

Abstract
Tetrapyrroles like hemes, chlorophylls, and cobalamin are complex macrocycles which play essential roles in almost all living organisms. Heme serves as prosthetic group of many proteins involved in fundamental biological processes like respiration, photosynthesis, and the metabolism and transport of oxygen. Further, enzymes such as catalases, peroxidases, or cytochromes P450 rely on heme as essential cofactors. Heme is synthesized in most organisms via a highly conserved biosynthetic route. In humans, defects in heme biosynthesis lead to severe metabolic disorders called porphyrias. The elucidation of the 3D structures for all heme biosynthetic enzymes over the last decade provided new insights into their function and elucidated the structural basis of many known diseases. In terms of structure and function several rather unique proteins were revealed such as the V-shaped glutamyl-tRNA reductase, the dipyrromethane cofactor containing porphobilinogen deaminase, or the “Radical SAM enzyme” coproporphyrinogen III dehydrogenase. This review summarizes the current understanding of the structure–function relationship for all heme biosynthetic enzymes and their potential interactions in the cell.
Keywords: heme biosynthesis, enzymes, structure, metabolic pathway, porphyrin
Introduction
Tetrapyrroles like hemes and chlorophylls are molecules of central importance to essential metabolic processes, including electron transfer during respiration, photosynthesis, and enzyme catalysis. In this context, the iron-containing porphyrin heme fulfils widely diverse biological functions as prosthetic group for many enzymes, transporters, and receptors in most living organisms. Heme-containing cytochromes are integral components of various respiratory and photosynthetic electron transport chains. Heme serves as a prosthetic group in hemoglobin, myoglobin, catalases, peroxidases, cytochromes P450, and in sensor proteins for diatomic gases such as O2 or NO.1 In all eukaryotes and most prokaryotes, except the archaea and some eubacteria, heme is synthesized via a well established conserved biosynthetic pathway.2 In humans, malfunction of this pathway leads to severe metabolic disorders, termed porphyrias.3,4 The determination of the three-dimensional structures of all 11 heme biosynthetic enzymes within the last 15 years has led to a renaissance in the field of heme biosynthesis. Besides a significant gain in the overall understanding of this metabolic pathway, a number of novel enzyme mechanisms have been unraveled. Examples include the unusual reduction of tRNA-bound glutamate by glutamyl-tRNA reductase, the dipyrromethane cofactor dependent oligomerization of porphobilinogen by porphobilinogen deaminase, and the employment of radical chemistry by coproporphyrinogen III dehydrogenase.
This review will focus on the structures and catalytic mechanisms of the following heme biosynthetic enzymes (Fig. 1): 5-Aminolevulinate synthase (ALAS), glutamyl-tRNA reductase (GluTR), glutamate-1-semialdehyde-2,1-aminomutase (GSAM), porphobilinogen synthase (PBGS), porphobilinogen deaminase (PBGD), uroporphyrinogen III synthase (UROS), uroporphyrinogen III decarboxylase (UROD), oxygen-dependent coproporphyrinogen III oxidase (CPO), coproporphyrinogen III dehydrogenase (CPDH, also known as oxygen-independent coproporphyrinogen III oxidase), oxygen-dependent protoporphyrinogen IX oxidase (PPO), and ferrochelatase (FC).
Figure 1.
Heme biosynthesis. A: The first cyclic tetrapyrrole uroporphyrinogen III is formed from the precursor 5-aminolevulinic acid in three enzymatic steps via the intermediates porphobilinogen and pre-uroporphyrinogen. Depending on the organism, ALA is either synthesized by condensation of glycine with succinyl-CoA or from tRNA-bound glutamate via glutamate-1-semialdehyde. B: Uroporphyrinogen III is converted into heme in four consecutive enzymatic steps via the intermediates coproporphyrinogen III, protoporphyrinogen IX, and protoporphyrin IX. Structures of all heme biosynthesis enzymes have been determined with the exception of oxygen-independent PPO (n.d., structure not determined).
For each enzyme a brief historical overview on important discoveries is followed by the presentation of their three-dimensional structures and catalytic mechanisms. Amino acid exchanges caused by gene mutations implicated in human porphyrias are discussed in the light of the available structural information. Finally, the importance of interactions between a number of heme biosynthetic enzymes to achieve efficient transfer of metabolites is outlined.
Heme Biosynthesis
The biosynthesis of heme (Fig. 1) sets off with the formation of 5-aminolevulinic acid (ALA) as the common precursor for all naturally occurring tetrapyrroles. ALA represents the sole source of carbon and nitrogen atoms necessary for heme formation. Two ALA molecules are then condensed to the pyrrole porphobilinogen (PBG). Oligomerization of four PBG molecules to the linear tetrapyrrole intermediate pre-uroporphyrinogen (1-hydroxymethylbilane) followed by ring closure leads to the first cyclic tetrapyrrole intermediate uroporphyrinogen III (UROGEN). Next, the side chains of the macrocycle are modified resulting in the intermediates coproporphyrinogen III (COPROGEN) and protoporphyrinogen IX (PROTOGEN). Subsequently, the ring system is aromatized giving protoporphyrin IX (PROTO). Finally, iron is inserted to yield heme. Variations from this canonical pathway5 occur in some bacteria and the archaea.6–8
Structures and Mechanisms of Heme Biosynthetic Enzymes
Heme biosynthesis can be divided into three parts: (i) formation of the precursor molecule ALA, (ii) formation of the first cyclic tetrapyrrole uroporphyrinogen III, and (iii) conversion of uroporphyrinogen III into heme.
Two ways lead to 5-aminolevulinic acid
5-Aminolevulinic acid (ALA) is synthesized in nature by two different, unrelated biosynthetic routes. The first one, discovered by Shemin, involves the condensation of glycine and succinyl-CoA by 5-aminolevulinic acid synthase (ALAS) and occurs in mammals, fungi, and α-proteobacteria. In plants, archaea and most bacteria the alternative course, termed “C5-pathway,” builds ALA from tRNA-bound glutamate in two steps. The initial substrate of the C5-pathway, glutamyl-tRNA, is provided by glutamyl-tRNA synthetase and is shared between heme and protein biosyntheses. Glutamyl-tRNA reductase (GluTR) then converts glutamyl-tRNA in an NADPH-dependent reaction into the labile intermediate glutamate-1-semialdehyde which is then further transformed into ALA by glutamate-1-semialdehyde-2,1-aminomutase (GSAM).9
Shemin pathway
5-Aminolevulinic acid synthase (ALAS; EC 2.3.1.37) catalyzes the condensation of glycine and succinyl-CoA to yield ALA. During this reaction CO2 and free coenzyme A are released. The first report about the incorporation of nitrogen atoms originating from glycine into heme was published in 1945, by Shemin and Rittenberg.10 Ingestion of 15N-labeled glycine over a 3-day-period led to 15N-incorporation into Shemin's own heme. Further incorporation studies using radioactively labeled acetate performed by the Shemin and Neuberger groups led to the identification of succinyl-CoA as the second source of carbon atoms for heme formation in 1952.11–13 After the identification of ALA as a potential precursor for heme biosynthesis,14,15 ALA synthase (ALAS) activity in crude cell extracts was simultaneously described by the Shemin and Neuberger groups in 1958.16–19 Since these early studies ALAS was purified from various eukaryotic and bacterial sources and biochemically characterized.20 Whereas there is usually only one isoform of bacterial ALAS proteins, mammals carry two different ALAS isoforms, one of them fulfilling a housekeeping function (ALAS1) and the other sustaining high levels of heme biosynthesis in erythrocytes (ALAS2).21 Mutations in the human erythroid-specific ALAS isoform lowering its catalytic activity are the cause of X-linked sideroblastic anemia which manifests in iron accumulation in erythroblast mitochondria.22 On the other hand, an extremely high expression level of ALAS2 due to genetic alterations leads to the accumulation of protoporphyrin IX and causes X-linked dominant protoporphyria.23 The only crystal structure of ALAS, from the phototrophic bacterium Rhodobacter capsulatus, was elucidated in 2005.24
5-Aminolevulinic acid synthase
ALAS functions as a homodimer and requires the cofactor pyridoxal 5′-phosphate (PLP) for catalysis.25,26 Within the family of PLP-dependent enzymes ALAS belongs to the subfamily of α-oxoamine synthases which catalyze the condensation of a carboxylic acid CoA thioester and small amino acids with concomitant decarboxylation of the latter.27 The ALAS reaction mechanism is unusual for a PLP-dependent enzyme in that two α-carbon bonds are cleaved during the reaction cycle. The current knowledge on the ALAS reaction mechanism was derived from various studies using specifically labeled substrates and extensive kinetic investigations using stopped-flow techniques, in combination with the available crystal structure of ALAS from R. capsulatus.28–34 In the active form the PLP cofactor is covalently linked to an essential active site lysine residue via an internal aldimine. Catalysis begins with the binding of glycine to the active site and formation of an external aldimine between glycine and the PLP cofactor [Fig. 2 (A)]. Subsequently, the pro-R proton of glycine is removed by the active site lysine, succinyl-CoA is attached to the glycine α-carbon, and coenzyme A is released leading to an α-amino-β-keto adipate intermediate. Decarboxylation of this intermediate is achieved by a protonation step for which an active site histidine residue acts as general acid [Fig. 2(B)]. The formed enol intermediate is in equilibrium with a spectroscopically traceable quinonoid intermediate and with the ALA bound external aldimine upon protonation of the C-5 position. Finally, ALA is released and the internal aldimine restored.35
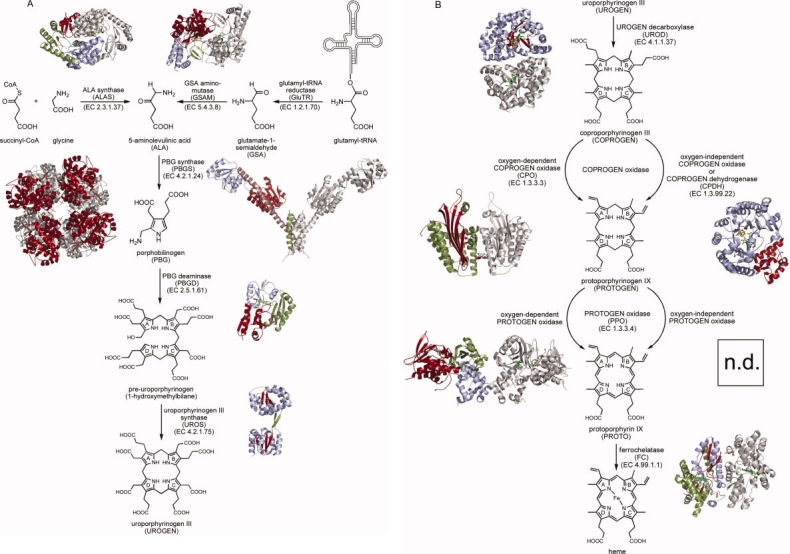
Figure 2.
Active site architectures and catalytic steps of enzymes involved in ALA formation. A + B, ALAS; C + D, GluTR; E + F, GSAM. A: Active site of ALAS from R. capsulatus modeled from the glycine- and succinyl-CoA-bound structures showing the external aldimine between PLP and glycine in close proximity to the succinyl-CoA CS1 position. B: During the ALAS reaction cycle, the decarboxylation of the α-amino-β-keto adipate intermediate is promoted by His-mediated protonation. C: Active site of GluTR from M. kandleri with bound glutamycin and the catalytically essential Cys(Ser)48. D: During catalysis Cys48 nucleophilically attacks the activated α-carboxylate of the glutamyl-tRNA. E: Active site of GSAM from Synechococcus showing the inhibitor gabaculine covalently bound to the PLP cofactor resulting in the stable m-carboxyphenylpyridoxalamine phosphate (mCPP). F: The potential reaction intermediate 4,5-diaminovalerate covalently bound to PLP as an external aldimine.
The crystal structures of R. capsulatus ALAS holoenzyme, in its free form as well as in complex with glycine and succinyl-CoA have provided detailed insights into the active site architecture [Fig. 2(A)].24 Each subunit of the tightly interlocked ALAS homodimer consists of three domains (Fig. 1). The central catalytic domains of each monomer harbor the active site clefts located at the subunit interface. The active site pockets are deeply buried in the dimeric ALAS structure excluding water from the reaction to take place. Glycine and succinyl-CoA can enter the active site pocket through a narrow channel. PLP is precisely held in its binding site by various interactions with the surrounding amino acid residues. In the ALAS-glycine complex structure the substrate glycine replaces the active site lysine by forming the external aldimine with PLP. However, the lysine remains in a position suitable for proton abstraction from the PLP bound glycine. The structure of the ALAS-succinyl-CoA complex revealed that the 3′-phosphate ADP moiety of the stretched co-substrate binds to a hydrophobic pocket at the entrance of the active site channel on the surface of the enzyme and the succinate carboxylate group close to the PLP. An overlay of both substrate bound structures revealed a distance of ∼2.8 Å between the Cα of PLP bound glycine and the CS1 of succinyl-CoA being poised for the required nucleophilic attack for C—C bond formation [Fig. 2(A)]. In all substrate bound structures a histidine, located directly above the PLP ring, could act as an acid promoting the decarboxylation of the α-amino-β-keto adipate intermediate.
Because of the high sequence conservation (49% identity) between R. capsulatus and human ALAS2 the structure of the bacterial enzyme can be used as a model for the mapping and analysis of point mutations causing X-linked sideroblastic anemia (XLSA). Some of the XLSA mutations are directly or indirectly affecting PLP or substrate binding. For example, Thr245 (Thr388 in human ALAS2) is involved in hydrogen bonding of the PLP phosphate group. When this residue is replaced by serine the patients suffer from a form of XLSA which can be treated by supplemental pyridoxine. In another XLSA mutation Arg517 (human ALAS2), which is crucial for glycine recognition and discrimination, is replaced by a cysteine.
C5-Pathway
The second pathway leading to the formation of ALA was not discovered until the 1970s. By that time the Shemin pathway was well established, however all attempts to detect ALAS activity in extracts of green plants had failed. Furthermore, experiments with chlorophyll synthesizing tissues using labeled glycine did not result in the typical incorporation pattern as it was observed before for avian heme biosynthesis. In 1973, Beale and Castelfranco used various 14C-labeled compounds to study the incorporation of the label into ALA by greening cucumber cotyledons. They observed that the metabolically related compounds glutamate, α-ketoglutarate and glutamine gave rise to 14C-labeled ALA.36 Two years later, it was found that the intact five-carbon skeleton of glutamate was incorporated into ALA.37 It was shown only a few years later that glutamate was transformed into ALA via the reduced intermediate glutamate-1-semialdehyde (GSA) by detection of GSA aminotransferase activity in barley and the subsequent purification and characterization of the enzyme.38,39 The next major discovery concerning the C5-pathway in the mid-1980s was the finding that an RNA moiety was involved in the transformation of glutamate into ALA.40 Finally, it was established that the initial metabolite of the C5-pathway is Glu-tRNAGlu which is reduced by glutamyl-tRNA reductase (GluTR, EC 1.2.1.70) to GSA which is then converted into ALA by glutamate-1-semialdehyde-2,1-aminomutase (GSAM, EC 5.4.3.8).41,42 Since these early studies both enzymes were purified and characterized from various plant and bacterial sources.43–52 The first crystal structure of GluTR was solved for the enzyme from Methanopyrus kandleri revealing an unusual V-shaped structure.53 Crystal structures for GSAM were solved for the Synechococcus (GSAMSyn) and the Thermosynechococcus elongatus enzyme (GSAMTel).54,55
Glutamyl-tRNA reductase
GluTR functions as a homodimeric, NADPH-dependent protein. The large tRNA substrate is recognized by its overall shape and NADPH binds to the enzyme with all its major determinants.53,56,57 During catalysis a highly conserved cysteine residue located in the active site of the enzyme acts as a nucleophile attacking the activated α-carboxyl group of tRNA-bound glutamate [Fig. 2(C,D)]. This results in the release of the tRNA and formation of an enzyme-bound thioester intermediate.58 NADPH mediated hydride transfer to the thioester intermediate leads to the formation of glutamate-1-semialdehyde, while in the absence of NADPH the thioester bond is slowly hydrolyzed producing free glutamate.
