

Abstract
Background
Residual chronic myeloid leukemia disease following imatinib treatment has been attributed to the presence of quiescent leukemic stem cells intrinsically resistant to imatinib. Mesenchymal stromal cells in the bone marrow may favor the persistence and progression of leukemia by preserving the proliferation and self-renewal capacities of the malignant progenitor cells.
Design and Methods
BV173 or primary chronic myeloid leukemia cells were co-cultured with human mesenchymal stromal cells and imatinib-induced cell death was then measured. The roles of pro-and anti-apoptotic proteins and chemokine CXCL12 in this context were evaluated. We also studied the ability of BV173 cells to repopulate NOD/SCID mice following in vitro exposure to imatinib and mesenchymal stromal cells.
Results
Whilst imatinib induced dose-dependent apoptosis of BV173 cells and primary chronic myeloid leukemia cells, co-culture with mesenchymal stromal cells protected both types of chronic myeloid leukemia cells. Molecular analysis indicated that mesenchymal stromal cells reduced caspase-3 activation and modulated the expression of the anti-apoptotic protein Bcl-XL. Furthermore, chronic myeloid leukemia cells exposed to imatinib in the presence of mesenchymal stromal cells retained the ability to engraft into NOD/SCID mice. We observed that chronic myeloid leukemia cells and mesenchymal stromal cells express functional levels of CXCR4 and CXCL12, respectively. Finally, the CXCR4 antagonist, AMD3100 restored apoptosis by imatinib and the susceptibility of the SCID leukemia repopulating cells to the tyrosine kinase inhibitor.
Conclusions
Human mesenchymal stromal cells mediate protection of chronic myeloid leukemia cells from imatinib-induced apoptosis. Disruption of the CXCL12/CXCR4 axis restores, at least in part, the leukemic cells’ sensitivity to imatinib. The combination of anti-CXCR4 antagonists with tyrosine kinase inhibitors may represent a powerful approach to the treatment of chronic myeloid leukemia.
Keywords: chronic myeloid leukemia, mesenchymal stromal cells, chemokines, imatinib
Introduction
Mesenchymal stromal cells (MSC) of bone marrow origin contain a large proportion of self-renewing cells capable of differentiating into tissues of mesodermal origin. For this reason they are often referred to as mesenchymal stem cells. A unique feature of MSC is their ability to produce anti-proliferative and anti-apoptotic effects, which non-specifically suppress immune responses in a non-cognate-dependent fashion. We have recently shown that MSC-mediated immunosuppression is the result of cyclin D2 inhibition and leads to cell cycle arrest at the G0/G1 phase.1
The anti-proliferative effect of MSC is not confined to immune cells but is also extended to target cells of different tissue origin, including hematopoietic stem cells. In the bone marrow “niche” MSC play a crucial role in supporting hematopoiesis by providing hematopoietic progenitor cells the necessary cytokine and cell contact-mediated signals to self-renew and differentiate.2 Malignant hematopoiesis appears to be similarly affected by the presence of MSC, which confer tumor cells a better survival by preserving their proliferative capacity and self-renewal ability. There is evidence that leukemic cells grow and accumulate in close association with bone marrow MSC which might regulate their differentiation.3 In the case of acute lymphoblastic leukemia, MSC have been shown to regulate response to cytotoxic agents by directly interfering with their mechanisms of action.4
Chronic myeloid leukemia (CML) is caused by the malignant transformation of a hematopoietic stem cell and is characterized by the Philadelphia chromosome (Ph1), a reciprocal translocation of chromosomes 9 and 22, which generates the BCR/ABL fusion gene encoding a constitutively active tyrosine kinase. Imatinib, an ATP-competitive inhibitor of BCR/ABL kinase, has transformed the therapy of CML because the drug induces durable responses in a high proportion of patients.5 However, most patients continue to have low levels of residual disease independently of the presence of BCR/ABL mutations responsible for drug resistance. The inherent difficulty in eradicating the disease appears to be related to the inability of imatinib to target the CML stem cell. A quiescent population of BCR-ABL+ leukemic stem cells in primary Lin−CD34+CD38− CML stem cells can be detected at diagnosis.6 This cell fraction is particularly resistant to apoptosis induced by a wide range of pro-apoptotic stimuli, including tyrosine kinase inhibitors6–9 and persists following imatinib treatment in vivo. In contrast, leukemic CD34+ cells that are actively cycling die when exposed to imatinib in vitro.
Here we studied the effect of MSC on the intrinsic resistance of leukemic cells to imatinib, and also investigated an antagonist of the CXCR4/CXCL12 axis.
Design and Methods
Generation of mesenchymal stromal cells
Three milliliters of bone marrow cells were obtained from normal donors aged between 20 and 50 years old. All samples were obtained with written, informed consent in accordance with the Hammersmith Hospital and Queen Charlotte’s Hospital ethical committee requirements. To isolate MSC, bone marrow mononuclear cells separated by Ficoll-Paque (Amersham-Pharmacia, Piscataway, NJ, USA) were plated in 75 cm2 flasks (Costar, Cambridge, MA, USA) at a concentration of 50,000 mononuclear cells/cm2 in alpha modified Eagle’s medium (αMEM), with high glucose concentration (Gibco, UK), 10% fetal bovine serum (FBS, Lonza, UK), and 1% penicillin - streptomycin – amphotericin (Gibco BRL). After 72 h incubation at 37°C in a 5% CO2 atmosphere, non-adherent cells were removed. When 70–80% confluent, adherent cells were trypsinised and expanded for 3–5 weeks. Before their use in the experiments, MSC were checked for positivity of CD105, CD106, CD73, HLA-class I, and the lack of expression of CD45.
Mice
Non-obese diabetic – severe combined immunodeficient (NOD/SCID) mice used for the in vivo studies were obtained from Harlan-Olac Ltd. (Bicester, UK) and bred and maintained in a pathogen-free environment at Hammersmith Centre for Biological Services. The mice were between 6 and 10 weeks of age and all procedures were carried out in accordance with the Home Office Animal (Scientific Procedures) Act of 1986. Mice received 250 cGy total body irradiation from a 137Cs radiation source (0.57 Gy/min) before being intravenously injected with the cells in a total volume of 0.1 mL sterile phosphate-buffered saline (PBS). After 6 weeks, the mice were sacrificed by CO2 asphyxiation; bone marrow and spleen were collected and processed for FACS analysis.
