

Abstract
Protein association in lipid membranes is a complex process with thermodynamics directed by a multitude of different factors. Amino-acid sequence is a molecular parameter that affects dimerization as shown by limited directed mutations along the transmembrane domains. Membrane-mediated interactions are also important although details of such contributions remain largely unclear. In this study, we probe directly the free energy of association of Glycophorin A by means of extensive parallel Monte Carlo simulations with recently developed methods and a model that accounts for sequence-specificity while representing lipid membranes faithfully. We find that lipid-induced interactions are significant both at short and intermediate separations. The ability of molecules to tilt in a specific hydrophobic environment extends their accessible interfaces, leading to intermittent contacts during protein recognition. The dimer with the lowest free energy is largely determined by the favorable lipid-induced attractive interactions at the closest distance. Finally, the coarse-grained model employed herein, together with the extensive sampling performed, provides estimates of the free energy of association that are in excellent agreement with existing data.
Abbreviations used: GpA, Glycophorin A; TM, transmembrane; CG, coarse-grain; DPPC, di-palmitoyl phosphatidylcholine; DOPC, di-oleoyl phosphatidylcholine; DLPC, di-lauroyl phosphatidylcholine; PO4,, phosphate group; GLY, glycerol group; PMF, potential of mean force; RC, reaction coordinate (ξ: lateral separation, τ: tilt angle); Ω, crossing angle; (MW)2–XDOS, multiple-walkers multiple-windows expanded density of states; MC, Monte Carlo; MD, molecular dynamics; CM, center of mass; NPT, constant number of particles, pressure, temperature ensemble
Introduction
The association of transmembrane proteins to a larger functional structure is a complex process of particular physiological significance. Major signaling mechanisms are directed by events that are stimulated by ligand-binding to extramembrane domains. However, mutations in the transmembrane (TM) sequence lead to changes in the association thermodynamics underlining the role of the TM domains (1–3). Furthermore, specific motifs along the TM amino-acid sequence prevail in the interface formed in the dimer state, supporting the sequence-specific character of the process. In the case of Glycophorin A (GpA), a well-studied protein with a single TM domain that undergoes dimerization, a pattern of seven amino acids (LIxxGVxxGVxxT) is considered important for the formation of a stable dimer (4–11). Van der Waals interactions at the dimer interface are suggested to be a major factor of GpA dimerization (6,12–14).
GpA is used as a model system for extensive experimental, theoretical, and simulation studies focusing on TM protein association. The motifs critical to GpA stability are often employed to analyze the association affinity of other TM proteins (15). Extensive computational studies with atomistic and coarse-grain models have examined the GpA dimer in agreement with experimental findings (5,16–20). Petrache et al. (18) performed atomistic molecular dynamics (MD) simulations of preformed dimers in different lipid environments (di-myristoyl phosphatidylcholine, DMPC; di-palmitoyl phosphatidylcholine, DPPC, and di-oleoyl phosphatidylcholine, DOPC) verifying their stability. While structure was maintained, different fluctuations indicated that the range of accessible conformations can be modulated by membrane thickness. Hénin et al. (21) performed a thorough study using biased MD simulations to extract the free energy profile as a function of lateral separation between two GpA helices in a dodecane layer. This information is of paramount interest, as it provides direct insight into the association mechanism that is not experimentally accessible. It was supported that the process presents high complexity. Association in a detergent-like environment could be divided into two regimes: a short-range where a configuration characteristic to a dimer is found, and a long-range where several residues form occasionally interhelical contacts. Furthermore, this configuration-dependent formation of contacts resulted in an additional minimum in the free energy profile (21). Such early intermittent contacts could provide significant contributions to dimer formation; to our knowledge, their presence and characteristics in a lipid bilayer has not been thoroughly investigated.
Helix-association in detergent-like environments or implicit membranes is often the method of choice to study protein association due to moderate demand of computational time (22–25). However, such methods do not allow a complete characterization of membrane-induced effects on protein assembly and activity; the latter can depend highly on details of the lipidic environment in proximity to the proteins (26–29). In the case of GpA association, experiments suggested that hydrophobic mismatch holds a principal role on dimerization (30) while other studies have supported the idea that protein-protein interactions can be separable from lipid-protein and lipid-lipid contributions (31). Simulations that account explicitly for the membrane environment have led to mixed results. Integral equation theory predicts that lipids induce a (nonmonotonic with distance) attractive interaction on two cylinders embedded perpendicularly in a lipid membrane. This attraction can be several kBT, yet at intermediate distances, a repulsion was observed (32–34). Similar results were obtained with mesoscopic simulations (35). The atomistic study of GpA in dodecane resulted in exclusively attractive, induced interactions, in contrast to theoretical predictions (24), although no separate contributions by the hydrophobic environment and the solvent were provided in the former (21).
Recently, Bond and Sansom (36) have studied GpA association in explicit membranes with a coarse-grain (CG) representation that is capable of accounting for sequence-specificity; such studies have been performed for other proteins systems as well (37). A subsequent study addressed mutations and provided estimates of the free energy difference between monomer and dimer state using a set of seven independent MD simulations (38). Although coarse-graining at this resolution appears appealing, unfortunately the level of detail included is coupled with a decrease in efficiency. Therefore, the study of multiple association phenomena remains a challenging task.