The crystal structure of M. kandleri GluTR was solved in complex with the antibiotic inhibitor glutamycin and revealed an unusual V-shaped structure for the dimeric protein (Fig. 1).53 Each monomer consists of three distinct domains which are arranged along a curved “spinal” α-helix linking domains II and III. The N-terminal domain I is located at the central part of the spinal helix and represents the catalytic domain. Domain II is located at the top of the extended helix and adopts a classical nucleotide-binding fold containing characteristic βαβ motifs and a conserved glycine rich loop. The dimerization domain III is located at the C-terminal end of the spinal helix and together with the corresponding domain of the second monomer forms an unusual six-helix bundle. Glutamycin was found to bind in a deep pocket of the catalytic domain. Several highly conserved amino acid residues specifically hold the inhibitor in its position. The glutamycin γ-carboxylate group is specifically recognized by a bidentate salt bridge with an arginine and two additional hydrogen bonds. Both the inhibitor α-amino group as well as the amide group of glutamycin are engaged in hydrogen bonds with the enzyme. Mutational analysis of these active site residues and kinetic characterization of the enzyme variants revealed their functional importance.57 During catalysis Cys48 nucleophilically attacks the activated α-carboxylate of the glutamyl-tRNA. In the GluTR crystal structure Cys48 (replaced by a serine for technical reasons) is located in close proximity (3.9 Å) to the α-carbonyl carbon of glutamycin. The NADPH cofactor was modeled into the enzyme's nucleotide binding domain which was observed to be slightly compressed compared to other NADPH binding domains. From this model it was concluded that the observed GluTR conformation probably represented an open, pre-active state of the enzyme since NADPH was ∼21 Å away from the glutamate binding pocket. It was suggested that upon glutamyl-tRNA binding a tipping of domain II towards domain I with a concomitant opening of the nucleotide binding pocket could be induced. The authentic substrate glutamyl-tRNA was also modeled into the GluTR structure revealing a striking surface complementarity between the enzyme and its unusual substrate. Functional identification of tRNAGlu identity elements for GluTR recognition showed the importance of the unique overall structure of the tRNA molecule.56
Glutamate-1-semialdehyde-2,1-aminomutase
GSAM belongs to the α-family of pyridoxal 5′-phosphate (PLP)-dependent enzymes and can be classified structurally and mechanistically as an aminotransferase. However, in contrast to other aminotransferases GSAM functions as aminomutase acting on a single substrate molecule. Two alternative mechanistic routes are conceivable for GSAM catalysis. The reaction starts either with enzyme bound pyridoxamine 5′-phosphate (PMP) or PLP and proceeds via the intermediates 4,5-diaminovalerate (DAVA) and PLP or 4,5-dioxovalerate (DOVA) and PMP, respectively.59–61 All GSAM proteins studied so far are able to catalyze both reactions.51,62 However, the first mechanism was found to be kinetically preferred and, therefore, DAVA was proposed to be the true reaction intermediate during GSAM catalysis.63–65 GSAM acts as a functional homodimer as confirmed by X-ray crystallography (Fig. 1). Each monomer of the enzyme consists of three domains. The large central domains harbor the active sites which are located at the dimer interface. For the GSAMSyn structure an asymmetric homodimer was observed in which one subunit contained the cofactor in its PMP form whereas the second subunit contained PLP covalently linked to the active site lysine residue. Because of a lack of order for about 30 amino acid residues the PLP containing subunit was described to adopt an “open” conformation. In contrast, the same region was well ordered in the PMP binding subunit and covered the active site leading to a “closed” conformation. In contrast to these observations a symmetric homodimer was observed for the GSAMTel structure in which both subunits were observed to adopt the “open” PLP bound form with the disordered region covering the active site. The PLP phosphate group is recognized via hydrogen bonds to surrounding amino acid residues and a water molecule. The PLP pyridinium ring is sandwiched between a valine and a tyrosine residue. In both GSAM structures a highly conserved arginine was observed ideally positioned to bind the carboxylate group of the substrate GSA. In a recent study the suicide inhibitor gabaculine was observed to bind covalently to the PLP cofactor resulting in a stable m-carboxyphenylpyridoxalamine phosphate form (mCPP).66 The structure of the GSAM-inhibitor complex revealed that the cofactor rotates ∼15° away from the active site lysine residue when compared to the PLP bound form of the enzyme. Further, the carboxylate group of gabaculine forms hydrogen bonds with the side chain of an active site serine and interacts with a nearby tyrosine and the conserved arginine via ordered water molecules [Fig. 2(E,F)].
Uroporphyrinogen III is formed from ALA in three consecutive enzymatic steps
Uroporphyrinogen III (UROGEN) represents the first cyclic intermediate of tetrapyrrole biosynthesis and at the same time the first major branch point for the diverging routes leading to formation of different classes of tetrapyrroles.5 This important intermediate is built from eight ALA molecules in three consecutive enzymatic steps. First, two ALA molecules are asymmetrically condensed by the enzyme porphobilinogen synthase (PBGS, EC 4.2.1.24, also known as ALA dehydratase) to yield the pyrrole derivative PBG. Next, four PBG molecules are linked together in a linear fashion by the enzyme porphobilinogen deaminase (PBGD, EC 2.5.1.61, also called hydroxymethylbilane synthase) yielding the linear tetrapyrrole pre-uroporphyrinogen (1-hydroxymethylbilane). Finally, UROGEN is formed from pre-uroporphyrinogen by the action of the enzyme uroporphyrinogen III synthase (UROS, EC 4.2.1.75). The heme precursor PBG was detected for the first time in the urine of a patient suffering from acute porphyria in 1931.67 Shortly after its isolation and structure determination in 1952,68,69 it was shown that PBG serves as an effective precursor for heme formation in avian red cells.70 The hypothesis arose that an early step during heme biosynthesis could be the condensation of two ALA molecules to PBG14,15 and finally the first purifications and characterizations of PBGS proteins from different sources in the mid-1950s provided proof for this proposal.71,72 At about the same time it was found by Bogorad and Granick that the conversion of PBG into uroporphyrin III required the cooperative action of two enzymes and later these were identified as PBGD and UROS.73,74 In the beginning of these early studies of heme biosynthesis the porphyrins uroporphyrin III, coproporphyrin III, and protoporphyrin IX were believed to be biosynthetic intermediates. However, in the mid-1950s it was established that the reduced forms of these porphyrins, the “porphyrinogens,” are the true intermediates during heme biosynthesis.75 Since these early studies PBGS, PBGD, and UROS proteins were purified from many different eukaryotic and prokaryotic sources and biochemically characterized.76 Crystal structures of PBGS were solved much later for the enzymes from yeast, E. coli and Pseudomonas aeruginosa and Chlorobium vibrioforme with many of them containing inhibitors or reaction intermediates.77–80 Structures for PBGD are available for the E. coli and human proteins.81–83 The structure of human UROS was elucidated in 2001 followed by the Thermus thermophilus enzyme structure in complex with UROGEN.84,85
Porphobilinogen synthase
Porphobilinogen synthases usually function as homooctamers with the exception of the hexameric enzyme from Rhodobacter capsulatus.86 Despite the high degree of amino acid sequence similarity between PBGSs from different organisms they are divided into subgroups according to their metal dependency. Human and yeast PBGS contain two zinc ions, one of them playing a role in catalysis, which are coordinated by highly conserved cysteine residues. E. coli PBGS also contains the active site zinc and an additional magnesium ion. P. aeruginosa PBGS does not require any zinc but contains magnesium ions at an allosteric site and monovalent cations in the active site. Finally, it was reported that some PBGS proteins are metal-independent.86 In a series of biochemical and structural studies of PBGS using various substrate analogs and inhibitors the enzyme mechanism was elucidated.87–96 In the first step, the ALA molecule which ultimately forms the propionate group of PBG (P-side ALA) binds to a highly conserved lysine residue within the so-called “P-site” of the enzyme via Schiff base formation. This is followed by the binding of the second ALA molecule (A-side ALA) giving rise to the acetate group of PBG to the “A-site,” also covalently linked to another strictly conserved active site lysine residue [Fig. 3 (A,B)]. This second Schiff base at the A-site then undergoes transformation into an enamine as a prerequisite for the next catalytic step. In an aldole addition reaction the C3 atom of A-side ALA attacks the C4 of P-side ALA which results in formation of the C—C bond between the two ALA molecules. Next, the amino group of P-side ALA attacks the Schiff base at the C4 atom of A-side ALA which leads to C—N bond formation and release of the A-site lysine. Lysis of the remaining bond between the P-site lysine and the reaction intermediate is triggered by protonation of the former and possibly by conformational changes of the enzyme. Finally, proton abstraction leads to the aromatization and release of PBG.
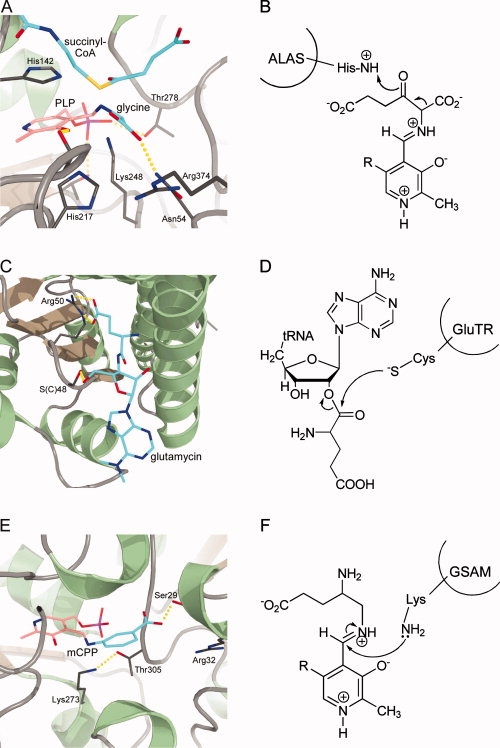
Figure 3.
Active site architectures and catalytic steps of enzymes involved in uroporphyrinogen III formation. A + B, PBGS; C + D, PBGD; E + F, UROS. A: Active site of P. aeruginosa PBGS with bound inhibitor 5-fluorolevulinic acid (5F-LA). B: At the beginning of the PBGS reaction cycle both ALA molecules are covalently bound to the enzyme via Schiff bases to conserved lysine residues. C: Active site of human PBGD showing the unique dipyrromethane cofactor (DPM) covalently bound to an invariant cysteine residue. D: The oligomerization of PBG molecules proceeds via deaminated azafulvene intermediates. E: Active site of T. thermophilus UROS with bound uroporphyrinogen III (UROGEN) showing a puckered “two-up, two-down” conformation of the reaction product. F: Dehydration of pre-uroporphyrinogen results in the first azafulvene intermediate which further reacts to form a spirocyclic pyrrolenine intermediate as indicated by the arrows.
The crystal structures of yeast, P. aeruginosa, and E. coli PBGS revealed that the octameric enzymes are composed of four dimers which are arranged around a central four-fold axis. Each monomer adopts the classical TIM-barrel fold with the active sites located at the C-terminal ends of the β-barrel. The active sites are covered by a flexible loop and thereby shielded from the surrounding medium. PBGS monomers contain an “N-terminal arm” which wraps around its dimeric partner. Recently, this “hugging” of the monomers was observed to be important for the formation of the functional octameric quarternary structure of PBGS, since human PBGS with a detached N-terminal arm was only able to form hexameric PBGS with low catalytic activity.97 In the crystal structure of P. aeruginosa PBGS in complex with the inhibitor 5-fluorolevulinic acid (5F-LA) both 5F-LA molecules were observed to bind covalently to the lysine residues of the A- and P-sites, respectively [Fig. 3(A)].93 In addition, the inhibitors are precisely positioned in the P- and A-site, respectively, through hydrogen bonds and hydrophobic interactions. Overall, the two active site lysine residues with the covalently bound inhibitor molecules are oriented roughly parallel to each other giving rise to two stacked T-shaped structures. A comparison of the 5F-LA conformations around their C3 atoms implies that enamine formation is facilitated for the A-site inhibitor molecule suggesting that the C—C bond formation through aldole condensation occurs prior to C—N bond formation.
In humans there are three distinct disorders due to decreased levels of PBGS activity of which only one is caused by mutations of the PBGS gene. PBGS porphyria is a very rare recessive autosomal disorder due to PBGS gene mutations and manifests with accumulation and urinary excretion of ALA and acute neurovisceral attacks. In cases of hepatorenal tyrosinemia a gene defect affecting the last step of tyrosine catabolism leads to the accumulation of succinylacetone which is a potent inhibitor of PBGS. Finally, lead poisoning also leads to inhibition of PBGS due to the replacement of the catalytically essential zinc ions of human PBGS by the toxic metal.3 Bacterial PBGS provides a target for antibiotic therapy as revealed by the recent structure of P. aeruginosa PBGS in complex with the antibiotic alaremycin.98
Porphobilinogen deaminase
It was found around 1980 in a series of studies using labeled PBG, that first a linear tetrapyrrole is formed followed by ring closure with rearrangement of ring D.99,100 At that time it was also observed that the oligomerization of PBG starts with ring A followed by rings B, C, and D.101,102 Finally, 1-hydroxymethylbilane (pre-uroporphyrinogen) was identified by the groups of Scott and Battersby to be the reaction product of the enzyme now known as porphobilinogen deaminase.103–105 Another major progress for the understanding of the PBGD protein was the discovery by Jordan and Warren and Battersby's group that the enzyme contains a covalently attached unique dipyrromethane cofactor consisting of two linked PBG molecules [Fig. 3(C,D)].106–108 This cofactor serves as a primer for the oligomerization of another four PBG molecules resulting in the formation of a protein bound linear hexapyrrole. From this precursor the linear tetrapyrrole pre-uroporphyrinogen is then cleaved off leaving the free enzyme with its dipyrromethane cofactor behind.109 During catalysis, a highly conserved aspartate residue plays an important role probably by assisting in the deamination of incoming PBG molecules.81,110 The formed reactive azafulvene intermediate can then react with the protein bound oligopyrrole [Fig. 3(D)]. Although it is not yet clear how exactly the enzyme deals with the growing oligopyrrole chain during catalysis a number of conserved arginine residues seems to be involved in the correct positioning of the dipyrromethane cofactor and the incoming PBG molecules. Further, it was observed that the protein undergoes conformational changes during catalysis which might help the enzyme in “pulling” the oligopyrrole through the active site or alternatively being caused by the reorganization of the oligopyrrole as it is formed.111 Mutations in human PBGD can cause acute intermittent porphyria (AIP), an inherited autosomal dominant disorder which manifests with attacks of abdominal pain and neurological symptoms accompanied by elevated levels of ALA and PBG in the patients' urine.
The crystal structures of E. coli and human PBGD revealed a monomeric protein consisting of three equally sized α/β domains with a high degree of flexibility in terms of domain movements.81–83 The N-terminal domain I and the central domain II adopt a fold similar to transferrins and some bacterial periplasmic binding proteins. Domain III consists of a three-stranded, anti-parallel β-sheet decorated on one side by three α-helices and interacts both with domains I and II. The large active site cleft is located at the interface between domains I and II and is predominantly positively charged. The dipyrromethane cofactor is covalently bound to the enzyme via a thioether linkage to a conserved cysteine which resides on a loop of domain III. The cofactor is positioned in the active site by various ionic interactions between the carboxylate groups of its acetate and propionate side chains to conserved arginine and lysine residues. The catalytically essential aspartate forms hydrogen-bonds to both pyrrole NH-groups of the cofactor [Fig. 3(C)]. The proposed active site of PBGD was occupied by an acetate ion in the E. coli PBGD structure and a sulfate ion in the human PBGD structure. The sulfate ion was located in hydrogen-bonding distance to an arginine and a serine which were proposed to be involved in substrate binding. Indeed, a mutation involving this arginine results in AIP in humans. Further mutations leading to AIP involve arginine residues being involved in cofactor assembly and positioning or substrate binding. In total, there are more than 100 known mutations causing AIP in humans either due to less stable or truncated forms of the protein or defects in cofactor and substrate binding.