Chronic myeloid leukemia cells and cell lines
The BV173 cell line is derived from a patient with lymphoid blast crisis of CML. Apheresis products of peripheral blood from four patients with chronic-phase CML were obtained after informed consent in accordance with institutional guidelines and the Declaration of Helsinki. In some experiments, CD34+ cells were separated using a magnetic cell sorting system (miniMACS; Miltenyi Biotec, Bergisch Gladbach, Germany) in accordance with the manufacturer’s recommendations. All cells were grown in Roswell’s Park Memorial Institute (RPMI) medium (Gibco, BRL) supplemented with 10% FBS and antibiotic/antimycotic solution. Cells were incubated at 37°C in 5% CO2 in a humidified cell culture incubator and fed every 2 days.
Treatment of cells
To study the effect of bone marrow stroma on CML cells, BV173 or primary CML cells were cultured at a density of 5×104 cells/well with and without an underlying confluent layer of MSC in 48-well plates for 48 h. Co-cultured leukemia cells were separated from the MSC monolayer by careful pipetting with ice-cold PBS (repeated twice), preserving the MSC monolayers. MSC contamination, assessed by FACS as the fraction of CD19-negative cells, was always less than 1%.
To study the effects of the imatinib and/or the CXCR4 antagonist, AMD3100, BV173 or CML cells were plated in 48-well plates containing subconfluent MSC (10:1 ratio). After 48 h, each single drug or their combination was added to cultures for a further 48 h.
To evaluate the role of soluble factors, BV173 or primary CML cells were cultured for 48 h physically separated from MSC using a transwell system (24-well plate, 3 mM pore filter, Corning, VWR International Ltd., Leicestershire, UK) and imatinib was then added for another 48 h.
For in vivo experiments, BV173 cells (8×106) were co-cultured with MSC in 25 cm2 flasks. Imatinib (1 μM) with or without AMD3100 (5 μM) was added after 48 h and cells incubated for an additional 48 h. BV173 cells were then harvested as described above, incubated for 4 h to remove any adherent cells, washed and then resuspended in PBS for intravenous injection. This method minimized contamination of BV173 cells by MSC (the fraction of CD19-negative cells before injection was always less than 0.1%, as quantified by FACS).
Flow cytometry analysis of CXCR4 expression
Monoclonal antibodies against human CD19-PE (BD PharMingen), and CXCR4-PE (clone 12G5, BD PharMingen, DAKO Cytomation) were used for flow cytometry analysis. PE-conjugated IgG1 and IgG2a control monoclonal antibodies were from BD Biosciences. Cell death was quantified by double staining with propidium iodide and annexin V (BD PharMingen).
Transmigration assays
For quantitative transmigration assays, BV173 cells (5×104 cells) were placed in the upper chamber of a transwell system (24-well plate, 5 μM-pore filter, Corning) in a total volume of 150 μL DMEM medium. Six hundred microliters of supernatants concentrated 10-fold, undiluted or diluted in DMEM containing 0.5% FCS, were placed in the lower chamber of the transwell. Further control wells assessed the chemotactic activity of recombinant CXCL12 at concentrations from 10 ng/mL to 1 μg/mL. Cells were harvested from the lower chamber after 3 h, and counted using a hemocytometer.
Western blotting
Western blotting was performed on whole cell extracts prepared by lysing cells in Nonidet P-40 lysis buffer (1% Nonidet P-40, 100 mM NaCl and 20 mM Tris-HCl, pH 7.4) or 200 mM NaCl, 50 mM Tris-HCl, pH 7.4, 2mM EDTA, 1% Triton X-100, 10% glycerol both with the addition of 10 mM NaF, 1 mM sodium orthovanadate, 30 mM Na β-glycerophosphate, and protease inhibitors (“Complete” protease inhibitor mixture, as instructed by the manufacturer, Roche Applied Science, Lewes, UK) on ice for 15 min. Insoluble material was removed by centrifugation, and protein concentration was determined by the Bio-Rad Dc protein assay (Bio-Rad, Hemel Hempstead, UK). Twenty micrograms of protein were size-fractionated using sodium dodecylsulfate poly-acrylamide gele electrophoresis, and electro-transferred onto Protran nitrocellulose membranes (Schliecher and Schuell, Dassel, Germany). Membranes were blocked in 5% BSA or 5% milk in Tris-buffered saline plus 0.5% Tween for 30 min at room temperature and then incubated with specific antibodies. Antibodies used were Bcl-XL, Bad, Bax, CXCL12 (BD Biosciences, Oxford), Caspase-3, Bim, Akt (New England Biolabs, Hitchin), and CXCR4 (Millipore Chemicon, Watford). Total Akt expression was assessed to confirm the equivalent loading of all samples.
Results
Mesenchymal stromal cells reduce imatinib-induced apoptosis of BV173 and primary chronic myeloid leukemia cells
We previously observed that when BV173 cells are cultured in the presence of MSC, they predominantly accumulate in the G0–G1 phase of the cell cycle and are protected from starvation-induced cell death.10 To assess whether MSC could also protect the cells from imatinib-induced apoptosis, BV173 cells were exposed to serial concentrations of imatinib in the presence or absence of MSC and after 48 h were analyzed using annexin-V antibody and propidium iodide staining (Figure 1A). While exposure of BV173 cells to imatinib for 48 h in control cultures induced cell death in a dose-dependent fashion (31±8.4 and 55±9.9% of annexin/propidium iodide+ cells, for imatinib 1 and 5 μM, respectively; Figure 1A–B), the proportion of dying BV173 cells in the presence of MSC was significantly reduced (8.2±3 and 29.6±10.6%, for imatinib 1 and 5 μM, respectively; P=0.013 and P=0.004, BV173 versus BV173+MSC with 1 and 5 μM imatinib, respectively) (Figure 1A, B).
Figure 1.

