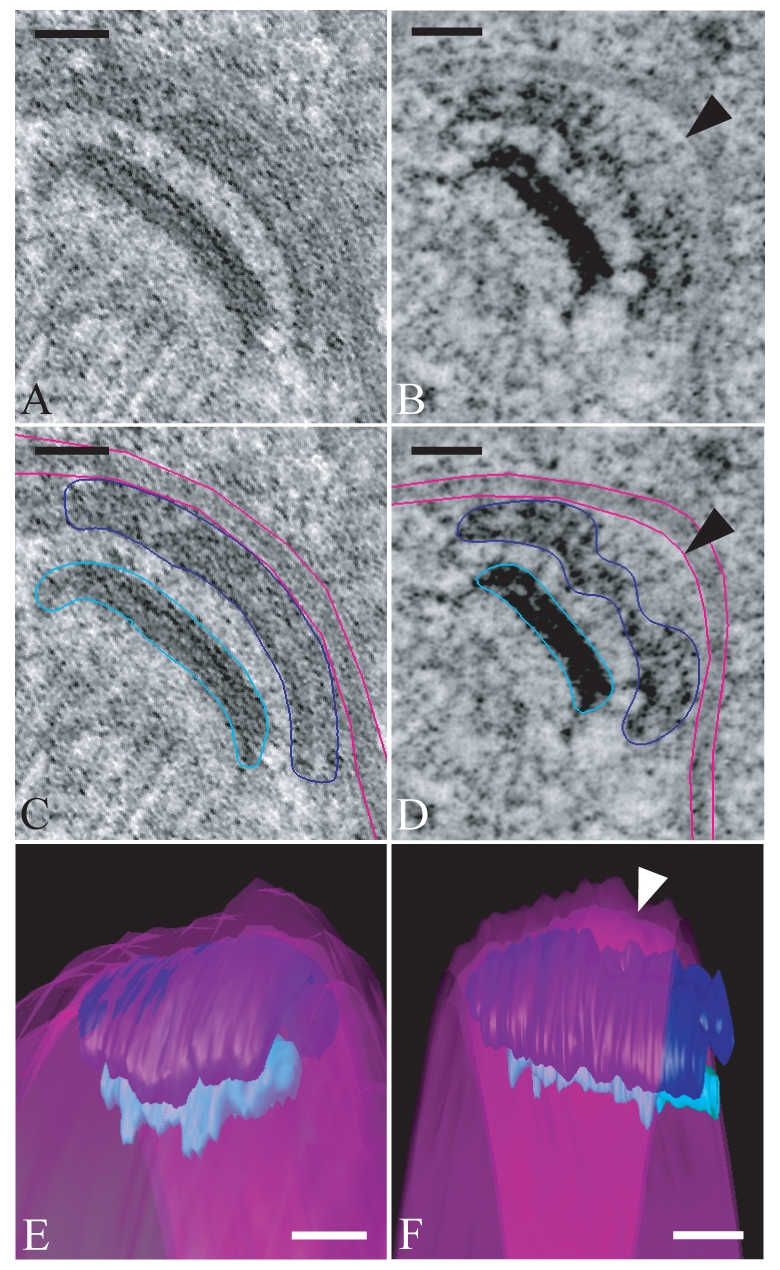
Localized disruptions of MOP structure are seen in an ady4Δ mutant. (A) A 10-nm tomographic slice of a meiosis II spindle-pole body in a wild-type cell showing the MOP in close association with the prospore membrane. (B) A 10-nm tomographic slice of a meiosis II spindle-pole body in an ady4Δ cell. Arrowhead indicates an area in which the connection between the prospore membrane and the MOP is lost. (C) Image from A with graphic overlay delineating the position of elements shown in the 3D reconstruction. The MOP (dark blue), prospore membrane (pink) and central plaque (light blue) are shown. (D) Image from B with graphic overlay delineating the position of elements shown in 3D reconstruction. The arrowhead indicates an area in which the connection between the prospore membrane and the MOP is lost. Color assignments are as in C. (E) Reconstruction based on the tomogram in A demonstrating the close association of the prospore membrane across the entire MOP surface. Color assignments are as in C. (F) Reconstruction based on the tomogram in B. The area of separation of the prospore membrane from the MOP surface is indicated by an arrowhead. Color assignments are as in (C). Scale bars: 50 nm. For movies of the complete tomograms and models available see supplementary material Movies 1-6.
