

Abstract
Cells lacking functional NF-κB die after ligation of some tumor necrosis factor (TNF) receptor family members through failure to express NF-κB-dependent anti-apoptotic genes. NF-κB activation requires the IκB kinase (IKK) complex containing two catalytic subunits named IKKα and IKKβ that regulate distinct NF-κB pathways. IKKβ is critical for classical signaling that induces pro-inflammatory and anti-apoptotic gene profiles, whereas IKKα regulates the non-canonical pathway involved in lymphoid organogenesis and B-cell development. To determine whether IKKα and IKKβ differentially function in rescuing cells from death induced by activators of the classical and non-canonical pathways, we analyzed death after ligation of the TNF and lymphotoxin-β receptors, respectively. Using murine embryonic fibroblasts (MEFs) lacking each of the IKKs, the caspase inhibitor benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone, and dominant negative Fas-associated death domain protein, we found that deletion of these kinases sensitized MEFs to distinct cell death pathways. MEFs lacking IKKα were sensitized to death in response to both cytokines that was entirely caspase-dependent, demonstrating that IKKα functions in this process. Surprisingly, death of IKKβ−/− MEFs was not blocked by caspase inhibition, demonstrating that IKKβ negatively regulates caspase-independent cell death (CICD). CICD was strongly activated by both TNF and lymphotoxin-β receptor ligation in IKKβ−/− MEFs and was accompanied by loss of mitochondrial membrane potential and the generation of reactive oxygen species. CICD was inhibited by the anti-oxidant butylated hydroxyanosole and overexpression of Bcl-2, neither of which blocked caspase-dependent apoptosis. Our findings, therefore, demonstrate that both IKKα and IKKβ regulate cytokine-induced apoptosis, and IKKβ additionally represses reactive oxygen species- and mitochondrial-dependent CICD.
Cytokines of the TNF2 superfamily induce pro-inflammatory responses in many cell types via activation of the NF-κB family of transcription factors (1). In addition to inducing pro-inflammatory gene transcription, NF-κB also regulates the transcriptional activation of many anti-apoptotic genes including the caspase-8 inhibitor c-FLIP, the caspase inhibitors c-IAP1 and -2, the Bcl-2 homologue A1, and the signal inhibitor A20 (2–4). Induction of this anti-apoptotic gene profile is pivotal for the prevention of programmed cell death initiated in response to TNF and ligation of other death receptors.
Ligation of the TNF receptor 1 (TNFR1) by TNF initiates its ligand-induced trimerization leading to the recruitment of adapter proteins named TNFR-1-associated death domain protein and Fas-associated death domain protein (FADD). FADD initiates the recruitment and autocatalytic activation of pro-caspase-8 via its death effector domain, or alternatively, it may bind the caspase-8 inhibitor c-FLIP, thereby preventing the activation of this protease (5, 6). In the absence of NF-κB activation and induction of c-FLIP, caspase-8 activation results in proteolytic activation of effector caspases including caspase-3. This process occurs either directly or via a mitochondrial amplification process dependent upon the cleavage of Bcl-2 homology domain (BH)3-only-containing proteins such as Bid that bind and oligomerize the “multidomain” proteins Bax or Bak (7, 8). This binding results in mitochondrial permeability, release of cytochrome c, and activation of the apoptosome.
The mechanisms through which the TNF family member lymphotoxin-β (LTβ) activates NF-κB and initiates apoptosis are less well defined. The LTβ receptor does not contain a death domain; however, the death process is dependent upon self-association of the receptor, TRAF3, TRAF2, c-IAP1, and SMAC (9–12). It is clear, however, that for both TNF receptor- and LTβ receptor-induced signaling, the precise series of biochemical events upstream of NF-κB activation is critical for both the control of inflammation and the regulation of apoptosis.
NF-κB proteins are retained inactive in the cytosol through interaction with members of the IκB family of proteins typified by IκBα (1). Activation of NF-κB requires the signal-induced phosphorylation, ubiquitination, and degradation of IκBs, thereby allowing NF-κB to translocate to the nucleus. IκB phosphorylation is facilitated by the IκB kinase (IKK) complex that contains two catalytic subunits named IKKα and IKKβ and a regulatory subunit named NEMO (NF-κB essential modulator) (13). Genetic studies manipulating components of the IKK complex revealed that NF-κB activation induced by the rapid and transient degradation of IκB family members in response to pro-inflammatory cytokines is dependent upon IKKβ and NEMO (13). This “classical” NF-κB pathway is largely independent of IKKα (14). In contrast, IKKα is critical for activation of the “non-canonical” NF-κB pathway that involves IKKα-dependent processing of NF-κB2/p100 to generate p52 (15, 16). Activation of the non-canonical pathway occurs in response to a limited number of ligands including lymphotoxin α1β2 (LTα1β2), CD40L, and BAFF and functions primarily in the maturation of B cells and the development of organized secondary lymphoid tissue (14). In addition to its role in the non-canonical pathway, IKKα functions in the nucleus to regulate NF-κB-dependent target genes by a number of mechanisms including promoter-associated histone H3 phosphorylation (17, 18). A kinase-independent role for IKKα in epidermal differentiation (19) and a negative-regulatory role in controlling the function of IKKβ (20) have also been described.
Current evidence, therefore, indicates that signals induced by distinct cytokines utilize separate components of the IKK complex. In this regard TNF strongly activates the classical pathway but does not induce non-canonical signaling, suggesting that this cytokine predominately activates IKKβ. However, LTα1β2 only moderately activates the classical pathway but strongly stimulates the non-canonical pathway, suggesting that it predominantly signals via IKKα. We, therefore, sought to determine whether IKKα and IKKβ play separate or overlapping roles in regulating apoptosis in response to TNF and LTα1β2. Using IKKα−/− and IKKβ−/− mouse embryonic fibroblasts (MEFs), the broad range caspase inhibitor zVADfmk, and transduction with dominant negative FADD to block receptor-induced caspase activation, we found that deletion of these kinases sensitized MEFs to distinct cell death pathways. Thus, MEFs lacking IKKα were sensitized to death that was entirely caspase-mediated, whereas the death of IKKβ−/− cells was not blocked by caspase inhibition. Interestingly, we did not observe similar caspase-independent cell death (CICD) in NEMO-deficient cells, demonstrating that IKKβ normally represses CICD in a NEMO-independent manner. We further found that CICD in IKKβ−/− MEFs was dependent upon the generation of reactive oxygen species (ROS) and could be blocked by overexpression of Bcl-2. Our findings, therefore, demonstrate that both IKKα and IKKβ regulate cytokine-induced apoptosis, and IKKβ additionally represses ROS and mitochondrial-dependent CICD.