Uroporphyrinogen III synthase
UROS catalyzes the cyclization of the linear tetrapyrrole pre-uroporphyrinogen to uroporphyrinogen III under inversion of ring D. Although the first purification of an UROS protein dates back to 1957 it took another 20 years until the actual substrate, pre-uroporphyrinogen, of this enzyme was identified. However, in 1961 a mechanism for the formation of UROGEN was proposed by Mathewson and Corwin which has stood the test of time.112 Studies using 13C-labeled PBG and NMR spectroscopy, investigations employing synthetic spirocyclic inhibitors as well as the trapping of an azafulvene reaction intermediate contributed to the current understanding of the UROS catalytic mechanism.113–115 In the first steps of catalysis the hydroxyl group on ring A of pre-uroporphyrinogen is lost as a water molecule resulting in formation of the first azafulvene intermediate [Fig. 3(F)]. Next, the spirocyclic pyrrolenine intermediate is formed by reaction of the azafulvene with the substituted α-position of ring D. Bond breakage between rings C and D then leads to formation of the second azafulvene intermediate on ring C. In the final steps of catalysis the enzyme has to orientate the ring D pyrrole in such a way that the C ring azafulvene can react with its free α-position. Finally, deprotonation and bond rearrangements lead to the formation of UROGEN.
In addition to the crystal structure of human UROS the structure of T. thermophilus UROS in complex with its reaction product UROGEN was obtained only recently.84,85 Despite a rather low level of amino acid sequence identity of only 14% between human and T. thermophilus UROS the overall fold of the enzymes from the two different organisms was found conserved. UROS is a monomeric protein which consists of two α/β domains connected by a flexible, two-stranded linker (Fig. 1). Domain I belongs to the flavodoxin-like fold family and domain II adopts a DNA glycosylase-like fold. In the human UROS structure the linker connecting the two domains adopts an antiparallel β-ladder conformation. In contrast, T. thermophilus UROS crystallized in several different conformations in which the linker is much less ordered than in the human UROS structure. The observed remarkable array of different domain-domain orientations suggests the linker being highly mobile in solution in the absence of the substrate.85 In the crystal structure of the T. thermophilus UROS-product complex it was observed that uroporphyrinogen III binds at the interface between domains I and II and that the two domains approach each other more closely than in the different apo-UROS structures. The porphyrinogen adopts a puckered “two-up, two-down” conformation in which the pyrrole NH-groups of rings A and C point in one direction and those of rings B and D in the opposite direction [Fig. 3(E)]. Only few hydrophobic interactions between the enzyme and the pyrrole rings D and B were observed. The pyrrole NH-groups do not directly interact with the enzyme but rather form hydrogen bonds with ordered water molecules, while the carboxylate groups of the acetate and propionate substituents form hydrogen bonds with main chain NH-groups. Most of these hydrogen bonds involve the acetate and propionate side chains of rings A and B which are thus precisely positioned in a constrained conformation within the “closed” form of the active site. This is consistent with the observation that substrate binding is abolished for porphyrinogens in which the ring A or B substituents are altered.116,117 For the ring C substituents there are hydrogen bonds between the propionate carboxylate group and main chain NH-groups whereas the acetate side chain does not directly interact with the protein. Likewise, the carboxylate groups of the ring D side chains do not interact with the enzyme facilitating the ring flip during the reaction.
Mutations in human UROS cause congenital erythropoietic porphyria (CEP) which is characterized by the accumulation of uroporphyrin I in the patients' urines and severe photosensitivity. Most of the known CEP mutations reduce the enzyme's activity by reducing its stability by disrupting the overall structural integrity. For a long time identification of plant UROS failed due to the low degree of amino acid sequence conservation. Recently, the Arabidopsis thaliana enzyme was identified and characterized as a typical UROS.118
Conversion of uroporphyrinogen III into heme requires four enzymatic steps
During the conversion of uroporphyrinogen III into heme the side chains of the macrocycle are modified, the tetrapyrrole ring system is oxidized, and finally iron is inserted. Uroporphyrinogen III decarboxylase (UROD, EC 4.1.1.37) catalyzes the stepwise decarboxylation of the four acetic acid chains of UROGEN to yield the corresponding methyl groups of coproporphyrinogen III. COPROGEN is then oxidatively decarboxylated yielding protoporphyrinogen IX by the action of either oxygen-dependent coproporphyrinogen III oxidase (CPO, EC 1.3.3.3) or oxygen-independent CPO which should rather be named coproporphyrinogen III dehydrogenase (CPDH, EC 1.3.99.22). In the penultimate step of heme biosynthesis, PROTOGEN is oxidized to PROTO by oxygen-dependent or -independent protoporphyrinogen IX oxidase (PPO, EC 1.3.3.4) before, finally, iron is inserted into the macrocycle by ferrochelatase (FC, EC 4.99.1.1) yielding heme.119 The oxidized forms of the pathway intermediates, uroporphyrin III, coproporphyrin III, and protoporphyrin IX, are found in unusual high amounts in the urines of patients suffering from different porphyrias. Early descriptions of such porphyrin accumulations date back to the middle of the 19th century and were followed by the isolation and characterization of these porphyrins providing first hints for heme biosynthetic intermediates. The enzymes UROD, CPO and CPDH, PPO, and FC were subsequently purified from various eukaryotic and prokaryotic sources and the corresponding genes were cloned and sequenced.120–122 Crystal structures are available for human UROD in its apo and COPROGEN bound forms, as well as for Nicotiana tabacum and Bacillus subtilis UROD.123–126 CPO was crystallized and the structures solved for the yeast and human enzymes.127,128 The structure of E. coli CPDH was reported in 2003.129 The first structure of PPO was obtained for the tobacco protein followed by structures of Myxococcus xanthus and B. subtilis PPOs.130–132 Ferrochelatase was structurally characterized for the water soluble B. subtilis protein, the membrane attached human enzyme and the yeast FC followed by various structures of FC variants and of FC in complex with substrate and inhibitors.133–136
Uroporphyrinogen III decarboxylase
UROD activity was first described in 1958 by Mauzerall and Granick.137 In 1976, it was proposed that the four decarboxylation reactions occur sequentially in a clockwise manner starting with the acetate side chain on ring D followed by those on rings A, B, and C.138 This was substantiated by studies in which different partially decarboxylated intermediates accumulating in the urines of patients suffering from porphyria cutanea tarda (PCT) as a result of diminished UROD activity were analyzed.139 PCT is the most common form of porphyria in humans which occurs with an estimated frequency of 1 in 20,000 Caucasians. UROD proteins were purified from many species including bacteria, yeast, and human.140 It was found that UROD accepts a wide variety of different substrates including uroporphyrinogens I and III as well as all 14 possible intermediates between UROGEN and COPROGEN.141 Accordingly, at high substrate concentrations the four decarboxylation reactions take place randomly.142 UROD represents an unusual decarboxylase in that it is not dependent on any prosthetic group or cofactor. In a recent study the decarboxylation reaction catalyzed by UROD was described as a “benchmark for the catalytic proficiency of enzymes” due to the enormous rate enhancement of substrate decarboxylation by this enzyme.143 The solved crystal structures for UROD proteins revealed a dimeric enzyme in which the two single-domain subunits are orientated head-to-head with the active site clefts facing each other at the dimer interface (Fig. 1). This arrangement of the two monomers results in one large active site well shielded from the surrounding solvent. On the basis of these structural observations two possible scenarios for the sequential decarboxylation of the four acetate side chains of UROGEN have been put forward. In one of these proposals, the acetate side chain of ring D is first decarboxylated in the active site of one monomer, the resulting intermediate then passed to the active site of the other monomer where the three remaining decarboxylation reactions on rings A, B, and C take place, making the 180° flipping of the substrate superfluous.125 However, recently it was shown using a dimeric, single-chain UROD construct and variants thereof, that substrate shuttling between the active sites of UROD is not required to generate COPROGEN,144 suggesting a reorientation of the substrate by only 90° for each decarboxylation which is in agreement with the crystal structure of human UROD in complex with its reaction product COPROGEN bound in a dome-shaped conformation.124 This conformation placed the four central NH-groups of the tetrapyrrole in hydrogen bonding distance to a highly conserved aspartate residue being proposed to be involved in substrate binding and catalysis [Fig. 4 (A)]. According to this mechanism, the substrate pyrrole ring is first protonated at the α-position adjacent to the C atom bearing the acetate side chain. This protonation is promoted by the aspartate residue by stabilizing the positively charged reaction intermediate and possibly by acting as the conjugate acid directly donating the required proton [Fig. 4(B)].143 Decarboxylation of the acetate side chain yields another reaction intermediate which has to be protonated at the methylene carbon and deprotonated at the α-C atom to yield the reaction product. It was suggested that a highly conserved arginine residue within the active site could be involved in the final protonation step.143 After reorientation of the substrate by 90° the next decarboxylation takes place in the same active site following the same mechanistic strategy.
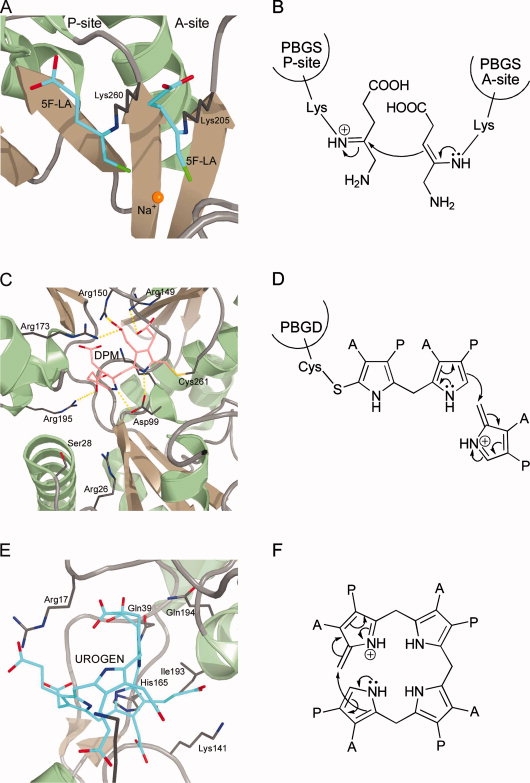
Figure 4.
Active site architectures and catalytic steps of enzymes involved in protoporphyrinogen IX formation. A + B, UROD; C + D, CPO; E + F, CPDH. A: Active site of human UROD with bound coproporphyrinogen III (COPROGEN) in a dome-shaped conformation showing the catalytically essential aspartate in hydrogen bonding distance to the pyrrole NH-groups. B: The aspartate residue stabilizes the protonated, positively charged reaction intermediate. C: Active site of human CPO with bound citrate showing several arginine residues possibly involved in substrate binding and the catalytically essential aspartate. D: During catalysis, a pyrrole peroxide anion is formed which further reacts via proton abstraction through the peroxide to form an intermediate containing an exocyclic double bond. E: Active site of E. coli CPDH showing the catalytically essential [4Fe-4S] cluster and the two bound SAM molecules. F: During the initial reaction steps the reduced iron-sulfur cluster transfers an electron to SAM, which is thereby cleaved into methionine and a 5′-deoxyadenosyl radical. This radical then abstracts a hydrogen atom from the substrate propionate side chain resulting in the formation of 5′-deoxyadenosine and a substrate radical. R = tetrapyrrole.
The UROD-product complex represented the first crystal structure which contained a highly oxygen-sensitive porphyrinogen.124 The UROD reaction product COPROGEN was observed to bind in the active site cleft in a dome-shaped conformation with one face of the tetrapyrrole nestled up against a ring of conserved hydrophobic amino acid residues. In the observed conformation, the pyrrole rings A, B, and D are tilted 35–60° from the plane whereas ring C lies approximately in the plane of the four central NH-groups. As a result thereof the ring A, B, and D amines are positioned in an optimal distance for hydrogen-bonding with one of the γ-carboxylate oxygen atoms of the catalytically essential aspartate which is precisely positioned above the tetrapyrrole. The propionate side chains of COPROGEN are coordinated by conserved arginine residues within the active site. These arginines do, however, rather provide a favorable positively charged environment to accommodate the negatively charged substrate instead of precisely orienting the porphyrinogen, thus explaining the enzyme's ability to accept a large array of different substrates.
Coproporphyrinogen III oxidase
The conversion of coproporphyrinogen III into protoporphyrinogen IX requires the oxidative decarboxylation of the propionate side chains on rings A and B of the tetrapyrrole to yield the corresponding vinyl groups. This reaction is catalyzed by enzymes which are generally named coproporphyrinogen III oxidases. However, depending on the requirement for molecular oxygen as terminal electron acceptor there are two distinct types of CPO which are structurally and mechanistically completely unrelated proteins. In this review, we will refer to the oxygen-dependent enzyme as CPO whereas we will name the oxygen-independent protein coproporphyrinogen III dehydrogenase (CPDH).
CPO is mainly found in eukaryotes with only few described bacterial representatives.145 Decreased activity of CPO in humans manifests in the autosomal dominant hereditary coproporphyria.146 The first partial purification of CPO was reported in 1961 by Sano and Granick for the bovine liver enzyme.147 This was followed 4 years later by the partial isolation of the enzyme from rat liver by Battle et al. and in the mid-1970s by the purification of yeast CPO by Poulson and Polglase.148,149 Since these early studies a number of native and recombinant CPO proteins were purified to homogeneity and biochemically characterized.150–155 CPO functions as a dimeric protein which requires molecular oxygen as the terminal electron acceptor during catalysis. It was shown that CPO catalyzes the decarboxylation of the ring A propionate side chain prior to that on ring B under formation of the reaction intermediate harderoporphyrinogen.156,157 Early stereochemical and mechanistic studies using deuterium and tritium-labeled substrates showed that only the pro-S-hydrogen atom at the β-carbon of the substrate propionate side chain is removed during the reaction and that the overall conversion of propionate side chains into vinyl side chains occurs stereochemically by antiperiplanar elimination.158–160 Recently, a putative reaction mechanism was proposed for the oxygen-dependent decarboxylation of COPROGEN and evaluated by quantum chemical treatments.161,162 In this proposal the first step is a base catalyzed deprotonation of the pyrrole NH-group yielding an azacyclopentadienyl anion. This anion then reacts with molecular oxygen at the α-position to form a pyrrole peroxide anion [Fig. 4(D)]. Next, a proton at the β-position of the substrate propionate side chain is abstracted by the peroxide via a six-membered ring transition state resulting in an exocyclic double bond. Finally, elimination of CO2 and H2O2 and bond rearrangements lead to formation of the vinyl group. Indeed, it was shown in a recent study that H2O2 is generated during CPO catalysis.155 Unfortunately, the two solved crystal structures for CPO did not contain substrate or product precluding a precise assignment of amino acid residues directly involved in catalysis.127,128 However, in a recent biochemical study employing mutant human CPOs it was suggested that a highly conserved aspartate residue might serve as the base for NH proton abstraction, and two conserved arginine residues were proposed to coordinate the substrate carboxylate groups.163
Both yeast and human CPO structures confirmed the dimeric state of the protein.127,128 Each monomer consists of a single domain adopting an unprecedented fold containing a large seven-stranded antiparallel β-sheet covered on both sides by α-helices. In the homodimer the two β-sheets of the monomers face each other and the subunits are rotated by ∼40° relative to one another (Fig. 1). A putative active site cleft was localized matching the size of the substrate [Fig. 4(C)]. Interestingly, yeast CPO crystallized in two different crystal forms. The structures revealed that in one of these forms the active site cleft was in an open, accessible conformation whereas in the second form the active site was covered by one of the α-helices leading to a cavity well shielded from the solvent. Two invariant arginine residues were found to point their side chains into the active site cavity suggesting interactions with the substrate's propionate side chains. An invariant aspartate residue which exposes its side chain to the cavity near the centre of one face was proposed to coordinate the pyrrole NH-groups similar to the UROD-product complex structure.