Discussion
The prospore membrane stabilizes the MOP structure
Earlier studies examining the stability of the MOP component Mpc54p-GFP demonstrated that the protein is unable to exchange from a mature MOP but exchanges readily prior to the assembly of an organized MOP structure (Taxis et al., 2005). We report here that the full stability of components within the MOP requires not only the assembly of an organized structure but the initiation of prospore-membrane synthesis. Transmission electron microscopy studies have shown that the structure of the MOP seems to be the same when unfused vesicles are docked and when the prospore membrane is present (Nakanishi et al., 2006). However, we report that the behavior of individual MOP proteins is altered in these two conditions. FRAP analysis reveals that, in sso1Δ cells, in which vesicles never fuse, MOP components are dynamic and exchange freely. By contrast, MOP components do not exchange in wild-type cells, in which prospore membranes form adjacent to the surface of the MOP. This change is unique to the MOP because the constitutive spindle-pole-body components Cnm67p-GFP and Spc42p-GFP were stably localized in both conditions.
In our assays, the fluorescence recovery of Mpc54p, Spo21p and Spo74p only reached ~25% of the initial fluorescence. We determined that, over the 5-minute course of the experiment, fluorescence of unbleached MOPs dropped to ~75% of the initial fluorescence due to nonspecific photobleaching (E.M.M., unpublished data). Therefore, for complete exchange of the GFP-tagged MOP components, we would expect a recovery to 75% of their initial fluorescence, but we only observed a maximal recovery of 25% for these proteins, or 33% of their potential. Thus, only about one third of the MOP subunits are exchangeable prior to membrane formation. Why is exchange limited in this way? It seems likely that exchange involves the lateral movement of proteins in and out of the edge of the MOP, particularly for the coiled-coil proteins Mpc54p and Spo21p. If one conceives the MOP as a disc that is only exchanging at its edges, then exchange of 33% of the proteins components corresponds to exchange in a ring on the outside of the structure with a width that is 20% of the radius of the MOP. In this view, prior to membrane fusion, the MOP components found in the more central region of the complex fail to exchange because they cannot exchange directly from the interior. Alternatively, it is worth noting that, in the sso1Δ and spo14Δ mutants, unfused vesicles are docked across the surface of the MOP (Nakanishi et al., 2006). It is possible that interactions between vesicle membranes and the MOP at those sites of docking stabilize the MOP underneath the vesicles. Thus, the docking of vesicles at multiple points on the surface of the MOP in an sso1Δ cell would reduce the pool of exchangeable MOP components. Distinguishing between these possibilities will require the identification of mutants in which vesicle association with the MOP is abolished.
How does vesicle fusion confer stability to the MOP structure?
We determined that, in wild-type cells, the change in the stability of the MOP structure corresponds with the timing of the initial fusion of vesicles into a small prospore-membrane cap. Our data indicate that the MOP structure then remains static and stable throughout prospore-membrane formation.