MSC protect imatinib-induced cell death of BV173 cells. BV173 cells were cultured alone or in the presence of MSC. After 48 h imatinib was added at different concentrations and apoptosis was quantified after a further 48 h by annexin-V and propidium iodide staining. A representative FACS plot for propidium iodide-gated annexin-positive cells (A) and histograms representing the mean of three or four individual experiments (B–C) are shown. (BV173 treated with imatinib 1 or 5 μM, with or without MSC: P=0.042 and P=0.025, respectively; CML treated with imatinib 0.1, 1 or 5 μM: P=0.012, P=0.036 and P=0.029, respectively).
Sensitivity to imatinib in the presence of MSC was also studied in primary cells from four patients with CML in chronic phase. Clinical and laboratory data are shown in Online Supplementary Table S1.
As shown in Figure 1C, the protective effect of MSC was also observed on primary CML cells obtained from patients’ peripheral blood. In fact, the proportion of apoptotic CML cells was reduced in presence of MSC from 47.2±15 to 16±6.7% and from 59±11.5 to 25.6±5% (imatinib 1 μM and 5 μM, respectively; P=0.029 and P=0.011). No significant protection was observed when CML cells were cultured and treated on a monolayer of endothelial cells (data not shown).
Mesenchymal stromal cells mediate the protection of all CD34+ chronic myeloid leukemia progenitors from imatinib
Only a small subpopulation of CML cells in the patients’ peripheral blood is formed of hematopoietic precursors and it has been proposed that it is the CD34+ fraction, which contains the leukemic stem cells resistant to imatinib.6 We, therefore, evaluated the sensitivity of the CD34+ population isolated from the peripheral blood of CML patients to imatinib in the presence or absence of MSC. A significant proportion of CD34+ progenitors from CML patients underwent apoptosis in a dose-dependent fashion in response to imatinib. As observed with unfractionated CML peripheral blood mononuclear cells, when CD34+ CML cells were exposed to imatinib in the presence of MSC, they showed a significant reduction in cell death compared to the cultures in which CD34+ were treated with imatinib without MSC (from 43.4±4.5 to 26.9±6.1 at imatinib 1 μM, P=0.0058; from 76.8±6.3 to 56.5±5 at imatinib 5 μM, P=0.004; Online Supplementary Figure S1).
Apoptotic pathways in chronic myeloid leukemia cells cultivated with mesenchymal stromal cells
Pro- and anti-apoptotic proteins of the Bcl-2 family play a pivotal role in the regulation of apoptotic cell death in mammalian cells. Modulation of proteins such as Bax and Bcl-XL can cause permeabilization of the mitochondrial outer membrane and hence the release of soluble molecules responsible for activation of the caspase cascade.11 To assess their role in the MSC-mediated protection from imatinib, we analyzed BV173 cells following treatment with imatinib. As shown in Figure 2, imatinib-induced death of CML cells was associated with strong activation of caspase-3, which was significantly inhibited when CML cells were treated in the presence of MSC. Interestingly, MSC were also effective in triggering the expression of Bcl-XL, thus reversing the inhibitory effect of imatinib on this anti-apoptotic protein.
Figure 2.
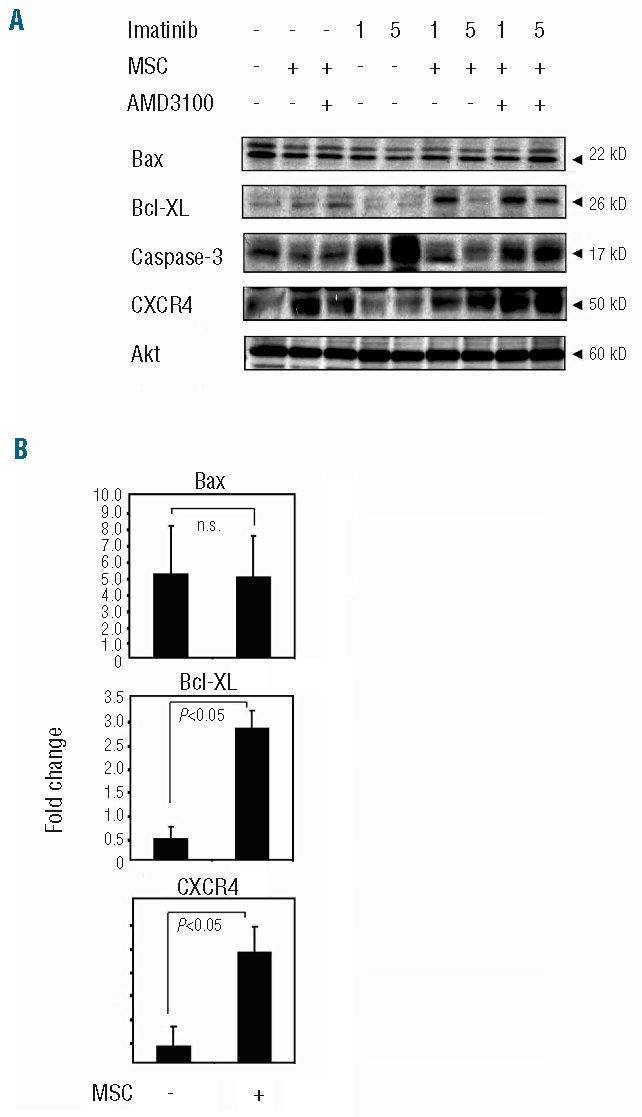
Changes in signal transduction pathways induced by MSC. Protein cell lysates were prepared from BV173 cell cultures after treatment with 1 or 5 μM imatinib for 72 h, in the presence or absence or MSC. Protein expression of Bcl-XL, Bax, CXCR4, caspase-3 and Akt was measured by western blot analysis (A). Densitometric data (B) are expressed as mean fold change from three independent experiments. All densitometric data were normalized for total Akt signals.