In this article, we examine the association of GpA by providing free-energy profiles in explicit lipid bilayers. To the best of our knowledge, such estimates for GpA in lipid bilayers have not been reported. A second aim is to investigate how the characteristics of the membrane environment affect the association process, a subject of recent studies (35,39). Our simulations examine the effect of both the hydrophobic environment and amino-acid sequence; this is not feasible without sufficient resolution and rigorous sampling of protein conformations. We employ CG models capable of representing the properties of lipid bilayers and maintain sequence-specificity (40,41). Furthermore, we apply efficient large-scale parallel Monte Carlo (MC) simulations involving hundreds of pairs of proteins over extended ranges of separation. We find that the stability of the dimer state depends largely on lipid-induced interactions; the lipid membrane does not only modulate the final structure but has a significant impact on the formation of early contacts. Helix-tilting enhances the extent of accessible interfaces with multiple free energy-minima appearing as a function of lateral separation. Finally, our estimates on the free energy of association are in excellent agreement with experimental data and theoretical predictions.
Methods and Model
Model
The model of GpA was composed of a sequence of 27 amino acids that included the transmembrane domain
We employed 27 residues to ensure that the helix maintains contact with the water-lipid interface. The CG model was built as described in the literature preserving an α-helical secondary structure between amino acids 73 and 95 (41). As common with such CG models, the secondary structure is imposed through specific potential terms. This approximation follows the two-stage model by Popot and Engelman (42) and is supported by experimental data on GpA (43). The charged amino acids close to the interface (residues 70, 72, and 96) were neutralized by two sodium ions and one chlorine ion. The model is similar to past successful studies (36,38) although interaction potentials were based on a more recent version of the force field with no modifications applied (40,41). These effective interactions account for the characteristics (such as hydrogen-bonding) of each amino acid. The association of GpA is assessed in three lipid bilayers: DPPC (di-16:0 PC), DOPC (di-18:1 PC), and di-lauroyl phosphatidylcholine (DLPC, di-12:0 PC) that exhibit different properties (Table 1). Due to tail length and unsaturation (for DOPC) the liquid-ordered to liquid-disordered phase transition temperatures are experimentally reported as 314 K for DPPC, 251 K for DOPC, and 273 K for DLPC (44). All our simulations were performed at 323 K while in the liquid-disordered phase.
Table 1.
Lipid bilayer properties with thickness increasing from DLPC to DPPC and DOPC
| Lipid | Area/lipid (nm2) | Thickness (PO4–PO4 and GLY-GLY) (nm) |
|---|---|---|
| DLPC | 0.63 | 3.37, 2.49 |
| DPPC | 0.63 | 4.09, 3.16 |
| DOPC | 0.70 | 4.36, 3.48 |
Lipid bilayer properties. Thickness (average separation along the normal between characteristic groups on the two leaflets) increases from DLPC to DPPC and DOPC.
Simulations
(MW)2-XDOS potential of mean force calculations
Potential of mean force (PMF) calculations provide the free energy along a generalized reaction coordinate (RC) with ξ = lateral separation and τ = tilt angle. We use the multiple-walkers, multiple-windows expanded density of states (MW)2-XDOS method (45), an enhanced, parallel version of the EXEDOS method (46,47) based on the Wang-Landau density-of-states algorithm (48). The method allows continuous uniform sampling of the RC without a priori knowledge of the free energy profile. This is achieved by an iterative computation of weighting factors g(ξ). Upon convergence, the PMF can be extracted using the weights as U(ξ) = –kBT ln g(ξ) + C. Alternatively, the PMF can be calculated by integrating the mean projected force along the RC 〈F〉ξ = −∂U(ξ)/∂ξ. In this study, decomposition of PMF profiles is performed by integrating separate average forces by ions, water, lipid, and intermolecular protein atoms.
The RC employed was the projection of the distance between the center-of-mass (CM) of the two helices on the midbilayer plane, denoted as ξ. For reasons discussed further below, the range of ξ in DPPC and DOPC bilayers was 0.4–4.6 nm while in DLPC it was increased to 5.4 nm. Systems consisted of two protein helices, 400 lipid molecules, 4000 CG water beads, four sodium ions, and two chlorine ions. Simulations were performed over 16 windows with eight walkers in each window (total of 128) for each system (45). For DPPC and DOPC, the first five windows (up to 1.3 nm) were 0.3 nm in size, with 0.1 nm overlap at each end to allow for frequent successful configuration exchanges (45). The remaining 11 windows were 0.4 nm large with 0.12 nm overlap. Due to the increased RC range in the DLPC system, window sizes were increased to 0.35 nm for the first 5, 0.45 nm for the next 10, and 0.55 nm for the last one.
(MW)2-XDOS simulations involved
-
1.
MC moves on helices with 0.8% probability. These moves were further distributed to 50% directional displacement (translation along ξ (45)) of both proteins, 30% rotations of one GpA around helix axis, and 20% changes in τ. Both angular moves were centered at the CM, keeping ξ unaltered.
-
2.
A total of 99.1% MC moves are on lipid, water, or ion beads. Selections were performed in a preferential scheme using two centers defined by the CM of the two proteins (45). Ninety-five percent of these moves were simple random translations of individual beads; 3% corresponded to preferential selection (based on the phosphate group, or PO4, bead) and displacement of a lipid and 2% rotation.
-
3.
A total of 0.1% hybrid 1-ps MD moves (5 fs time step), which altered the whole configuration (49). The acceptance ratio included the change of the total Hamiltonian with a prefactor ξold/ξnew (45).
Additional calculations
Additional supplemental simulations performed are discussed briefly herein:
-
1.