EXPERIMENTAL PROCEDURES
Materials
Alexa Fluor 488-conjugated annexin V, propidium iodide, JC-1, and H2DCFDA were all purchased from Molecular Probes (Eugene, OR). Butylated hydroxyanosole (BHA) was purchased from Sigma. zVADfmk and zVDVADfmk were purchased from Calbiochem. Polyclonal anti-Mn-SOD and anti-Bax were purchased from Upstate (Temecula, CA), anti-catalase was purchased from Abcam (Cambridge, MA), and anti-Bcl-2 was from BD Bioscience. Phoenix cells and the LZRS expression construct were gifts from Dr. Gary Nolan (Stanford University, CA). WT, IKKα−/−, and IKKβ−/− cells were provided by Dr. Inder Verma, and NEMO−/− cells were from Dr. Michael Karin (both of the University of California, San Diego, CA).
Cell Culture
MEFs were cultured in Dulbecco’s modified essential media (Invitrogen) supplemented with 10% fetal calf serum (Sigma) and 100 units/ml penicillin and 100 μg/ml streptomycin (Invitrogen). Cells were passaged by washing with phosphate-buffered saline (PBS) before brief treatment with trypsin/EDTA (0.05%, Invitrogen). Human umbilical vein endothelial cells (HUVEC) were isolated from discarded tissue in accordance with a protocol approved by the University of Pennsylvania Internal Review Board. After collagenase digestion (1 mg/ml in PBS) of the canulated umbilical vein, endothelial cells were serially cultured on gelatin (J. T. Baker Inc.)-coated tissue culture plastic (Falcon, Lincoln Park, NJ) in Medium 199 (M199) (Invitrogen) supplemented with 20% fetal calf serum (Sigma), 200 μM L-glutamine (Invitrogen), 50 μg/ml EC growth factor (Collaborative Biomedical Products, Bedford, MA), 100 μg/ml porcine heparin (Sigma), 100 units/ml penicillin, and 100 μg/ml streptomycin (Invitrogen). Cells were passaged using trypsin/EDTA (0.05%, Invitrogen), and all experiments were performed using cells at passage 2.
Retroviral Transduction
Ecotropic phoenix cells were transfected with retroviral expression constructs LZRS-EGFP, LZRS-FADDDN, LZRS-IKKβ, and LZRS-Bcl-2 using FuGENE 6 (Invitrogen). The FADDDN construct encodes a protein lacking the N-terminal 80 amino acids encompassing the death effector domain and does not associate with caspases (21). Twenty-four hours after transfection, phoenix cells were selected using growth media supplemented with puromycin (1 μg/ml). After expansion of the cell population, media supplemented with puromycin was removed, and cells were washed then returned to normal growth media for a period of 12–24 h. This media was collected, supplemented with Polybrene (8 μg/ml), filtered, and used immediately or stored at −80 °C. MEFs were retrovirally transduced by 2–3 rounds of incubation with retroviral-conditioned media for 6–12 h at 37 °C. This procedure resulted in infection of at least 95% of the population.
Cell Viability Assays
In all assays floating cells were collected and pooled with remaining attached cells that were harvested using trypsin/EDTA.
Annexin V Staining
Cells were washed in PBS, and each sample was re-suspended in 100 μl of annexin V staining buffer (10 mM HEPES, 140 mM NaCl, 2.5 mM CaCl2, pH 7.4) containing Alexa Fluor 488-conjugated annexin V (5 μl/sample). Samples were incubated at room temperature for 15–20 min before the addition of a further 400 μl of annexin V staining buffer and immediate analysis by FACS using Cell Quest software (FACSort, BD Biosciences).
Propidium Iodide (PI) Permeabilization Assay
Cells were washed once in PBS before the addition of PBS containing PI (25 μg/ml). After incubation for 20 min at 37 °C, the cells were analyzed by FACS.
Cell Cycle and Hypodiploid DNA Analysis
Harvested cells were washed in PBS and fixed for 30–60 min in 70% ethanol at 4 °C. After fixation, samples were washed once more before incubation in PBS containing PI (50 μg/ml) and RNase A (1 μg/ml) for at least 1 h. DNA analysis was performed by FACS.
Mitochondrial Membrane Potential (ΔΨ) Analysis
Floating and attached cells were harvested following the procedures described above. Cells were washed once in PBS containing 1% bovine serum albumin before re-suspension in 200 μl of PBS/bovine serum albumin containing JC-1 (10 μg/ml). After 15 min of incubation at 37 °C MEFs were washed, re-suspended in PBS, and analyzed by FACS.
Western Blotting
For all immunoblots, each well of a 12-well plate was treated as described. After experimental manipulation, any detached cells were collected, washed in PBS, and pooled with the lysate from the same well prepared by washing the adherent cells twice in ice-cold PBS before the addition of 100–200 μl of TNT lysis buffer (50 mM Tris-Cl, pH 7.5, 150 mM NaCl, and 1% Triton X-100) containing complete protease inhibitor mixture (Roche Applied Science), NaF (2 mM), and β-glycerophosphate (2 mM). Protein content was determined using Coomassie Plus Reagent (Pierce), and for each sample an equal amount of protein (10–20 μg) was fractionated by SDS-PAGE then transferred electrophoretically to polyvinylidene difluoride membranes (Immobilon P, Millipore, Milford, MA) and immunoblotted with the appropriate primary and species-specific horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch, Westgrove, PA). Detection of the bound antibody by chemiluminescence was performed using Luminol reagent according to the manufacturer’s instructions (Santa Cruz).
ROS Measurement
After experimental manipulation, floating and attached cells were harvested and pooled, then washed once in PBS before incubation in prewarmed PBS containing H2DCFDA (10 μM) for 20 min at room temperature. Samples were then washed once in PBS before re-suspension in PBS. After incubation at room temperature for a further 10–15 min, samples were analyzed by FACS.
RESULTS
TNF and LTα1β2 Induce Cell Death in IKKα−/− and IKKβ−/− MEFs
To compare the mechanisms of cell death induced by TNF and LTα1β2, we utilized MEFs lacking either IKKα or IKKβ. Although IKKβ critically regulates the classical NF-κB pathway and induction of anti-apoptotic genes, we found that deletion of either kinase sensitized MEFs to cell death in response to both TNF and LTα1β2. Death was detected by detachment of MEFs from the culture dish (not shown), an increase in annexin V staining (Figs. 1, A and B), and an increase in membrane permeability determined by uptake of propidium iodide (Fig. 1, C and D). Interestingly, MEFs lacking IKKβ were less sensitive to death in response to LTα1β2 than TNF. In contrast, MEFs lacking IKKα were equally sensitive to either TNF or LTα1β2.
