

Abstract
Cell-penetrating peptides like the cationic human immunodeficiency virus-1 trans-acting activator of transcription (TAT) peptide have the capability to traverse cell membranes and to deliver large molecular cargoes into the cellular interior. We used optical sectioning and state-of-the-art single-molecule microscopy to examine the passive membrane permeation of fluorescently labeled TAT peptides across the membranes of giant unilamellar vesicles (GUVs). In GUVs formed by phosphatidylcholine and cholesterol only, no translocation of TAT up to a concentration of 2 μM into the GUVs could be observed. At the same peptide concentration, but with 40 mol % of anionic phosphatidylserine in the membrane, rapid translocation of TAT peptides across the bilayers was detected. Efficient translocation of TAT peptides was observed across GUVs containing 20 mol % of phosphatidylethanolamine, which is known to induce a negative curvature into membranes. We discovered that TAT peptides are not only capable of penetrating membranes directly in a passive manner, but they were also able to form physical pores with sizes in the nanometer range, which could be passed by small dye tracer molecules. Lipid topology and anionic charge of the lipid bilayer are decisive parameters for pore formation.
Introduction
The discovery of small peptides with the ability to rapidly translocate across the plasma membrane, designated as cell-penetrating peptides (CPPs) or protein transduction domains (PTDs), and often with the potential to reach the nucleus of mammalian cells, has enthusiastically been considered in biomedical research (1,2). CPPs have been shown to enter a large number of cell types (3) and to function as vectors for various macromolecular cargoes such as fluorophores, nucleotides, drugs, proteins, DNA, peptide-nucleic acids, and also particles like liposomes and magnetic nanoparticles (4).
Descriptions of the internalization process range from energy-independent cell penetration of membranes—designated here as passive translocation—to active endocytic uptake: clathrin-independent endocytosis (5,6), caveolae-mediated endocytosis (7,8), or macropinocytosis (9–11). The mechanisms behind passive translocation of the CPPs are still mostly unknown, although it is clear that the molecular details of peptide–membrane interaction are important for the internalization process. The role of any particular secondary peptide structure in the process, however, remains disputed. Although the chemical composition of real biomembranes varies, their physical properties are governed mainly by the amphiphilic nature of the phospholipids forming a bilayer in water.
Three mechanisms were suggested for the charged peptides to cross the hydrophobic barrier of a biological membrane without using an active transport system (12). One possibility is direct membrane penetration. An alternative hypothesis invokes the binding of PTDs to negatively charged lipids and disruption of the bilayer structure by formation of inverted micelles resulting in their traversing into the cytoplasm (13). Third, a translocation by means of pore formation, similar to the action of antibiotic peptides, has been suggested and also been shown for the case of nona-arginine (14–16).
The trans-acting activator of transcription (TAT) protein of human immunodeficiency virus-1 (HIV1) provides a powerful tool for biologically active molecule delivery. The so-called HIV1 TAT peptide corresponds to the amino acids 48–57 of the TAT protein, and translocates in bulk quantities across the plasma membrane of living cells within 3–5 min (17). The TAT-PTD comprises two lysines and six arginines making the peptide strongly cationic. It has been shown that the amount and rate of cellular uptake depends strongly on the number of basic residues present, specifically the number of arginines (18,19). Proteins with a mass exceeding 100 kDa, 40 nm nanoparticles, and even 200 nm liposomes have been delivered to the cellular interior by TAT peptides (20,21).
It is consensus that electrostatic forces between the positively charged TAT-PTD and negative charges of phospholipids play a major role in the membrane binding of the peptide (22). In addition, nonelectrostatic forces, such as hydrogen bonding and hydrophobic or van der Waals forces support binding of TAT-PTD to the membrane by contributing ∼20% to the binding energy (23). It is unlikely that anionic lipids alone play a major part in the translocation of TAT-PTD across cell membranes. The binding of TAT-PTD to glycosaminoglycans on the outer cell surface is distinctly more probable for thermodynamic and statistical reasons (24).
A parameter, which should provide information about the internalization process, is the peptide mobility on the membrane surface (25). Aggregation, pore nucleation, or micelle formation could be reflected by distinct changes of peptide mobility in the membrane plane. In membrane systems peptide mobility can conveniently be studied by single molecule tracking using high-speed video fluorescence microscopy (26). Single fluorescently labeled molecules can be imaged with millisecond time resolution by sensitive light microscopy as diffraction-limited spots, which can be localized by image processing with high spatial precision. Thereby, the trajectories of single molecules can be determined by fluorescence microscopy. The technique has often been applied to analyze the movement of single lipid molecules and receptors in biological and cell membranes (reviewed by Saxton and Jacobson (27) and Schütz et al. (28)), but can also be used to study single molecules in physiological solution and living cells (26).