One model through which vesicle fusion might stabilize the MOP is that the prospore membrane acts as a steric barrier to the exchange between proteins in the plaque and proteins in the soluble cytoplasmic pool. However, the observation that both Sec7p-GFP and Ady4p-GFP are able to exchange in the presence of a prospore membrane argues against this model. A second model is that the conversion of vesicles into a membrane stabilizes the MOP. There are several ways that the fusion of precursor vesicles could lead to a change in MOP stability: (1) the presence of a continuous sheet of membrane provides a large number of binding sites for MOP proteins, resulting in a decrease in the rate of exchange of those components that bind to the membrane. It should be noted, however, that no MOP protein(s) have yet been shown to directly interact with the prospore membrane. (2) Proteins involved in and stabilized by binding to the membrane might also stabilize other MOP components that surround them, through their interactions. In fact, each MOP component has been shown to interact with other spindle-pole-body components by two-hybrid assays (Knop and Strasser, 2000; Nickas et al., 2003). (3) The prospore membrane might stabilize the MOP by subtly altering the organization of the MOP components. Coincident with the fusion of vesicles, proteins might rearrange within the MOP, thereby locking them into a more stable conformation. The mechanism by which the presence of the prospore membrane stabilizes the MOP remains to be determined; however, our analysis of the ady4Δ mutant demonstrates the necessity of maintaining a stable MOP complex throughout prospore-membrane growth to ensure efficient spore formation.
Ady4p stabilizes MOP components at specific stages of prospore-membrane formation
Our results with ADY4 also support the idea of a structural change within the MOP coincident with membrane formation. The results presented here provide direct evidence that Ady4p is an auxiliary subunit that stabilizes the MOP complex. Furthermore, Ady4p enhances the binding of different MOP proteins during different stages of prospore-membrane formation. Prior to vesicle fusion, ADY4 overexpression decreases the exchange of Spo21p and Spo74p. Previously, Ady4p was shown to interact with the N-terminus of Spo21p by yeast two-hybrid, whereas Spo21p and Spo74p were shown to be co-dependent for recruitment to the spindle-pole body (Nickas et al., 2003). Perhaps the physical interaction of Ady4p with Spo21p allows Ady4p to directly influence the recruitment of both Spo21p and Spo74p. By contrast, after vesicles fuse into the prospore membrane, the loss of ADY4 does not have an effect on the exchange of Spo21p or Spo74p. However, the rate at which Mpc54p exchanges is increased in ady4Δ mutants. This alteration in MOP-component sensitivity to Ady4p dosage before and after vesicle coalescence is consistent with the idea that some rearrangement of the MOP occurs at the time of vesicle fusion into the prospore membrane and that this rearrangement leads to alterations in ADY4 dependence. Moreover, the increased exchange of Mpc54p from mature MOPs and the localized disorganization of the MOP revealed in the EM tomography of ady4Δ cells provide direct evidence for a role for Ady4p in maintaining the integrity of the mature MOP.
Ady4p maintains the interface between the MOP and the prospore membrane
Seven out of ten 3D reconstructions of MOPs from the ady4Δ strain displayed defects in the connection between the MOP and the prospore membrane or more severe structural defects. For those MOPs with attachment defects, the prospore membrane was seen to bubble away from small areas of the MOP surface, as if the connection between the MOP and the prospore membrane was lost in these areas. In the regions of the MOP opposite these prospore-membrane bubbles, the regular order of the MOP structure seemed to be disrupted. In most cases, this disordered region represented a small fraction of an otherwise intact MOP. It might be that the limited increase in exchange of Mpc54p-GFP that is seen by FRAP in the ady4Δ mutant represents a rapid exchange of Mpc54p from these small, disordered regions of the MOP.
Cells lacking ADY4 form asci that frequently contain fewer than four spores owing to the failure of a fraction of the prospore membranes to form properly (Nickas et al., 2003). In this study we observed that the majority of MOPs in ady4Δ cells analyzed by tomography had structural defects. Therefore, it seems likely that most spindle-pole bodies in ady4Δ cells have at least a modest structural defect. Perhaps if this structural defect remains limited then spore formation is not impaired. However, if these defects expand they might lead to a broader dissociation from the prospore membrane (e.g. supplementary material Movie 1) and a failure to properly form a spore. This interpretation implies that a minor loss of structural integrity within the MOP has the potential to cascade into more severe structural deficiencies in the absence of a stabilizing factor such as Ady4p.