FIGURE 1. TNF and LTα1β2 induce cell death in IKKα−/− and IKKβ−/− MEFs.

A, MEFs were untreated (shaded) or treated with TNF (10 ng/ml; solid line) or LTα1β2 (100 ng/ml; dotted line) for 18 h. Cells were harvested, stained with Alexa Fluor-conjugated annexin V, and analyzed by FACS. The percentage of positively stained cells gated in region M1 were determined and plotted in the histogram shown in B. Ctr, control. C, MEFs were treated as in A then incubated in PBS containing PI and analyzed by FACS. The percentage of membrane-permeable cells that took up PI gated in region M1 was determined, and these data are shown in the histogram in panel D.
Caspase Inhibition Does Not Prevent Death in IKKβ−/− MEFs
To determine whether the cell death we observed in response to cytokine treatment of IKKα- or IKKβ-deficient MEFs was caspase-dependent apoptosis, we employed the broad range caspase inhibitor zVADfmk. To confirm its activity, we determined the effects of zVADfmk on cell death in HUVEC induced by TNF plus cycloheximide as this death is well characterized as apoptosis (22). Treatment of HUVEC with TNF plus cycloheximide resulted in apoptosis measured by increased uptake of PI that was completely blocked by co-treatment with zVADfmk (25 μM) (Figs. 2, A and B). zVADfmk also effectively blocked DNA fragmentation in HUVEC (Fig. 2C). MEFs lacking IKKα behaved in a similar manner to HUVEC. Thus, LTα1β2 induced death characterized by PI uptake that was completely blocked by incubation with zVADfmk (Figs. 2, D and E). Similar effects were observed when IKKα−/− MEFs were treated with TNF (not shown).
FIGURE 2. Caspase inhibition blocks cytokine-induced cell death in HUVEC and IKKα−/− MEFs.

A, HUVEC were untreated (shaded) or treated with TNF (10 ng/ml; dashed line), cycloheximide (2.5 μg/ml; solid line), or a combination of TNF plus cycloheximide (dotted line). Each treatment was performed in the absence of presence of zVADfmk (25 μM). Cells were harvested, incubated in PBS containing PI, and analyzed by FACS. The population percentage that was membrane-permeable and took up PI was determined from region M1 and illustrated in B. Ctl, control. C, HUVEC were treated as described in A, then the percentage of the population with hypodiploid DNA was determined by FACS. D, WT and IKKα−/− MEFs were either untreated (shaded) or treated with LTα1β2 (solid line) either alone or in combination with zVADfmk (25 μM) as shown. Cells were harvested, incubated in PBS containing PI, and analyzed by FACS. The membrane-permeable population percentage that took up PI was determined from region M1 and is shown in the histogram in panel E.
Surprisingly, however, the death of IKKβ-deficient cells in response to TNF was unaffected by co-treatment with zVADfmk. We did not observe any inhibition of cell detachment from the culture plate (not shown) or uptake of PI (Fig. 3, A and open bars in B) at any concentration of zVADfmk tested (up to 50 μM). Remarkably, the extent of LTα1β2-induced cell death in IKKβ−/− MEFs actually increased in the presence of zVADfmk (Fig. 3, A and gray bars in B). Despite failing to inhibit cell death, zVADfmk prevented the generation of hypodiploid DNA in IKKβ−/− MEFs (Fig. 3C), indicating that the death mechanism was not apoptosis. Because zVADfmk is a relatively poor inhibitor of caspase-2, we also used the caspase-2 inhibitor zVDVADfmk. Treatment of IKKβ−/− MEFs with TNF in the presence of zVDVADfmk (25 μM) either alone or in combination with zVADfmk (25 μM) did not prevent cell death (data not shown). Hence, a lack of caspase-2 inhibition and escape from protease inhibition was unlikely to account for the failure of zVADfmk to prevent cell death.
FIGURE 3. Caspase inhibition does not block TNF- and LTα1β2-induced cell death in IKKβ−/− MEFs.

A, WT or IKKβ−/− MEFs were either untreated (shaded) or treated overnight with TNF (10 ng/ml; solid line) or LTα1β2 (100 ng/ml; dotted line) in the absence (control (Ctr)) or presence of zVADfmk at the concentrations indicated (left). Cells were harvested, incubated in PBS containing PI, and then analyzed by FACS. Membrane-permeable cells that took up PI were determined in region M1. The % PI-positive MEFs treated with 25 μM zVADfmk are shown in panel B (+) compared with cells that received no inhibitor (−). C, WT or IKKβ−/− MEFs were either untreated or treated with TNF in the presence (+) or absence (−) of zVADfmk (25 μM). Cells were harvested, fixed, permeabilized, and incubated with PI, then the percentage of the population with hypodiploid DNA was determined by FACS.
IKKβ Represses Death in zVADfmk-treated MEFs in a NEMO-independent Manner
To determine whether the TNF-induced death of zVADfmk-treated IKKβ−/− cells could be repressed by exogenous IKKβ, we reconstituted IKKβ-deficient MEFs by retroviral transduction with LZRS-IKKβ. As shown in Figs. 4, A and B, transduction with IKKβ but not LZRS alone markedly reduced the TNF-induced cell death observed in the presence of zVADfmk. The activity of IKKβ in regulating the classical NF-κB pathway is critically dependent upon its interaction with NEMO. We, therefore, questioned whether the death observed in zVADfmk-treated IKKβ−/− MEFs also occurred in cells lacking NEMO. Interestingly, TNF-induced death of NEMO−/− MEFs was completely blocked by ZVADfmk (Fig. 4, C and D), demonstrating that IKKβ functions independently of NEMO to repress cell death in zVADfmk-treated cells.
FIGURE 4. Repression of CICD by IKKβ does not require NEMO.

A, WT and IKKβ−/− MEFs transduced with LZRS or IKKβ as shown (parentheses) were either untreated (shaded) or treated overnight with TNF (10 ng/ml; unshaded) in the presence of zVADfmk (25 μM). Cells were harvested, incubated in PBS containing PI, and then analyzed by FACS. Membrane-permeable cells that took up PI were determined in region M1, and the percentage PI-positive MEFs is shown in panel B. C, WT and NEMO-deficient MEFs were either untreated (shaded) or treated overnight with TNF (10 ng/ml; solid line) in the absence (Control) or presence of zVADfmk (25 μM). PI uptake was determined as described for panel A, and the percentage of PI-positive cells is shown in D.
