

Abstract
To increase read-out speed, sensitivity or specificity, an often applied strategy in fluorescence-based biomolecular spectroscopy and imaging is to simultaneously record two or more of the fluorescence parameters: intensity, lifetime, polarization or wavelength. This review highlights how additional, to-date largely unexploited, information can be extracted by monitoring long-lived, photo-induced transient states of organic dyes and their dynamics. Two major approaches are presented, where the transient state information is obtained either from fluorescence fluctuation analysis or by recording the time-averaged fluorescence response to a time-modulated excitation. The two approaches combine the detection sensitivity of the fluorescence signal with the environmental sensitivity of the long-lived transient states. For both techniques, proof-of-principle experiments are reviewed, and advantages, limitations and possible applications for biomolecular cellular biology studies are discussed.
Keywords: fluorescence, transient state imaging, triplet state, trans–cis isomerization, modulated excitation
1. Introduction
Fluorescence spectroscopy and imaging have developed very strongly in the last 10–20 years, not the least for biomolecular studies. Fluorescence-based read-outs can offer high specificity and sensitivity and lend themselves well for miniaturization and automation (Eggeling et al. 2003; de Jong et al. 2005). Strong fluorescence signal levels can be combined with low background levels, suppressed by spatial confinement of the excitation volume and by separation of the spectrally (Stokes)-shifted fluorescence signal from the scattered excitation light. These high signal-to-background levels make fluorescence spectroscopy one of the major means by which single-molecule detection (SMD) and analysis for fundamental biomolecular studies are performed (see Haustein & Schwille 2004; Joo et al. 2008; Schuler & Eaton 2008 for reviews).
In fluorescence applications, where molecules are to be monitored at low concentrations or within short interrogation times, simultaneous registration of several independent parameters is an important means to increase the information content per amount of sample and time. For both high-throughput screening (de Jong et al. 2005) and SMD-based fundamental biomolecular studies (Eggeling et al. 2001; Widengren et al. 2006), this has proven to be very useful to minimize reagent consumption and increase throughput, and to increase accuracy and precision, respectively. Simultaneous read-out of several fluorescence parameters has also been successfully applied to a range of other fluorescence-based techniques, including read-out of DNA and protein microarrays (Schäferling & Nagl 2006), to flow cytometry as an analysis platform for high-throughput, high-content biological testing and drug discovery (Edwards et al. 2004), as well as within both wide-field and confocal fluorescence microscopy (Suhling et al. 2005).
In most applications, multiple parameter read-out is based on the recording of two or more of the traditional fluorescence parameters: intensity, emission wavelength, lifetime and polarization. However, the clear rationale for simultaneous registration of several parameters in biomolecular imaging also makes it interesting to consider additional independent read-out parameters. In this review, it will be discussed how parameters related to the population dynamics of photo-induced, long-lived, non- or weakly fluorescent transient states of fluorophores, generated by trans–cis isomerization, intersystem crossing or photo-induced charge transfer can be monitored and provide additional independent information in fluorescence-based biomolecular studies. Figure 1a shows a schematic drawing of the ground singlet, first excited singlet and the lowest triplet state of a typical organic dye molecule. An attractive feature of triplet states, as well as several other dark, or weakly emitting, photo-induced transient states is their long lifetimes. While the fluorescence lifetime of a singlet excited-state of a fluorophore is approximately 10−9 s, the lifetimes of these transient states are approximately 10−6 to 10−3 s. Consequently, these states have a factor of approximately 103 to 106 more times to interact with the immediate environment of the fluorophore, rendering them highly sensitive to the local environment. Their kinetics can thus change considerably because of small changes in the accessibility of quencher molecules or microviscosities, reflecting, e.g., a biomolecular interaction. Interestingly, this information has to-date only been exploited to a limited extent for biomolecular studies, mainly due to methodological constraints.
Figure 1.

(a) Jablonski diagram showing a three-state electronic model of a fluorophore, including the ground singlet state (S0), the first excited singlet state (S1) and the lowest triplet state (T1). k01, k10, kISC and kT are the rate constants for excitation from S0 to S1, relaxation of S1 to S0, intersystem crossing from S1 to T1 and relaxation of T1 to S0, respectively. (b) Experimental set-up for FCS. A continuous wave (CW) laser is fed into a confocal detection unit. The laser beam is deflected by a dichroic mirror (DM) and then focused by the microscope objective down to the diffraction limit. The dimensions of the laser beam, together with the collection efficiency function of the microscope define the detection volume (lower left) from which fluorescence is collected. The fluorescence passes the DM, is spectrally and spatially filtered by an emission filter (EM) and a pinhole (PH), located in the image plane. A beam-splitter divides the fluorescence into two paths, and fluorescence photons are eventually detected by avalanche photodiodes (APDs). The photocurrents of the APDs are fed into a PC-based correlator. (c) Calculated FCS curve, given by equation (1.3), for a solute undergoing three-dimensional diffusion through an observation volume (b) as well as transitions to and from a non-fluorescent triplet state (a). τAB and τT denote the anti-bunching and the triplet state relaxation times. Here, a diffusion time of τD = 50 µs and on average one molecule in a three-dimensional Gaussian detection volume were assumed. The dotted line shows the diffusion-generated part of the FCS curve, denoted as GD(τ) in equation (1.3).
A range of techniques indeed exist to characterize these transient states and their population kinetics. Transient absorption spectroscopy is a well-established technique that has been extensively used to characterize fluorophore photodynamics (Korobov & Chibisov 1983; Chen & Chance 1993; van Amerongen & van Grondelle 1995). However, the technique is relatively technically complicated, lacks the sensitivity for measurements at low (less than micromolar) concentrations and is mainly restricted to cuvette experiments. This makes the technique less suited for biomolecular studies. Emission originating either directly (phosphorescence) or indirectly (delayed fluorescence) from the long-lived first excited triplet state can also be used for monitoring (Jovin & Vaz 1989; Cioni & Strambini 2002) and has been exploited for microscopic imaging (Marriott et al. 1991). However, coupled to the long-lived emission is also the susceptibility of the triplet state to dynamic quenching by oxygen and trace impurities, which can be circumvented only after elaborate and careful sample preparation, or by creating oxygen diffusion barrier shields around the phosphorescent probes (Lebedev et al. 2009). This quenching not only shortens the triplet lifetime but also makes the luminescence practically undetectable. Biomolecular monitoring by this read-out is thus largely restricted to deoxygenized, carefully prepared samples, or to larger, shielded and therefore less environment-sensitive probes, which restricts the applicability.