It has been shown that imatinib can modulate the chemokine receptor CXCR4 on CML cells, thus increasing interactions with the stroma.12 As illustrated in Figure 2 AB, BV173 cells showed significantly higher expression of CXCR4 when co-cultured with MSC, whereas this modulation was not noted in the presence of imatinib. To further define the role of the CXCL12/CXCR4 interaction in MSC/CML cross-talk, we analyzed the effect of the CXCR4 antagonist, AMD3100, on the pattern of expression of apoptotic proteins. Consistently with our FACS data, incubation of CML/MSC cells with AMD3100 was effective in restoring a higher level of expression of caspase 3 (Figure 2A). However, AMD3100 did not significantly affect Bcl-XL expression induced by MSC (Figure 2A) and there were no significant changes in the expression of Bax (Figure 2A–B) or in Bim and Bad (data not shown).
These data indicate that MSC protect CML cells from caspase-3-dependent cell death through a CXCR4-mediated mechanism. MSC protection of CML cells is also associated with Bcl-XL expression but in a CXCR4-independent manner.
Mesenchymal stromal cell-mediated protection of chronic myeloid leukemia cells from imatinib-induced cell death requires cell contact
It has been shown that the immunosuppressive effect of human MSC does not require cell contact and is mediated by several soluble factors. To investigate this issue in the protection of tumor cells from imatinib we used a transwell system in which BV173 or CML cells were physically separated from MSC and exposed to imatinib. In these conditions, the spontaneous cell death of BV173 cells was not reduced in the presence of MSC (BV173 alone 26±4%, BV173/MSC 23±3.6%; P=0.56, data not shown). The differences in the proportion of apoptotic BV173 cells produced by imatinib in the presence or absence of MSC, in the trans well system, were negligible (BV173/MSC, 46±8 and 69±7.5 at imatinib 1 and 5 μM, respectively as compared to BV173 cells treated alone, 48±6.5 % and 66±6 % at imatinib 1 and 5 μM, respectively) and the comparisons were not statistically significant (BV173 versus BV173/MSC, P=0.18 and P=0.09 at imatinib 1 and 5 μM, respectively). The transwell system was also used to assess protection of primary CML cells by MSC. Again, in absence of physical contact, MSC failed to protect primary CML cells from imatinib-induced cell death as the rate of cell death did not differ significantly between MSC/CML co-cultures (50.5±10.6 and 72.6±9.7 at imatinib 1 and 5 μM, respectively) as compared to CML cells treated alone (47.2±8.2% and 70±9.1 % at imatinib 1 and 5 μM, respectively; not shown). AMD3100 added to BV173 or primary CML co-cultured with MSC in transwells did not significantly change the rate of cell death induced by imatinib (data not shown). MSC must, therefore, be in physical contact with leukemic cells to protect them from imatinib-mediated cell death.
Mesenchymal stromal cells produce functional CXCL12
As a consequence of the evidence that the MSC-mediated protective effect requires cell contact, we focused on the CXCR4/CXCL12 axis because it is one of the central regulators of cell survival in solid tumors.13 BV173 and primary CML cells were first evaluated for CXCR4 expression by FACS analysis. To compare expression between the samples exactly, we defined the mean channel fluorescence ratio as the ratio between mean channel fluorescence for CXCR4 and the respective negative control. In agreement with previous studies,14 CXCR4 expression was demonstrated in BV173 cells (ratio 9.25±3.7) and, at lower levels, in peripheral blood CML cells (ratio from 2.2 to 4, mean ratio 3.1±0.73) as well as in CD34+ progenitors from all four patients (ratio from 1.4 to 3.3, mean ratio 1.9±0.9) (Figure 3A; Online Supplementary Table S1).
Figure 3.
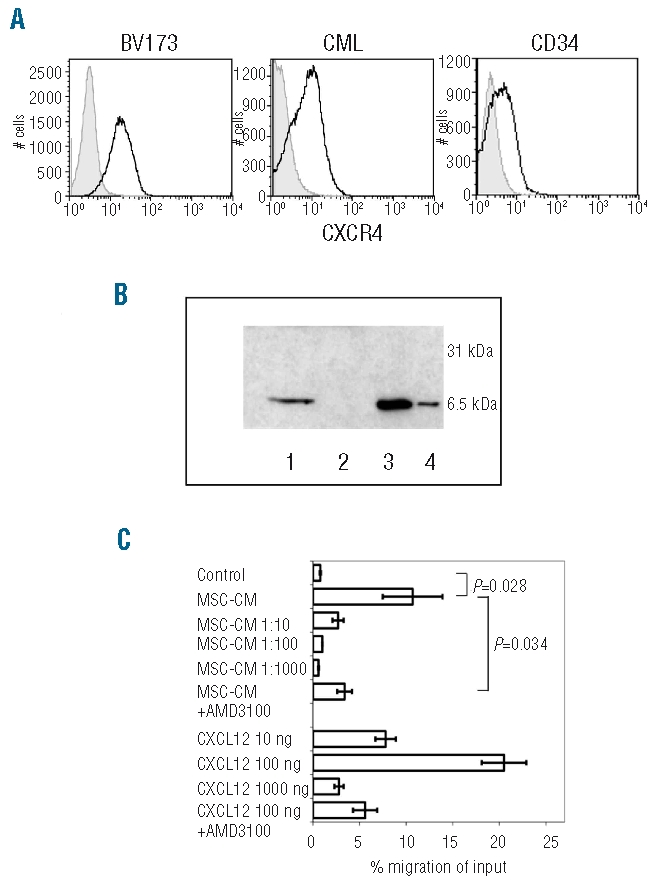
BV173 cells express the functional CXCL12 receptor, CXCR4 and migrate toward conditioned medium from MSC. (A) FACS plot of CXCR4 expression in BV173 cells, peripheral blood CML, and CD34+ CML cells. (B) Detection of CXCL12 in concentrated supernatant from MSC by immunoblot. MSC were cultured in 75 cm2 flasks until confluent. After 24 h culture in low-serum media, supernatants were harvested and concentrated by ultrafiltration. 1: MSC supernatant; 2: BV173 cell supernatant; 3: rCXCL12 50 ng; 4: rCXCL12 10 ng. (C) Migration of BV173 cells toward supernatant collected from MSC culture or toward rCXCL12 in a transwell system. 5x104 BV173 cells were added to the upper chamber of each well in a total volume of 150 mL DMEM 0.5% FBS. Undiluted or diluted supernatants from MSC were added in the lower chamber of the transwell. Cells that had migrated into the lower chamber after 3 h were collected and quantified using a hemocytometer. As a control, the chemotactic activity of rCXCL12 at different concentrations was tested. CM= supernatant from MSC cultures.