Dominant Negative FADD Recapitulates the Effects of zVADfmk in IKKβ−/− MEFs
Our findings strongly suggest that the cell death observed in zVADfmk-treated cells lacking IKKβ is caspase-independent. However, to ensure that the effects were not an artifact of the inhibitor and could be recapitulated by non-pharmacological caspase inhibition, we transduced IKKα−/− and IKKβ−/− cells with a dominant negative version of FADD (FADDDN) to prevent receptor-mediated caspase activation. Similar to zVADfmk, FADDDN prevented TNF-induced death of IKKα−/− MEFs but exacerbated the death observed in IKKβ−/− cells (Figs. 5, A and B). Taken together, the findings in Figs. 3–5 lead us to conclude that cell death induced in IKKβ−/− cells by TNF and LTα1β2 in the presence of ZVADfmk is caspase-independent cell death (CICD).
FIGURE 5. FADDDN blocks cell death in IKKα−/− but not IKKβ−/− MEFs.

A, WT, IKKα−/−, or IKKβ−/− MEFs were either untreated (shaded) or treated overnight with TNF (10 ng/ml; unshaded) in the absence (Control) or presence of zVADfmk (25 μM). FADD DN-transduced IKKα−/− and IKKβ−/− MEFs were also either untreated or treated overnight with TNF. Cells were harvested, incubated in PBS containing PI, and then analyzed by FACS. Membrane-permeable cells that took up PI were determined in region M1, and the percentage of PI-positive cells is shown in B.
CICD in IKKβ−/− MEFs Is Dependent on the Mitochondria
To characterize the CICD induced in the absence of IKKβ, we explored the effects of TNF on mitochondrial membrane potential (ΔΨ). Using the ΔΨ-sensitive dye JC-1, we found that treatment of IKKβ−/− MEFs with TNF caused a loss of ΔΨ that could not be prevented by co-incubation with zVADfmk (Fig. 6A). Treatment with LTα1β2 and zVADfmk led to a similar loss of ΔΨ (not shown). We next questioned whether TNF and/or zVADfmk treatment affected the levels of the mitochondrial regulatory proteins Bcl-2, Bax, and Bcl-2-binding protein beclin-1. The expression levels of each of these proteins were unaffected by treatment of either WT or IKKβ−/− MEFs with TNF in the absence or presence of zVADfmk (data not shown).
FIGURE 6. CICD in IKKβ−/− MEFs results from mitochondrial dysfunction.

A, WT and IKKβ−/− MEFs were either untreated (shaded) or treated overnight with TNF (10 ng/ml; solid line) in the absence (Ctr) or presence of zVADfmk (25 μM). Cells were harvested and loaded with JC-1 (10 μg/ml), and mitochondrial membrane potential (ΔΨ) was determined by FACS. Loss of ΔΨ is indicated by a shift in the histogram to the right. B, LZRS and Bcl-2 transduced IKKβ−/− MEFs were either untreated (Ctr) or treated with etoposide (50 μM) overnight. Cells were harvested, incubated in PBS containing PI, and analyzed by FACS. The percentage of the population that was PI-positive is shown. C, LZRS or Bcl-2 transduced MEFs were either untreated (shaded), or treated overnight with either LTα1β2 (100 ng/ml; dotted line) or TNF (10 ng/ml; solid line) in the absence (Control) or presence of zVADfmk (25 μM). Cells were harvested, incubated in PBS containing PI, and analyzed by FACS, and the percentage that took up PI was determined from region M1. The data from the TNF with and without zVAD-treated MEFs are summarized in D.
Overexpression of Bcl-2 prevents mitochondria-dependent cell death in response to a wide range of stimuli and conditions including treatment with the DNA-damaging chemotherapeutic agent etoposide (23, 24). We confirmed that etoposide induced death in IKKβ−/− MEFs and that Bcl-2 overexpression effectively inhibited this process (Fig. 6B). We, therefore, investigated the effect of Bcl-2 overexpression on the death of IKKβ−/− MEFs in response to TNF and LTα1β2 plus ZVADfmk. As shown in Fig. 6, C and D, Bcl-2 overexpression did not affect the death of IKKβ−/− MEFs treated with LTα1β2 or TNF alone; however, it did prevent death in response to these cytokines in the presence of zVADfmk (Figs. 6, C and D). This, therefore, suggests that the death pathway in IKKβ−/− MEFs treated with cytokines changes in the presence of zVADfmk to one that is dependent upon the mitochondria.
CICD in IKKβ−/− MEFs Is Dependent on the Generation of ROS
Because mitochondrial function is closely linked to the generation of ROS, we questioned whether TNF treatment induced ROS in IKKβ−/− MEFs and whether scavenging such species could inhibit death. Using the dye H2DCFDA, we found that TNF treatment of WT MEFs did not lead to ROS generation, whereas a significant increase was observed in MEFs lacking IKKβ (Figs. 7, A and B). In WT MEFs, treatment with zVADfmk caused an increase in the generation of ROS in that it was reduced by co-treatment with TNF (Figs. 7, A and B). In contrast, treatment of IKKβ−/− MEFs with zVADfmk alone had no effect on ROS, and co-treatment with zVADfmk plus TNF still resulted in increased ROS levels (Figs. 7, A and B). To determine whether the generation of ROS contributed to TNF-induced cell death in IKKβ−/− MEFs, we utilized the ROS scavenger BHA. Pretreatment of IKKβ−/− MEFs with BHA significantly diminished the death of IKKβ−/− cells treated with TNF plus ZVADfmk but did not affect the death induced by TNF treatment alone (Figs. 7, C and D). Similar effects were observed when cells were treated with LTα1β2 and zVADfmk (not shown). Together with the data in Fig. 6, C and D, these findings further support a model in which the death pathway activated in IKKβ−/− MEFs by TNF or LTα1β2 alters the phenotype after the addition of zVADfmk to become dependent on the mitochondria and the generation of ROS.
FIGURE 7. The death of IKKβ−/− cells treated with TNF and zVADfmk is dependent on the generation of ROS.

A, WT and IKKβ−/− cells were either untreated (shaded) or treated overnight with TNF (10 ng/ml; solid line) in the absence (Control) or presence of zVADfmk (25 μM). After treatment, cells were harvested, and ROS levels were determined by FACS using H2DCFDA (10 μM. The generation of ROS relative to the levels observed in untreated WT or IKKβ−/− cells was determined and is shown in panel B. C, WT or IKKβ−/− MEFs were either untreated (Control) or pretreated for 60 min with BHA at the concentrations indicated (left) before either no further treatment (shaded) or overnight treatment with TNF (10 ng/ml; dashed line), zVADfmk (25 μM; dotted line), or TNF plus zVADfmk (solid line). Cells were harvested, incubated in PBS containing PI, and analyzed by FACS. The percentage of the population that were PI positive was determined from region M1 and is shown in panel D.