Like triplet states, most transient long-lived states of fluorophore molecules show very weak emission signals, or practically no emission at all (isomerized or photo-ionized states). This puts a restriction on the possibilities to monitor these states. On the other hand, it also opens for the monitoring of these states by the characteristic changes in the detected fluorescence that occur when the fluorophores transit to and from the non- or very weakly emitting transient states. By using the highly sensitive fluorescence signal as a read-out it is possible to combine a high signal level (given by the read-out of fluorescence photons) with a very prominent environmental sensitivity (given by the long lifetimes of the TRASTs). In the following, two different fluorescence-based approaches will be discussed based on this strategy.
1.1. Transient state monitoring by FCS
In fluorescence correlation spectroscopy (FCS), fluorescence fluctuations are recorded from a sparse average number of molecules passing through a focused laser beam in a confocal arrangement (figure 1b; Widengren & Mets 2002; Gösch & Rigler 2005; Haustein & Schwille 2007). Information on the translational diffusion coefficients and the absolute concentrations of the fluorescent molecules can be extracted from the average duration and the inverted relative amplitudes of the fluorescence fluctuations, respectively. Apart from translational diffusion, information can in principle be extracted from any molecular dynamic process in the nanosecond time range and longer that manifests itself as a change in fluorescence intensity. Transitions to and from a long-lived, dark state of the investigated fluorescent molecules generate fluorescence fluctuations, superimposed on those due to their translational diffusion into and out of the detection volume. Consequently, from these superimposed fluctuations, the population and the population kinetics of the transient state can be determined by FCS.
The emitted fluorescence, 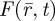 , from a fluorescent molecule observed at time t in an FCS experiment, and located at position
, from a fluorescent molecule observed at time t in an FCS experiment, and located at position 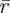 is proportional to the probability that the excited singlet state, S1, of the fluorophore is occupied at time t:
is proportional to the probability that the excited singlet state, S1, of the fluorophore is occupied at time t:
| 1.1 |
Here, Φf is the fluorescence quantum yield, k10 is the deactivation rate from S1 to the singlet ground state S0 and 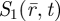 denote the occupation probability of S1 at time t and at location
denote the occupation probability of S1 at time t and at location  . The total detected fluorescence from the detection volume can then be expressed as:
. The total detected fluorescence from the detection volume can then be expressed as:
| 1.2 |
Here, ΦD is the detection quantum yield of the instrument, 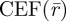 is the collection efficiency function and
is the collection efficiency function and 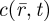 is the concentration of fluorescent molecules. The population kinetics is extracted from the fluorescence intensity fluctuations and analysed in terms of a normalized auto-covariance function, G(τ). For molecules undergoing electronic state transitions between their two lowest singlet states (S0, S1) and a dark triplet state (T1), as well as diffusion into and out of the FCS detection volume, G(τ) can be expressed as:
is the concentration of fluorescent molecules. The population kinetics is extracted from the fluorescence intensity fluctuations and analysed in terms of a normalized auto-covariance function, G(τ). For molecules undergoing electronic state transitions between their two lowest singlet states (S0, S1) and a dark triplet state (T1), as well as diffusion into and out of the FCS detection volume, G(τ) can be expressed as:
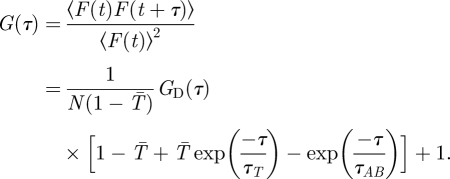 |
1.3 |
Here, N is the average number of fluorescent molecules in the detection volume. GD(τ) represents the diffusion-dependent part of G(τ), which decays with a time τD, corresponding to the average dwell times of the fluorescent molecules in the detection volume. 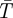 is the average steady-state probability for the fluorescent molecules within the detection volume to be in their triplet states. A schematic correlation curve given by equation (1.3) is depicted in figure 1c. In FCS, the time-dependent part of G(τ) (i.e. G(τ)−1) is proportional to the probability that a fluorescence photon is detected from a molecule at time t = τ, given that a fluorescence photon has been detected from the same molecule at time t = 0. This means, since a fluorescence photon was emitted at time t = 0, then the molecule has to be in its ground electronic state, S0. Thus, since F(t) is proportional to S1(t) (equation (1.1)), G(τ)−1 reflects the time development of S1(τ), with the initial condition that S0(0) = 1, S1(0) = 0 and T1(0) = 0. The relaxation times τAB and τT represent the equilibration times between the singlet states (typically occurring in the nanosecond time range, and referred to as the anti-bunching time), and between the two singlet states and the triplet state (microsecond time range), respectively. Eventually, as the time after the photon detection approaches τD, the fluorescent molecule diffuses out of the detection volume. The probability to detect a fluorescence photon from that particular molecule then approaches zero, and so does G(τ)−1 (figure 1c).
is the average steady-state probability for the fluorescent molecules within the detection volume to be in their triplet states. A schematic correlation curve given by equation (1.3) is depicted in figure 1c. In FCS, the time-dependent part of G(τ) (i.e. G(τ)−1) is proportional to the probability that a fluorescence photon is detected from a molecule at time t = τ, given that a fluorescence photon has been detected from the same molecule at time t = 0. This means, since a fluorescence photon was emitted at time t = 0, then the molecule has to be in its ground electronic state, S0. Thus, since F(t) is proportional to S1(t) (equation (1.1)), G(τ)−1 reflects the time development of S1(τ), with the initial condition that S0(0) = 1, S1(0) = 0 and T1(0) = 0. The relaxation times τAB and τT represent the equilibration times between the singlet states (typically occurring in the nanosecond time range, and referred to as the anti-bunching time), and between the two singlet states and the triplet state (microsecond time range), respectively. Eventually, as the time after the photon detection approaches τD, the fluorescent molecule diffuses out of the detection volume. The probability to detect a fluorescence photon from that particular molecule then approaches zero, and so does G(τ)−1 (figure 1c).