Mutations in human CPO cause the autosomal dominant disease hereditary coproporphyria (HCP). Most of the >20 known clinically identified HCP mutations cause a decreased overall stability of the protein. Some mutations located in the active site cavity, like that of an invariant arginine and serine, are thought to have deleterious effects due to altering the size, shape and polarity of the active site.128
Coproporphyrinogen III dehydrogenase
In most bacteria, the conversion of COPROGEN into PROTOGEN is catalyzed by coproporphyrinogen III dehydrogenase. CPDH uses a terminal electron acceptor other than oxygen for the oxidative decarboxylation reaction. The first reports describing CPDH activity in cell free extracts prepared from Rhodobacter sphaeroides were published around 1970 by Tait.164,165 Ten years later, it was found that the oxygen-independent conversion of COPROGEN into PROTOGEN proceeds with the same stereochemistry as the oxygen-dependent reaction and that only the pro-S-hydrogen atom at the β-carbon of the substrate propionate side chain is lost.159 However, all these studies did not result in the isolation or characterization of the protein(s) responsible for the reaction. In the 1990s, genetic approaches finally led to the identification of CPDH encoding genes from various bacteria and these were cloned and sequenced.166–170 Subsequently, recombinant CPDH from E. coli was purified to homogeneity and biochemically and structurally characterized. CPDH is a monomeric protein which belongs to the so-called “Radical SAM” protein family. As such it contains an oxygen-labile [4Fe-4S] cluster in which three iron atoms are coordinated by three highly conserved cysteine residues of the characteristic CX3CX2C motif.171 The fourth iron atom of the FeS cluster is ligated by an S-adenosyl-l-methionine (SAM) molecule which serves as a cofactor by initiating radical based catalysis.129 The CPDH reaction cycle starts with the reduction of the FeS centre by an as yet unidentified electron donor system such as flavodoxin/flavodoxin reductase. The reduced iron-sulfur cluster then transfers the electron to bound SAM which is thereby homolytically cleaved into methionine and a 5′-deoxyadenosyl radical [Fig. 4(F)]. This highly reactive radical then abstracts a hydrogen atom from the substrate propionate side chain resulting in the formation of 5′-deoxyadenosine and an allylic substrate radical.172 Finally, elimination of CO2 and transfer of the remaining single electron to an as yet unidentified terminal electron acceptor completes the CPDH reaction cycle. It was found that during formation of one PROTOGEN molecule two SAM molecules are cleaved.173 Interestingly, two SAM molecules were bound in the crystal structure of the E. coli enzyme providing the possibility that both decarboxylation reactions can take place without the need for release of a tricarboxylic acid intermediate from the active site.
The overall structure of CPDH revealed a monomeric, two domain protein (Fig. 1).129 The larger N-terminal domain consists of a curved, twelve-stranded, largely parallel β-sheet which is decorated at its outer surface by α-helices. The central core of this domain is formed by the six N-terminal β-strands which are part of a curved (β/α)6 repeat. This (β/α)6 repeat structurally resembles known TIM barrel domains, however, in the CPDH structure the β-strands are less strongly inclined relative to the barrel axis and the curvature is not as tight as in known TIM barrel structures leading to a three-quarter barrel with a lateral opening. The C-terminal domain consists of a bundle of four α-helices and a small three-stranded antiparallel β-sheet. The first 35 amino acid residues of the protein belong to neither of the two domains and are only partially ordered. Since several conserved amino acid residues are located within this N-terminal stretch it was proposed that it might adopt a fully ordered conformation upon substrate binding. The [4Fe-4S] cluster is located at the centre of the three-quarter barrel near the C-terminal ends of the β-strands. Two SAM molecules are bound in close proximity to the iron-sulfur cluster with one of them acting as the fourth ligand to the FeS-cluster [Fig. 4(E)]. Based on a modeled CPDH-substrate complex several conserved arginine residues were proposed to interact with the substrate's propionate substituents. Any definite conclusions concerning the sequence of the two successive decarboxylation events were not possible. However, very recently it was reported that CPDH, like CPO, catalyzes the decarboxylation of the ring A propionate side chain prior to that on ring B via the reaction intermediate harderoporphyrinogen.174
Protoporphyrinogen IX oxidase
In the penultimate step of heme biosynthesis protoporphyrinogen IX is oxidized to protoporphyrin IX. Although this oxidation also occurs on its own, it was found in early studies in the early 1960s that in vivo this reaction is catalyzed by protoporphyrinogen IX oxidase.147,175,176 As is the case for CPO there are also two distinct, unrelated PPO proteins catalyzing the six electron oxidation of PROTOGEN. Oxygen-dependent PPO uses molecular oxygen as the terminal electron acceptor whereas oxygen-independent PPO shuttles the electrons to anaerobic respiratory chains with alternative terminal electron acceptors such as nitrate or fumarate.177,178 In the late 1980s, native oxygen-dependent PPO proteins were purified to homogeneity from bovine liver, mouse liver, and yeast later followed by recombinant PPOs from eukaryotic sources.179–183 Bacterial oxygen-dependent PPOs were purified and characterized from Bacillus subtilis, Myxococcus Xanthus, and the thermophilic bacterium Aquifex aeolicus.184–186 Mutations in the ppo gene in humans lead to the dominantly inherited disorder variegate porphyria (VP). Oxygen-dependent PPOs are usually homodimeric, membrane-associated proteins with the exception of monomeric PPO from B. subtilis and A. aeolicus.132,186 In addition, B. subtilis PPO was found to be soluble in the cytoplasm.187 In all cases oxygen-dependent PPOs contain an FAD cofactor which transfers the electrons from the substrate to the electron acceptor oxygen. During the whole reaction three O2 molecules are thus reduced to three H2O2 molecules. On the basis of the results of biochemical studies using tritium-labeled substrates a reaction mechanism was proposed in which three meso-carbon hydride ions are removed successively from the same face of the macrocyclic plane, each accompanied by the loss of an NH proton. The final tautomerization leading to PROTO then occurs through the stereospecific loss of the remaining meso-carbon hydrogen as a proton from the opposite face of the tetrapyrrole plane.121 In a recent mechanistic proposal based on the modeled enzyme-substrate complex of tobacco PPO all three hydride abstractions occur from the C20-meso-carbon followed by hydrogen rearrangements via enamine-imine tautomerizations which take place over the whole ring system.130
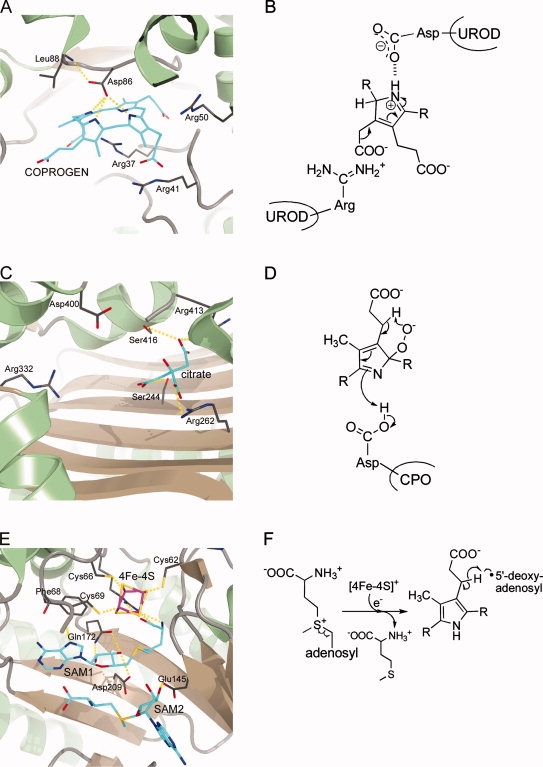
The crystal structures for mitochondrial tobacco PPO and the PPOs from M. xanthus and B. subtilis revealed that the overall fold of the protein is conserved despite a low degree of amino acid sequence conservation (∼27% identity). Each monomer of dimeric tobacco and M. xanthus PPO and monomeric B. subtilis PPO consists of three domains, an FAD-binding, a substrate-binding and a third domain involved in dimerization in the case of tobacco and M. xanthus PPO (Fig. 1). The FAD-binding and substrate-binding domains show a p-hydroxybenzoate-hydroxylase fold-like topology. The dimerization domain is exclusively α-helical and responsible for membrane-binding in the case of the tobacco and M. xanthus enzymes. Conformational differences were observed for this domain in the B. subtilis protein explaining the different oligomerization states of the different PPOs. On the basis of the localization of inhibitor molecules in the PPO structures the active site of PPO was identified [Fig. 5 (A)]. Whereas in tobacco and M. xanthus PPO the substrate binding site is a shallow cavity, it is much wider in the B. subtilis PPO structure explaining the ability of the latter enzyme to accept both PROTOGEN and COPROGEN as substrates. In the modeled tobacco PPO-substrate complex pyrrole ring A of protoporphyrinogen IX stacks against a conserved phenylalanine residue and ring B is sandwiched between two conserved leucine residues. A highly conserved arginine within the active site cavity provides a counter ion for the ring C propionate carboxylate group. Recent biochemical analysis of tobacco PPO variants showed the importance of these amino acid residues for substrate binding and catalysis.188 In this PPO-substrate model the C20 methylene bridge of the substrate is oriented towards the reactive N5 atom of the FAD cofactor suggesting that all hydride abstractions occur from this position. The most common mutation in human PPO causing VP is an exchange of Arg59 to tryptophane. According to the tobacco PPO structure this arginine is located on a loop between the isoalloxazine ring of FAD and the substrate-binding cavity and its replacement by a tryptophane in the human protein at this position probably disturbs the architecture of the active site and may also interfere with FAD and substrate binding.
Figure 5.

Active site architectures and catalytic steps of enzymes involved in the transformation of protoporphyrinogen IX into heme. A + B, PPO; C + D, FC. A: Active site of M. xanthus PPO with bound inhibitor acifluorfen (AF). B: PPO catalyzes the oxidation of protoporphyrinogen IX to protoporphyrin IX. C: Active site of human FC with bound protoporphyrin IX (PROTO). D: The insertion of ferrous iron into PROTO proceeds via a “sitting-atop” complex between the porphyrin and the metal ion.
Much less is known about the oxygen-independent PPO occurring in anaerobic bacteria and facultative anaerobes. In the mid-1970s, it was shown by Jacobs and Jacobs using cell free extracts prepared from E. coli that the oxygen-independent PPO activity was associated with the membrane fraction and could be solubilized by detergents. Further, it was observed that the PPO activity was coupled to the respiratory chain.177,178,189 Later, an E. coli mutant strain was found which was deficient in PPO activity. The mutated gene was subsequently mapped and named hemG.190,191 Only recently recombinant E. coli HemG was purified, characterized and shown to be an oxygen-independent PPO.192 The protein belongs to the protein family of so-called long chain flavodoxins and contains an FMN cofactor. HemG was reported to be able to oxidize PROTOGEN to PROTO in the presence of menadione as the terminal electron acceptor substituting for the physiological, membrane-bound electron acceptor menaquinone.
Ferrochelatase
Ferrochelatase catalyzes the terminal step of heme biosynthesis, namely the insertion of ferrous iron into protoporphyrin IX. The first report describing ferrochelatase activity in avian erythrocytes was published in 1956 by Ashenbrucker et al. and was followed by descriptions of the same enzyme activity in a variety of cell types.193,194 However, it took another 25 years until the first ferrochelatase was purified to homogeneity from rat liver mitochondria still with low yields.195 In the early 1990s, the genes encoding ferrochelatase from several eukaryotic organisms were cloned and sequenced. The production of recombinant ferrochelatases finally resulted in high protein yields for functional and structural characterization. Today, ferrochelatase is one of the best characterized enzymes of the heme biosynthesis pathway.122,194 In humans, deficiency of ferrochelatase results in the metabolic disorder erythropoietic protoporphyria (EPP). In eukaryotes ferrochelatase is a membrane-associated homodimer, while it is monomeric in bacteria.134,186,196 In addition, in Gram-positive bacteria such as B. subtilis FC is a soluble protein located in the bacterial cytoplasm.197 Eukaryotic FC proteins, with the exception of the plant enzymes, contain one [2Fe-2S] cluster per subunit.198,199 Bacterial ferrochelatases have been described with and without FeS cluster.200,201 However, the role of the iron-sulfur cluster is at present unknown. FC accepts a variety of different porphyrins as substrates showing that FC is rather tolerant towards different substituents on rings A and B.122,202 In contrast the propionate side chains on rings C and D are more critical for catalysis. Some porphyrins which are not accepted as substrates like N-alkyl porphyrins act as competitive inhibitors.203 FC is also able to insert divalent metal ions like Ni2+ and Zn2+ into porphyrins whereas others like Mn2+, Hg2+, or Pb2+ have inhibitory effects.204,205 During catalysis, the planar porphyrin macrocycle has to be distorted by the enzyme into a saddle conformation to facilitate metal chelation.136,206–208 Desolvation of ferrous iron and bond formation between the metal and two of the pyrrole nitrogens lead to a so-called “sitting-atop” complex [Fig. 5(D)]. Finally, the deprotonation of the remaining two NH-groups results in the formation of the metalated porphyrin.207
Although ferrochelatases from different species share little amino acid sequence identity the crystal structures of human, yeast and B. subtilis FC show a high degree of structural conservation. Each subunit of the dimeric eukaryotic enzymes consists of two similar domains, each containing a four-stranded parallel β-sheet surrounded by α-helices (Fig. 1). A C-terminal extension which is not present in the B. subtilis FC adopts a helix-turn-helix structure and is involved in coordination of the [2Fe-2S] cluster. In fact, the iron-sulfur cluster appears to contribute to anchoring the C-terminal extension to the rest of the monomer. In the FC homodimer this C-terminal extension is involved in interactions between the monomers and its absence in the B. subtilis enzyme might explain the monomeric structure of the latter. The active site pockets of dimeric human FC are both located on the same surface which is believed to be embedded within the membrane. The entrance of the active site pocket is lined by mostly hydrophobic residues whereas the base of the pocket is populated with several hydrophilic residues. The crystal structure of human FC in complex with PROTO revealed that the active site closes upon porphyrin binding and that the substrate is completely engulfed by the protein. Several key interactions between FC and the substrate position the porphyrin in the active site pocket [Fig. 5(C)]. The propionate carboxylate group of ring D forms a salt bridge with an arginine and that of ring C is involved in hydrogen bonding with the side chains of a serine and a tyrosine. In addition, interactions with nearby histidine and methionine, located on opposite sites of the macrocycle, also contribute to the positioning of the substrate. The FC-substrate complex structure also revealed that PROTO is distorted by ∼11.5° to adopt a modest saddle conformation which is consistent with theoretical calculations.209 After metal insertion it was observed in a FC-product complex structure that a histidine swings away from the metallated porphyrin and that the interactions between the product ring C propionate side chain and the serine and tyrosine residues are broken. In the FC-product structure it was also observed that a structural reorganization takes place in a conserved π-helix in human FC which facilitates product release from the active site.210 Mutations in the human ferrochelatase gene that cause EPP include missense mutations and exon deletions. Among the missense mutations which cause amino acid exchanges many lead to decreased levels of enzyme activity, although only two of them directly involve active site residues.211
Coordination of Heme Biosynthesis in the Cell
Several intermediates on the biosynthetic road to heme are highly unstable or reactive molecules. For example, the GluTR reaction product, GSA, is a reactive aldehyde which easily undergoes auto-cyclization or other side reactions. The linear tetrapyrrole pre-uroporphyrinogen rapidly undergoes cyclization to the non-physiological uroporphyrinogen I. Further, all porphyrinogens are easily oxidized to the corresponding porphyrins and these in turn are extremely sensitive to light, a feature exploited by photodynamic cancer therapy. As a consequence one can imagine that a highly concerted action of the heme biosynthesis enzymes is required in order to assure the efficient synthesis of this essential molecule. Such a coordinated action is achieved by (i) the co-transcription of heme biosynthesis genes and (ii) by protein-protein interactions allowing for direct product-substrate channeling between the enzymes. Indeed, the structures of GluTR and GSAM revealed a striking degree of surface complementarity and it was possible to build a model complex between the two proteins in which the active site “back door” of GluTR lies directly at the active site entrance of GSAM. In addition, complex formation between GluTR and GSAM was shown biochemically for the enzymes from E. coli and C. reinhardtii.212,213 Therefore, the reactive GSA is directly handed over from GluTR to GSAM avoiding any contact with the aqueous medium. Complex formation was also proposed to occur between PBGD and UROS to avoid formation of uroporphyrinogen I. Although such a complex has not been shown to exist at present on the biochemical level, the genes encoding PBGD and UROS are often tandemly organized in an operon in bacteria. Finally, as for GluTR/GSAM and PBGD/UROS a direct product/substrate channeling via protein–protein interactions between PPO and ferrochelatase was proposed to occur. Indeed, it was possible to dock the structures of tobacco PPO and human FC in such a way that product/substrate channeling could take place between the two proteins.130 In addition, it was observed that the presence of PPO is required for the iron insertion into PROTO by FC214 and complex formation in vivo was shown for PPO and FC from T. elongatus.215
Conclusions
Heme is an essential molecule for all eukaryotic and most prokaryotic organisms. The enzymes involved in the multistep heme biosynthetic pathway were identified and characterized. In humans, decreased activities of these enzymes due to genetic defects are responsible for various porphyrias and X-linked sideroblastic anemia. The fact that certain enzymes are involved in heme biosynthesis exclusively in plants or bacteria makes them attractive targets for new herbicides and antimicrobial compounds. Both biochemical and structural characterization of the heme biosynthesis enzymes help to understand the molecular basis of porphyria-causing mutations and provide the means to develop new strategies for the treatment of these severe metabolic disorders as well as to design new antibiotics. Structural characterization of the enzymes proved very valuable for the current understanding of the enzyme mechanisms. In the future, additional structures of enzyme-substrate and enzyme-inhibitor complexes will certainly provide even more insight into the mechanisms of enzyme catalyzed heme biosynthesis.