The production of CXCL12 by MSC was analyzed in MSC supernatants and detected by western blot analysis (Figure 3B). To test whether the CXCL12/CXCR4 axis exhibited functional activity, we investigated the chemotactic properties of MSC on BV173 cells in a transmigration assay. The BV173 cells migrated towards a CXCL12 gradient, demonstrating that CXCR4 was fully functional. As shown in Figure 3C, MSC supernatant exhibited potent chemoattractive activity on BV173 cells as 10.7 ± 3.2% of BV173 cells migrated towards undiluted supernatant compared to 0.8±0.06% of cells which migrated in the presence of media (P=0.028). Migration was also concentration-dependent as diluted supernatants were less chemoattractive (2.7±0.6% migration at a 1:10 dilution of the supernatant). To formally evaluate the role of CXCL12 in directing the transmigration of BV173 cells, the experiment was repeated in the presence of the CXCR4 antagonist AMD3100. The blockade of CXCR4 significantly reduced the fraction of BV173 cells (3.4±0.8%) migrating in response to MSC as well as the control chemokine CXCL12 (P=0.034; Figure 3C). All these data indicate the presence of a functional CXCR4/CXCL12 axis between CML cells and MSC and that this axis is prominent in driving migration.
The CXCR4 antagonist AMD 3100 restores sensitivity of chronic myeloid leukemia cells to imatinib in vitro
Having found that imatinib-induced apoptosis of CML cells was severely impaired in the presence of MSC, we tested the hypothesis that blocking CXCR4 could restore the effect of imatinib on CML cells. We did this by using the specific non-peptide CXCR4 antagonist, AMD3100. AMD3100 was added to co-cultures of BV173 or primary CML cells and MSC that were then exposed to imatinib. The addition of AMD3100 to primary CML cells (Figure 4A–B) or BV173 cells (Figure 4C) co-cultured in the presence of MSC significantly reduced the viability of leukemic cells treated with increasing concentrations of imatinib as compared to the cultures in which leukemic cells were exposed to imatinib in the presence of MSC but no AMD3100 (P=0.013, P=0.043, P=0.036 for BV173/MSC treated with imatinib in the presence or absence of AMD3100; P=0.047, P=0.009, P=0.042 for CML/MSC treated with imatinib in the presence or absence of AMD3100; Figure 4B–C). Disruption of the CXCR4/CXCL12 axis by AMD3100 was effective also when CD34+ CML progenitors were evaluated (Figure 5A). In fact, sensitivity to imatinib was significantly restored when the CXCR4 antagonist AMD3100 was added to CD34+ CML/MSC co-cultures (P=0.018 and P=0.025, imatinib 1 and 5 μM, respectively; Figure 5B).
Figure 4.

The CXCR4 antagonist, AMD3100 potentiates imatinib-induced cell death in BV173 and primary CML cell-MSC co-cultures: 5x104 primary CML (A and B) or BV173 (C) cells were cultured in 48-well plates alone or in the presence of a MSC monolayer. After 48 h, imatinib was added at different concentrations. Where indicated, AMD3100 (5 μg/mL) was added after the first 2 h of imatinib treatment. Apoptosis was quantified after a further 48 h incubation by annexin-V and propidium iodide staining. Histograms represent the mean of three independent experiments.
Figure 5.

CXCR4 blockade restores sensitivity of CD34+ CML progenitors to imatinib overcoming MSC protection: 5x104 CD34+ CML cells were cultured alone or in the presence of MSC. After 48 h imatinib was added at different concentrations and the fraction of annexin-V-positive cells was quantified by FACS after a further 48 h. A representative dot plot for annexin-positive cells (A) and average histograms from three individual experiments (B) are shown.
These data demonstrate that, by interfering with the CXCR4/CXCL12, AMD3100 is able to restore the sensitivity of leukemic cells, cultivated with MSC, to imatinib.
Mesenchymal stromal cells protect SCID leukemia-repopulating cells from imatinib via the CXCR4/CXCL12 axis
The data obtained thus far indicate that MSC protect leukemic cells from imatinib-induced apoptosis. To refine the target of their activity, we evaluated the effect of MSC on repopulating cells and tested this in a NOD/SCID model.15 BV173 cells were exposed to imatinib in the presence or absence of MSC and, after 48 h, harvested and injected into sublethally irradiated (250 cGy) NOD/SCID mice. Leukemic engraftment was monitored every week by enumerating human CD19+ cells in the peripheral blood of the recipients. After 6 weeks, mice were sacrificed and bone marrow and spleen analyzed for leukemic cells. The proportion of CD19+ cells in both the bone marrow and spleen of mice was significantly increased after 6 weeks when BV173 cells were treated with imatinib in the presence of MSC (BV173 + imatinib, 3±0.8%; BV173 + MSC + imatinib 13.3±4.07%; P=0.017; Figure 6A–B). These data support the notion that MSC may protect leukemia-repopulating cells from the effect of imatinib.
Figure 6.