Cell cultures, tissues, and whole organisms are good systems for examining the action of CPPs under in vivo conditions, but the use of a simple and well-defined model system provides a better chance to show the molecular mechanism of passive protein transduction. We used high-speed single-molecule tracking of lipid molecules and TAT peptides, and confocal laser scanning microscopy (CLSM) to systematically examine factors that affect membrane binding and penetration of fluorescence labeled TAT-PTDs, namely charge and geometric properties of lipids forming the membranes.
In giant unilamellar vesicles (GUVs) formed by phosphatidylcholine (PC) and cholesterol no translocation of TAT peptides across the membrane whatsoever could be observed. Introduction of a significant amount (40 mol %) of anionic lipids or lipids inducing locally a negative curvature into the membranes (20 mol %) effected TAT translocation across these membranes. Notably, we discovered that TAT peptides were not only capable of directly penetrating such membranes in a passive manner, but they were also capable of forming physical pores, which could be passed by small but not large tracer molecules.
Materials and Methods
Lipids
The phospholipids 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1,2-dipalmitoyl-sn-glycero-3-phospho-L-serine (DPPS), 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (DOPS), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), 1,2-dipalmitoleoyl-sn-glycero-3-phosphoethanolamine (DPPE) and cholesterol were purchased from Sigma-Aldrich (Karlsruhe, Germany). All phospholipids were dissolved in chloroform/methanol (2:1 v/v) at a concentration of 1.3 mM. The fluorescent probes Texas Red-labeled 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (TR-DHPE) and 2-(4,4-difluoro-5,7-dimethyl-4-bora-3a, 4a-diaza-s-indacene-3-pentanoyl)-1-hehadecanoyl-sn- glicero-3-phosphocholine (BodipyPC) were obtained from Invitrogen GmbH (Karlsruhe, Germany) and Sigma-Aldrich (Schnelldorf, Germany), respectively.
GUVs
GUVs were created by electroformation (29,30). Lipids were used in various ratios and compositions (Table 1). Cholesterol content was always 20 mol %. Thirty microliters of the respective lipid mixture in chloroform were deposited on an indium tin oxide coated coverslip (SPI Supplies, West Chester, PA), which was dried in a desiccator under vacuum. The dried lipid film was hydrated with 250 mM sucrose in bi-distilled water as described previously (25). Vesicles in the bulk solution were transferred into a microscope observation chamber containing 250 mM glucose solution or PBS/glucose (Biochrom AG, Berlin, Germany). The density difference caused GUV sedimentation, so that they could be examined in an inverted light microscope. Electroformation of GUVs was checked by differential interference contrast (DIC) or by CLSM.
Table 1.
Composition of GUVs
| GUVs (cholesterol 20 mol %) | Lipids, mol % |
|||||
|---|---|---|---|---|---|---|
| Saturated |
Unsaturated |
|||||
| DPPC | DPPS | DPPE | DOPC | DOPS | DOPE | |
| 1 | 80 | — | — | 80 | — | — |
| 2 | 70 | 10 | — | 70 | 10 | — |
| 60 | 20 | — | 60 | 20 | — | |
| 50 | 30 | — | 50 | 30 | — | |
| 3 | 70 | — | 10 | 70 | — | 10 |
| 60 | — | 20 | 60 | — | 20 | |
| 50 | — | 30 | 50 | — | 30 | |
DOPC, 1,2-dioleoyl-sn-glycero-3-phosphocholine; DOPE, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine; DOPS, 1,2-dioleoyl-sn-glycero-3-phospho-L-serine; DPPC, 1,2-dipalmitoyl-sn-glycero-3-phosphocholine; DPPE, 1,2-dipalmitoleoyl-sn-glycero-3-phosphoethanolamine; DPPS, 1,2-dipalmitoyl-sn-glycero-3-phospho-L-serine; GUVs, giant unilamellar vesicles.
Peptides
We used a fluorescently labeled TAT peptide corresponding to amino acids 48–57 of the HIV1-TAT protein (5,6-TAMRA-RRRQRRKKRG; 1.81 kDa, abbreviated as R-TAT and AlexaFluor 647-RRRQRRKKRG, 2.2 kDa, abbreviated as AF-TAT). All peptides were purchased from Peptide Specialty Laboratories GmbH (Heidelberg, Germany). AlexaFluor 647-maleimide, AlexaFluor 546 and dextrans (3 and 70 kDa) were purchased from Invitrogen GmbH.
Sample preparation for light microscopy
For measurements focusing on the interaction of TAT with model membranes, the glucose solution was mixed with TAT peptides before GUV addition. For single molecule experiments, fluorescent R-TAT or AF-TAT was used at 250 pM. Incubation time of GUVs in the peptide-glucose solution before image acquisition was 30 min. The molar ratio of lipids to TAT in the sample chamber was ∼220,000:1. For CLSM AF-TAT was used at a concentration of 2 μM.