Acknowledgments
The authors thank all their current and former coworkers in the field of heme biosynthesis for their invaluable contributions to the current understanding of enzyme mechanisms. Special thanks go to Prof. Wolf-Dieter Schubert (Cape Town, South Africa) and Drs. Jürgen Moser and Martina Jahn (Braunschweig, Germany) for their longstanding collaboration and work in our labs. Funding by the Deutsche Forschungsgemeinschaft and the Fonds der chemischen Industrie to G.L., D.J., and D.W.H. is gratefully acknowledged.
References
- 1.Munro AW, Girvan HM, McLean KJ, Cheesman MR, Leys D. Heme and hemoproteins. In: Warren MJ, Smith AG, editors. Tetrapyrroles: birth, life and death. Austin: Landes Bioscience; 2009. pp. 160–183. [Google Scholar]
- 2.Heinemann IU, Jahn M, Jahn D. The biochemistry of heme biosynthesis. Arch Biochem Biophys. 2008;474:238–251. doi: 10.1016/j.abb.2008.02.015. [DOI] [PubMed] [Google Scholar]
- 3.Ajioka RS, Phillips JD, Kushner JP. Biosynthesis of heme in mammals. Biochim Biophys Acta. 2006;1763:723–736. doi: 10.1016/j.bbamcr.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 4.Poblete-Gutierrez P, Wiederholt T, Merk HF, Frank J. The porphyrias: clinical presentation, diagnosis and treatment. Eur J Dermatol. 2006;16:230–240. [PubMed] [Google Scholar]
- 5.Battersby AR. Tetrapyrroles: the pigments of life. Nat Prod Rep. 2000;17:507–526. doi: 10.1039/b002635m. [DOI] [PubMed] [Google Scholar]
- 6.Ishida T, Yu L, Akutsu H, Ozawa K, Kawanishi S, Seto A, Inubushi T, Sano S. A primitive pathway of porphyrin biosynthesis and enzymology in Desulfovibrio vulgaris. Proc Natl Acad Sci USA. 1998;95:4853–4858. doi: 10.1073/pnas.95.9.4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lobo SA, Brindley A, Warren MJ, Saraiva LM. Functional characterization of the early steps of tetrapyrrole biosynthesis and modification in Desulfovibrio vulgaris Hildenborough. Biochem J. 2009;420:317–325. doi: 10.1042/BJ20090151. [DOI] [PubMed] [Google Scholar]
- 8.Buchenau B, Kahnt J, Heinemann IU, Jahn D, Thauer RK. Heme biosynthesis in Methanosarcina barkeri via a pathway involving two methylation reactions. J Bacteriol. 2006;188:8666–8668. doi: 10.1128/JB.01349-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jahn D, Heinz DW. Biosynthesis of 5-aminolevulinic acid. In: Warren MJ, Smith AG, editors. Tetrapyrroles: birth, life and death. Austin: Landes Bioscience; 2009. pp. 29–42. [Google Scholar]
- 10.Shemin D, Rittenberg D. The utilization of glycine for the synthesis of a porphyrin. J Biol Chem. 1945;159:567–568. [Google Scholar]
- 11.Radin NS, Rittenberg D, Shemin D. The role of acetic acid in the biosynthesis of heme. J Biol Chem. 1950;184:755–767. [PubMed] [Google Scholar]
- 12.Shemin D, Kumin S. The mechanism of porphyrin formation: the formation of a succinyl intermediate from succinate. J Biol Chem. 1952;198:827–837. [PubMed] [Google Scholar]
- 13.Muir HM, Neuberger A. The biogenesis of porphyrins. 2. The origin of the methyne carbon atoms. Biochem J. 1950;47:97–104. doi: 10.1042/bj0470097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neuberger A, Scott JJ. Aminolaevulinic acid and porphyrin biosynthesis. Nature. 1953;172:1093–1094. doi: 10.1038/1721093a0. [DOI] [PubMed] [Google Scholar]
- 15.Shemin D, Russell CS. Aminolevulinic acid, its role in the biosynthesis of porphyrins and purines. J Am Chem Soc. 1953;75:4873–4874. [Google Scholar]
- 16.Kikuchi G, Kumar A, Talmage P, Shemin D. The enzymatic synthesis of delta-aminolevulinic acid. J Biol Chem. 1958;233:1214–1219. [PubMed] [Google Scholar]
- 17.Kikuchi G, Shemin D, Bachmann BJ. The enzymic synthesis of delta-aminolevulinic acid. Biochim Biophys Acta. 1958;28:219–220. doi: 10.1016/0006-3002(58)90461-x. [DOI] [PubMed] [Google Scholar]
- 18.Gibson KD, Laver WG, Neuberger A. Initial stages in the biosynthesis of porphyrins. 2. The formation of delta-aminolaevulic acid from glycine and succinyl-coenzyme A by particles from chicken erythrocytes. Biochem J. 1958;70:71–81. doi: 10.1042/bj0700071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Laver WG, Neuberger A, Udenfriend S. Initial stages in the biosynthesis of porphyrins. I. The formation of delta-aminolaevulic acid by particles obtained from chicken erythrocytes. Biochem J. 1958;70:4–14. doi: 10.1042/bj0700004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ferreira GC, Gong J. 5-Aminolevulinate synthase and the first step of heme biosynthesis. J Bioenerg Biomembr. 1995;27:151–159. doi: 10.1007/BF02110030. [DOI] [PubMed] [Google Scholar]
- 21.Bishop DF, Henderson AS, Astrin KH. Human delta-aminolevulinate synthase: assignment of the housekeeping gene to 3p21 and the erythroid-specific gene to the X chromosome. Genomics. 1990;7:207–214. doi: 10.1016/0888-7543(90)90542-3. [DOI] [PubMed] [Google Scholar]
- 22.Camaschella C. Recent advances in the understanding of inherited sideroblastic anaemia. Br J Haematol. 2008;143:27–38. doi: 10.1111/j.1365-2141.2008.07290.x. [DOI] [PubMed] [Google Scholar]
- 23.Whatley SD, Ducamp S, Gouya L, Grandchamp B, Beaumont C, Badminton MN, Elder GH, Holme SA, Anstey AV, Parker M. C-terminal deletions in the ALAS2 gene lead to gain of function and cause X-linked dominant protoporphyria without anemia or iron overload. Am J Hum Genet. 2008;83:408–414. doi: 10.1016/j.ajhg.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Astner I, Schulze JO, van den Heuvel J, Jahn D, Schubert WD, Heinz DW. Crystal structure of 5-aminolevulinate synthase, the first enzyme of heme biosynthesis, and its link to XLSA in humans. Embo J. 2005;24:3166–3177. doi: 10.1038/sj.emboj.7600792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fanica-Gaignier M, Clement-Metral J. 5-Aminolevulinic-acid synthetase of Rhodopseudomonas spheroides Y. Purification and some properties. Eur J Biochem. 1973;40:13–18. doi: 10.1111/j.1432-1033.1973.tb03163.x. [DOI] [PubMed] [Google Scholar]
- 26.Nandi DL. Delta-aminolevulinic acid synthase of Rhodopseudomonas spheroides. Binding of pyridoxal phosphate to the enzyme. Arch Biochem Biophys. 1978;188:266–271. doi: 10.1016/s0003-9861(78)80008-3. [DOI] [PubMed] [Google Scholar]
- 27.Alexander FW, Sandmeier E, Mehta PK, Christen P. Evolutionary relationships among pyridoxal-5′-phosphate-dependent enzymes. Regio-specific alpha, beta and gamma families. Eur J Biochem. 1994;219:953–960. doi: 10.1111/j.1432-1033.1994.tb18577.x. [DOI] [PubMed] [Google Scholar]
- 28.Akhtar M, Jordan PM. The mechanism of the action of delta-aminolaevulate synthetase and the synthesis of stereospecifically tritiated glycine. J Chem Soc Chem Commun. 1968:1691–1692. [Google Scholar]
- 29.Zaman Z, Jordan PM, Akhtar M. Mechanism and stereochemistry of the 5-aminolaevulinate synthetase reaction. Biochem J. 1973;135:257–263. doi: 10.1042/bj1350257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abboud MM, Jordan PM, Akhtar M. Biosynthesis of 5-aminolevulinic acid: involvement of a retention–inversion mechanism. J Chem Soc Chem Commun. 1974:643–644. [Google Scholar]
- 31.Hunter GA, Ferreira GC. A continuous spectrophotometric assay for 5-aminolevulinate synthase that utilizes substrate cycling. Anal Biochem. 1995;226:221–224. doi: 10.1006/abio.1995.1217. [DOI] [PubMed] [Google Scholar]
- 32.Hunter GA, Ferreira GC. Lysine-313 of 5-aminolevulinate synthase acts as a general base during formation of the quinonoid reaction intermediates. Biochemistry. 1999;38:3711–3718. doi: 10.1021/bi982390w. [DOI] [PubMed] [Google Scholar]
- 33.Hunter GA, Ferreira GC. Pre-steady-state reaction of 5-aminolevulinate synthase. Evidence for a rate-determining product release. J Biol Chem. 1999;274:12222–12228. doi: 10.1074/jbc.274.18.12222. [DOI] [PubMed] [Google Scholar]
- 34.Hunter GA, Zhang J, Ferreira GC. Transient kinetic studies support refinements to the chemical and kinetic mechanisms of aminolevulinate synthase. J Biol Chem. 2007;282:23025–23035. doi: 10.1074/jbc.M609330200. [DOI] [PubMed] [Google Scholar]
- 35.Hunter GA, Ferreira GC. 5-Aminolevulinate synthase: catalysis of the first step of heme biosynthesis. Cell Mol Biol (Noisy-le-grand) 2009;55:102–110. [PMC free article] [PubMed] [Google Scholar]
- 36.Beale SI, Castelfranco PA. 14C incorporation from exogenous compounds into delta-aminolevulinic acid by greening cucumber cotyledons. Biochem Biophys Res Commun. 1973;52:143–149. doi: 10.1016/0006-291x(73)90966-2. [DOI] [PubMed] [Google Scholar]
- 37.Beale SI, Gough SP, Granick S. Biosynthesis of delta-aminolevulinic acid from the intact carbon skeleton of glutamic acid in greening barley. Proc Natl Acad Sci USA. 1975;72:2719–2723. doi: 10.1073/pnas.72.7.2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kannangara CG, Gough SP. Biosynthesis of delta-aminolevulinate in greening barley leaves: glutamate-1-semialdehyde aminotransferase. Carlsberg Res Commun. 1978;43:185–194. doi: 10.1007/BF02908411. [DOI] [PubMed] [Google Scholar]
- 39.Kannangara CG, Gough SP, Girnth CR. Delta-aminolevulinate synthesis in greening barley. 2. Purification of enzymes. In: Akoyunoglou G, editor. Vol. 5. Philadelphia: Balaban Int Sci Ser; 1981. pp. 117–127. Proceedings of Fifth International Congress in Photosynthesis. [Google Scholar]
- 40.Huang DD, Wang WY, Gough SP, Kannangara CG. Delta-aminolevulinic acid synthesizing enzymes need an RNA moiety for activity. Science. 1984;225:1482–1484. doi: 10.1126/science.6206568. [DOI] [PubMed] [Google Scholar]
- 41.Kannangara CG, Gough SP, Bruyant P, Hoober JK, Kahn A, von Wettstein D. tRNA(Glu) as a cofactor in delta-aminolevulinate biosynthesis: steps that regulate chlorophyll synthesis. Trends Biochem Sci. 1988;13:139–143. doi: 10.1016/0968-0004(88)90071-0. [DOI] [PubMed] [Google Scholar]
- 42.Jahn D, Verkamp E, Soll D. Glutamyl-transfer RNA: a precursor of heme and chlorophyll biosynthesis. Trends Biochem Sci. 1992;17:215–218. doi: 10.1016/0968-0004(92)90380-r. [DOI] [PubMed] [Google Scholar]
- 43.Chen MW, Jahn D, O'Neill GP, Soll D. Purification of the glutamyl-tRNA reductase from Chlamydomonas reinhardtii involved in delta-aminolevulinic acid formation during chlorophyll biosynthesis. J Biol Chem. 1990;265:4058–4063. [PubMed] [Google Scholar]
- 44.Rieble S, Beale SI. Purification of glutamyl-tRNA reductase from Synechocystis sp. PCC 6803. J Biol Chem. 1991;266:9740–9745. [PubMed] [Google Scholar]
- 45.Pontoppidan B, Kannangara CG. Purification and partial characterization of barley glutamyl-tRNA(Glu) reductase, the enzyme that directs glutamate to chlorophyll biosynthesis. Eur J Biochem. 1994;225:529–537. doi: 10.1111/j.1432-1033.1994.00529.x. [DOI] [PubMed] [Google Scholar]
- 46.Moser J, Lorenz S, Hubschwerlen C, Rompf A, Jahn D. Methanopyrus kandleri glutamyl-tRNA reductase. J Biol Chem. 1999;274:30679–30685. doi: 10.1074/jbc.274.43.30679. [DOI] [PubMed] [Google Scholar]
- 47.Wang WY, Huang DD, Stachon D, Gough SP, Kannangara CG. Purification, characterization, and fractionation of the delta-aminolevulinic acid synthesizing enzymes from light-grown Chlamydomonas reinhardtii cells. Plant Physiol. 1984;74:569–575. doi: 10.1104/pp.74.3.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hoober JK, Kahn A, Ash DE, Gough S, Kannangara CG. Biosynthesis of delta-aminolevulinate in greening barley leaves. IX. Structure of the substrate, mode of gabaculine inhibition, and the catalytic mechanism of glutamate 1-semialdehyde aminotransferase. Carlsberg Res Commun. 1988;53:11–25. doi: 10.1007/BF02908411. [DOI] [PubMed] [Google Scholar]
- 49.Grimm B. Primary structure of a key enzyme in plant tetrapyrrole synthesis: glutamate 1-semialdehyde aminotransferase. Proc Natl Acad Sci USA. 1990;87:4169–4173. doi: 10.1073/pnas.87.11.4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jahn D, Chen MW, Soll D. Purification and functional characterization of glutamate-1-semialdehyde aminotransferase from Chlamydomonas reinhardtii. J Biol Chem. 1991;266:161–167. [PubMed] [Google Scholar]
- 51.Smith MA, Grimm B, Kannangara CG, von Wettstein D. Spectral kinetics of glutamate-1-semialdehyde aminomutase of Synechococcus. Proc Natl Acad Sci USA. 1991;88:9775–9779. doi: 10.1073/pnas.88.21.9775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Palmieri G, Di Palo M, Scaloni A, Orru S, Marino G, Sannia G. Glutamate-1-semialdehyde aminotransferase from Sulfolobus solfataricus. Biochem J. 1996;320(Part 2):541–545. doi: 10.1042/bj3200541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moser J, Schubert WD, Beier V, Bringemeier I, Jahn D, Heinz DW. V-shaped structure of glutamyl-tRNA reductase, the first enzyme of tRNA-dependent tetrapyrrole biosynthesis. Embo J. 2001;20:6583–6590. doi: 10.1093/emboj/20.23.6583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hennig M, Grimm B, Contestabile R, John RA, Jansonius JN. Crystal structure of glutamate-1-semialdehyde aminomutase: an alpha2-dimeric vitamin B6-dependent enzyme with asymmetry in structure and active site reactivity. Proc Natl Acad Sci USA. 1997;94:4866–4871. doi: 10.1073/pnas.94.10.4866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schulze JO, Schubert WD, Moser J, Jahn D, Heinz DW. Evolutionary relationship between initial enzymes of tetrapyrrole biosynthesis. J Mol Biol. 2006;358:1212–1220. doi: 10.1016/j.jmb.2006.02.064. [DOI] [PubMed] [Google Scholar]