ROS levels can be regulated by the expression of scavenging enzymes. We, therefore, questioned whether treatment of IKKβ−/− MEFs with TNF and zVADfmk affected the expression of Mn-SOD and catalase that have been implicated recently in the regulation of cell death in response to both of these mediators (25, 26). Mn-SOD expression was induced by TNF in WT MEFs, and this was not affected by the presence of zVADfmk (Fig. 8A; lanes 1– 4). In MEFs lacking IKKβ, basal Mn-SOD expression was significantly reduced and was not modulated by TNF either alone or in combination with zVADfmk (Fig. 8A; lanes 5– 8). Despite the recent report that zVADfmk decreases catalase levels by a post-transcriptional mechanism (26), we did not observe any clear effect of TNF, zVADfmk, or TNF plus zVADfmk on its expression in either WT or IKKβ−/− MEFs (Fig. 8B). We did observe modification of catalase resulting in the appearance on immunoblots of bands both above and below the catalase band (lanes 3 and 4). The nature of these modifications remains unclear; however, no such modified catalase was detected in IKKβ−/− cells treated with zVADfmk (lanes 7 and 8), suggesting that this is dependent upon functional IKKβ.
FIGURE 8. Effects of TNF and zVADfmk on Mn-SOD and catalase levels in WT and IKKβ−/− MEFs.
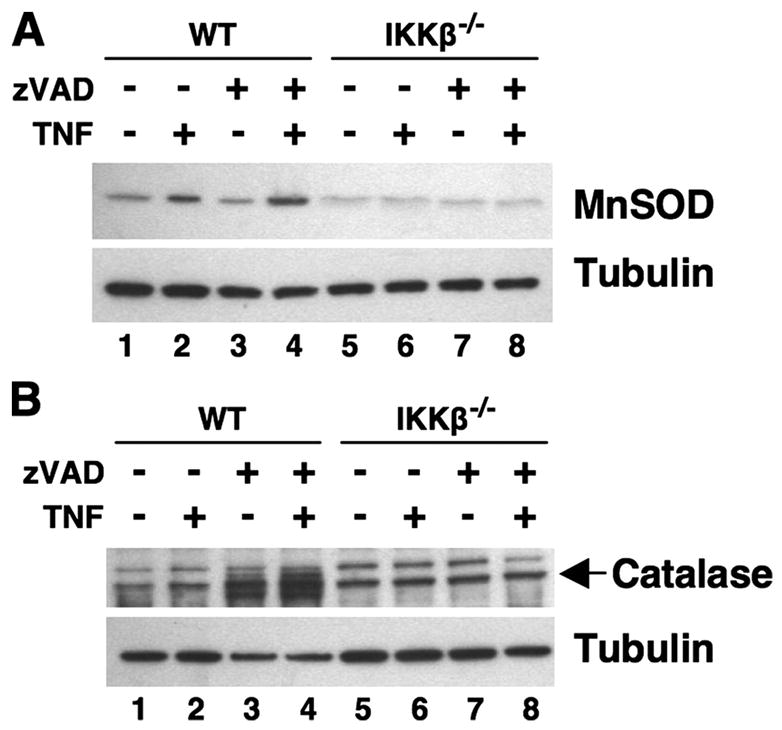
WT and IKKβ−/− MEFs were either untreated or incubated overnight with TNF (10 ng/ml) in the absence or presence of zVADfmk (25 μM) as shown. Cell lysates were separated by SDS-PAGE (10%), and the resulting immunoblots were probed using either anti-Mn-SOD (A) or anti-catalase (B). Anti-tubulin was used to ensure equal protein loading.
DISCUSSION
Cell death is a highly regulated process necessary for development. Dysregulation of the signals that control cell death can result in either failure to die or enhanced death that in turn supports tumor growth or leads to excessive tissue damage, respectively. To develop a clearer picture of the death signals induced by TNF family members, we utilized MEFs lacking either IKKα or IKKβ to determine whether these kinases played distinct roles in regulating death induced by TNF or LTα1β2. We found that cells lacking either kinase were sensitized to death in response to both cytokines, although more death occurred in IKKβ−/− MEFs incubated with TNF than those treated with LTα1β2. Conversely, LTα1β2 treatment caused greater cell death in IKKα−/− MEFS than TNF. We, therefore, conclude that signaling through IKKα and IKKβ differentially predominates to counteract the cell death signaling pathways induced by the separate cytokines.
Our finding that both TNF and LTα1β2 induce apoptosis in IKKα−/− cells was surprising in light of previous studies of mice lacking each of the IKK subunits. Animals lacking either NEMO or IKKβ die during development from massive hepatocyte apoptosis (27, 28), whereas mice lacking IKKα survive but die shortly after birth due to morphogenic defects (19, 29). IKKα is, therefore, thought to play no role in the induction of classical NF-κB-dependent anti-apoptotic genes (e.g. cIAPs, c-FLIP, A20) and protection from TNF-induced liver apoptosis. However, it has been reported recently that hepatocyte-specific deletion of NEMO but not IKKβ sensitizes mice to TNF-induced liver apoptosis (30), suggesting that IKKα can substitute for IKKβ and activate NF-κB to protect hepatocytes from death. Separate studies have also linked IKKα to classical NF-κB target gene expression (31, 32), but we do not yet know whether IKKα controls anti-apoptotic genes by a direct regulatory function in the classical pathway or by another mechanism such as histone phosphorylation (17, 18). Although further studies are required to determine its precise mechanism of action, our findings clearly demonstrate that IKKα plays a crucial role in protecting MEFs from apoptosis in response to TNF and LTα1β2.
Cell death occurs by various mechanisms including apoptosis, autophagy, and necrosis; however, only apoptosis is dependent upon caspase activation. To determine whether the death of IKKα- and IKKβ-deficient MEFs was caspase-dependent apoptosis or a separate non-apoptotic mechanism, we utilized the broad range caspase inhibitor zVADfmk. This inhibitor has been extensively characterized and shown to block the processes that characterize apoptosis including membrane blebbing, outer membrane inversion and exposure of phosphatidylserine, cell shrinkage, nuclear condensation, generation of nucleosomes, and DNA breakdown. Death of both IKKα−/− and IKKβ−/− MEFs in response to TNF and LTα1β2 displayed several of these characteristics including nuclear condensation and DNA fragmentation. Consistent with this, caspase inhibition blocked the cell death induced by both TNF and LTα1β2 in IKKα-deficient MEFs, demonstrating that this occurs via caspase-dependent apoptosis. Surprisingly however, caspase inhibition with either ZVADfmk or non-pharmacologically via transduction of FADDDN did not block death in IKKβ-deficient MEFs, providing further evidence that IKKα and IKKβ regulate separate cell death processes. Intriguingly, TNF-induced death of NEMO-deficient MEFs was blocked by caspase inhibition, demonstrating that the repression of CICD by IKKβ occurs independent of its typical functional association with NEMO. Although further studies are required to determine the targets of IKKβ in repressing CICD, our findings establish a novel NEMO-independent role for IKKβ in regulating cell death.