An atomistic, NPT ensemble, 60 ns long MD simulation of a single GpA helix in a DPPC bilayer with 236 lipids using the GROMOS 53a6 force field (50,51) and GROMACS (52).
-
2.
CG MD simulations of a single GpA helix in DLPC, DPPCN, and DOPC.
-
3.
EXEDOS PMF calculations along τ of a single helix in each of the three systems. The free energy as a function of τ was calculated by the weights required to perform uniform sampling (Fig. 1 B) with a profile similar to recent studies (53).
Figure 1.

(A) Distribution of GpA amino acids (CM) along the membrane-normal in DOPC, DPPC, and DLPC centered with respect to the bilayer midplane. Results from atomistic simulations are plotted with a line, while symbols correspond to the CG model. The average thickness of each CG bilayer is shown based on the PO4-PO4 distance (Table 1). (B) PMF as a function of τ for a single helix in each lipid bilayer. Points are extracted in equilibrium MD simulations by accumulation of probabilities (Inset). Lines are estimates extracted in MC PMF simulations by the weights required to perform uniform sampling along τ.
Results
Single helix in a lipid bilayer
Fig. 1 A presents the mean positions of the CM of the amino acids with respect to the membrane normal extracted by unbiased equilibrium MD simulations. The continuous line is the result of atomistic simulations, providing evidence for an α-helix secondary structure throughout the hydrophobic domain of the membrane. Data from the CG MD simulations are in fair agreement, supporting the simpler model (size of points represent the model's resolution; one bead is 0.47 nm) (40,41). We examined the membrane thickness in proximity of the protein but no appreciable deviation was observed from the average (Table 1). For these small molecules, tilting is the major mechanism to compensate for any hydrophobic mismatch. In addition, entropic contributions induce tilting even when there is no mismatch (53).
To quantify fluctuations around the average positions we examined the distribution of τ observed during CG MD simulations of a single helix as shown in the inset of Fig. 1 B. The most probable angle is ≈33° in DLPC, ≈16° in DPPC, and ≈13° in DOPC, with averages of ≈32°, ≈18°, and ≈15°, respectively. A more comprehensive measure is the free energy change along τ (53). A first estimate from the histograms is given by U = –kBT ln p where p is the probability of observing a value of τ. Such estimates (shifted to zero at the minimum) are shown by points in Fig. 1 B. Any value above kBT by equilibrium simulations is highly uncertain due to low number of states sampled. Therefore, we performed MC PMF calculations choosing τ as our RC. The profiles (lines in Fig. 1 B) are in good agreement with the MD simulations for low values of the PMF. Overall, we find that τ can fluctuate between 16° and 46° for DLPC. In contrast, in DPPC, values range from 3° to 30° and in DOPC from 3° to 26°. The free energy changes are similar to atomistic studies of a WALP peptide (53) providing further support for the simpler CG model used herein.
Association: subtleties of PMF calculations
We chose the lateral separation of the CM of the two helices ξ for our RC in association studies (as, e.g., in (21,35)); fluctuations along the normal of the membrane were <0.5 nm (21). Several shortcomings exist with any choice of a single RC, e.g., ξ is not entirely descriptive of the range of direct protein-protein interaction because, for a specific value, tilting can alter the number of interhelical contacts. The minimum interhelical separation between any amino acids on the two helices provides an alternate RC (54,55). Fig. 2 A presents the distribution of minimum distance as a function of ξ for GpA in DLPC. We noticed several features:
-
1.
For ξ < 1.6 nm there is always a contact (≈0.47 nm).
-
2.
For 1.6 nm < ξ < 3.5 nm, contacts are occasionally formed, depending on the relative orientation and configuration of the two helices.
-
3.
Although no contacts are apparent for ξ > 3.5 nm, the distribution of minimum distances becomes symmetric (at a constant ξ value with respect to the mean) at ≈5 nm.
Figure 2.

(A) Distribution of minimum interhelical distance as a function of ξ in a DLPC bilayer. The darker color denotes higher probability. At low separations, a contact at ≈0.47 nm (size of beads) is certain. (B) Evolution of ξ between two helices throughout the MC simulation (shown for four replicas) for GpA in DPPC. One-hundred-and-twenty-eight pairs were considered for each system. (C) PMF of GpA association in a DLPC bilayer as calculated by integrating the mean force projected along ξ (dashed line) and by the weights estimated to perform a random walk along this RC (symbols). The mean force 〈F〉ξ is shown by the solid line and in contrast to the PMF is calculated in absolute scale (with uniform sampling and error approximately the size of fluctuations at large ξ as 〈F〉ξ→∞ = 0).
As the lipid membrane can impose indirect correlations, we extended the maximum ξ to 5.4 nm for the DLPC bilayer (higher tilting).
Our parallel MC algorithm enhances sampling only along a single RC (ξ), therefore it is essential to perform rigorous canonical sampling of all remaining degrees of freedom at a value of ξ. Designed MC moves (preferential sampling) and the ability of proteins to experience large separations (where no correlations persist) are critical. The latter is achieved in (MW)2-XDOS simulations (Fig. 2 B) by employing 128 unconstrained pairs for each system.
Finally, the free energy profiles can be extracted by the weights iteratively estimated or by integrating the forces accumulated as a function of ξ. Mean forces along RC are known in absolute scale (in contrast to the unknown constant in PMFs due to integration). The flat-histogram method provides mean force samples of equal quality along ξ. Fig. 2 C presents the PMF profile as extracted by the weights and by integrating the forces, for GpA in DLPC. The free energy change of ≈6.3 kcal/mol supports the formation of a stable dimer. As discussed later, this estimate is consistent with the literature (6,21,24) The position of the minimum agrees well with atomistic studies in dodecane with free energy values appearing higher, closer to experimental data (21). Additionally, distinct features at short distances are attributed to intermittent contacts (21). In DLPC, we observed more than one oscillation in the mean force profile with a periodicity Δξ ≈ 0.4 nm; in DPPC and DOPC, separations between minima decrease considerably and their presence was not clear.