- 56.Randau L, Schauer S, Ambrogelly A, Salazar JC, Moser J, Sekine S, Yokoyama S, Soll D, Jahn D. tRNA recognition by glutamyl-tRNA reductase. J Biol Chem. 2004;279:34931–34937. doi: 10.1074/jbc.M401529200. [DOI] [PubMed] [Google Scholar]
- 57.Luer C, Schauer S, Virus S, Schubert WD, Heinz DW, Moser J, Jahn D. Glutamate recognition and hydride transfer by Escherichia coli glutamyl-tRNA reductase. Febs J. 2007;274:4609–4614. doi: 10.1111/j.1742-4658.2007.05989.x. [DOI] [PubMed] [Google Scholar]
- 58.Schauer S, Chaturvedi S, Randau L, Moser J, Kitabatake M, Lorenz S, Verkamp E, Schubert WD, Nakayashiki T, Murai M. Escherichia coli glutamyl-tRNA reductase. Trapping the thioester intermediate. J Biol Chem. 2002;277:48657–48663. doi: 10.1074/jbc.M206924200. [DOI] [PubMed] [Google Scholar]
- 59.Grimm B, Smith MA, von Wettstein D. The role of Lys272 in the pyridoxal 5-phosphate active site of Synechococcus glutamate-1-semialdehyde aminotransferase. Eur J Biochem. 1992;206:579–585. doi: 10.1111/j.1432-1033.1992.tb16962.x. [DOI] [PubMed] [Google Scholar]
- 60.Tyacke RJ, Harwood JL, John RA. Properties of the pyridoxaldimine form of glutamate semialdehyde aminotransferase (glutamate-1-semialdehyde 2,1-aminomutase) and analysis of its role as an intermediate in the formation of aminolaevulinate. Biochem J. 1993;293(Part 3):697–701. doi: 10.1042/bj2930697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brody S, Andersen JS, Kannangara CG, Meldgaard M, Roepstorff P, von Wettstein D. Characterization of the different spectral forms of glutamate 1-semialdehyde aminotransferase by mass spectrometry. Biochemistry. 1995;34:15918–15924. doi: 10.1021/bi00049a006. [DOI] [PubMed] [Google Scholar]
- 62.Ilag LL, Jahn D. Activity and spectroscopic properties of the Escherichia coli glutamate 1-semialdehyde aminotransferase and the putative active site mutant K265R. Biochemistry. 1992;31:7143–7151. doi: 10.1021/bi00146a016. [DOI] [PubMed] [Google Scholar]
- 63.Pugh CE, Harwood JL, John RA. Mechanism of glutamate semialdehyde aminotransferase. Roles of diamino- and dioxo-intermediates in the synthesis of aminolevulinate. J Biol Chem. 1992;267:1584–1588. [PubMed] [Google Scholar]
- 64.Friedmann HC, Duban ME, Valasinas A, Frydman B. The enantioselective participation of (S)- and (R)-diaminovaleric acids in the formation of delta-aminolevulinic acid in cyanobacteria. Biochem Biophys Res Commun. 1992;185:60–68. doi: 10.1016/s0006-291x(05)80955-6. [DOI] [PubMed] [Google Scholar]
- 65.Smith MA, King PJ, Grimm B. Transient-state kinetic analysis of Synechococcus glutamate 1-semialdehyde aminotransferase. Biochemistry. 1998;37:319–329. doi: 10.1021/bi9717587. [DOI] [PubMed] [Google Scholar]
- 66.Orriss GL, Patel TR, Sorensen J, Stetefeld J. Absence of a catalytic water confers resistance to the neurotoxin gabaculine. Faseb J. 2010;24:404–414. doi: 10.1096/fj.09-138297. [DOI] [PubMed] [Google Scholar]
- 67.Sachs P. Ein Fall von akuter Porphyrie mit hochgradiger Muskelatropie. Klin Wschr. 1931;10:1123–1125. [Google Scholar]
- 68.Westall RG. Isolation of porphobilinogen from the urine of a patient with acute porphyria. Nature. 1952;170:614–616. doi: 10.1038/170614a0. [DOI] [PubMed] [Google Scholar]
- 69.Cookson GH, Rimington C. Porphobilinogen: chemical constitution. Nature. 1953;171:875–876. doi: 10.1038/171875a0. [DOI] [PubMed] [Google Scholar]
- 70.Falk JE, Dresel EI, Rimington C. Porphobilinogen as a porphyrin precursor, and interconversion of porphyrins, in a tissue system. Nature. 1953;172:292–294. doi: 10.1038/172292a0. [DOI] [PubMed] [Google Scholar]
- 71.Gibson KD, Neuberger A, Scott JJ. The enzymic conversion of delta-aminolaevulic acid to porphobilinogen. Biochem J. 1954;58:xli–xlii. [PubMed] [Google Scholar]
- 72.Schmid R, Shemin D. The enzymatic formation of porphobilinogen from delta-aminolevulinic acid and its conversion to protoporphyrin. J Am Chem Soc. 1955;77:506–507. [Google Scholar]
- 73.Bogorad L, Granick S. The enzymatic synthesis of porphyrins from porphobilinogen. Proc Natl Acad Sci USA. 1953;39:1176–1188. doi: 10.1073/pnas.39.12.1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bogorad L. The enzymatic synthesis of porphyrins from porphobilinogen. II. Uroporphyrin III. J Biol Chem. 1958;233:510–515. [PubMed] [Google Scholar]
- 75.Neve RA, Labbe RF, Aldrich RA. Reduced uroporphyrin III in the biosynthesis of heme. J Am Chem Soc. 1956;78:691–692. [Google Scholar]
- 76.Schubert HL, Erskine PT, Cooper JB. 5-Aminolevulinic acid dehydratase, porphobilinogen deaminase and uroporphyrinogen III synthase. In: Warren MJ, Smith AG, editors. Tetrapyrroles: birth, life and death. Austin: Landes Bioscience; 2009. pp. 43–73. [Google Scholar]
- 77.Erskine PT, Senior N, Awan S, Lambert R, Lewis G, Tickle IJ, Sarwar M, Spencer P, Thomas P, Warren MJ. X-ray structure of 5-aminolaevulinate dehydratase, a hybrid aldolase. Nat Struct Biol. 1997;4:1025–1031. doi: 10.1038/nsb1297-1025. [DOI] [PubMed] [Google Scholar]
- 78.Erskine PT, Norton E, Cooper JB, Lambert R, Coker A, Lewis G, Spencer P, Sarwar M, Wood SP, Warren MJ. X-ray structure of 5-aminolevulinic acid dehydratase from Escherichia coli complexed with the inhibitor levulinic acid at 2.0 A resolution. Biochemistry. 1999;38:4266–4276. doi: 10.1021/bi982137w. [DOI] [PubMed] [Google Scholar]
- 79.Frankenberg N, Erskine PT, Cooper JB, Shoolingin-Jordan PM, Jahn D, Heinz DW. High resolution crystal structure of a Mg2+-dependent porphobilinogen synthase. J Mol Biol. 1999;289:591–602. doi: 10.1006/jmbi.1999.2808. [DOI] [PubMed] [Google Scholar]
- 80.Coates L, Beaven G, Erskine PT, Beale SI, Avissar YJ, Gill R, Mohammed F, Wood SP, Shoolingin-Jordan P, Cooper JB. The X-ray structure of the plant like 5-aminolaevulinic acid dehydratase from Chlorobium vibrioforme complexed with the inhibitor laevulinic acid at 2.6 A resolution. J Mol Biol. 2004;342:563–570. doi: 10.1016/j.jmb.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 81.Louie GV, Brownlie PD, Lambert R, Cooper JB, Blundell TL, Wood SP, Warren MJ, Woodcock SC, Jordan PM. Structure of porphobilinogen deaminase reveals a flexible multidomain polymerase with a single catalytic site. Nature. 1992;359:33–39. doi: 10.1038/359033a0. [DOI] [PubMed] [Google Scholar]
- 82.Song G, Li Y, Cheng C, Zhao Y, Gao A, Zhang R, Joachimiak A, Shaw N, Liu ZJ. Structural insight into acute intermittent porphyria. Faseb J. 2009;23:396–404. doi: 10.1096/fj.08-115469. [DOI] [PubMed] [Google Scholar]
- 83.Gill R, Kolstoe SE, Mohammed F, Al DBA, Mosely JE, Sarwar M, Cooper JB, Wood SP, Shoolingin-Jordan PM. Structure of human porphobilinogen deaminase at 2.8 A: the molecular basis of acute intermittent porphyria. Biochem J. 2009;420:17–25. doi: 10.1042/BJ20082077. [DOI] [PubMed] [Google Scholar]
- 84.Mathews MA, Schubert HL, Whitby FG, Alexander KJ, Schadick K, Bergonia HA, Phillips JD, Hill CP. Crystal structure of human uroporphyrinogen III synthase. Embo J. 2001;20:5832–5839. doi: 10.1093/emboj/20.21.5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schubert HL, Phillips JD, Heroux A, Hill CP. Structure and mechanistic implications of a uroporphyrinogen III synthase-product complex. Biochemistry. 2008;47:8648–8655. doi: 10.1021/bi800635y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bollivar DW, Clauson C, Lighthall R, Forbes S, Kokona B, Fairman R, Kundrat L, Jaffe EK. Rhodobacter capsulatus porphobilinogen synthase, a high activity metal ion independent hexamer. BMC Biochem. 2004;5:17. doi: 10.1186/1471-2091-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jaffe EK. The porphobilinogen synthase catalyzed reaction mechanism. Bioorg Chem. 2004;32:316–325. doi: 10.1016/j.bioorg.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 88.Chaudhry AG, Jordan PM. Stereochemical studies on the formation of porphobilinogen. Biochem Soc Trans. 1976;4:760–761. doi: 10.1042/bst0040760. [DOI] [PubMed] [Google Scholar]
- 89.Jordan PM, Gibbs PN. Mechanism of action of 5-aminolaevulinate dehydratase from human erythrocytes. Biochem J. 1985;227:1015–1020. doi: 10.1042/bj2271015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Erskine PT, Coates L, Newbold R, Brindley AA, Stauffer F, Wood SP, Warren MJ, Cooper JB, Shoolingin-Jordan PM, Neier R. The X-ray structure of yeast 5-aminolaevulinic acid dehydratase complexed with two diacid inhibitors. FEBS Lett. 2001;503:196–200. doi: 10.1016/s0014-5793(01)02721-1. [DOI] [PubMed] [Google Scholar]
- 91.Kervinen J, Jaffe EK, Stauffer F, Neier R, Wlodawer A, Zdanov A. Mechanistic basis for suicide inactivation of porphobilinogen synthase by 4,7-dioxosebacic acid, an inhibitor that shows dramatic species selectivity. Biochemistry. 2001;40:8227–8236. doi: 10.1021/bi010656k. [DOI] [PubMed] [Google Scholar]
- 92.Jaffe EK, Kervinen J, Martins J, Stauffer F, Neier R, Wlodawer A, Zdanov A. Species-specific inhibition of porphobilinogen synthase by 4-oxosebacic acid. J Biol Chem. 2002;277:19792–19799. doi: 10.1074/jbc.M201486200. [DOI] [PubMed] [Google Scholar]
- 93.Frere F, Schubert WD, Stauffer F, Frankenberg N, Neier R, Jahn D, Heinz DW. Structure of porphobilinogen synthase from Pseudomonas aeruginosa in complex with 5-fluorolevulinic acid suggests a double Schiff base mechanism. J Mol Biol. 2002;320:237–247. doi: 10.1016/S0022-2836(02)00472-2. [DOI] [PubMed] [Google Scholar]
- 94.Erskine PT, Coates L, Butler D, Youell JH, Brindley AA, Wood SP, Warren MJ, Shoolingin-Jordan PM, Cooper JB. X-ray structure of a putative reaction intermediate of 5-aminolaevulinic acid dehydratase. Biochem J. 2003;373:733–738. doi: 10.1042/BJ20030513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Goodwin CE, Leeper FJ. Stereochemistry and mechanism of the conversion of 5-aminolaevulinic acid into porphobilinogen catalysed by porphobilinogen synthase. Org Biomol Chem. 2003;1:1443–1446. doi: 10.1039/b302509h. [DOI] [PubMed] [Google Scholar]
- 96.Frere F, Nentwich M, Gacond S, Heinz DW, Neier R, Frankenberg-Dinkel N. Probing the active site of Pseudomonas aeruginosa porphobilinogen synthase using newly developed inhibitors. Biochemistry. 2006;45:8243–8253. doi: 10.1021/bi052611f. [DOI] [PubMed] [Google Scholar]
- 97.Breinig S, Kervinen J, Stith L, Wasson AS, Fairman R, Wlodawer A, Zdanov A, Jaffe EK. Control of tetrapyrrole biosynthesis by alternate quaternary forms of porphobilinogen synthase. Nat Struct Biol. 2003;10:757–763. doi: 10.1038/nsb963. [DOI] [PubMed] [Google Scholar]
- 98.Heinemann IU, Schulz C, Schubert WD, Heinz DW, Wang YG, Kobayashi Y, Awa Y, Wachi M, Jahn D, Jahn M. Structure of the heme biosynthetic Pseudomonas aeruginosa porphobilinogen synthase in complex with the antibiotic alaremycin. Antimicrob Agents Chemother. 2009;54:267–272. doi: 10.1128/AAC.00553-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Battersby AR, Hodgson GL, Hunt E, McDonald E, Saunders J. Biosynthesis of porphyrins and related macrocycles. Part VI. Nature of the rearrangement process leading to the natural type III porphyrins. J Chem Soc Perkin Trans. 1976;1:273–282. doi: 10.1039/p19760000273. [DOI] [PubMed] [Google Scholar]
- 100.Battersby AR, Fookes CJR, Meegan MJ, McDonald E, Wurziger HKW. Biosynthesis of porphyrins and related macrocycles. Part 16. Proof that the single intramolecular rearrangement leading to natural porphyrins (type-III) occurs at the tetrapyrrole level. J Chem Soc Perkin Trans. 1981;1:2786–2799. [Google Scholar]
- 101.Battersby AR, Fookes CJR, Matcham GWJ, McDonald E. Order of assembly of the four pyrrole rings during biosynthesis of the natural porphyrins. J Chem Soc Chem Commun. 1979:539–541. [Google Scholar]
- 102.Jordan PM, Seehra JS. The biosynthesis of uroporphyrinogen III: order of assembly of the four porphobilinogen molecules in the formation of the tetrapyrrole ring. FEBS Lett. 1979;104:364–366. doi: 10.1016/0014-5793(79)80853-4. [DOI] [PubMed] [Google Scholar]
- 103.Burton G, Fagerness PE, Hosozawa S, Jordan PM, Scott AI. 13C NMR evidence for a new intermediate, pre-uroporphyrinogen, in the enzymic transformation of porphobilinogen into uroporphyrinogens I and III. J Chem Soc Chem Commun. 1979:202–204. [Google Scholar]
- 104.Battersby AR, Fookes CJR, Gustafson-Potter KE, McDonald E, Matcham GWJ. Biosynthesis of the natural porphyrins: experiments on the ring-closure steps and with the hydroxy-analogue of porphobilinogen. J Chem Soc Chem Commun. 1979:316–319. [Google Scholar]
- 105.Battersby AR, Fookes CJR, Gustafson-Potter KE, McDonald E, Matcham GWJ. Biosynthesis of porphyrins and related macrocycles. Part 18. Proof by spectroscopy and synthesis that unrearranged hydroxymethylbilane is the product from deaminase and the substrate for cosynthetase in the biosynthesis of uroporphyrinogen-III. J Chem Soc Perkin Trans. 1982;1:2427–2444. [Google Scholar]
- 106.Hart GJ, Miller AD, Leeper FJ, Battersby AR. Biosynthesis of the natural porphyrins: proof that hydroxymethylbilane synthase (porphobilinogen deaminase) uses a novel binding group in its catalytic action. J Chem Soc Chem Commun. 1987:1762–1765. [Google Scholar]
- 107.Jordan PM, Warren MJ. Evidence for a dipyrromethane cofactor at the catalytic site of E. coli porphobilinogen deaminase. FEBS Lett. 1987;225:87–92. doi: 10.1016/0014-5793(87)81136-5. [DOI] [PubMed] [Google Scholar]
- 108.Warren MJ, Jordan PM. Investigation into the nature of substrate binding to the dipyrromethane cofactor of Escherichia coli porphobilinogen deaminase. Biochemistry. 1988;27:9020–9030. doi: 10.1021/bi00425a021. [DOI] [PubMed] [Google Scholar]