FCS measurements have proven useful for monitoring several different photo-induced transient states, including triplet states (Widengren et al. 1995), isomerized states (Widengren & Schwille 2000) and states generated by photo-induced charge transfer (Widengren et al. 1997). These states in turn reflect a range of environmental properties, including oxygen and other quencher molecule concentrations, viscosity, local redox environments and temperature. Potentially, there are many applications where a combination of the environmental sensitivity of these long-lived transient states and the signal strength of fluorescence would be of particular benefit. In figure 2a–c, an example is given showing how population dynamics of photo-induced transient states, as monitored by FCS, can provide additional and highly sensitive information about low-frequency molecular interactions. In the presented example, collisional interactions in a lipid membrane between a fluorophore-marked lipid molecule and lipids marked with an electron spin resonance (ESR) probe are measured via the influence of the ESR probe on the triplet state transitions of the fluorophore. The collisional interactions occur on a time scale too slow to essentially affect the fluorophores within the lifetimes of their excited states (approx. ns). However, because of the long lifetimes of the transient (here, triplet) state (approx. microseconds to milliseconds), a relatively large influence on its population and kinetic properties can be detected. As an additional example, figure 3a–c illustrates how transient state parameters, monitored by FCS, can be used as a measure of the extent to which fluorescence (or Förster) resonance energy transfer (FRET) occurs between two fluorophores, reflecting intra- or intermolecular distances (Widengren et al. 2001). Since the population and kinetics of the long-lived transient state is strongly dependent on the applied excitation rates, no matter the mode of excitation, also FRET-mediated excitation is clearly reflected in the transient state build-up and kinetics. In particular, given the relatively slow kinetics of these states, also fairly slow excitation rates, i.e. low FRET efficiencies, reflect themselves in the TRAST dynamics and can be detected. Thus, for both low-frequency collisional interactions and for low efficiency, long-range FRET, transient state read-outs can potentially provide a more sensitive read-out than can be offered by traditional fluorescence parameters.
Figure 2.

Low-frequency molecular collisions in lipid membranes monitored by FCS (Strömqvist et al. in preparation). (a) Fluorophore (Rhodamine Lissamine B)-labelled lipids were introduced into 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) liposomes (approx. 30 µm in diameter) with a density of less than 1 fluorophore per liposome. Lipids with the ESR-label TEMPO were introduced at different concentrations. (b) Three FCS curves, recorded under de-oxygenized conditions (568 nm, 0.07 MW cm−2 excitation), and from liposomes (approx. 30 nm in diameter) with different fractions of TEMPO-labelled lipids included (0% (filled squares), 0.13% (filled circles), 0.5% (filled triangles)). One per cent of TEMPO-labelled lipids corresponds to approximately 20 labelled lipid molecules per liposome. In the liposomes, the triplet state population of the lipid-anchored fluorophore was markedly influenced by the local surface concentration of TEMPO. Analysis of the excitation power-dependence of the triplet relaxation rate and population (τT and 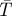 , in equation (1.3); Widengren et al. 1995) reveals that TEMPO increases both the formation and deactivation rates of the lowest triplet state (lower part of a). In the absence of molecular oxygen that acts as a triplet state quencher, the presence of a few TEMPO-labelled lipids in the liposomes is more clearly reflected in the triplet state parameters. Inset: triplet state parameters recorded from the same set of liposomes, at varying excitation intensities (from 0.03 to 0.2 MW cm−2).
, in equation (1.3); Widengren et al. 1995) reveals that TEMPO increases both the formation and deactivation rates of the lowest triplet state (lower part of a). In the absence of molecular oxygen that acts as a triplet state quencher, the presence of a few TEMPO-labelled lipids in the liposomes is more clearly reflected in the triplet state parameters. Inset: triplet state parameters recorded from the same set of liposomes, at varying excitation intensities (from 0.03 to 0.2 MW cm−2).
Figure 3.

Fluorescence (or Förster) resonance energy transfer (FRET) measurements via transient state parameters (trans–cis isomerization; Widengren et al. 2001). Oligonucleotides labelled with a donor (Rhodamine 110) and an acceptor (Cy5) fluorophore (b) were studied by FCS. The distance between the dyes varied from one sample to the other. In the FCS experiments, the oligonucleotide samples were subject to excitation of the donor, and the fluctuations of the acceptor fluorescence, generated by FRET from the donor, were analysed. In the acceptor dye, photo-induced isomerization (c, lower left) between a fluorescent all-trans state and a non-fluorescent cis state can occur (Widengren & Schwille 2000). Since this isomerization is light-driven in both directions (a, right, black, dashed lines), the rate of isomerization is proportional to the excitation rate. For the oligonucleotides, the excitation rate of both the trans and the cis state of the acceptor dye is FRET-mediated. The sum of the transition rates between the isomers, via the excited singlet states, S1, and by the rates of isomerization and back-isomerization, kISO and kBISO, thus directly reflects the FRET efficiency (a). (c) Three FCS curves recorded from oligonucleotides with 4, 13 and 18 base-pairs separation between the donor and acceptor dyes. The isomerization relaxation rates (i.e. the inverse isomerization relaxation times of the FCS curves) are proportional to the FRET efficiency of the oligonucleotide sample. Inset: Isomerization rates for different oligonucleotides and at different excitation intensities of the donor. For further details, see Widengren et al. (2001).