How Ady4p promotes MOP stability remains unknown. Ady4p behaves quite differently than the other MOP components in that, on the basis of GFP fluorescence intensity, it is present in substoichiometric levels (A.M.N., unpublished observations) and it rapidly exchanges out of the MOP even in wild-type cells. Through interactions with other MOP components, Ady4p might simply act as a glue to hold the structure together, even if an individual molecule of Ady4p is not tightly associated with the structure. Alternatively, Ady4p might act catalytically to maintain the stability of the MOP, perhaps as a modifier of one of the subunits.
Materials and Methods
Yeast strains and media
Standard S. cerevisiae genetic methods and media were used (Rose and Fink, 1990). The strains used in this study are listed in supplementary material Table S4. All strains used were in the fast-sporulating SK-1 strain background (Kane and Roth, 1974). Gene insertions and replacements were performed using cassettes amplified by PCR (Longtine et al., 1998) and verified by PCR or phenotype. EMD85 (SPC42-GFP/SPC42-GFP) was constructed by crossing AN117-4B with ESM440, a MATa SPC42-GFP haploid obtained from Elmar Scheibel (Pereira et al., 2001), followed by mating of two of the SPC42-GFP segregants. EMD3 (sso1Δ/sso1Δ SPC42-GFP/SPC42-GFP) was made by crossing HI1, a MATα sso1Δ haploid (Nakanishi et al., 2006), with ESM440, followed by mating of two of the sso1Δ SPC42-GFP segregants. EMD4 (CNM67-GFP/CNM67-GFP) was made by inserting GFP-HIS3MX6 at the 3′ end of the CNM67 open reading frame (ORF) of AN117-16D and AN117-4B (Neiman et al., 2000) and then mating the resulting haploids. EMD6 (sso1Δ/sso1Δ CNM67-GFP/CNM67-GFP) was constructed by crossing HI1 with the MATa CNM67-GFP haploid, followed by mating two of the sso1ΔCNM67-GFP segregants. To construct EMD10 (sso1Δ/sso1Δ ady4Δ/ady4Δ), HI1 was crossed to an ady4Δ haploid, AN1119 (Nickas et al., 2003), followed by mating of two of the sso1Δady4Δ segregants. HI60 (spo14Δ/spo14Δ) was constructed as follows: an XbaI-ClaI DNA fragment from pKR466 (Rose et al., 1995) was used for targeted SPO14 disruption in two haploid wild-type cells, AN117-4B and AN117-16D. The resulting MATα haploid was then exposed to 5-FOA and a resistant spo14Δ::ura3 haploid was isolated. This haploid was mated to the MATa spo14Δ::URA3 haploid and the resulting diploid was plated on 5-FOA medium to select for spo14Δ::ura3/spo14Δ::ura3 convertants. To construct MND46 (ady4Δ/ady4Δ MPC54-GFP/MPC54-GFP), the following chromosomal insertions were made in AN117-16D and AN117-4B, respectively: HIS3MX6 replacing the ADY4 ORF and GFP-HIS3MX6 at the 3′ end of the MPC54 ORF. The resulting haploids were crossed, followed by the mating of two of the ady4Δ MPC54-GFP segregants. MND48 was constructed similarly, but with GFP-HISMX6 inserted into the 3′ end of the SPO21 ORF of AN117-4B.
Plasmids
The plasmids used in this study are listed in supplementary material Table S5. pRS426TEF-mRFP was constructed by cloning a HindIII-XhoI fragment carrying the gene for monomeric red fluorescent protein (RFP) (Campbell et al., 2002) into similarly digested pRS426TEF (Mumberg et al., 1995). The RFP gene was amplified by PCR using pTiKmRFP (Gao et al., 2005) as a template and YSO33 and HNO944 as primers (primer sequences are available upon request). pRS424-DTR1-RFP was constructed by replacing the GFP coding region in pRS424-DTR1-GFP (Nakanishi et al., 2006) with the gene for RFP using an EcoRI-XhoI fragment carrying the RFP of pRS426TEF-mRFP. pRS424-ADY4-DTR1-RFP was constructed by first amplifying ADY4 by PCR using AN117-16D genomic DNA as template and EMO76 and EMO78 as primers. This PCR product was digested with SacI and NotI and cloned into similarly digested pRS424-DTR1-RFP. To generate pRS426-ADY4-DTR1-RFP, a SacI-KpnI fragment containing ADY4 and DTR1-RFP from pRS424-ADY4-DTR1-RFP was cloned into similarly digested pRS426. pRS314-SPO21-GFP was a gift from Hiroyuki Tachikawa (University of Tokyo, Tokyo, Japan). pRS426-MPC54 was constructed by first amplifying MPC54 by PCR using AN117-16D genomic DNA as template and EMO75 and MNO185 as primers. This PCR product was digested with BamHI and XhoI and cloned into similarly digested pRS426.