Potential of mean force in different lipid environments
The extracted free energy profiles are summarized in Fig. 3. In all three bilayers a stable dimer is formed with DPPC providing the most favorable environment (≈7.3 kcal/mol) followed by DOPC and DLPC (both ≈6.3 kcal/mol). By analyzing the free energy profile we find that the total free energy is a result of a competitive or synergistic effect of lipid-induced and protein-protein contributions. Water induces a repulsion between the two proteins corresponding to dewetting of hydrophilic residues at the interfaces, whereas ion-induced contributions were negligible and omitted for clarity. The most favorable environment for dimerization is provided by DPPC due to extensive attractive interactions between the helices and synergistic lipid-induced effects at similar values of ξ.
Figure 3.

(A) PMF for GpA association in three different bilayers with decomposition to separate contributions: total (continuous line), protein-protein (dashed), lipid-protein (dash-dotted), and water-protein (dotted). (B) Comparison of protein-protein and lipid-induced contributions.
Lipid-induced interactions
Membrane-mediated interactions in all three systems present a nonmonotonic profile with ξ. At intermediate ranges, desorption of lipids is unfavorable, leading to a repulsion that grows with membrane thickness. However, when lipids in-between the proteins are depleted, there is an induced attraction due to entropic gain of lipid tails. Zhang and Lazaridis (24) predicted a positive contribution, whereas Hénin's (21) results in dodecane were always negative (sum of solvent and dodecane), accounting for almost 50% of the total. We attribute differences to the highly anisotropic ordered lipid bilayer environment. Our estimations agree with calculations for 0.5 nm cylinders in membranes by a combination of integral equation theory and atomistic simulations (32–34). Dissipative particle dynamics with mesoscopic models resulted in similar predictions (35). Given the increased complexity of our systems, we find the agreement very favorable.
The range of the attractive contributions is different due to tilting effects. In DLPC at a separation below 1.5 nm, no significant lipid atoms are located in-between helices. In DOPC, this distance is lowered to 0.85 nm with a clear repulsive peak at larger distances. At the total PMF minimum, the lipids provide for up to 2/3 of the free energy of association. The actual value is not merely a function of thickness of the bilayer; although similar values at the lowest separation are found between DPPC and DLPC (difference in the slope attributed to the volume between the proteins as a function of their tilting), for DOPC, contributions were weaker (Fig. 3 B). We attribute this to the reduced structural ordering due to oleoyl-tail content which lowers the gel-to-liquid transition temperature (44). Experiments support stronger protein dimerization in liquid-ordered domains (27).
Protein-protein interactions
Fig. 3 B suggests that DOPC presents the most extensive protein-protein interactions as a result of low tilting and the larger surface of the interface formed. A very intriguing finding: the oscillations in the mean force and the nonmonotonic dependence of the free energy with ξ, mostly apparent for DLPC. As described by Hénin et al. (21) these features could have a critical role in recognition and dimerization. To examine their origin, it is first noted that protein-protein interactions are indirectly affected by the environment. Helices form different configurations at each value of ξ with respect to properties as, e.g., tilt angle and crossing angle. As the proteins approach, their entropy (e.g., rotational entropy) is reduced and the ensemble of these structures is drastically changing.
Fig. 4 presents contact maps for DLPC and DPPC at approximately the free energy minimum and two snapshots of sampled protein configurations. GpA association displays specific packing that is in fair agreement to the LIxxGVxxGVxxT motif extensively reported in the literature (4–11). However, for this range of ξ, an ensemble of structures is probed in DLPC, consisting of both symmetric and asymmetric dimers across the same face of the helices with the small Gly residues. In Fig. 4 C, Gly-79 packs against Leu-75, Gly-83 against Gly-79, and Gly-86 with Gly-83. This asymmetric configuration has a separation at the free energy minimum in DLPC; because significant tilting is required to form such structures, symmetric dimers as shown in Fig. 4 D are preferred in DPPC. Most configurations differ by a shift along the helical axis of the other protein moving along the same interface with the Gly residues. These shifts (≈0.65 nm in the CG model) produce a change in lateral separation that is dependent on tilting. For τ = 30°, these changes along the lateral separation will be ≈0.33 nm. Despite the simplicity of this calculation (as helical turns fluctuate in distance and proteins assert different tilt angles and orientations), it is enlightening that this agrees well with Δξ values between minima in Fig. 2 C and Fig. 3. For GpA in DPPC and DOPC, the interface was significantly more well defined. In addition, for proteins nearly parallel to the normal of the membrane, contacts can shift along this interface without any direct noticeable effect on our PMF profiles (ξ is the lateral CM separation).
Figure 4.

(A) Average interfaces formed upon dimerization of GpA in DLPC at 0.9 nm < ξ < 1.0 nm. (B) Same in DPPC and 0.7 nm < ξ < 0.8 nm. (C) Asymmetric dimer sampled in DLPC. Higher tilting is required to sample this conformation, which remains along the same interface of Gly residues (in blue; Leu beads in red). (D) Symmetric dimer sampled in DPPC.