The FCS concept for transient state monitoring is applicable to a wide range of samples. However, FCSs as well as other forms of fluorescence fluctuation spectroscopy, relies on spontaneous fluctuations of individual molecules. Thus, only very few molecules (less than approx. 200) can be detected at a time, and the approaches depend on a high fluorescence brightness of the molecules investigated as well as on a high-detection sensitivity (Koppel 1974). Moreover, since the fluctuations of the transient states to be investigated typically take place in the microsecond time range, also a relatively high-time resolution of the detection is required. Total internal reflection (TIR)-mediated evanescent field excitation, combined with a highly sensitive fluorescence detection by an electron multiplied charge-coupled device (EM-CCD) camera, has been shown to allow FCS measurements with both autocorrelation (Kannan et al. 2006; Kolin et al. 2006) and more recently also cross-correlation fluctuation analyses of molecular diffusion and transport in molecular membranes (Sankaran et al. 2009). However, while sufficient for relatively slow dynamics, transient state fluctuations typically take place in the microsecond time range and require a time resolution far beyond that of an EM-CCD camera. Such a time resolution in combination with high detection sensitivity allows to date only a limited number of spots to be measured simultaneously (Blom & Gösch 2004). Alternatively, a compromise between the temporal analysis of FCS and fluorescence fluctuation analysis in the spatial domain (Petersen et al. 1993) can be obtained by exploiting the time structure of sample/laser scanning confocal microscope (LSCM) images (Digman et al. 2005; Xiao et al. 2005). Thereby, spatial correlation analysis of the emitted fluorescence is combined with temporal characterization of the fluorescence emission from the serial data stream of subsequently scanned pixels. This means, however, that the temporal characterization refers to the average of a sample distributed over a relatively large part of the image recorded, rather than to individual pixels. Taken together, FCS provides a relatively simple and widely applicable means to monitor long-lived transient states, at equilibrium and combining a strong fluorescence-based signal with the environmental sensitivity of the long-lived transient states. On the other hand, FCS-based approaches are essentially limited to dilute samples, place high demands on molecular brightness of the sample and require a combination of high sensitivity, time-resolution and noise suppression of the detection. It is therefore of interest to consider complementary approaches to FCS for transient state monitoring.
1.2. Transient state (TRAST) monitoring by time-modulated excitation
Recently, we presented a concept for monitoring photo-induced long-lived transient states that circumvents the above limitations of FCS, yet maintains the favourable combination of high-detection sensitivity (given by the read-out of fluorescence photons) and a strong environmental responsiveness (given by the long lifetimes of the transient states) (Sandén et al. 2007). The concept is based on modulation of the excitation source. The excitation source is typically modulated in the range of the relaxation time of the transient state (τT for the case of a triplet state, see figure 1a,c) of a fluorescent dye, and has an intensity that is high enough to drive the dye into this transient state under continuous wave (CW) excitation. With this modulated excitation, the transient state will be populated to significantly different degrees depending on the repetition rate, duration and height of the pulses. Given the variation of the transient state population, also the time-averaged fluorescence will vary systematically with the pulse characteristics. From the systematic variation of the plain time-averaged fluorescence, reflecting the corresponding population variations of the electronic states under modulated excitation, the transient state parameters can be extracted. In general, the pulse train characteristics can be varied in many ways (including variation of repetition rate, pulse duration, height and shape), but preferentially in such a manner that a sufficient fluorescence signal is maintained, yet allowing large contrasts in transient state populations to arise. Like FCS, the transient state information is in principle extracted from the probability of detecting a fluorescent photon from a fluorescent species at a certain time after the fluorescent molecule was found in its ground state, S0, i.e. at the onset of an excitation pulse. The concept is illustrated in figure 4a, for the case that the kinetics of singlet-triplet state transitions are to be determined.
Figure 4.

Modulated excitation approach for transient state (TRAST) imaging (Sandén et al. 2007). (a) Jablonski diagram, including the ground singlet (S0), excited singlet (S1) and the lowest triplet (T1) state. (b) Principal time development of the fluorescence intensity F(t) as a function of time after onset of excitation at time t = 0. Typically, the populations of S0 and S1 equilibrate in the nanoseconds time range (τAB, the anti-bunching relaxation time), while equilibration between the singlet states and the triplet state (τT, the triplet relaxation time) occurs in the time range of microseconds after onset of excitation. Finally, a steady-state is established with a constant population of T1, denoted 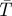 . (c) Variation of average fluorescence intensity with pulse duration: for excitation pulses with a duration pw1 < τT, the average recorded fluorescence intensity during excitation (
. (c) Variation of average fluorescence intensity with pulse duration: for excitation pulses with a duration pw1 < τT, the average recorded fluorescence intensity during excitation ( ) can be significantly higher than that recorded during a longer pulse with duration pw2 > τT (
) can be significantly higher than that recorded during a longer pulse with duration pw2 > τT (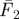 ). Ftot1 and Ftot2 denote the corresponding fluorescence intensity averaged over the total duration of the excitation pulse train (i.e. also including the time when the excitation is idle). (d) Typical variation with pulse width of the time-averaged fluorescence intensity (Ftot, left) and the fluorescence normalized by the excitation pulse train duty cycle (
). Ftot1 and Ftot2 denote the corresponding fluorescence intensity averaged over the total duration of the excitation pulse train (i.e. also including the time when the excitation is idle). (d) Typical variation with pulse width of the time-averaged fluorescence intensity (Ftot, left) and the fluorescence normalized by the excitation pulse train duty cycle (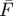 , right). (e) Example of an experimental realization of the excitation modulation using a stationary CW laser and an acousto-optical modulator.
, right). (e) Example of an experimental realization of the excitation modulation using a stationary CW laser and an acousto-optical modulator.