Sporulation assays
Cells were induced to sporulate in liquid medium essentially as described previously (Neiman, 1998).
Live-cell imaging
Cells were induced to sporulate in liquid medium and were analyzed in early meiosis II. Glass-bottom tissue-culture dishes (MatTek Corporation, Ashland, MA) were prepared with agarose (1% KOAc, 2% agarose, 2 mM NaHCO3). A square of agarose was cut from the agarose plate so that the glass bottom was exposed. A total of 10 μl of cell culture was placed onto the agarose square and allowed to dry on its surface for 2 minutes. The agarose square was then inverted and returned to the glass bottom of the dish, so that the cells were flush with the glass. The time-series images for all photobleaching techniques were performed using a Zeiss LSM 510 META NLO two-photon laser-scanning microscope system (Central Microscopy Imaging Center, Stony Brook, NY). The culture plates were mounted on a Zeiss inverted Axiovert 200M microscope. All analyses were performed using a 100× oil objective (Plan-Neofluar numerical aperture 1.46). GFP was excited with an argon laser at 488 nm, and emission was collected using a 505-nm long-pass filter. During meiosis II, GFP-tagged MOP components localize to the four spindle-pole bodies, forming four GFP foci (Knop and Strasser, 2000). For each cell analyzed by FRAP, a single GFP-labeled MOP was bleached with 100% power of an argon laser at 488 nm for 10 seconds. The recovery of the bleached spindle-pole body was measured over a period of 5 minutes. Images were acquired using LSM 510 Meta version 3.2 imaging software. The images were processed and presented using Adobe Photoshop. The fluorescence intensity of the bleached area was quantified using the mean region of interest (ROI) function of the LSM imaging software and graphically depicted using Microsoft Excel. The initial fluorescence was normalized to 100% fluorescence. The first time point after bleaching was designated time zero and was set to 0% fluorescence. Spindle-pole bodies are mobile in the cell and occasionally move outside the focal plane. To prevent these focal anomalies from affecting the results, FRAP values that were less than 35% of the preceding time point were removed. For Ady4p-GFP, fluorescence recovered so rapidly in both wild-type and sso1Δ cells that time zero did not achieve baseline fluorescence. Time zero was set to 0% nonetheless.
Cell lysis
The lysis protocol was adapted from previously described methods (Knop et al., 1999). Briefly, cells were grown in sporulation medium. When 25-40% of the cells were observed by DAPI staining to be in meiosis II, 1×108 cells were harvested. Cells were resuspended in 25% glycerol and frozen at −80°C. Prior to lysis, cells were thawed on ice for 15 minutes, washed and then resuspended in 1 ml of cold water. The cell suspensions were mixed with 150 μl of 1.85 M NaOH, 7.5% β-mercaptoethanol (freshly prepared) and placed on ice for 15 minutes. Then 150 μl of 55% trichloroacetic acid (TCA) (w/v; stored in the dark at 4°C) was added and the mixture was incubated for 10 minutes on ice. The cells were pelleted (10 minutes at 16,000 g, at 4°C or at room temperature) and the supernatant was removed. This step was repeated to remove all residual traces of TCA. The pellet was resuspended in 100 μl HU-buffer (8 M urea, 5% SDS, 200 mM Tris pH 6.8, 1 mM EDTA, 0.01% bromophenol blue, 1.5% DTT; the buffer is stored without DTT at −20°C). Cells were resuspended using a sonicator. If the buffer capacity of the HU-buffer was not high enough to neutralize the remaining traces of TCA (yellow color), 4 μl of 2 M Tris-base was added. The samples were then heated for 10 minutes at 65°C and centrifuged for 2.5 minutes at 16,000 g at room temperature. Samples were stored at −20°C.