Dimer characterization
Fig. 5 A presents the average τ of helices as a function of ξ. At large separations, the proteins assert their monomeric state with τ as in Fig. 1 B. As the proteins approach, approximately at 2 nm in all three systems, τ increases. As shown in Fig. 2 A, below this distance, contacts persist. There is an additional interesting feature: the extent of tilting is highly dependent on the lipid environment. In DLPC, helices tilt up to 42°, which in a single monomeric state corresponds to ≈ 0.5 kBT per protein. This change is diminished as we move to DPPC and DOPC systems. At even shorter separations, close to the minimum of the PMF, τ-values found were close to the single-helix unperturbed angle. Favorable lipid-induced interactions force the helices to approach further, with values of 25° ± 3°. The process is intrinsically different: in DLPC, there is a continuous decrease of τ to ≈ 27°, whereas for DPPC and DOPC, a nonmonotonic dependence is found.
Figure 5.
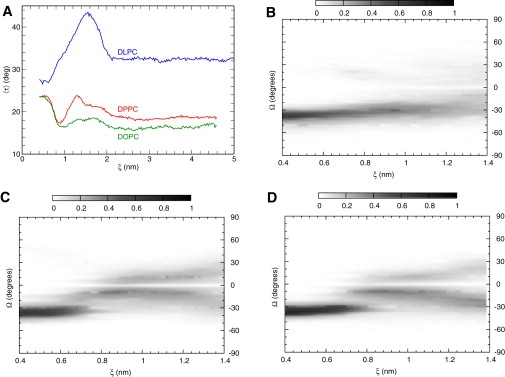
(A) Average tilt τ of helices as a function of ξ. (B–D) Crossing angle Ω as a function of ξ in (B) DLPC, (C) DPPC, and (D) DOPC. Darker color represents higher probability.
This nonmonotonic dependence is coupled to another property that describes the configuration of a dimer, the crossing angle Ω (54). The experimentally determined Ω for GpA in detergents is −40° (12), while in membranes, a lower value of −35° was reported (56); both correspond to a right-handed conformation (16,4,5). Hénin et al. (21) reported a steep change of Ω during dimerization in dodecane. Fig. 5, C and D, presenting the entire distribution of sampled Ω as a function of ξ in DPPC and DOPC, are in agreement. In contrast, in DLPC, the proteins approach in configurations that monotonically decrease Ω. We emphasize that Fig. 5, B–D, represent a conditional probability of Ω for a specific separation. An equilibrium simulation will result in an ensemble of structures of various ξ-values with a probability prescribed by the PMF (Fig. 3). Therefore a bimodal distribution can be observed (as in (38)) that we attribute to dimers with different lateral separations. The overall population will depend on the balance between lipid-induced and protein-protein contributions, as shown in Fig. 3.
Comparison to experiments
Experimentally, protein association is characterized by an overall observed free energy change ΔG. Due to various factors (e.g., concentration of hydrophobic environment), comparing ΔG among different experiments, theories, or models is not straightforward (24,57). To derive the free energy, we integrated over RC and extracted an association constant K. Different calculation formulas are available (58,59); herein we employed the following definition (21,60):
| (1) |
The above expression (with ξmax the value where the PMF converges to zero at ≈3.3–4.0 nm) and
| (2) |
yields −6.7, −7.5, and −6.6 kcal/mol in DLPC, DPPC, and DOPC, respectively. Because this estimate implies a standard concentration of one molecule/nm2 (58,24), we calculated the standard concentration 1 M in terms of area of hydrophobic phase. Note that 1 M corresponds to 1.660 nm3 per molecule; given the thickness reported in Table 1 (lipid tail thickness as in (24) for the glycerol group GLY-GLY beads), this corresponds to a concentration of 1 molecule per 0.67, 0.53, and 0.47 nm2. Adding the term RT ln C will result to a change to −6.9, −7.9, and −7.0 kcal/mol. The values reported by experiments in detergents ranged from −3.8 to −7.5 kcal/mol (57,61). Our estimates are in quantitative agreement and a significant improvement over past predictions of −11.5 ± 0.4 (21) or −9 kcal/mol if standardized appropriately (24). We also note the excellent agreement to the theoretical predictions by Zhang and Lazaridis (24) of −7.7 ± 1.8 kcal/mol.
Conclusions
We studied the association of GpA in lipid membranes using recently developed parallel MC simulations (45) and models. Despite the application of efficient algorithms, extended computational time was required for our systems (studied on 128 processors for over a month). It would be highly desirable to use data extracted for further development of efficient implicit membrane methods or theoretical estimations (23,24).
Although excellent agreement is found to existing data, our results provide further insight into the mechanism of protein dimerization. Specifically, we have shown that association is assisted by the lipid-induced interactions with the most favorable contributions rising from the bilayer presenting the highest structural order. However, at intermediate distances, repulsive lipid-induced interactions are present and most significant for the thickest membranes. In addition, proteins tilt to a different extent, depending both on membrane properties and amino-acid sequence. This leads to the formation of multiple favorable dimers along the same interface that all contribute to dimerization.
Clearly our study did not account for ectodomains; however, given the agreement with experiments, we believe that relative TM contributions were accurately predicted. Furthermore, we note that the PMF profiles are a result of coupled competitive or synergistic lipid-induced and protein-protein interactions. The former depend on membrane composition as well as on the ability of the helices to efficiently dimerize and increase the available volume to the lipids; this effect is directly related to amino-acid sequence. Protein-protein interactions depend on the interfaces formed which, as shown in our study, can be modulated by the lipid environment. The importance of stabilization of the dimer in terms of interface (and not only proximity) is paramount for TM receptors (62).