Unlike fluorescence-based single-molecule or fluctuation approaches like FCS, the presented approach does not rely on the ability to accurately record fluorescence time traces reflecting the spontaneous blinking behaviour of individual or a very low number of fluorescent molecules. The approach is therefore not restricted to samples displaying a high molecular fluorescence brightness, which is required for detection and monitoring of the fluctuation behaviour of individual molecules. This opens for the possibility to use a wide range of fluorophores. In particular, fluorophores with much higher triplet quantum yields than rhodamine dyes (approx. 1%) are thus from a fluorescence brightness point of view not a problem, but can instead provide higher triplet state populations and thus a better contrast and environment sensitivity. Further, the TRAST modulation approach is not restricted to low concentration samples, but can also analyse samples at higher concentrations, where the spontaneous fluorescence fluctuations tend to average out and therefore would not be detectable with FCS. Instead, limitations are set by the influence high concentrations may have on the fluorescence properties of the molecules themselves. Such effects (such as self-quenching and dye aggregation) are often observed at concentrations of 10 µM or higher. This is far beyond the typical concentration range of FCS and other fluorescence fluctuation techniques. Finally, in the TRAST approach, the time information is entirely kept in the modulation of the excitation. It is therefore fully compatible with low time-resolution detection, for instance by a charge-coupled device (CCD) camera, and with the possibility of a massively parallel read-out.
A first experimental realization of this approach was made by inserting an acousto-optic modulator into the stationary laser excitation source of a home-built FCS instrument (Sandén et al. 2007; figure 4e). Thereby, the TRAST data could be verified against FCS data, obtained under close to identical conditions.
Evidently, the time-modulated excitation experienced by a stationary sample can also be generated by translation of the sample with respect to the excitation, or vice versa. In this way, the excitation need not be idle, and in particular for low duty cycle excitation, a larger sample volume can be interrogated within the same period of time. Based on this notion, we recently established the TRAST imaging concept using a laser scanning confocal microscope (LSCM) (Sandén et al. 2008). The concept is outlined in figure 5a, together with two images displaying the triplet state population of the dye Rhodamine 110 in lipid vesicles (figure 5b,c).
Figure 5.

(a) Principle of the modulated excitation approach in an LSCM (Sandén et al. 2008). In an LSCM, variation of scanning speed/pixel dwell time is from the sample point of view equivalent to varying the duration of the excitation pulse of a stationary excitation field. With the imaging of the triplet state as an example, within pixel dwell times less than τT, only a minor build-up of T1 takes place (red). For slow scanning on the other hand, with the pixel dwell time greater than τT, a more prominent T1 build-up can take place (black). This leads to a lowered average fluorescence intensity recorded from each pixel within their pixel dwell times. (b,c) Triplet contrast images obtained by a conventional LSCM, showing liopsomes immobilized in a 1 per cent agarose gel. The liposomes contain the dye Rhodamine 110, and are kept under air (b) and argon (c) atmospheres, respectively. The triplet contrast image is generated by taking the ratio of the intensities from a fast scanned image with a pixel dwell time less than τT, divided by those from an image recorded with a slow scan, with pixel dwell times greater than τT. The colour scale shows this fluorescence intensity ratio, which scales with 1/(1−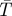 ). The brightness of the images scales with the estimated inverse relative error of these ratios (see Sandén et al. 2008 for further details). Upon deoxygenation (c), the triplet ratios are increased, as a consequence of a higher triplet state build-up in the absence of oxygen, which acts as a potent triplet state quencher.
). The brightness of the images scales with the estimated inverse relative error of these ratios (see Sandén et al. 2008 for further details). Upon deoxygenation (c), the triplet ratios are increased, as a consequence of a higher triplet state build-up in the absence of oxygen, which acts as a potent triplet state quencher.
As long as sufficient transient state populations can be induced, and proper corrections for possible fluorescence decay owing to photodegradation can be asserted, the proposed approach is compatible with a range of modalities for excitation time-modulation (including time-modulated wide-field excitation, moving arrays of laser foci or laser excitation fringe patterns), as well as various spatial confinement strategies of the excitation and/or the detection. Very recently, TRAST imaging by modulated wide-field excitation was applied to monitor the oxygen consumption upon contraction of individual smooth muscle cells (Geissbühler et al. 2010). Recently, we introduced TRAST monitoring by evanescent-field excitation via total internal reflection (TIR) (Spielmann et al. 2010). The experimental set-up, as well as the principle of measurement is shown in figure 6. TIR-mediated evanescent field excitation confines the excitation to a region close to a dielectric surface. This reduction of the excitation volume also reduces the overall photobleaching in the sample. This is particularly advantageous for measurements in closed reservoirs, e.g. in biological cells, where the replenishment of bleached molecules into the excitation volume is limited by the small volumes of the cells. Apart from the triplet state kinetics, also the population and kinetics of photo-oxidized states of the dye molecules could be extracted by the TIR set-up of figure 6. This illustrates that the approach is applicable not only to triplet states, but to several other photo-induced transient states, including photo-isomerized states and states generated by photo-induced charge transfer.
Figure 6.