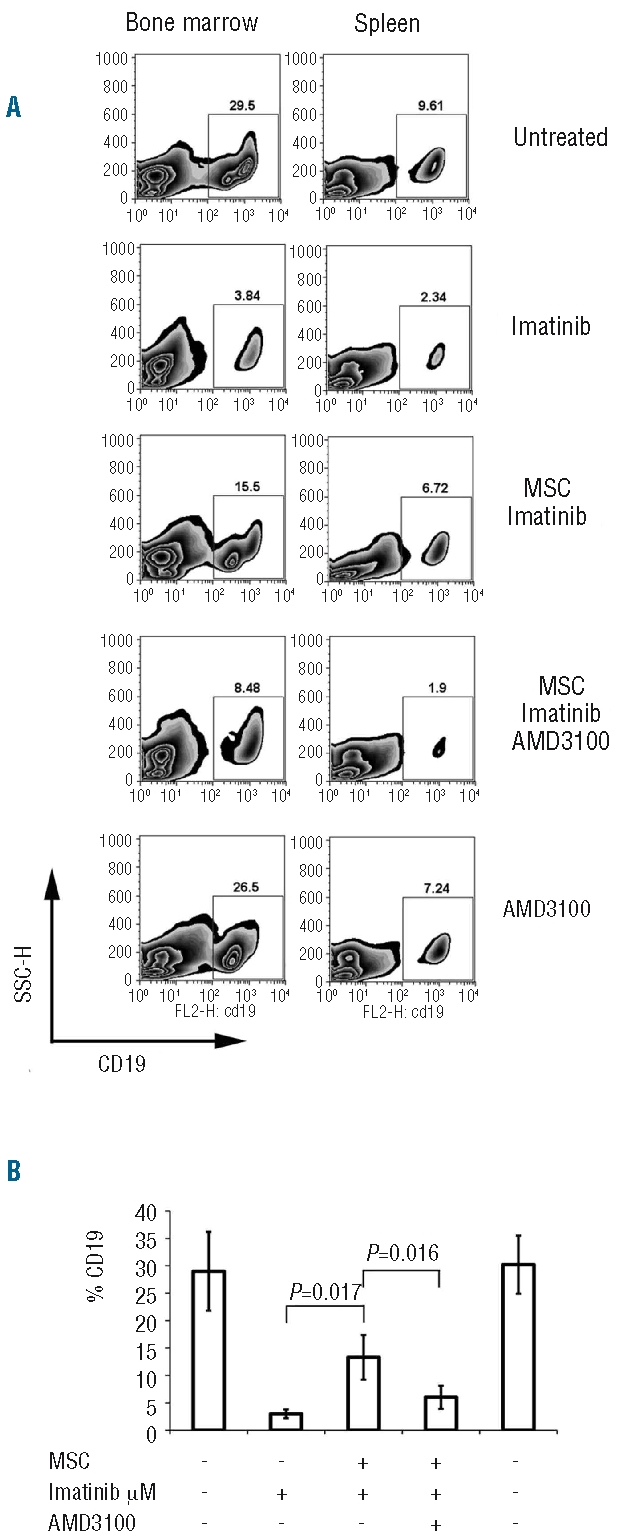
Bone marrow and spleen and SP engraftment of BV173 cells in NOD/SCID mice: 8x106 BV173 cells alone or with a monolayer of confluent MSC were cultured for 48 h before imatinib treatment (1 μM) for an additional 48 h. In some co-cultures, AMD3100 (5 μg/mL) was added after the first 24 h of culture. At the end of incubation, cells were gently harvested by pipetting with cold PBS and intravenously injected into mice (n=3–4 in each group). Bone marrow and spleens were harvested after 6 weeks. A representative FACS plot of CD19+ cells in the bone marrow and spleen (A) and cumulative number of CD19+ cells in bone marrow (B) quantified by FACS are shown.
Since our in vitro findings advocate that the CXCR4/CXCL12 interaction may be responsible for protecting CML cells from imatinib, BV173 cells, co-cultivated with MSC, were incubated with AMD3100 before being exposed to imatinib. After 48 h, BV173 cells were harvested from the cultures and injected into irradiated NOD/SCID recipients. The pretreatment with AMD3100 was associated with significant reductions in the percentages of human CD19+ cells engrafting in bone marrow and spleen (6.01±2.1%; P=0.016 BV173 + MSC + imatinib versus BV173 + MSC + imatinib + AMD3100; Figure 6 A–B), thus indicating that the CXCR4 antagonist was able to restore the sensitivity of leukemia-repopulating cells to imatinib. When BV173 cells were pretreated with AMD3100 in the absence of MSC, the fraction of CD19+ cells in the bone marrow and spleen of mice did not differ significantly from that in the untreated controls (Figure 6 A–B).
Discussion
The contribution of the stromal microenvironment to the development of a wide variety of tumors is supported by extensive clinical evidence and the use of experimental mouse models of cancer pathogenesis.16–19 It has been shown that tumor cells actively recruit stromal cells, including inflammatory cells, vascular cells, and MSC into the tumor, and that this recruitment is essential for the generation of a microenvironment that actively fosters tumor growth.20–22 In contrast to solid tumors that invade the bone marrow, leukemia originates in the marrow in which it is in close contact with stromal cells that provide growth and survival signals. We have previously shown that human MSC preserve the proliferative capacity and self-renewal ability of tumor cells by inhibiting cell cycling and protecting the tumor cells from apoptosis.10 The concept of MSC interactions is of particular importance in CML. Although a small population of non-cycling CD34+ cells appears to be intrinsically resistant to imatinib, most of the remaining CD34+ CML progenitors seem susceptible to the drug.6 However, the role of MSC in the response of CML cells to imatinib has not been clearly dissected.
In this study we provide evidence that a cell-contact-mediated interaction between MSC and CML cells effectively protects leukemia progenitors from imatinib-induced cell death. When CML cells are exposed to imatinib while in contact with MSC, their ability to engraft NOD/SCID mice is preserved, thus indicating that MSC protect SCID leukemia-repopulating cells – and probably CML progenitor cells – from the effect of imatinib. Protection from imatinib-induced cell death by MSC involves caspase-3 activation in a CXCR4-dependent manner. We found that the interaction between MSC and CML cells triggered the expression of Bcl-XL, although the role of this anti-apoptotic protein in the protection from imatinib-induced cell death has not been fully clarified. In fact, Bcl-XL did not correlate with the restored sensitivity to imatinib following CXCR4 blockade by AMD3100. It is possible that CXCR4 may affect the expression of other BCL family members, although this remains speculative.
We previously demonstrated that the reduction of spontaneous tumor cell apoptosis by MSC is associated with inhibition of cyclin D2 expression and upregulation of p27.1,10 In this study, we add further insights into MSC-mediated protection of CML cells by showing that a physical interaction between MSC and CML cells protects the tumor cells from drug-induced cell death in a CXCR4/CXCL12-dependent manner.