Previous studies have established that IKKβ regulates classical NF-κB-dependent expression of negative regulators of caspase-dependent apoptosis (2–4). Our finding that IKKβ−/− MEFs treated with zVADfmk and, therefore, lacking functional caspase activity continue to die in response to TNF and LTα1β2 therefore demonstrates that IKKβ also represses a CICD process. A potential model of the interplay between IKKβ and caspases in regulating cell death is shown in Fig. 9. Remarkably, we consistently observed that cytokine-induced death in IKKβ -deficient MEFs was greater in the presence of zVADfmk than in untreated cells, and this was particularly notable in response to LTα1β2. We noted a similarly enhanced death response in IKKβ−/− MEFs transduced with FADDDN that blocks receptor-induced caspase activation. This, therefore, suggests that CICD is normally limited by caspases in addition to being repressed by IKKβ. Previous studies have described CICD in the fibrosarcoma L929 cell line after TNF stimulation (33) and in caspase-8−/− Jurkat cells (34), and CICD has been reported in vivo where zVADfmk exacerbates the lethal toxicity of TNF shock in mice (35). Our findings, therefore, extend these previous studies and demonstrate that IKKβ is critical for repression of CICD induced by TNF and LTβ receptor ligation.
FIGURE 9. Model of apoptosis and CICD in WT and IKKβ−/− MEFs.

In this model activation is denoted by an arrowhead and inhibition by a straight line. A, in WT MEFs, TNF and LTα1β2 activate IKKβ that inhibits both caspase-dependent apoptosis and CICD. Caspases are blocked by the classical NF-κB-dependent induction of caspase inhibitors including c-FLIP and c-IAP2, and ROS are blocked by the up-regulation of antioxidant enzymes such as Mn-SOD (Fig. 8A). B, in the absence of IKKβ cytokines activate caspase-dependent apoptosis. ROS levels are increased possibly via a lack of induced Mn-SOD expression (Fig. 8A), and mitochondrial membrane potential is lost (Fig. 6A). However, in the presence of active caspases, CICD does not occur, and neither Bcl-2 (Figs. 6, C and D) nor BHA (Figs. 7, C and D) can block cell death. C, caspase-dependent apoptosis is blocked in IKKβ−/− MEFs stimulated with cytokines in the presence of zVADfmk. These cells die by CICD that can be inhibited by mitochondrial regulation via Bcl-2 overexpression (Figs. 6, C and D) or by ROS scavenging via BHA treatment (Figs. 7, C and D).
The cell death in cytokine-stimulated IKKβ−/− MEFs treated with zVADfmk was typical of necrosis; we observed membrane leakiness, phosphatidylserine exposure, and loss of mitochondrial membrane potential that occurred in the absence of the DNA and nuclear breakdown seen in apoptosis. Autophagic death is hallmarked by accumulation of vacuoles that sequester and target cytoplasmic components for lysosomal degradation in a process dependent on Atg genes including Beclin-1 (36). We did not observe any changes in Beclin-1 levels upon TNF stimulation of either WT or IKKβ−/− MEFs in the presence of zVADfmk (data not shown). Furthermore treatment with the autophagy inhibitor 3-MA (36, 37) did not block cytokine plus zVADfmk-induced death of IKKβ−/− MEFs (data not shown). Consequently, we conclude that in contrast to the CICD previously reported in L929 cells (26, 36), the death of IKKβ-deficient MEFs treated with cytokines and zVADfmk is not autophagic but is instead necrotic.
The continued loss of mitochondrial membrane potential in TNF-treated IKKβ-deficient MEFs after caspase inhibition led us to test whether exogenous expression of mitochondrial regulators might affect CICD. Overexpression of the anti-apoptotic mitochondrial protein Bcl-2 did not affect cytokine-induced death of IKKβ-deficient MEFs, indicating that death occurs by apoptosis via a mitochondrial-independent process. Activation of caspase-8 and the mitochondrial-independent activation of caspase-3 in a so-called “extrinsic pathway” after death receptor ligation have been described in various cell types (22). Bcl-2 has also been shown previously to inhibit CICD (22, 38, 39) most likely by preventing the loss of mitochondrial membrane potential, and consistent with this, we found that in the presence of zVADfmk, overexpressed Bcl-2 diminished death in cytokine-stimulated IKKβ−/− MEFs. Our data, therefore, demonstrate that in cells lacking IKKβ, a switch in the cytokine-induced death phenotype from mitochondrial-independent apoptosis to caspase-independent, mitochondrial-dependent cell death occurs in the presence of zVADfmk.
It has been proposed that mitochondria and the generation of ROS contribute to TNF-mediated CICD (33, 35, 40). We, therefore, questioned whether cytokine stimulation of IKKβ-deficient cells led to increased ROS and whether the anti-oxidant BHA affected cell death. Similar to Kamata et al. (25), we found that TNF increased ROS in IKKβ−/− but not WT MEFs, and this is also consistent with the effects observed in TRAF2/5 double knock-out, p65−/−, and ΔN-IκBα-transduced cells (41, 42). It is, therefore, likely that induction of classical NF-κB-dependent anti-oxidant enzymes such as Mn-SOD that we (Fig. 8A), and others (25, 43) have observed contributes to the rescue by IKKβ from TNF-induced ROS generation. Despite the increased ROS generation in TNF-stimulated IKKβ-deficient MEFs, BHA did not protect against cell death. Similarly, treatment of WT MEFs with zVADfmk increased ROS but did not cause death. We found that BHA only protected against death in IKKβ−/− MEFs in response to cytokines in the presence of zVADfmk and conclude that ROS only cause death in cells lacking both IKKβ and functional caspases. It is not clear why our findings conflict with those of with Kamata et al. (25) who reported that BHA alone blocked IKKβ-deficient cell death; however, our data strongly suggest that ROS-independent, caspase-dependent apoptosis remains functional and maintains cell death in the face of ROS inhibition. Consistent with this model we also found that Bcl-2 overexpression inhibited TNF-induced death in IKKβ-deficient MEFs only when caspases were blocked. It is, therefore, likely that Bcl-2 and BHA inhibit the same CICD pathway involving premitochondrial or mitochondrial generation of ROS (Fig. 9).