TRAST monitoring by total internal reflection fluorescence microscopy (Spielmann et al. 2010). (a) Simplified state diagram of a typical organic dye molecule, including the ground singlet state (S0), the first excited singlet state (S1), the lowest triplet state (T1) and the oxidized radical state (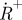 ). The parameters kisc, kT, kox and kred denote the rate constants for intersystem crossing from S1 to T1, relaxation of T1 to S0, oxidation of T1 to
). The parameters kisc, kT, kox and kred denote the rate constants for intersystem crossing from S1 to T1, relaxation of T1 to S0, oxidation of T1 to 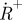 and reduction from
and reduction from 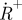 to S0, respectively. (b) Set-up and measurement principle for TRAST imaging with TIR-based evanescent wave excitation: laser beam for excitation passes through an AOM (green dashed lines). By the AOM modulation, pulse trains with increasing pulse widths but the same duty cycle, η, are generated, as shown in the upper left inset. The pulsed laser excitation is fed into a TIR microscope, and is focused off axis on the back focal plane of a microscope objective. After passing the objective, an evanescent excitation field is generated at the dielectric interface between the cover glass and the aqueous sample, which decays exponentially in the axial direction and forms a two-dimensional Gaussian profile in the radial dimension. The fluorescence generated is recorded by a charge-coupled device (CCD) camera in the image plane. The image of the fluorescence generated by this excitation field can be divided into regions of similar excitation intensities as shown on the CCD. For simplicity, only three regions have been represented, in reality the Gaussian is divided into more than 20 regions. CCD images are recorded for different excitation pulse widths. The fluorescence intensity within a certain region is normalized to 1 for short pulse widths (less than τT), and fluorescence response curves can then be generated for that particular region. In these curves, the fluorescence intensity decreases with increasing pulsewidths because of an increased population build-up in T1 and thereafter in
to S0, respectively. (b) Set-up and measurement principle for TRAST imaging with TIR-based evanescent wave excitation: laser beam for excitation passes through an AOM (green dashed lines). By the AOM modulation, pulse trains with increasing pulse widths but the same duty cycle, η, are generated, as shown in the upper left inset. The pulsed laser excitation is fed into a TIR microscope, and is focused off axis on the back focal plane of a microscope objective. After passing the objective, an evanescent excitation field is generated at the dielectric interface between the cover glass and the aqueous sample, which decays exponentially in the axial direction and forms a two-dimensional Gaussian profile in the radial dimension. The fluorescence generated is recorded by a charge-coupled device (CCD) camera in the image plane. The image of the fluorescence generated by this excitation field can be divided into regions of similar excitation intensities as shown on the CCD. For simplicity, only three regions have been represented, in reality the Gaussian is divided into more than 20 regions. CCD images are recorded for different excitation pulse widths. The fluorescence intensity within a certain region is normalized to 1 for short pulse widths (less than τT), and fluorescence response curves can then be generated for that particular region. In these curves, the fluorescence intensity decreases with increasing pulsewidths because of an increased population build-up in T1 and thereafter in 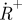 (lower left). (c) Measured fluorescence response curves of the dye Rhodamine 110 in aqueous solution, showing two distinct relaxation processes with increasing pulse widths, which can be attributed to triplet state build-up and photo-oxidation, respectively. (d) CCD images showing the triplet state build-up (left) and build-up of the photo-oxidized state
(lower left). (c) Measured fluorescence response curves of the dye Rhodamine 110 in aqueous solution, showing two distinct relaxation processes with increasing pulse widths, which can be attributed to triplet state build-up and photo-oxidation, respectively. (d) CCD images showing the triplet state build-up (left) and build-up of the photo-oxidized state 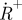 . In analogy to the images of figure 5b,c, these are formed as ratio images. The triplet state image (left) is formed as a ratio of two images recorded with excitation pulse widths w1 <τT and w2 > τT, but shorter than the build-up time of
. In analogy to the images of figure 5b,c, these are formed as ratio images. The triplet state image (left) is formed as a ratio of two images recorded with excitation pulse widths w1 <τT and w2 > τT, but shorter than the build-up time of 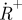 . The radical state image is formed as a ratio image from images recorded with the pulse width w2, and a pulse width, w3, longer than the build-up time of
. The radical state image is formed as a ratio image from images recorded with the pulse width w2, and a pulse width, w3, longer than the build-up time of 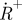 . The pulse widths w1, w2 and w3 are indicated in figure 6c.
. The pulse widths w1, w2 and w3 are indicated in figure 6c.
1.3. Concluding remarks
Photo-induced transient states of fluorescent dye molecules have in the last years received increased attention for several reasons. Photophysical properties of the fluorescently labelled molecules set the fundamental limits for the fluorescence flux, and the total number of photons emitted per molecule, which are highly relevant for all applications of fluorescence spectroscopy and imaging, where a high sensitivity or a fast read-out is important. However, rather than mere limiting factors for fluorescence signal strength and for generating blinking artefacts, photo-induced transient states of fluorophores and fluorescent proteins have also in the last few years found key roles in several biomolecular and cellular biology applications. Photo-switching into long-lived transient states can be exploited for protein transport and localization studies in cells (Lippincott-Schwartz et al. 2003). In general, photo-switching also provides a core mechanism in practically all recently developed approaches for fluorescence-based ultra-high resolution microscopy (Hell & Wichmann 1994; Betzig et al. 2006; Rust et al. 2006; Hell 2007). Moreover, and as emphasized in this review, the population kinetics of long-lived photo-induced transient states can contribute with additional dimensions of fluorescence-based information. Practically, all organic dye molecules are prone to undergo transitions into long-lived transient states upon excitation. However, many other fluorescent or luminescent molecules show excitation-driven transitions into various long-lived transient states, which are also of interest to be exploited in this context. In particular, most fluorescent proteins display a wealth of light-driven transitions occurring over a broad range of time scales (Haupts et al. 1998; Widengren et al. 1999; Jung et al. 2005; Blum & Subramaniam 2009). Simultaneous recording of two or more of the traditional fluorescence parameters: intensity, emission wavelength, lifetime and polarization is widely used as a means to increase the information content in fluorescence spectroscopy and imaging. A particular strength of the TRAST monitoring as suggested here is the long lifetimes of the monitored states, which make them interesting to exploit for micro-environmental monitoring, or for analysing rare molecular events or slow rate processes. There is a range of biomolecular events and processes that are too slow to significantly affect the excited states of fluorophore molecules, which typically decay in the nanosecond time range. Still, to date, these long-lived transient states have been used to a very limited extent for biomolecular studies. In this review, two approaches are discussed that use the characteristic changes of the strong fluorescence signal to monitor these states. Use of modulated excitation has been shown to make the read-out of these states more widely applicable. It seems reasonable to predict that the exploitation of these states as a read-out in biomolecular or cellular biology studies will increase in the future.
Acknowledgements
The development of TRAST imaging by modulated excitation is supported by the European Union 7th Framework Programme (FLUODIAMON 201837) and by the Swedish Research Council (60346701). Many persons have contributed to the work presented in this review. In particular, I would like to thank all the authors, colleagues and co-worker behind the work presented in Widengren et al. (2001), Sandén et al. (2007, 2008), Spielmann et al. (2010) and Strömqvist et al. (in preparation).