Cell membranes are multicomponent; red cell membranes contain numerous distinct lipid species (e.g., with choline and ethanolamine headgroups). Any of the studied bilayers herein is a simple model of the complex native lipid environment of GpA. Efficient MC moves (e.g., identity exchanges) could be introduced in our algorithms to extend our studies to such direction. Another area of particular interest is the study of protein concentration effects (63). Herein, we remained at the limit of infinitely-low protein concentration and it is unclear how the presence of many protein molecules would affect association. Such subjects would be addressed in future research.
Acknowledgments
We are deeply grateful to the University of Houston Research Computing Center for the generous allocation of CPU time on the Maxwell cluster.
Financial support was provided by University of Houston grant No. GEAR IO96691.
References
- 1.Cao H., Bangalore L., Stern D.F. A subdomain in the transmembrane domain is necessary for p185neu∗ activation. EMBO J. 1992;11:923–932. doi: 10.1002/j.1460-2075.1992.tb05131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lemmon M.A., Engelman D.M. Helix-helix interactions inside lipid bilayers. Curr. Opin. Struct. Biol. 1992;2:511–518. [Google Scholar]
- 3.Lemmon M.A., Flanagan J.M., Engelman D.M. Glycophorin A dimerization is driven by specific interactions between transmembrane α-helices. J. Biol. Chem. 1992;267:7683–7689. [PubMed] [Google Scholar]
- 4.Lemmon M.A., Treutlein H.R., Engelman D.M. A dimerization motif for transmembrane α-helices. Nat. Struct. Biol. 1994;1:157–163. doi: 10.1038/nsb0394-157. [DOI] [PubMed] [Google Scholar]
- 5.Adams P.D., Engelman D.M., Brünger A.T. Improved prediction for the structure of the dimeric transmembrane domain of glycophorin A obtained through global searching. Proteins. 1996;26:257–261. doi: 10.1002/(SICI)1097-0134(199611)26:3<257::AID-PROT2>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 6.Fleming K.G., Ackerman A.L., Engelman D.M. The effect of point mutations on the free energy of transmembrane α-helix dimerization. J. Mol. Biol. 1997;272:266–275. doi: 10.1006/jmbi.1997.1236. [DOI] [PubMed] [Google Scholar]
- 7.Brosig B., Langosch D. The dimerization motif of the glycophorin A transmembrane segment in membranes: importance of glycine residues. Protein Sci. 1998;7:1052–1056. doi: 10.1002/pro.5560070423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Russ W.P., Engelman D.M. The GxxxG motif: a framework for transmembrane helix-helix association. J. Mol. Biol. 2000;296:911–919. doi: 10.1006/jmbi.1999.3489. [DOI] [PubMed] [Google Scholar]
- 9.Mackenzie K.R. Folding and stability of α-helical integral membrane proteins. Chem. Rev. 2006;106:1931–1977. doi: 10.1021/cr0404388. [DOI] [PubMed] [Google Scholar]
- 10.MacKenzie K.R., Fleming K.G. Association energetics of membrane spanning α-helices. Curr. Opin. Struct. Biol. 2008;18:412–419. doi: 10.1016/j.sbi.2008.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moore D.T., Berger B.W., DeGrado W.F. Protein-protein interactions in the membrane: sequence, structural, and biological motifs. Structure. 2008;16:991–1001. doi: 10.1016/j.str.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.MacKenzie K.R., Prestegard J.H., Engelman D.M. A transmembrane helix dimer: structure and implications. Science. 1997;276:131–133. doi: 10.1126/science.276.5309.131. [DOI] [PubMed] [Google Scholar]
- 13.MacKenzie K.R., Engelman D.M. Structure-based prediction of the stability of transmembrane helix-helix interactions: the sequence dependence of glycophorin A dimerization. Proc. Natl. Acad. Sci. USA. 1998;95:3583–3590. doi: 10.1073/pnas.95.7.3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doura A.K., Fleming K.G. Complex interactions at the helix-helix interface stabilize the glycophorin A transmembrane dimer. J. Mol. Biol. 2004;343:1487–1497. doi: 10.1016/j.jmb.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 15.Mendrola J.M., Berger M.B., Lemmon M.A. The single transmembrane domains of ErbB receptors self-associate in cell membranes. J. Biol. Chem. 2002;277:4704–4712. doi: 10.1074/jbc.M108681200. [DOI] [PubMed] [Google Scholar]
- 16.Treutlein H.R., Lemmon M.A., Brünger A.T. The glycophorin A transmembrane domain dimer: sequence-specific propensity for a right-handed supercoil of helices. Biochemistry. 1992;31:12726–12732. doi: 10.1021/bi00166a003. [DOI] [PubMed] [Google Scholar]
- 17.Braun R., Engelman D.M., Schulten K. Molecular dynamics simulations of micelle formation around dimeric glycophorin A transmembrane helices. Biophys. J. 2004;87:754–763. doi: 10.1529/biophysj.104.040279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Petrache H.I., Grossfield A., Woolf T.B. Modulation of glycophorin A transmembrane helix interactions by lipid bilayers: molecular dynamics calculations. J. Mol. Biol. 2000;302:727–746. doi: 10.1006/jmbi.2000.4072. [DOI] [PubMed] [Google Scholar]