Western blot analysis
A total of 1 μl from each fraction was loaded, separated on 10% SDS-PAGE and transferred to a nitrocellulose membrane. GFP-tagged MOP components were blotted with a monoclonal anti-GFP antibody (Clontech/BD Biosciences, Palo Alto, CA) and horseradish peroxidase (HRP)-conjugated anti-mouse secondary antibody (GE Healthcare, Buckinghamshire, UK), and detected with Amersham ECL Plus Western Blotting Detection Reagents (GE Healthcare). Porin expression was used as a loading control in this experiment and was blotted with monoclonal anti-porin (Molecular Probes, Eugene, OR) and HRP-conjugated anti-mouse secondary antibody (GE Healthcare). Detection was performed using the Fujifilm Fluorescent Image Analysis (FLA-7000) system and Fujifilm Image Reader program (Fujifilm, Tokyo, Japan). Band intensities were analyzed using Fujifilm Multi Gauge Analysis Software (Fujifilm).
Electron microscopy
Cells were prepared for transmission electron microscopy as described elsewhere (Straight et al., 2000). Briefly, cells from sporulating cultures were collected by vacuum filtration to form a yeast paste. Cells were then rapidly frozen by high-pressure freezing (BAL-TEC HPM-010, Technotrade International, Manchester, NH) and freeze-substituted at −80°C in 2% osmium tetroxide plus 0.1% uranyl acetate in acetone for 3 days. The cells were gradually warmed to room temperature then infiltrated with Epon/Araldite resin over a period of 5 days. Sections (300 nm) of embedded cells were cut for electron tomography.
Electron tomography
10-nm colloidal gold (BBI International, Cardiff, UK) used as fiducials was adsorbed to 300-nm sections of sporulating cells, which were imaged using a Tecnai TF30 transmission electron microscope (Philips, Eindhoven, the Netherlands). Tilt-series (±60° with a tilt increment of 1°) were acquired using the automated tilt-series acquisition program SerialEM (Mastronarde, 2005). Tomographic reconstructions were computed by weighted back projection using the IMOD software program (Kremer et al., 1996). Models were generated using IMOD.
Supplementary Material
Acknowledgments
The authors wish to thank Elmar Schiebel (University of Heidelberg) for strains, Hiroyuki Tachikawa (University of Tokyo) for plasmids, and Nancy Hollingsworth (Stony Brook University) for comments on the manuscript and use of the Fujifilm FLA-7000 system. We would also like to thank Guo-Wei Tian and the Stony Brook Center for Microscopy for his instruction and assistance with the FRAP work. This work was supported by NIH grant GM062184 to A.M.N. This work was supported in part by National Institutes of Health Biotechnology Resources grant RR000592 to A. Hoenger. Deposited in PMC for release after 12 months.