References
- Betzig E., Patterson G. H., Sougrat R., Lindwasser O. W., Olenych S., Bonifacino J. S., Davidson M. W., Lippincott-Schwartz J., Hess H. F. 2006. Imaging intracellular fluorescent proteins at nanometer resolution. Science 313, 1642–1645. ( 10.1126/science.1127344) [DOI] [PubMed] [Google Scholar]
- Blom H., Gösch M. 2004. Parallel confocal detection of single biomolecules using diffractive optics and integrated detector units. Curr. Pharm. Biotechnol. 5, 231–241. ( 10.2174/1389201043377011) [DOI] [PubMed] [Google Scholar]
- Blum C., Subramaniam V. 2009. Single-molecule spectroscopy of fluorescent proteins. Anal. Bioanal. Chem. 393, 527–541. ( 10.1007/s00216-008-2425-x) [DOI] [PubMed] [Google Scholar]
- Chen E., Chance M. R. 1993. Nanosecond transient absorption-spectroscopy. Metallobiochemistry, Part C, vol. 226, pp. 119–147. San Diego, CA: Academic Press Inc. [DOI] [PubMed] [Google Scholar]
- Cioni P., Strambini G. B. 2002. Tryptophan phosphorescence and pressure effects on protein structure. Biochim. Biophys. Acta-Protein Struct. Mol. Enzymol. 1595, 116–130. ( 10.1016/S0167-4838(01)00339-9) [DOI] [PubMed] [Google Scholar]
- de Jong L. A. A., Uges D. R. A., Franke J. P., Bischoff R. 2005. Receptor-ligand binding assays: technologies and applications. J. Chromatogr. B-Anal. Technol. Biomed. Life Sci. 829, 1–25. ( 10.1016/j.jchromb.2005.10.002) [DOI] [PubMed] [Google Scholar]
- Digman M. A., Brown C. M., Sengupta P., Wiseman P. W., Horwitz A. R., Gratton E. 2005. Measuring fast dynamics in solutions and cells with a laser scanning microscope. Biophys. J. 89, 1317–1327. ( 10.1529/biophysj.105.062836) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards B. S., Oprea T., Prossnitz E. R., Sklar L. A. 2004. Flow cytometry for high-throughput, high-content screening. Curr. Opin. Chem. Biol. 8, 392–398. ( 10.1016/j.cbpa.2004.06.007) [DOI] [PubMed] [Google Scholar]
- Eggeling C., Berger S., Brand L., Fries J. R., Schaffer J., Volkmer A., Seidel C. A. M. 2001. Data registration and selective single-molecule analysis using multi-parameter fluorescence detection. J. Biotechnol. 86, 163–180. ( 10.1016/S0168-1656(00)00412-0) [DOI] [PubMed] [Google Scholar]
- Eggeling C., Brand L., Ullman D., Jäger S. 2003. Highly sensitive fluorescence detection technology currently available for HTS. Drug Discovery Today 8, 632–641. ( 10.1016/S1359-6446(03)02752-1) [DOI] [PubMed] [Google Scholar]
- Geissbühler M., et al. 2010. Triplet imaging of oxygen consumption during the contraction of a single smooth muscle cell (A7r5). Biophys. J. 98, 339–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gösch M., Rigler R. 2005. Fluorescence correlation spectroscopy of molecular motions and kinetics. Adv. Drug Deliv. Rev. 57, 169–190. ( 10.1016/j.addr.2004.07.016) [DOI] [PubMed] [Google Scholar]
- Haupts U., Maiti S., Schwille P., Webb W. W. 1998. Dynamics of fluorescence fluctuations in green fluorescent protein observed by fluorescence correlation spectroscopy. Proc. Natl Acad. Sci. USA 95, 13 573–13 578. ( 10.1073/pnas.95.23.13573) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haustein E., Schwille P. 2004. Single-molecule spectroscopic methods. Curr. Opin. Struct. Biol. 14, 531–540. ( 10.1016/j.sbi.2004.09.004) [DOI] [PubMed] [Google Scholar]
- Haustein E., Schwille P. 2007. Fluorescence correlation spectroscopy: novel variations of an established technique. Ann. Rev. Biophys. Biomol. Struct. 36, 151–169. ( 10.1146/annurev.biophys.36.040306.132612) [DOI] [PubMed] [Google Scholar]
- Hell S. W. 2007. Far-field optical nanoscopy. Science 316, 1153–1158. ( 10.1126/science.1137395) [DOI] [PubMed] [Google Scholar]
- Hell S. W., Wichmann J. 1994. Breaking the diffraction resolution limit by stimulated-emission–stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 19, 780–782. ( 10.1364/OL.19.000780) [DOI] [PubMed] [Google Scholar]
- Joo C., Balci H., Ishitsuka Y., Buranachai C., Ha T. 2008. Advances in single-molecule fluorescence methods for molecular biology. Ann. Rev. Biochem. 77, 51–76. ( 10.1146/annurev.biochem.77.070606.101543) [DOI] [PubMed] [Google Scholar]
- Jovin T. M., Vaz W. L. C. 1989. Rotational and translational diffusion in membranes measured by fluorescence and phosphorescence methods. Methods Enzymol. 172, 471–513. ( 10.1016/S0076-6879(89)72030-9) [DOI] [PubMed] [Google Scholar]
- Jung G., Wiehler J., Zumbusch A. 2005. The photophysics of green fluorescent protein: influence of the key amino acids at positions 65, 203, and 222. Biophys. J. 88, 1932–1947. ( 10.1529/biophysj.104.044412) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan B., Har J. Y., Liu P., Maruyama I., Ding J. L., Wohland T. 2006. Electron multiplying charge-coupled device camera based fluorescence correlation spectroscopy. Anal. Chem. 78, 3444–3451. ( 10.1021/ac0600959) [DOI] [PubMed] [Google Scholar]
- Kolin D. L., Ronis D., Wiseman P. W. 2006. k-Space image correlation spectroscopy: a method for accurate transport measurements independent of fluorophore photophysics. Biophys. J. 91, 3061–3075. ( 10.1529/biophysj.106.082768) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppel D. E. 1974. Statistical accuracy in fluorescence correlation spectroscopy. Phys. Rev. A 10, 1938–1945. ( 10.1103/PhysRevA.10.1938) [DOI] [Google Scholar]
- Korobov V. E., Chibisov A. K. 1983. Primary photoprocesses in dye molecules. Uspekhi Khimii 52, 43–71. [Google Scholar]