- 109.Warren MJ, Scott AI. Tetrapyrrole assembly and modification into the ligands of biologically functional cofactors. Trends Biochem Sci. 1990;15:486–491. doi: 10.1016/0968-0004(90)90304-t. [DOI] [PubMed] [Google Scholar]
- 110.Woodcock SC, Jordan PM. Evidence for participation of aspartate-84 as a catalytic group at the active site of porphobilinogen deaminase obtained by site-directed mutagenesis of the hemC gene from Escherichia coli. Biochemistry. 1994;33:2688–2695. doi: 10.1021/bi00175a043. [DOI] [PubMed] [Google Scholar]
- 111.Warren MJ, Gul S, Aplin RT, Scott AI, Roessner CA, O'Grady P, Shoolingin-Jordan PM. Evidence for conformational changes in Escherichia coli porphobilinogen deaminase during stepwise pyrrole chain elongation monitored by increased reactivity of cysteine-134 to alkylation by N-ethylmaleimide. Biochemistry. 1995;34:11288–11295. doi: 10.1021/bi00035a038. [DOI] [PubMed] [Google Scholar]
- 112.Mathewson JH, Corwin AH. Biosynthesis of pyrrole pigments: a mechanism for porphobilinogen polymerization. J Am Chem Soc. 1961;83:135–137. [Google Scholar]
- 113.Shoolingin-Jordan PM. Porphobilinogen deaminase and uroporphyrinogen III synthase: structure, molecular biology, and mechanism. J Bioenerg Biomembr. 1995;27:181–195. doi: 10.1007/BF02110033. [DOI] [PubMed] [Google Scholar]
- 114.Spivey AC, Capretta A, Frampton CS, Leeper FJ, Battersby AR. Biosynthesis of porphyrins and related macrocycles. Part 45. Determination by a novel X-ray method of the absolute configuration of the spiro lactam which inhibits uroporphyrinogen III synthase (cosynthetase) J Chem Soc Perkin Trans. 1996;1:2091–2102. [Google Scholar]
- 115.Silva PJ, Ramos MJ. Comparative density functional study of models for the reaction mechanism of uroporphyrinogen III synthase. J Phys Chem B. 2008;112:3144–3148. doi: 10.1021/jp076235f. [DOI] [PubMed] [Google Scholar]
- 116.Battersby AR, Fookes CJR, Pandey PS. Linear tetrapyrrolic intermediates for biosynthesis of the natural porphyrins. Experiments with modified substrates. Tetrahedron. 1983;39:1919–1926. [Google Scholar]
- 117.Pichon C, Atshaves BP, Xue T, Stolowich NJ, Scott AI. Studies on uro'gen III synthase with modified bilanes. Bioorg Med Chem. 1994;4:1105–1110. [Google Scholar]
- 118.Tan FC, Cheng Q, Saha K, Heinemann IU, Jahn M, Jahn D, Smith AG. Identification and characterization of the Arabidopsis gene encoding the tetrapyrrole biosynthesis enzyme uroporphyrinogen III synthase. Biochem J. 2008;410:291–299. doi: 10.1042/BJ20070770. [DOI] [PubMed] [Google Scholar]
- 119.Cornah JE, Smith AG. Transformation of uroporphyrinogen III into protohaem. In: Warren MJ, Smith AG, editors. Tetrapyrroles: birth, life and death. Austin: Landes Bioscience; 2009. pp. 74–88. [Google Scholar]
- 120.Shoolingin-Jordan PM. The biosynthesis of coproporphyrinogen III. In: Kadish KM, Smith KM, Guilard R, editors. The porphyrin handbook, Vol. 12, The iron and cobalt pigments: biosynthesis, structure and degradation. New York: Elsevier; 2003. pp. 33–74. [Google Scholar]
- 121.Akhtar M. Coproporphyrinogen III and protoporphyrinogen IX oxidases. In: Kadish KM, Smith KM, Guilard R, editors. The porphyrin handbook, Vol. 12, The iron and cobalt pigments: biosynthesis, structure and degradation. New York: Elsevier; 2003. pp. 75–92. [Google Scholar]
- 122.Dailey H, Dailey T. Ferrochelatase. In: Kadish KM, Smith KM, Guilard R, editors. The porphyrin handbook, Vol. 12, The iron and cobalt pigments: biosynthesis, structure and degradation. New York: Elsevier; 2003. pp. 93–121. [Google Scholar]
- 123.Whitby FG, Phillips JD, Kushner JP, Hill CP. Crystal structure of human uroporphyrinogen decarboxylase. Embo J. 1998;17:2463–2471. doi: 10.1093/emboj/17.9.2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Phillips JD, Whitby FG, Kushner JP, Hill CP. Structural basis for tetrapyrrole coordination by uroporphyrinogen decarboxylase. Embo J. 2003;22:6225–6233. doi: 10.1093/emboj/cdg606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Martins BM, Grimm B, Mock HP, Huber R, Messerschmidt A. Crystal structure and substrate binding modeling of the uroporphyrinogen-III decarboxylase from Nicotiana tabacum. Implications for the catalytic mechanism. J Biol Chem. 2001;276:44108–44116. doi: 10.1074/jbc.M104759200. [DOI] [PubMed] [Google Scholar]
- 126.Fan J, Liu Q, Hao Q, Teng M, Niu L. Crystal structure of uroporphyrinogen decarboxylase from Bacillus subtilis. J Bacteriol. 2007;189:3573–3580. doi: 10.1128/JB.01083-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Phillips JD, Whitby FG, Warby CA, Labbe P, Yang C, Pflugrath JW, Ferrara JD, Robinson H, Kushner JP, Hill CP. Crystal structure of the oxygen-dependent coproporphyrinogen oxidase (Hem13p) of Saccharomyces cerevisiae. J Biol Chem. 2004;279:38960–38968. doi: 10.1074/jbc.M406050200. [DOI] [PubMed] [Google Scholar]
- 128.Lee DS, Flachsova E, Bodnarova M, Demeler B, Martasek P, Raman CS. Structural basis of hereditary coproporphyria. Proc Natl Acad Sci USA. 2005;102:14232–14237. doi: 10.1073/pnas.0506557102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Layer G, Moser J, Heinz DW, Jahn D, Schubert WD. Crystal structure of coproporphyrinogen III oxidase reveals cofactor geometry of Radical SAM enzymes. Embo J. 2003;22:6214–6224. doi: 10.1093/emboj/cdg598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Koch M, Breithaupt C, Kiefersauer R, Freigang J, Huber R, Messerschmidt A. Crystal structure of protoporphyrinogen IX oxidase: a key enzyme in haem and chlorophyll biosynthesis. Embo J. 2004;23:1720–1728. doi: 10.1038/sj.emboj.7600189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Corradi HR, Corrigall AV, Boix E, Mohan CG, Sturrock ED, Meissner PN, Acharya KR. Crystal structure of protoporphyrinogen oxidase from Myxococcus xanthus and its complex with the inhibitor acifluorfen. J Biol Chem. 2006;281:38625–38633. doi: 10.1074/jbc.M606640200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Qin X, Sun L, Wen X, Yang X, Tan Y, Jin H, Cao Q, Zhou W, Xi Z, Shen Y. Structural insight into unique properties of protoporphyrinogen oxidase from Bacillus subtilis. J Struct Biol. 2010;170:76–82. doi: 10.1016/j.jsb.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 133.Al-Karadaghi S, Hansson M, Nikonov S, Jonsson B, Hederstedt L. Crystal structure of ferrochelatase: the terminal enzyme in heme biosynthesis. Structure. 1997;5:1501–1510. doi: 10.1016/s0969-2126(97)00299-2. [DOI] [PubMed] [Google Scholar]
- 134.Wu CK, Dailey HA, Rose JP, Burden A, Sellers VM, Wang BC. The 2.0 A structure of human ferrochelatase, the terminal enzyme of heme biosynthesis. Nat Struct Biol. 2001;8:156–160. doi: 10.1038/84152. [DOI] [PubMed] [Google Scholar]
- 135.Karlberg T, Lecerof D, Gora M, Silvegren G, Labbe-Bois R, Hansson M, Al-Karadaghi S. Metal binding to Saccharomyces cerevisiae ferrochelatase. Biochemistry. 2002;41:13499–13506. doi: 10.1021/bi0260785. [DOI] [PubMed] [Google Scholar]
- 136.Medlock A, Swartz L, Dailey TA, Dailey HA, Lanzilotta WN. Substrate interactions with human ferrochelatase. Proc Natl Acad Sci USA. 2007;104:1789–1793. doi: 10.1073/pnas.0606144104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Mauzerall D, Granick S. Porphyrin biosynthesis in erythrocytes. III. Uroporphyrinogen and its decarboxylase. J Biol Chem. 1958;232:1141–1162. [PubMed] [Google Scholar]
- 138.Jackson AH, Sancovich HA, Ferramola AM, Evans N, Games DE, Matlin SA, Elder GH, Smith SG. Macrocyclic intermediates in the biosynthesis of porphyrins. Philos Trans R Soc Lond B Biol Sci. 1976;273:191–206. doi: 10.1098/rstb.1976.0009. [DOI] [PubMed] [Google Scholar]
- 139.Lim CK, Rideout JM, Wright DJ. High-performance liquid chromatography of naturally occurring 8-, 7-, 6-, 5- and 4-carboxylic porphyrin isomers. J Chromatogr. 1983;282:629–641. doi: 10.1016/s0021-9673(00)91640-6. [DOI] [PubMed] [Google Scholar]
- 140.Akhtar M. Mechanism and stereochemistry of the enzymes involved in the conversion of uroporphyrinogen III into haem. In: Jordan PM, editor. New comprehensive biochemistry, Vol. 19, Biosynthesis of tetrapyrroles. Amsterdam: Elsevier; 1991. pp. 67–99. [Google Scholar]
- 141.Smith AG, Francis JE. Decarboxylation of porphyrinogens by rat liver uroporphyrinogen decarboxylase. Biochem J. 1979;183:455–458. doi: 10.1042/bj1830455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Luo J, Lim CK. Order of uroporphyrinogen III decarboxylation on incubation of porphobilinogen and uroporphyrinogen III with erythrocyte uroporphyrinogen decarboxylase. Biochem J. 1993;289(Part 2):529–532. doi: 10.1042/bj2890529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lewis CA, Jr, Wolfenden R. Uroporphyrinogen decarboxylation as a benchmark for the catalytic proficiency of enzymes. Proc Natl Acad Sci USA. 2008;105:17328–17333. doi: 10.1073/pnas.0809838105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Phillips JD, Warby CA, Whitby FG, Kushner JP, Hill CP. Substrate shuttling between active sites of uroporphyrinogen decarboxylase is not required to generate coproporphyrinogen. J Mol Biol. 2009;389:306–314. doi: 10.1016/j.jmb.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Cavallaro G, Decaria L, Rosato A. Genome-based analysis of heme biosynthesis and uptake in prokaryotic systems. J Proteome Res. 2008;7:4946–4954. doi: 10.1021/pr8004309. [DOI] [PubMed] [Google Scholar]
- 146.Martasek P. Hereditary coproporphyria. Semin Liver Dis. 1998;18:25–32. doi: 10.1055/s-2007-1007137. [DOI] [PubMed] [Google Scholar]
- 147.Sano S, Granick S. Mitochondrial coproporphyrinogen oxidase and protoporphyrin formation. J Biol Chem. 1961;236:1173–1180. [PubMed] [Google Scholar]
- 148.del Batlle AM, Benson A, Rimington C. Purification and properties of coproporphyrinogenase. Biochem J. 1965;97:731–740. doi: 10.1042/bj0970731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Poulson R, Polglase WJ. Aerobic and anaerobic coproporphyrinogenase activities in extracts from Saccharomyces cerevisiae. J Biol Chem. 1974;249:6367–6371. [PubMed] [Google Scholar]
- 150.Yoshinaga T, Sano S. Coproporphyrinogen oxidase. II. Reaction mechanism and role of tyrosine residues on the activity. J Biol Chem. 1980;255:4727–4731. [PubMed] [Google Scholar]
- 151.Yoshinaga T, Sano S. Coproporphyrinogen oxidase. I. Purification, properties, and activation by phospholipids. J Biol Chem. 1980;255:4722–4726. [PubMed] [Google Scholar]
- 152.Labbe P. Purification and properties of coproporphyrinogen III oxidase from yeast. Methods Enzymol. 1997;281:367–378. doi: 10.1016/s0076-6879(97)81044-0. [DOI] [PubMed] [Google Scholar]
- 153.Kohno H, Furukawa T, Tokunaga R, Taketani S, Yoshinaga T. Mouse coproporphyrinogen oxidase is a copper-containing enzyme: expression in Escherichia coli and site-directed mutagenesis. Biochim Biophys Acta. 1996;1292:156–162. doi: 10.1016/0167-4838(95)00188-3. [DOI] [PubMed] [Google Scholar]
- 154.Medlock AE, Dailey HA. Human coproporphyrinogen oxidase is not a metalloprotein. J Biol Chem. 1996;271:32507–32510. doi: 10.1074/jbc.271.51.32507. [DOI] [PubMed] [Google Scholar]
- 155.Breckau D, Mahlitz E, Sauerwald A, Layer G, Jahn D. Oxygen-dependent coproporphyrinogen III oxidase (HemF) from Escherichia coli is stimulated by manganese. J Biol Chem. 2003;278:46625–46631. doi: 10.1074/jbc.M308553200. [DOI] [PubMed] [Google Scholar]
- 156.Cavaleiro JAS, Kenner GW, Smith KM. Pyrroles and related compounds. Part XXXII. Biosynthesis of protoporphyrin-IX from coproporphyrinogen-III. J Chem Soc Perkin Trans. 1974;1:1188–1194. doi: 10.1039/p19740001188. [DOI] [PubMed] [Google Scholar]
- 157.Elder GH, Evans JO, Jackson JR, Jackson AH. Factors determining the sequence of oxidative decarboxylation of the 2- and 4-propionate substituents of coproporphyrinogen III by coproporphyrinogen oxidase in rat liver. Biochem J. 1978;169:215–223. doi: 10.1042/bj1690215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Zaman Z, Akhtar M. Mechanism and stereochemistry of vinyl-group formation in haem biosynthesis. Eur J Biochem. 1976;61:215–223. doi: 10.1111/j.1432-1033.1976.tb10014.x. [DOI] [PubMed] [Google Scholar]
- 159.Seehra JS, Jordan PM, Akhtar M. Anaerobic and aerobic coproporphyrinogen III oxidases of Rhodopseudomonas spheroides. Mechanism and stereochemistry of vinyl group formation. Biochem J. 1983;209:709–718. doi: 10.1042/bj2090709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Battersby AR, McDonald E, Wurziger HKW, James KJ. Stereochemistry of biosynthesis of the vinyl groups of protoporphyrin-IX: a short synthesis of porphobilinogen. J Chem Soc Chem Commun. 1975:493–494. [Google Scholar]
- 161.Lash TD. The enigma of coproporphyrinogen oxidase: how does this unusual enzyme carry out oxidative decarboxylations to afford vinyl groups? Bioorg Med Chem Lett. 2005;15:4506–4509. doi: 10.1016/j.bmcl.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 162.Silva PJ, Ramos MJ. A comparative density-functional study of the reaction mechanism of the O2-dependent coproporphyrinogen III oxidase. Bioorg Med Chem. 2008;16:2726–2733. doi: 10.1016/j.bmc.2008.01.008. [DOI] [PubMed] [Google Scholar]
- 163.Stephenson JR, Stacey JA, Morgenthaler JB, Friesen JA, Lash TD, Jones MA. Role of aspartate 400, arginine 262, and arginine 401 in the catalytic mechanism of human coproporphyrinogen oxidase. Protein Sci. 2007;16:401–410. doi: 10.1110/ps.062636907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Tait GH. Coproporphyrinogenase activity in extracts from Rhodopseudomonas spheroides. Biochem Biophys Res Commun. 1969;37:116–122. doi: 10.1016/0006-291x(69)90888-2. [DOI] [PubMed] [Google Scholar]