Footnotes
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/123/14/2481/DC1
References
- Adams I. R., Kilmartin J. V. (1999). Localization of core spindle pole body (SPB) components during SPB duplication in Saccharomyces cerevisiae. J. Cell Biol. 145, 809-823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajgier B. K., Malzone M., Nickas M., Neiman A. M. (2001). SPO21 is required for meiosis-specific modification of the spindle pole body in yeast. Mol. Biol. Cell 12, 1611-1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullitt E., Rout M. P., Kilmartin J. V., Akey C. W. (1997). The yeast spindle pole body is assembled around a central crystal of Spc42p. Cell 89, 1077-1086 [DOI] [PubMed] [Google Scholar]
- Campbell R. E., Tour O., Palmer A. E., Steinbach P. A., Baird G. S., Zacharias D. A., Tsien R. Y. (2002). A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. USA 99, 7877-7882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond A. E., Park J. S., Inoue I., Tachikawa H., Neiman A. M. (2009). The anaphase promoting complex targeting subunit Ama1 links meiotic exit to cytokinesis during sporulation in Saccharomyces cerevisiae. Mol. Biol. Cell 20, 134-145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito R. E., Klapholz S. (1981). Meiosis and ascospore development. In The Molecular Biology of the Yeast Saccharomyces: Life Cycle and Inheritance (ed. Strathern E. W. J. J. N., Broach J. R.), pp. 211-287 Cold Spring Harbor, NY: Cold Spring Harbor Press; [Google Scholar]
- Gao X. D., Tachikawa H., Sato T., Jigami Y., Dean N. (2005). Alg14 recruits Alg13 to the cytoplasmic face of the endoplasmic reticulum to form a novel bipartite UDP-N-acetylglucosamine transferase required for the second step of N-linked glycosylation. J. Biol. Chem. 280, 36254-36262 [DOI] [PubMed] [Google Scholar]
- Jaspersen S. L., Winey M. (2004). The budding yeast spindle pole body: structure, duplication, and function. Annu. Rev. Cell Dev. Biol. 20, 1-28 [DOI] [PubMed] [Google Scholar]
- Kane S. M., Roth R. (1974). Carbohydrate metabolism during ascospore development in yeast. J. Bacteriol. 118, 8-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knop M., Strasser K. (2000). Role of the spindle pole body of yeast in mediating assembly of the prospore membrane during meiosis. EMBO J. 19, 3657-3667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knop M., Siegers K., Pereira G., Zachariae W., Winsor B., Nasmyth K., Schiebel E. (1999). Epitope tagging of yeast genes using a PCR-based strategy: more tags and improved practical routines. Yeast 15, 963-972 [DOI] [PubMed] [Google Scholar]
- Kremer J. R., Mastronarde D. N., McIntosh J. R. (1996). Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 116, 71-76 [DOI] [PubMed] [Google Scholar]
- Longtine M. S., McKenzie A., 3rd, Demarini D. J., Shah N. G., Wach A., Brachat A., Philippsen P., Pringle J. R. (1998). Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14, 953-961 [DOI] [PubMed] [Google Scholar]
- Mastronarde D. N. (2005). Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36-51 [DOI] [PubMed] [Google Scholar]
- Moens P. B., Rapport E. (1971). Spindles, spindle plaques, and meiosis in the yeast Saccharomyces cerevisiae (Hansen). J. Cell Biol. 50, 344-361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller E. G., Snydsman B. E., Novik I., Hailey D. W., Gestaut D. R., Niemann C. A., O'Toole E. T., Giddings T. H., Jr, Sundin B. A., Davis T. N. (2005). The organization of the core proteins of the yeast spindle pole body. Mol. Biol. Cell 16, 3341-3352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumberg D., Muller R., Funk M. (1995). Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156, 119-122 [DOI] [PubMed] [Google Scholar]
- Nakanishi H., de los Santos P., Neiman A. M. (2004). Positive and negative regulation of a SNARE protein by control of intracellular localization. Mol. Biol. Cell 15, 1802-1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi H., Morishita M., Schwartz C. L., Coluccio A., Engebrecht J., Neiman A. M. (2006). Phospholipase D and the SNARE Sso1p are necessary for vesicle fusion during sporulation in yeast. J. Cell Sci. 119, 1406-1415 [DOI] [PubMed] [Google Scholar]