- 19.Kokubo H., Okamoto Y. Prediction of membrane protein structures by replica-exchange Monte Carlo simulations: case of two helices. J. Chem. Phys. 2004;120:10837–10847. doi: 10.1063/1.1712942. [DOI] [PubMed] [Google Scholar]
- 20.Cuthbertson J.M., Bond P.J., Sansom M.S.P. Transmembrane helix-helix interactions: comparative simulations of the glycophorin a dimer. Biochemistry. 2006;45:14298–14310. doi: 10.1021/bi0610911. [DOI] [PubMed] [Google Scholar]
- 21.Hénin J., Pohorille A., Chipot C. Insights into the recognition and association of transmembrane α-helices. The free energy of α-helix dimerization in glycophorin A. J. Am. Chem. Soc. 2005;127:8478–8484. doi: 10.1021/ja050581y. [DOI] [PubMed] [Google Scholar]
- 22.Mottamal M., Zhang J., Lazaridis T. Energetics of the native and non-native states of the glycophorin transmembrane helix dimer. Proteins. 2006;62:996–1009. doi: 10.1002/prot.20844. [DOI] [PubMed] [Google Scholar]
- 23.Bu L., Im W., Brooks C.L. Membrane assembly of simple helix homo-oligomers studied via molecular dynamics simulations. Biophys. J. 2007;92:854–863. doi: 10.1529/biophysj.106.095216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang J., Lazaridis T. Calculating the free energy of association of transmembrane helices. Biophys. J. 2006;91:1710–1723. doi: 10.1529/biophysj.106.081224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gervais C., Wüst T., Xu Y. Application of the Wang-Landau algorithm to the dimerization of glycophorin A. J. Chem. Phys. 2009;130:215106. doi: 10.1063/1.3148186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gil T., Ipsen J.H., Zuckermann M.J. Theoretical analysis of protein organization in lipid membranes. Biochim. Biophys. Acta. 1998;1376:245–266. doi: 10.1016/s0304-4157(98)00022-7. [DOI] [PubMed] [Google Scholar]
- 27.Mall S., Broadbridge R., Lee A.G. Self-association of model transmembrane α-helices is modulated by lipid structure. Biochemistry. 2001;40:12379–12386. doi: 10.1021/bi011075y. [DOI] [PubMed] [Google Scholar]
- 28.Lee A.G. How lipids affect the activities of integral membrane proteins. Biochim. Biophys. Acta. 2004;1666:62–87. doi: 10.1016/j.bbamem.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 29.Vigh L., Escribá P.V., Harwood J.L. The significance of lipid composition for membrane activity: new concepts and ways of assessing function. Prog. Lipid Res. 2005;44:303–344. doi: 10.1016/j.plipres.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 30.Orzáez M., Lukovic D., Mingarro I. Influence of hydrophobic matching on association of model transmembrane fragments containing a minimized glycophorin A dimerization motif. FEBS Lett. 2005;579:1633–1638. doi: 10.1016/j.febslet.2005.01.078. [DOI] [PubMed] [Google Scholar]
- 31.Fleming K.G., Engelman D.M. Specificity in transmembrane helix-helix interactions can define a hierarchy of stability for sequence variants. Proc. Natl. Acad. Sci. USA. 2001;98:14340–14344. doi: 10.1073/pnas.251367498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lagüe P., Zuckermann M.J., Roux B. Protein inclusion in lipid membranes: a theory based on the hypernetted chain integral equation. Faraday Discuss. 1998;111:165–172. doi: 10.1039/a807109h. discussion 225–246. [DOI] [PubMed] [Google Scholar]
- 33.Lagüe P., Zuckermann M.J., Roux B. Lipid-mediated interactions between intrinsic membrane proteins: a theoretical study based on integral equations. Biophys. J. 2000;79:2867–2879. doi: 10.1016/S0006-3495(00)76524-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lagüe P., Zuckermann M.J., Roux B. Lipid-mediated interactions between intrinsic membrane proteins: dependence on protein size and lipid composition. Biophys. J. 2001;81:276–284. doi: 10.1016/S0006-3495(01)75698-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Meyer F.J.-M., Venturoli M., Smit B. Molecular simulations of lipid-mediated protein-protein interactions. Biophys. J. 2008;95:1851–1865. doi: 10.1529/biophysj.107.124164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bond P.J., Sansom M.S.P. Insertion and assembly of membrane proteins via simulation. J. Am. Chem. Soc. 2006;128:2697–2704. doi: 10.1021/ja0569104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Periole X., Huber T., Sakmar T.P. G protein-coupled receptors self-assemble in dynamics simulations of model bilayers. J. Am. Chem. Soc. 2007;129:10126–10132. doi: 10.1021/ja0706246. [DOI] [PubMed] [Google Scholar]
- 38.Psachoulia E., Fowler P.W., Sansom M.S. Helix-helix interactions in membrane proteins: coarse-grained simulations of glycophorin A helix dimerization. Biochemistry. 2008;47:10503–10512. doi: 10.1021/bi800678t. [DOI] [PubMed] [Google Scholar]
- 39.Im W., Lee J., Rui H. Novel free energy calculations to explore mechanisms and energetics of membrane protein structure and function. J. Comput. Chem. 2009;30:1622–1633. doi: 10.1002/jcc.21320. [DOI] [PubMed] [Google Scholar]