Post-translational down-regulation of catalase expression by zVADfmk has been reported to cause increased ROS in L929 cells (26). We did not detect any reduction in catalase levels in zVADfmk-treated WT or IKKβ−/− MEFs, although we did observe its modification in WT cells. This, therefore, suggests that IKKβ plays an important role in regulating catalase in cells lacking functional caspases. We do not yet know the nature of this modification, but it is possible that it renders catalase inactive, leading to increased ROS in WT MEFs. This modification was not present in the IKKβ-deficient cells, although the failure to induce Mn-SOD in response to TNF (Fig. 7A) may play a predominant role in maintaining elevated ROS levels in these cells. A further possibility is that zVADfmk actively induces ROS generation and potential mechanisms for this include enhanced leakiness of the mitochondrial electron transport chain (44), decreased mitochondrial turnover (45), or enhanced cPLA2 activation (35). Further work will be required to determine whether any or all of these mechanisms occur in IKKβ-deficient cells.
Intriguingly, IKKβ negatively regulates JNK (25, 46), and we confirmed that the duration of JNK activity was increased in IKKβ−/− MEFs (not shown). Some studies have implicated the generation of ROS in cell death mediated by sustained JNK activity (25, 41, 42, 47). We did not observe any effect of zVADfmk on either the JNK response (data not shown) or generation of ROS in IKKβ−/− cells. Because JNK is not required for cell death mediated by ROS (42), our data suggest that ROS are generated in IKKβ−/− MEFs treated with zVADfmk by a mechanism that does not involve JNK. Consistent with this, a pharmacological inhibitor of JNK (JNK inhibitor II) failed to block death in TNF- and zVADfmk-treated IKKβ-deficient MEFs (data not shown). Consequently, we conclude that JNK does not control the CICD pathway activated in the absence of IKKβ and caspases.
In summary, we have shown that IKKα plays an anti-apoptotic role in death receptor signaling. The anti-apoptotic function of IKKα is most notable in response to LTα1β2, suggesting that distinct death-inducing cytokines preferentially utilize separate IKK subunits. We have also demonstrated that in addition to inhibiting caspase-dependent apoptosis, IKKβ negatively regulates CICD in a NEMO-independent manner. The function of IKKβ as a CICD inhibitor is revealed in the presence of zVADfmk that induces a change in the death phenotype from apoptosis to necrosis. Furthermore our data suggest that caspases normally inhibit CICD and that CICD depends on the mitochondria and the generation of ROS. Although further studies are required to determine the precise mechanism of ROS-induced cell death, our findings clearly identify an unanticipated interplay between IKKβ and caspases in regulating CICD (see model Fig. 9).
Footnotes
The abbreviations used are: TNF, tumor necrosis factor; CICD, caspase-independent cell death; IκB, inhibitor of NF-κB; IKK, IκB kinase; MEF, murine embryonic fibroblast; NF-κB, nuclear factor κB; NEMO, NF-κB essential modulator; ROS, reactive oxygen species; zVADfmk, benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone; zVDVADfmk, benzyloxycarbonyl-VD-VAD-fluoromethyl ketone; H2DCFDA, dichloro-dihydrofluorescein diacetate; FADD, Fas-associated death domain protein; LT, lymphotoxin; BHA, butylated hydroxyanosole; Mn-SOD, manganese superoxide dismutase; WT, wild type; PBS, phosphate-buffered saline; HUVEC, human umbilical vein endothelial cell; FACS, fluorescence-activated cell sorter; PI, propidium iodide; JNK, c-Jun NH2-terminal kinase.
This work was supported by National Institutes of Health Grant 1RO1HL080612-01A1.
References
- 1.Hayden MS, Ghosh S. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 2.Hu X, Yee E, Harlan JM, Wong F, Karsan A. Blood. 1998;92:2759–2765. [PubMed] [Google Scholar]
- 3.Kreuz S, Siegmund D, Scheurich P, Wajant H. Mol Cell Biol. 2001;21:3964–3973. doi: 10.1128/MCB.21.12.3964-3973.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS., Jr Science. 1998;281:1680–1683. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- 5.Boldin MP, Goncharov TM, Goltsev YV, Wallach D. Cell. 1996;85:803–815. doi: 10.1016/s0092-8674(00)81265-9. [DOI] [PubMed] [Google Scholar]
- 6.Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer JL, Schroter M, Burns K, Mattmann C, Rimoldi D, French LE, Tschopp J. Nature. 1997;388:190–195. doi: 10.1038/40657. [DOI] [PubMed] [Google Scholar]
- 7.Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zong WX, Lindsten T, Ross AJ, MacGregor GR, Thompson CB. Genes Dev. 2001;15:1481–1486. doi: 10.1101/gad.897601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Force WR, Cheung TC, Ware CF. J Biol Chem. 1997;272:30835–30840. doi: 10.1074/jbc.272.49.30835. [DOI] [PubMed] [Google Scholar]
- 10.Kuai J, Nickbarg E, Wooters J, Qiu Y, Wang J, Lin LL. J Biol Chem. 2003;278:14363–14369. doi: 10.1074/jbc.M208672200. [DOI] [PubMed] [Google Scholar]
- 11.VanArsdale TL, VanArsdale SL, Force WR, Walter BN, Mosialos G, Kieff E, Reed JC, Ware CF. Proc Natl Acad Sci U S A. 1997;94:2460–2465. doi: 10.1073/pnas.94.6.2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu MY, Wang PY, Han SH, Hsieh SL. J Biol Chem. 1999;274:11868–11873. doi: 10.1074/jbc.274.17.11868. [DOI] [PubMed] [Google Scholar]
- 13.Scheidereit C. Oncogene. 2006;25:6685–6705. doi: 10.1038/sj.onc.1209934. [DOI] [PubMed] [Google Scholar]