In fact, although imatinib has the ability to induce the apoptosis of a large proportion of leukemic cells – whether primary cells or a cell line – this effect is clearly impaired if the leukemic cells are in contact with MSC. This notion has fundamental implications in vivo, because it suggests that the persistence of leukemia following imatinib treatment may not be exclusively accounted for by the presence of an intrinsically resistance stem cell population but also by other leukemia stem cells that reside in the hematopoietic stem cell niche.
We found that the leukemia-stroma interaction and the consequences on tumor survival following exposure to imatinib involve the CXCR4-CXCL12 axis. The CXCR4 inhibitor, AMD3100, reduced the migration of CML cells in response to SDF-1 as well as to MSC, thus suggesting a role for SDF-1 in inducing CML cell migration to bone marrow MSC. More importantly, AMD3100 restored CML cell sensitivity to imatinib treatment in vitro and their content in SCID leukemia-repopulating cells.
The role of CXCL12/SDF-1 and its receptor CXCR4 has previously been investigated in other hematologic malignancies23 and, in accordance with our findings, AMD3100 disrupts the interaction of multiple myeloma cells with bone marrow stroma in vitro and in vivo, thus enhancing their sensitivity to bortezomib.24 Stroma has also been reported to play a role in the survival of other leukemias. Recently, Mishra et al. provided evidence that CXCL12 produced by stromal cells is responsible for protecting Bcr/Abl lymphoblasts from imatinib-induced cell death.25 In contrast to our findings, they found that the protective effect is mediated by soluble CXCL12 produced by MSC and independent of direct contact of lymphoma cells with stroma. However, biological differences between lymphoblasts and CML progenitors may account for this discrepancy as protection mediated by cell contact between stroma and other leukemias has been previously reported.26
Our and previous findings suggest that agents targeting this axis may be able to mobilize CML cells from their protective microenvironment and increase the therapeutic efficacy of imatinib.
The potential benefit of CXCR4 antagonist therapy in CML is supported by the observation that exposure of CML cells in stroma-free cultures to granulocyte colony-stimulating factor (G-CSF) enhances the effect of imatinib by increasing the proportion of cells in cell cycle.27 The recently reported failure of a clinical trial employing G-CSF to eradicate minimal residual disease following imatinib therapy28 may be related to a selective resistance of leukemic cells to be mobilized by G-CSF and/or to a regimen ineffective at doing so. It is well known that AMD3100 is more potent than G-CSF at inducing normal hematopoietic stem cell mobilization. The observed up-regulation of CXCR4 on CML cells induced by imatinib12 highlights the importance of concentrating on this axis with a view to interfering also with the migration of CML cells to the bone marrow mesenchymal niche.12,19 Our in vitro data demonstrate that migration of BV173 cells towards a CXCL12 gradient is impaired when CXCR4 is blocked by AMD3100. However, since the engraftment of BV173 cells pretreated with AMD3100 did not differ from that of controls, our data are more consistent with an effect of AMD3100 in disrupting CXCR4-mediated protection from the cytotoxic effect of imatinib.
Our data support the case for clinical trials aimed at mobilizing leukemic cells by disrupting the CXCR4-CXCL12 axis. Further investigations in pre-clinical models are warranted to identify the best administration schedule to maximize the sensitivity to imatinib.
Footnotes
Funding: this work was supported by the Leukaemia Research Fund.
DM: honoraria from Novartis, institutional research support by Novatis; JA: honoraria from Novartis, lecture fees from Novartis, institutional research support by Novatis.
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures
FD was the principal investigator and takes primary responsibility for the paper. FV, FV, VT, SL, KKH, and ARG performed the laboratory work for this study. FV and FD coordinated the research. FV and FD wrote the paper. DM, DB, JA and EWFL helped in the experimental design of the study.
The other authors reported no potential conflicts of interest.
References
- 1.Glennie S, Soeiro I, Dyson PJ, Lam EW, Dazzi F. Bone marrow mesenchymal stem cells induce division arrest anergy of activated T cells. Blood. 2005;105(7):2821–7. doi: 10.1182/blood-2004-09-3696. [DOI] [PubMed] [Google Scholar]
- 2.Arai F, Hirao A, Ohmura M, Sato H, Matsuoka S, Takubo K, et al. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell. 2004;118(2):149–61. doi: 10.1016/j.cell.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 3.Umiel T, Friedman S, Zaizov R, Cohen IJ, Gozes Y, Epstein N, et al. Long-term culture of infant leukemia cells: dependence upon stromal cells from the bone marrow and bilineage differentiation. Leuk Res. 1986;10(8):1007–13. doi: 10.1016/0145-2126(86)90253-5. [DOI] [PubMed] [Google Scholar]
- 4.Iwamoto S, Mihara K, Downing JR, Pui CH, Campana D. Mesenchymal cells regulate the response of acute lymphoblastic leukemia cells to asparaginase. J Clin Invest. 2007;117(4):1049–57. doi: 10.1172/JCI30235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Druker BJ, Guilhot F, O’Brien SG, Gathmann I, Kantarjian H, Gattermann N, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355(23):2408–17. doi: 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]
- 6.Graham SM, Jorgensen HG, Allan E, Pearson C, Alcorn MJ, Richmond L, et al. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood. 2002;99(1):319–25. doi: 10.1182/blood.v99.1.319. [DOI] [PubMed] [Google Scholar]