- Lebedev A. Y., Cheprakov A. V., Sakad S., Boas D. A., Wilson D. F., Vinogradov S. A. 2009. Dendritic phosphorescent probes for oxygen imaging in biological systems. ACS Appl. Mater. Interfaces 1, 1292–1304. ( 10.1021/am9001698) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott-Schwartz J., et al. 2003. Photobleaching and photoactivation: following protein dynamics in living cells. Nat. Cell Biol. (Suppl. 5), S7–S14. [PubMed] [Google Scholar]
- Marriott G., Clegg R. M., Arndt-Jovin D. J., Jovin T. M. 1991. Time resolved imaging microscopy: phosphorescence and delayed fluorescence imaging. Biophys. J. 60, 1374–1387. ( 10.1016/S0006-3495(91)82175-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen N. O., Höddelius P. L., Wiseman P. W., Seger O., Magnusson K. E. 1993. Quantitation of membrane-receptor distributions by image correlation spectroscopy: concept and application. Biophys. J. 65, 1135–1146. ( 10.1016/S0006-3495(93)81173-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rust M. J., Bates M., Zhuang X. 2006. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 3, 793–795. ( 10.1038/nmeth929) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandén T., Persson G., Thyberg P., Blom H., Widengren J. 2007. Monitoring kinetics of highly environment sensitive states of fluorescent molecules by modulated excitation and time-averaged fluorescence intensity recording. Anal. Chem. 79, 3330–3341. ( 10.1021/ac0622680) [DOI] [PubMed] [Google Scholar]
- Sandén T., Persson G., Widengren J. 2008. Transient state imaging for microenvironmental monitoring by laser scanning microscopy. Anal. Chem. 80, 9589–9596. ( 10.1021/ac8018735) [DOI] [PubMed] [Google Scholar]
- Sankaran J., Manna M., Guo L., Kraut R., Wohland T. 2009. Diffusion, transport, and cell membrane organization investigated by imaging fluorescence cross-correlation spectroscopy. Biophys. J. 97, 2630–2639. ( 10.1016/j.bpj.2009.08.025) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäferling M., Nagl S. 2006. Optical technologies for the read out and quality control of DNA and protein microarrays. Anal. Bioanal. Chem. 385, 500–517. ( 10.1007/s00216-006-0317-5) [DOI] [PubMed] [Google Scholar]
- Schuler B., Eaton W. A. 2008. Protein folding studied by single-molecule FRET. Curr. Opin. Struct. Biol. 18, 16–26. ( 10.1016/j.sbi.2007.12.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spielmann T., Blom H., Geissbuehler M., Lasser T., Wildengren J. 2010. Transient state monitoring by total internal reflection fluorescence microscopy. J. Phys. Chem. B. 114, 4035–4046. ( 10.1021/jp911034v) [DOI] [PubMed] [Google Scholar]
- Strömqvist J., Chmyrov A., Johansson S., Andersson A., Mäter L., Widengren J. In preparation Nitroxide spin-label quenching of fluorophore's triplet state as a tool for studying diffusion mediated reactions in lipid membranes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhling K., French P. M. W., Phillips D. 2005. Time-resolved fluorescence microscopy. Photochem. Photobiol. Sci. 4, 13–22. ( 10.1039/b412924p) [DOI] [PubMed] [Google Scholar]
- van Amerongen H., van Grondelle R. 1995. Transient absorption-spectroscopy in study of processes and dynamics in biology. Biochemical spectroscopy, vol. 246, pp. 201–226. San Diego, CA: Academic Press Inc. [DOI] [PubMed] [Google Scholar]
- Widengren J., Mets Ü. 2002. Conceptual basis of fluorescence correlation spectroscopy and related techniques as tools in bioscience. In Single molecule detection in solution, methods and applications (eds Zander C., Enderlein J., Keller R. A.), pp. 69–120. Viernheim, Germany: Wiley-VCH. [Google Scholar]
- Widengren J., Schwille P. 2000. Characterization of photoinduced isomerization and back-isomerization of the cyanine dye Cy5 by fluorescence correlation spectroscopy. J. Phys. Chem. A 104, 6416–6428. ( 10.1021/jp000059s) [DOI] [Google Scholar]
- Widengren J., Dapprich J., Rigler R. 1997. Fast interactions between Rh6G and dGTP in water studied by fluorescence correlation spectroscopy. Chem. Phys. 216, 417–426. ( 10.1016/S0301-0104(97)00014-1) [DOI] [Google Scholar]
- Widengren J., Kudryavtsev V., Antonik M., Berger S., Gerken M., Seidel C. A. M. 2006. Single-molecule detection and identification of multiple species by multiparameter fluorescence detection. Anal. Chem. 78, 2039–2050. ( 10.1021/ac0522759) [DOI] [PubMed] [Google Scholar]
- Widengren J., Mets Ü., Rigler R. 1995. Fluorescence correlation spectroscopy of triplet-states in solution: a theoretical and experimental-study. J. Phys. Chem. 99, 13 368–13 379. ( 10.1021/j100036a009) [DOI] [Google Scholar]
- Widengren J., Mets Ü., Rigler R. 1999. Photodynamic properties of green fluorescent proteins investigated by fluorescence correlation spectroscopy. Chem. Phys. 250, 171–186. ( 10.1016/S0301-0104(99)00255-4) [DOI] [Google Scholar]
- Widengren J., Schweinberger E., Berger S., Seidel C. A. M. 2001. Two new concepts to measure fluorescence resonance energy transfer via fluorescence correlation spectroscopy: theory and experimental realizations. J. Phys. Chem. A 105, 6851–6866. ( 10.1021/jp010301a) [DOI] [Google Scholar]
- Xiao Y., Buschmann V., Weston K. D. 2005. Scanning fluorescence correlation spectroscopy: a tool for probing microsecond dynamics of surface-bound fluorescent species. Anal. Chem. 77, 36–46. ( 10.1021/ac049010z) [DOI] [PubMed] [Google Scholar]