- 14.Bonizzi G, Karin M. Trends Immunol. 2004;25:280–288. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 15.Bonizzi G, Bebien M, Otero DC, Johnson-Vroom KE, Cao Y, Vu D, Jegga AG, Aronow BJ, Ghosh G, Rickert RC, Karin M. EMBO J. 2004;23:4202–4210. doi: 10.1038/sj.emboj.7600391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dejardin E, Droin NM, Delhase M, Haas E, Cao Y, Makris C, Li ZW, Karin M, Ware CF, Green DR. Immunity. 2002;17:525–535. doi: 10.1016/s1074-7613(02)00423-5. [DOI] [PubMed] [Google Scholar]
- 17.Anest V, Hanson JL, Cogswell PC, Steinbrecher KA, Strahl BD, Baldwin AS. Nature. 2003;423:659–663. doi: 10.1038/nature01648. [DOI] [PubMed] [Google Scholar]
- 18.Yamamoto Y, Verma UN, Prajapati S, Kwak YT, Gaynor RB. Nature. 2003;423:655–659. doi: 10.1038/nature01576. [DOI] [PubMed] [Google Scholar]
- 19.Hu Y, Baud V, Oga T, Kim KI, Yoshida K, Karin M. Nature. 2001;410:710–714. doi: 10.1038/35070605. [DOI] [PubMed] [Google Scholar]
- 20.Lawrence T, Bebien M, Liu GY, Nizet V, Karin M. Nature. 2005;434:1138–1143. doi: 10.1038/nature03491. [DOI] [PubMed] [Google Scholar]
- 21.Chinnaiyan AM, O’Rourke K, Tewari M, Dixit VM. Cell. 1995;81:505–512. doi: 10.1016/0092-8674(95)90071-3. [DOI] [PubMed] [Google Scholar]
- 22.Madge LA, Li JH, Choi J, Pober JS. J Biol Chem. 2003;278:21295–21306. doi: 10.1074/jbc.M212837200. [DOI] [PubMed] [Google Scholar]
- 23.Elliott MJ, Murphy KM, Stribinskiene L, Ranganathan V, Sturges E, Farnsworth ML, Lock RB. Cancer Chemother Pharmacol. 1999;44:1–11. doi: 10.1007/s002800050938. [DOI] [PubMed] [Google Scholar]
- 24.Murphy KM, Ranganathan V, Farnsworth ML, Kavallaris M, Lock RB. Cell Death Differ. 2000;7:102–111. doi: 10.1038/sj.cdd.4400597. [DOI] [PubMed] [Google Scholar]
- 25.Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Cell. 2005;120:649–661. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 26.Yu L, Wan F, Dutta S, Welsh S, Liu Z, Freundt E, Baehrecke EH, Lenardo M. Proc Natl Acad Sci U S A. 2006;103:4952–4957. doi: 10.1073/pnas.0511288103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li ZW, Chu W, Hu Y, Delhase M, Deerinck T, Ellisman M, Johnson R, Karin M. J Exp Med. 1999;189:1839–1845. doi: 10.1084/jem.189.11.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tanaka M, Fuentes ME, Yamaguchi K, Durnin MH, Dalrymple SA, Hardy KL, Goeddel DV. Immunity. 1999;10:421–429. doi: 10.1016/s1074-7613(00)80042-4. [DOI] [PubMed] [Google Scholar]
- 29.Takeda K, Takeuchi O, Tsujimura T, Itami S, Adachi O, Kawai T, Sanjo H, Yoshikawa K, Terada N, Akira S. Science. 1999;284:313–316. doi: 10.1126/science.284.5412.313. [DOI] [PubMed] [Google Scholar]
- 30.Luedde T, Assmus U, Wustefeld T, Meyer zu Vilsendorf A, Roskams T, Schmidt-Supprian M, Rajewsky K, Brenner DA, Manns MP, Pasparakis M, Trautwein C. J Clin Investig. 2005;115:849–859. doi: 10.1172/JCI23493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li X, Massa PE, Hanidu A, Peet GW, Aro P, Savitt A, Mische S, Li J, Marcu KB. J Biol Chem. 2002;277:45129–45140. doi: 10.1074/jbc.M205165200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Massa PE, Li X, Hanidu A, Siamas J, Pariali M, Pareja J, Savitt AG, Catron KM, Li J, Marcu KB. J Biol Chem. 2005;280:14057–14069. doi: 10.1074/jbc.M414401200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G, Declercq W, Grooten J, Fiers W, Vandenabeele P. J Exp Med. 1998;187:1477–1485. doi: 10.1084/jem.187.9.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kawahara A, Ohsawa Y, Matsumura H, Uchiyama Y, Nagata S. J Cell Biol. 1998;143:1353–1360. doi: 10.1083/jcb.143.5.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cauwels A, Janssen B, Waeytens A, Cuvelier C, Brouckaert P. Nat Immunol. 2003;4:387–393. doi: 10.1038/ni914. [DOI] [PubMed] [Google Scholar]
- 36.Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, Baehrecke EH, Lenardo MJ. Science. 2004;304:1500–1502. doi: 10.1126/science.1096645. [DOI] [PubMed] [Google Scholar]
- 37.Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, Tsujimoto Y. Nat Cell Biol. 2004;6:1221–1228. doi: 10.1038/ncb1192. [DOI] [PubMed] [Google Scholar]
- 38.Okuno S, Shimizu S, Ito T, Nomura M, Hamada E, Tsujimoto Y, Matsuda H. J Biol Chem. 1998;273:34272–34277. doi: 10.1074/jbc.273.51.34272. [DOI] [PubMed] [Google Scholar]
- 39.Thon L, Mohlig H, Mathieu S, Lange A, Bulanova E, Winoto-Morbach S, Schutze S, Bulfone-Paus S, Adam D. FASEB J. 2005;19:1945–1956. doi: 10.1096/fj.05-3726com. [DOI] [PubMed] [Google Scholar]
- 40.Yamashita K, Takahashi A, Kobayashi S, Hirata H, Mesner PW, Jr, Kaufmann SH, Yonehara S, Yamamoto K, Uchiyama T, Sasada M. Blood. 1999;93:674–685. [PubMed] [Google Scholar]
- 41.Sakon S, Xue X, Takekawa M, Sasazuki T, Okazaki T, Kojima Y, Piao JH, Yagita H, Okumura K, Doi T, Nakano H. EMBO J. 2003;22:3898–3909. doi: 10.1093/emboj/cdg379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ventura JJ, Cogswell P, Flavell RA, Baldwin AS, Jr, Davis RJ. Genes Dev. 2004;18:2905–2915. doi: 10.1101/gad.1223004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Delhalle S, Deregowski V, Benoit V, Merville MP, Bours V. Oncogene. 2002;21:3917–3924. doi: 10.1038/sj.onc.1205489. [DOI] [PubMed] [Google Scholar]
- 44.Schulze-Osthoff K, Bakker AC, Vanhaesebroeck B, Beyaert R, Jacob WA, Fiers W. J Biol Chem. 1992;267:5317–5323. [PubMed] [Google Scholar]
- 45.Fiers W, Beyaert R, Declercq W, Vandenabeele P. Oncogene. 1999;18:7719–7730. doi: 10.1038/sj.onc.1203249. [DOI] [PubMed] [Google Scholar]
- 46.Tang G, Minemoto Y, Dibling B, Purcell NH, Li Z, Karin M, Lin A. Nature. 2001;414:313–317. doi: 10.1038/35104568. [DOI] [PubMed] [Google Scholar]
- 47.Pham CG, Bubici C, Zazzeroni F, Papa S, Jones J, Alvarez K, Jayawardena S, De Smaele E, Cong R, Beaumont C, Torti FM, Torti SV, Franzoso G. Cell. 2004;119:529–542. doi: 10.1016/j.cell.2004.10.017. [DOI] [PubMed] [Google Scholar]