- 165.Tait GH. Coproporphyrinogenase activities in extracts of Rhodopseudomonas spheroides and Chromatium strain D. Biochem J. 1972;128:1159–1169. doi: 10.1042/bj1281159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Coomber SA, Jones RM, Jordan PM, Hunter CN. A putative anaerobic coproporphyrinogen III oxidase in Rhodobacter sphaeroides. I. Molecular cloning, transposon mutagenesis and sequence analysis of the gene. Mol Microbiol. 1992;6:3159–3169. doi: 10.1111/j.1365-2958.1992.tb01772.x. [DOI] [PubMed] [Google Scholar]
- 167.Xu K, Elliott T. Cloning, DNA sequence, and complementation analysis of the Salmonella typhimurium hemN gene encoding a putative oxygen-independent coproporphyrinogen III oxidase. J Bacteriol. 1994;176:3196–3203. doi: 10.1128/jb.176.11.3196-3203.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Troup B, Hungerer C, Jahn D. Cloning and characterization of the Escherichia coli hemN gene encoding the oxygen-independent coproporphyrinogen III oxidase. J Bacteriol. 1995;177:3326–3331. doi: 10.1128/jb.177.11.3326-3331.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Lieb C, Siddiqui RA, Hippler B, Jahn D, Friedrich B. The Alcaligenes eutrophus hemN gene encoding the oxygen-independent coproporphyrinogen III oxidase, is required for heme biosynthesis during anaerobic growth. Arch Microbiol. 1998;169:52–60. doi: 10.1007/s002030050540. [DOI] [PubMed] [Google Scholar]
- 170.Fischer HM, Velasco L, Delgado MJ, Bedmar EJ, Scharen S, Zingg D, Gottfert M, Hennecke H. One of two hemN genes in Bradyrhizobium japonicum is functional during anaerobic growth and in symbiosis. J Bacteriol. 2001;183:1300–1311. doi: 10.1128/JB.183.4.1300-1311.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Layer G, Verfurth K, Mahlitz E, Jahn D. Oxygen-independent coproporphyrinogen-III oxidase HemN from Escherichia coli. J Biol Chem. 2002;277:34136–34142. doi: 10.1074/jbc.M205247200. [DOI] [PubMed] [Google Scholar]
- 172.Layer G, Pierik AJ, Trost M, Rigby SE, Leech HK, Grage K, Breckau D, Astner I, Jansch L, Heathcote P. The substrate radical of Escherichia coli oxygen-independent coproporphyrinogen III oxidase HemN. J Biol Chem. 2006;281:15727–15734. doi: 10.1074/jbc.M512628200. [DOI] [PubMed] [Google Scholar]
- 173.Layer G, Grage K, Teschner T, Schunemann V, Breckau D, Masoumi A, Jahn M, Heathcote P, Trautwein AX, Jahn D. Radical S-adenosylmethionine enzyme coproporphyrinogen III oxidase HemN: functional features of the [4Fe-4S] cluster and the two bound S-adenosyl-l-methionines. J Biol Chem. 2005;280:29038–29046. doi: 10.1074/jbc.M501275200. [DOI] [PubMed] [Google Scholar]
- 174.Rand K, Noll C, Schiebel HM, Kemken D, Dulcks T, Kalesse M, Heinz DW, Layer G. The oxygen-independent coproporphyrinogen III oxidase HemN utilizes harderoporphyrinogen as a reaction intermediate during conversion of coproporphyrinogen III to protoporphyrinogen IX. Biol Chem. 2010;391:55–63. doi: 10.1515/BC.2010.006. [DOI] [PubMed] [Google Scholar]
- 175.Porra RJ, Falk JE. Protein-bound porphyrins associated with protoporphyrin biosynthesis. Biochem Biophys Res Commun. 1961;5:179–184. doi: 10.1016/0006-291x(61)90106-1. [DOI] [PubMed] [Google Scholar]
- 176.Porra RJ, Falk JE. The enzymic conversion of coproporphyrinogen 3 into protoporphyrin 9. Biochem J. 1964;90:69–75. doi: 10.1042/bj0900069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Jacobs NJ, Jacobs JM. Fumarate as alternate electron acceptor for the late steps of anaerobic heme synthesis in Escherichia coli. Biochem Biophys Res Commun. 1975;65:435–441. doi: 10.1016/s0006-291x(75)80112-4. [DOI] [PubMed] [Google Scholar]
- 178.Jacobs NJ, Jacobs JM. Nitrate, fumarate, and oxygen as electron acceptors for a late step in microbial heme synthesis. Biochim Biophys Acta. 1976;449:1–9. doi: 10.1016/0005-2728(76)90002-5. [DOI] [PubMed] [Google Scholar]
- 179.Siepker LJ, Ford M, de Kock R, Kramer S. Purification of bovine protoporphyrinogen oxidase: immunological cross-reactivity and structural relationship to ferrochelatase. Biochim Biophys Acta. 1987;913:349–358. doi: 10.1016/0167-4838(87)90146-4. [DOI] [PubMed] [Google Scholar]
- 180.Ferreira GC, Dailey HA. Mouse protoporphyrinogen oxidase. Kinetic parameters and demonstration of inhibition by bilirubin. Biochem J. 1988;250:597–603. doi: 10.1042/bj2500597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Camadro JM, Labbe P. Cloning and characterization of the yeast HEM14 gene coding for protoporphyrinogen oxidase, the molecular target of diphenyl ether-type herbicides. J Biol Chem. 1996;271:9120–9128. doi: 10.1074/jbc.271.15.9120. [DOI] [PubMed] [Google Scholar]
- 182.Dailey TA, Dailey HA. Expression, purification, and characteristics of mammalian protoporphyrinogen oxidase. Methods Enzymol. 1997;281:340–349. doi: 10.1016/s0076-6879(97)81041-5. [DOI] [PubMed] [Google Scholar]
- 183.Lermontova I, Kruse E, Mock HP, Grimm B. Cloning and characterization of a plastidal and a mitochondrial isoform of tobacco protoporphyrinogen IX oxidase. Proc Natl Acad Sci USA. 1997;94:8895–8900. doi: 10.1073/pnas.94.16.8895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Hansson M, Gustafsson MC, Kannangara CG, Hederstedt L. Isolated Bacillus subtilis HemY has coproporphyrinogen III to coproporphyrin III oxidase activity. Biochim Biophys Acta. 1997;1340:97–104. doi: 10.1016/s0167-4838(97)00030-7. [DOI] [PubMed] [Google Scholar]
- 185.Dailey HA, Dailey TA. Protoporphyrinogen oxidase of Myxococcus xanthus. Expression, purification, and characterization of the cloned enzyme. J Biol Chem. 1996;271:8714–8718. doi: 10.1074/jbc.271.15.8714. [DOI] [PubMed] [Google Scholar]
- 186.Wang KF, Dailey TA, Dailey HA. Expression and characterization of the terminal heme synthetic enzymes from the hyperthermophile Aquifex aeolicus. FEMS Microbiol Lett. 2001;202:115–119. doi: 10.1111/j.1574-6968.2001.tb10789.x. [DOI] [PubMed] [Google Scholar]
- 187.Corrigall AV, Siziba KB, Maneli MH, Shephard EG, Ziman M, Dailey TA, Dailey HA, Kirsch RE, Meissner PN. Purification of and kinetic studies on a cloned protoporphyrinogen oxidase from the aerobic bacterium Bacillus subtilis. Arch Biochem Biophys. 1998;358:251–256. doi: 10.1006/abbi.1998.0834. [DOI] [PubMed] [Google Scholar]
- 188.Heinemann IU, Diekmann N, Masoumi A, Koch M, Messerschmidt A, Jahn M, Jahn D. Functional definition of the tobacco protoporphyrinogen IX oxidase substrate-binding site. Biochem J. 2007;402:575–580. doi: 10.1042/BJ20061321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Jacobs NJ, Jacobs JM. Protoporphyrinogen oxidation in Rhodopseudomonas spheroides, a step in heme and bacteriochlorophyll synthesis. Arch Biochem Biophys. 1981;211:305–311. doi: 10.1016/0003-9861(81)90458-6. [DOI] [PubMed] [Google Scholar]
- 190.Sasarman A, Chartrand P, Lavoie M, Tardif D, Proschek R, Lapointe C. Mapping of a new hem gene in Escherichia coli K12. J Gen Microbiol. 1979;113:297–303. doi: 10.1099/00221287-113-2-297. [DOI] [PubMed] [Google Scholar]
- 191.Sasarman A, Letowski J, Czaika G, Ramirez V, Nead MA, Jacobs JM, Morais R. Nucleotide sequence of the hemG gene involved in the protoporphyrinogen oxidase activity of Escherichia coli K12. Can J Microbiol. 1993;39:1155–1161. doi: 10.1139/m93-174. [DOI] [PubMed] [Google Scholar]
- 192.Boynton TO, Daugherty LE, Dailey TA, Dailey HA. Identification of Escherichia coli HemG as a novel, menadione-dependent flavodoxin with protoporphyrinogen oxidase activity. Biochemistry. 2009;48:6705–6711. doi: 10.1021/bi900850y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Ashenbrucker H, Cartwright GE, Goldberg A, Wintrobe MM. Studies on the biosynthesis of heme in vitro by avian erythrocytes. Blood. 1956;11:821–833. [PubMed] [Google Scholar]
- 194.Dailey HA, Dailey TA, Wu CK, Medlock AE, Wang KF, Rose JP, Wang BC. Ferrochelatase at the millennium: structures, mechanisms and [2Fe-2S] clusters. Cell Mol Life Sci. 2000;57:1909–1926. doi: 10.1007/PL00000672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Taketani S, Tokunaga R. Rat liver ferrochelatase. Purification, properties, and stimulation by fatty acids. J Biol Chem. 1981;256:12748–12753. [PubMed] [Google Scholar]
- 196.Grzybowska E, Gora M, Plochocka D, Rytka J. Saccharomyces cerevisiae ferrochelatase forms a homodimer. Arch Biochem Biophys. 2002;398:170–178. doi: 10.1006/abbi.2001.2730. [DOI] [PubMed] [Google Scholar]
- 197.Hansson M, Hederstedt L. Purification and characterization of a water-soluble ferrochelatase from Bacillus subtilis. Eur J Biochem. 1994;220:201–208. doi: 10.1111/j.1432-1033.1994.tb18615.x. [DOI] [PubMed] [Google Scholar]
- 198.Dailey HA, Finnegan MG, Johnson MK. Human ferrochelatase is an iron-sulfur protein. Biochemistry. 1994;33:403–407. doi: 10.1021/bi00168a003. [DOI] [PubMed] [Google Scholar]
- 199.Day AL, Parsons BM, Dailey HA. Cloning and characterization of Gallus and Xenopus ferrochelatases: presence of the [2Fe-2S] cluster in nonmammalian ferrochelatase. Arch Biochem Biophys. 1998;359:160–169. doi: 10.1006/abbi.1998.0910. [DOI] [PubMed] [Google Scholar]
- 200.Dailey TA, Dailey HA. Identification of [2Fe-2S] clusters in microbial ferrochelatases. J Bacteriol. 2002;184:2460–2464. doi: 10.1128/JB.184.9.2460-2464.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Shepherd M, Dailey TA, Dailey HA. A new class of [2Fe-2S]-cluster-containing protoporphyrin (IX) ferrochelatases. Biochem J. 2006;397:47–52. doi: 10.1042/BJ20051967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Cornah JE, Roper JM, Pal Singh D, Smith AG. Measurement of ferrochelatase activity using a novel assay suggests that plastids are the major site of haem biosynthesis in both photosynthetic and non-photosynthetic cells of pea (Pisum sativum L.) Biochem J. 2002;362:423–432. doi: 10.1042/0264-6021:3620423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.De Matteis F, Gibbs AH, Harvey C. Studies on the inhibition of ferrochelatase by N-alkylated dicarboxylic porphyrins. Steric factors involved and evidence that the inhibition is reversible. Biochem J. 1985;226:537–544. doi: 10.1042/bj2260537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Medlock AE, Carter M, Dailey TA, Dailey HA, Lanzilotta WN. Product release rather than chelation determines metal specificity for ferrochelatase. J Mol Biol. 2009;393:308–319. doi: 10.1016/j.jmb.2009.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Hunter GA, Sampson MP, Ferreira GC. Metal ion substrate inhibition of ferrochelatase. J Biol Chem. 2008;283:23685–23691. doi: 10.1074/jbc.M803372200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Karlberg T, Hansson MD, Yengo RK, Johansson R, Thorvaldsen HO, Ferreira GC, Hansson M, Al-Karadaghi S. Porphyrin binding and distortion and substrate specificity in the ferrochelatase reaction: the role of active site residues. J Mol Biol. 2008;378:1074–1083. doi: 10.1016/j.jmb.2008.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Al-Karadaghi S, Franco R, Hansson M, Shelnutt JA, Isaya G, Ferreira GC. Chelatases: distort to select? Trends Biochem Sci. 2006;31:135–142. doi: 10.1016/j.tibs.2006.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Wang Y, Shen Y, Ryde U. QM/MM study of the insertion of metal ion into protoporphyrin IX by ferrochelatase. J Inorg Biochem. 2009;103:1680–1686. doi: 10.1016/j.jinorgbio.2009.09.013. [DOI] [PubMed] [Google Scholar]
- 209.Sigfridsson E, Ryde U. The importance of porphyrin distortions for the ferrochelatase reaction. J Biol Inorg Chem. 2003;8:273–282. doi: 10.1007/s00775-002-0413-8. [DOI] [PubMed] [Google Scholar]
- 210.Medlock AE, Dailey TA, Ross TA, Dailey HA, Lanzilotta WN. A pi-helix switch selective for porphyrin deprotonation and product release in human ferrochelatase. J Mol Biol. 2007;373:1006–1016. doi: 10.1016/j.jmb.2007.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Sellers VM, Dailey TA, Dailey HA. Examination of ferrochelatase mutations that cause erythropoietic protoporphyria. Blood. 1998;91:3980–3985. [PubMed] [Google Scholar]
- 212.Luer C, Schauer S, Mobius K, Schulze J, Schubert WD, Heinz DW, Jahn D, Moser J. Complex formation between glutamyl-tRNA reductase and glutamate-1-semialdehyde 2,1-aminomutase in Escherichia coli during the initial reactions of porphyrin biosynthesis. J Biol Chem. 2005;280:18568–18572. doi: 10.1074/jbc.M500440200. [DOI] [PubMed] [Google Scholar]
- 213.Nogaj LA, Beale SI. Physical and kinetic interactions between glutamyl-tRNA reductase and glutamate-1-semialdehyde aminotransferase of Chlamydomonas reinhardtii. J Biol Chem. 2005;280:24301–24307. doi: 10.1074/jbc.M502483200. [DOI] [PubMed] [Google Scholar]
- 214.Ferreira GC, Andrew TL, Karr SW, Dailey HA. Organization of the terminal two enzymes of the heme biosynthetic pathway. Orientation of protoporphyrinogen oxidase and evidence for a membrane complex. J Biol Chem. 1988;263:3835–3839. [PubMed] [Google Scholar]
- 215.Masoumi A, Heinemann IU, Rohde M, Koch M, Jahn M, Jahn D. Complex formation between protoporphyrinogen IX oxidase and ferrochelatase during haem biosynthesis in Thermosynechococcus elongatus. Microbiology. 2008;154:3707–3714. doi: 10.1099/mic.0.2008/018705-0. [DOI] [PubMed] [Google Scholar]