- 40.Marrink S.J., Risselada H.J., de Vries A.H. The MARTINI force field: coarse grained model for biomolecular simulations. J. Phys. Chem. B. 2007;111:7812–7824. doi: 10.1021/jp071097f. [DOI] [PubMed] [Google Scholar]
- 41.Monticelli L., Kandasamy S.K., Marrink S.-J. The MARTINI coarse-grained force field: extension to proteins. J. Chem. Theory Comput. 2008;4:819–834. doi: 10.1021/ct700324x. [DOI] [PubMed] [Google Scholar]
- 42.Popot J.L., Engelman D.M. Membrane protein folding and oligomerization: the two-stage model. Biochemistry. 1990;29:4031–4037. doi: 10.1021/bi00469a001. [DOI] [PubMed] [Google Scholar]
- 43.Fisher L.E., Engelman D.M., Sturgis J.N. Detergents modulate dimerization, but not helicity, of the glycophorin A transmembrane domain. J. Mol. Biol. 1999;293:639–651. doi: 10.1006/jmbi.1999.3126. [DOI] [PubMed] [Google Scholar]
- 44.Ladbrooke B.D., Chapman D. Thermal analysis of lipids, proteins and biological membranes. A review and summary of some recent studies. Chem. Phys. Lipids. 1969;3:304–356. doi: 10.1016/0009-3084(69)90040-1. [DOI] [PubMed] [Google Scholar]
- 45.Janosi L., Doxastakis M. Accelerating flat-histogram methods for potential of mean force calculations. J. Chem. Phys. 2009;131:054105. doi: 10.1063/1.3183165. [DOI] [PubMed] [Google Scholar]
- 46.Kim E.B., Faller R., de Pablo J.J. Potential of mean force between a spherical particle suspended in a nematic liquid crystal and a substrate. J. Chem. Phys. 2002;117:7781–7787. doi: 10.1103/PhysRevE.69.061703. [DOI] [PubMed] [Google Scholar]
- 47.Doxastakis M., Chen Y.-L., de Pablo J.J. Potential of mean force between two nanometer-scale particles in a polymer solution. J. Chem. Phys. 2005;123:034901. doi: 10.1063/1.1953575. [DOI] [PubMed] [Google Scholar]
- 48.Wang F.G., Landau D.P. Efficient, multiple-range random walk algorithm to calculate the density of states. Phys. Rev. Lett. 2001;86:2050–2053. doi: 10.1103/PhysRevLett.86.2050. [DOI] [PubMed] [Google Scholar]
- 49.Tuckerman M.E., Berne B.J., Klein M.L. Efficient molecular-dynamics and hybrid Monte-Carlo algorithms for path-integrals. J. Chem. Phys. 1993;99:2796–2808. [Google Scholar]
- 50.van Gunsteren W., Billeter S., Tironi I. Vdf; Zürich, Switzerland: 1996. Biomolecular Simulation: The GROMOS 96 Manual and User Guide. [Google Scholar]
- 51.Oostenbrink C., Villa A., van Gunsteren W.F. A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 2004;25:1656–1676. doi: 10.1002/jcc.20090. [DOI] [PubMed] [Google Scholar]
- 52.Lindahl E., Hess B., van der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Model. 2001;7:306–317. [Google Scholar]
- 53.Lee J., Im W. Transmembrane helix tilting: insights from calculating the potential of mean force. Phys. Rev. Lett. 2008;100:018103. doi: 10.1103/PhysRevLett.100.018103. [DOI] [PubMed] [Google Scholar]
- 54.Chothia C., Levitt M., Richardson D. Helix to helix packing in proteins. J. Mol. Biol. 1981;145:215–250. doi: 10.1016/0022-2836(81)90341-7. [DOI] [PubMed] [Google Scholar]
- 55.Lee J., Im W. Role of hydrogen bonding and helix-lipid interactions in transmembrane helix association. J. Am. Chem. Soc. 2008;130:6456–6462. doi: 10.1021/ja711239h. [DOI] [PubMed] [Google Scholar]
- 56.Smith S.O., Song D., Aimoto S. Structure of the transmembrane dimer interface of glycophorin A in membrane bilayers. Biochemistry. 2001;40:6553–6558. doi: 10.1021/bi010357v. [DOI] [PubMed] [Google Scholar]
- 57.Fleming K.G. Standardizing the free energy change of transmembrane helix-helix interactions. J. Mol. Biol. 2002;323:563–571. doi: 10.1016/s0022-2836(02)00920-8. [DOI] [PubMed] [Google Scholar]
- 58.Gilson M.K., Given J.A., McCammon J.A. The statistical-thermodynamic basis for computation of binding affinities: a critical review. Biophys. J. 1997;72:1047–1069. doi: 10.1016/S0006-3495(97)78756-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mihailescu M., Gilson M.K. On the theory of noncovalent binding. Biophys. J. 2004;87:23–36. doi: 10.1529/biophysj.103.031682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chandler D. Oxford University Press; Oxford, UK: 1987. Introduction to Modern Statistical Mechanics. [Google Scholar]
- 61.Fisher L.E., Engelman D.M., Sturgis J.N. Effect of detergents on the association of the glycophorin A transmembrane helix. Biophys. J. 2003;85:3097–3105. doi: 10.1016/S0006-3495(03)74728-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fleishman S.J., Schlessinger J., Ben-Tal N. A putative molecular-activation switch in the transmembrane domain of erbB2. Proc. Natl. Acad. Sci. USA. 2002;99:15937–15940. doi: 10.1073/pnas.252640799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yiannourakou M., Marsella L., Smit B. Towards an understanding of membrane-mediated protein-protein interactions. Faraday Discuss. 2010;144:359–367. doi: 10.1039/b902190f. discussion 445–481. [DOI] [PubMed] [Google Scholar]