- Neiman A. M. (1998). Prospore membrane formation defines a developmentally regulated branch of the secretory pathway in yeast. J. Cell Biol. 140, 29-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neiman A. M., Katz L., Brennwald P. J. (2000). Identification of domains required for developmentally regulated SNARE function in Saccharomyces cerevisiae. Genetics 155, 1643-1655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickas M. E., Schwartz C., Neiman A. M. (2003). Ady4p and Spo74p are components of the meiotic spindle pole body that promote growth of the prospore membrane in Saccharomyces cerevisiae. Eukaryotic Cell 2, 431-445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickas M. E., Diamond A. E., Yang M. J., Neiman A. M. (2004). Regulation of spindle pole function by an intermediary metabolite. Mol. Biol. Cell 15, 2606-2616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira G., Tanaka T. U., Nasmyth K., Schiebel E. (2001). Modes of spindle pole body inheritance and segregation of the Bfa1p-Bub2p checkpoint protein complex. EMBO J. 20, 6359-6370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinke C. A., Kozik P., Glick B. S. (2004). Golgi inheritance in small buds of Saccharomyces cerevisiae is linked to endoplasmic reticulum inheritance. Proc. Natl. Acad. Sci. USA 101, 18018-18023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedel C. G., Mazza M., Maier P., Korner R., Knop M. (2005). Differential requirement for phospholipase D/Spo14 and its novel interactor Sma1 for regulation of exocytotic vesicle fusion in yeast meiosis. J. Biol. Chem. 280, 37846-37852 [DOI] [PubMed] [Google Scholar]
- Rose K., Rudge S. A., Frohman M. A., Morris A. J., Engebrecht J. (1995). Phospholipase D signaling is essential for meiosis. Proc. Natl. Acad. Sci. USA 92, 12151-12155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose M. D., Winston F., Hieter P. (1990). Methods in Yeast Genetics Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; [Google Scholar]
- Rossanese O. W., Reinke C. A., Bevis B. J., Hammond A. T., Sears I. B., O'Connor J., Glick B. S. (2001). A role for actin, Cdc1p, and Myo2p in the inheritance of late Golgi elements in Saccharomyces cerevisiae. J. Cell Biol. 153, 47-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman J. E. (1994). Mechanisms of intracellular protein transport. Nature 372, 55-63 [DOI] [PubMed] [Google Scholar]
- Rout M. P., Kilmartin J. V. (1990). Components of the yeast spindle and spindle pole body. J. Cell Biol. 111, 1913-1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaerer F., Morgan G., Winey M., Philippsen P. (2001). Cnm67p is a spacer protein of the Saccharomyces cerevisiae spindle pole body outer plaque. Mol. Biol. Cell 12, 2519-2533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski R. S., Hieter P. (1989). A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122, 19-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogaard M., Tani K., Ye R. R., Geromanos S., Tempst P., Kirchhausen T., Rothman J. E., Sollner T. (1994). A rab protein is required for the assembly of SNARE complexes in the docking of transport vesicles. Cell 78, 937-948 [DOI] [PubMed] [Google Scholar]
- Sollner T., Whiteheart S. W., Brunner M., Erdjument-Bromage H., Geromanos S., Tempst P., Rothman J. E. (1993). SNAP receptors implicated in vesicle targeting and fusion. Nature 362, 318-324 [DOI] [PubMed] [Google Scholar]
- Straight P. D., Giddings T. H., Jr, Winey M. (2000). Mps1p regulates meiotic spindle pole body duplication in addition to having novel roles during sporulation. Mol. Biol. Cell 11, 3525-3537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suda Y., Nakanishi H., Mathieson E. M., Neiman A. M. (2007). Alternative modes of organellar segregation during sporulation in Saccharomyces cerevisiae. Eukaryotic Cell 6, 2009-2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taxis C., Keller P., Kavagiou Z., Jensen L. J., Colombelli J., Bork P., Stelzer E. H., Knop M. (2005). Spore number control and breeding in Saccharomyces cerevisiae: a key role for a self-organizing system. J. Cell Biol. 171, 627-640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickner W., Schekman R. (2008). Membrane fusion. Nat. Struct. Mol. Biol. 15, 658-664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder T. J., Pearson C. G., Bloom K., Davis T. N. (2003). The Saccharomyces cerevisiae spindle pole body is a dynamic structure. Mol. Biol. Cell 14, 3494-3505 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}