- 7.Holtz MS, Forman SJ, Bhatia R. Nonproliferating CML CD34+ progenitors are resistant to apoptosis induced by a wide range of proapoptotic stimuli. Leukemia. 2005;19(6):1034–41. doi: 10.1038/sj.leu.2403724. [DOI] [PubMed] [Google Scholar]
- 8.Copland M, Hamilton A, Elrick LJ, Baird JW, Allan EK, Jordanides N, et al. Dasatinib (BMS-354825) targets an earlier progenitor population than imatinib in primary CML but does not eliminate the quiescent fraction. Blood. 2006;107(11):4532–9. doi: 10.1182/blood-2005-07-2947. [DOI] [PubMed] [Google Scholar]
- 9.Jorgensen HG, Allan EK, Jordanides NE, Mountford JC, Holyoake TL. Nilotinib exerts equipotent antiproliferative effects to imatinib and does not induce apoptosis in CD34+ CML cells. Blood. 2007;109(9):4016–9. doi: 10.1182/blood-2006-11-057521. [DOI] [PubMed] [Google Scholar]
- 10.Ramasamy R, Lam EW, Soeiro I, Tisato V, Bonnet D, Dazzi F. Mesenchymal stem cells inhibit proliferation and apoptosis of tumor cells: impact on in vivo tumor growth. Leukemia. 2007;21(2):304–10. doi: 10.1038/sj.leu.2404489. [DOI] [PubMed] [Google Scholar]
- 11.Kim R, Emi M, Tanabe M. Role of mitochondria as the gardens of cell death. Cancer Chemother Pharmacol. 2006;57(5):545–53. doi: 10.1007/s00280-005-0111-7. [DOI] [PubMed] [Google Scholar]
- 12.Jin L, Tabe Y, Konoplev S, Xu Y, Leysath CE, Lu H, et al. CXCR4 up-regulation by imatinib induces chronic myelogenous leukemia (CML) cell migration to bone marrow stroma and promotes survival of quiescent CML cells. Mol Cancer Ther. 2008;7(1):48–58. doi: 10.1158/1535-7163.MCT-07-0042. [DOI] [PubMed] [Google Scholar]
- 13.Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121(3):335–48. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 14.Geay JF, Buet D, Zhang Y, Foudi A, Jarrier P, Berthebaud M, et al. p210BCR-ABL inhibits SDF-1 chemotactic response via alteration of CXCR4 signaling and down-regulation of CXCR4 expression. Cancer Res. 2005;65(7):2676–83. doi: 10.1158/0008-5472.CAN-04-2152. [DOI] [PubMed] [Google Scholar]
- 15.Bhatia M, Bonnet D, Murdoch B, Gan OI, Dick JE. A newly discovered class of human hematopoietic cells with SCID-repopulating activity. Nat Med. 1998;4(9):1038–45. doi: 10.1038/2023. [DOI] [PubMed] [Google Scholar]
- 16.Barcellos-Hoff MH, Ravani SA. Irradiated mammary gland stroma promotes the expression of tumorigenic potential by unirradiated epithelial cells. Cancer Res. 2000;60(5):1254–60. [PubMed] [Google Scholar]
- 17.Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature. 2004;432(7015):332–7. doi: 10.1038/nature03096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elenbaas B, Weinberg RA. Heterotypic signaling between epithelial tumor cells and fibroblasts in carcinoma formation. Exp Cell Res. 2001;264(1):169–84. doi: 10.1006/excr.2000.5133. [DOI] [PubMed] [Google Scholar]
- 19.Sieweke MH, Thompson NL, Sporn MB, Bissell MJ. Mediation of wound-related Rous sarcoma virus tumorigenesis by TGF-beta. Science. 1990;248(4963):1656–60. doi: 10.1126/science.2163544. [DOI] [PubMed] [Google Scholar]
- 20.Bhowmick NA, Chytil A, Plieth D, Gorska AE, Dumont N, Shappell S, et al. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science. 2004;303(5659):848–51. doi: 10.1126/science.1090922. [DOI] [PubMed] [Google Scholar]
- 21.Olumi AF, Grossfeld GD, Hayward SW, Carroll PR, Tlsty TD, Cunha GR. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999;59(19):5002–11. doi: 10.1186/bcr138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tlsty TD. Stromal cells can contribute oncogenic signals. Semin Cancer Biol. 2001;11(2):97–104. doi: 10.1006/scbi.2000.0361. [DOI] [PubMed] [Google Scholar]
- 23.Bradstock KF, Makrynikola V, Bianchi A, Shen W, Hewson J, Gottlieb DJ. Effects of the chemokine stromal cell-derived factor-1 on the migration and localization of precursor-B acute lymphoblastic leukemia cells within bone marrow stromal layers. Leukemia. 2000;14(5):882–8. doi: 10.1038/sj.leu.2401729. [DOI] [PubMed] [Google Scholar]
- 24.Azab AK, Runnels JM, Pitsillides C, Moreau AS, Azab F, Leleu X, et al. The CXCR4 inhibitor AMD3100 disrupts the interaction of multiple myeloma cells with the bone marrow microenvironment and enhances their sensitivity to therapy. Blood. 2009;113(18):4341–51. doi: 10.1182/blood-2008-10-186668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mishra S, Zhang B, Cunnick JM, Heisterkamp N, Groffen J. Resistance to imatinib of bcr/abl p190 lymphoblastic leukemia cells. Cancer Res. 2006;66(10):5387–93. doi: 10.1158/0008-5472.CAN-05-3058. [DOI] [PubMed] [Google Scholar]
- 26.Lagneaux L, Delforge A, Bron D, De Bruyn C, Stryckmans P. Chronic lymphocytic leukemic B cells but not normal B cells are rescued from apoptosis by contact with normal bone marrow stromal cells. Blood. 1998;91(7):2387–96. [PubMed] [Google Scholar]
- 27.Levesque JP, Hendy J, Takamatsu Y, Simmons PJ, Bendall LJ. Disruption of the CXCR4/CXCL12 chemotactic interaction during hematopoietic stem cell mobilization induced by GCSF or cyclophosphamide. J Clin Invest. 2003;111(2):187–96. doi: 10.1172/JCI15994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Drummond MW, Heaney N, Kaeda J, Nicolini FE, Clark RE, Wilson G, et al. A pilot study of continuous imatinib vs pulsed imatinib with or without G-CSF in CML patients who have achieved a complete cytogenetic response. Leukemia. 2009;23(6):1199–201. doi: 10.1038/leu.2009.43. [DOI] [PubMed] [Google Scholar]
- 29.Ramasamy R, Fazekasova H, Lam EW, Soeiro I, Lombardi G, Dazzi F. Mesenchymal stem cells inhibit dendritic cell differentiation and function by preventing entry into the cell cycle. Transplantation. 2007;83(1):71–6. doi: 10.1097/01.tp.0000244572.24780.54. [DOI] [PubMed] [Google Scholar]