CLSM
GUV formation and TAT-GUV interactions were examined by CLSM with an LSM 510 Meta (Carl Zeiss, Jena, Germany) equipped with a water immersion plan-apochromat 40× objective lens (NA 1.2) at room temperature. R-TAT was excited at 543 nm, and TR-DHPE and AF-TAT were excited at 633 nm. For fluorescence detection standard filter settings were used.
Single molecule microscopy
Single molecule imaging was carried out to visualize and track lipid tracer molecules (TR-DHPE), AF-TAT, and R-TAT peptides. The experiments were carried out at room temperature using an inverted custom-built single-molecule microscope equipped with a water immersion 63× objective lens (NA 1.2) (25,26). For peptide analysis a single frame integration time of 10 ms at a frame rate of 100 Hz was used, whereas an integration time of 33 ms at 30 Hz was used for lipid imaging. All single molecule tracking experiments were carried out on the upper surface of freshly prepared GUVs. Movies comprised 1000–2000 single frames. For each experimental situation a total of three independent experiments comprising 15 movies were acquired, and the trajectory data were pooled.
Trajectory and jump distance analysis
Identification and tracking of the single molecule signals were accomplished using Diatrack 3.02 (Semasopht, Chavannes, Switzerland). All further data processing was carried out using Origin 8.0 (OriginLab, Northampton, MA) and ImageJ (31).
The probability that a molecule starting at the origin will be found in a distance r within a shell of width dr at time t is given by (32):
| (1) |
Eq. 1 is valid for a single particle species diffusing in two dimensions. Experimentally, this probability distribution can be approximated by a frequency distribution, determined by counting the jump distances within respective intervals [r, r + dr] traveled by single particles from frame to frame. The jump distance distribution cannot be fitted by Eq. 1 with a single diffusion coefficient, when several particle species with different mobilities exist. Such data can be analyzed by curve fitting using more than one term according to Eq. 2, for a jump distance distribution containing contributions from two different species,
| (2) |
where M designates the total number of jumps considered in the analysis, and f1 and f2 are the relative fractions with diffusion constants D1 and D2, respectively.
Results
We examined lipid mixtures mimicking mammalian plasma membrane composition, but kept the system as simple as possible. PC, PE, PS, and cholesterol are all present in mammalian cell membranes (33). Therefore we used those lipids, but varied the mixing ratio to analyze the effect of lipid structure and headgroup charge on TAT translocation efficiency. To focus onto the interaction between TAT peptide and lipids the experiments were mostly carried out in sucrose/glucose solution with all ions excluded from the media. A set of control experiments, however, was carried out with GUVs transferred into PBS containing glucose. PC and PS have a cylindrical shape with zero intrinsic curvature, but PE is cone-shaped and thus produces a negative curvature in membranes. In addition, it has been shown that PE is ∼30% negatively charged despite the fact that it is structurally similar to the zwitter-ionic phospholipid PC (34). Cholesterol was added to mimic mammalian cell membranes and because it contributes to a better distribution of domains in GUVs (35).
Interaction of TAT peptides with neutral model membranes containing PC
To obtain a reference for the measurements on anionic and negatively curved membranes we started out to examine neutral membranes with zero intrinsic curvature. Thus, we had to partly repeat experiments published previously, however, with different compositions of the GUV membranes (25). Electro-formation of GUVs comprising cholesterol, DOPC or DPPC was visually controlled by DIC microscopy. In some experiments vesicle formation was followed by confocal laser scanning microscopy after addition of 0.1 mol % fluorescent lipid analogs DHPE-TR or Bodipy-PC to the lipid mixture.
Imaging and tracking of single lipid analogs within neutral GUVs
To study the internal dynamics of the specific chosen model bilayer systems we measured lipid mobility in GUVs by imaging and tracking single dye-coupled lipid analogs, e.g., TR-DHPE (36,37). The experiments were carried out on liquid-ordered GUVs formed by DPPC and cholesterol or liquid-disordered GUVs formed by DOPC and cholesterol. The exact lipid mixtures were summarized in Table 1. DHPE-TR tracer molecules were added in a ratio of 10−7 mol % to the lipids before preparation of GUVs. Single diffraction-limited spots of fluorescence could be observed in the single-molecule microscope, when the focal plane was adjusted to the upper GUV surface. Acquisition of movies at a frame rate of 30 Hz showed that these fluorescence spots were rapidly moving in the GUV surface (see Movie S1 in Supporting Material). The mobility of the tracer molecules within the model bilayers was evaluated by a jump distance analysis according to Eq. 1 (38). In this analysis, the distances moved by the single molecules from frame to frame are histogramed and evaluated. Fig. S1, A and B, show the jump distance distributions for neutral liquid-ordered (DPPC) and liquid-disordered (DOPC) GUVs, respectively. The higher mobility of the tracer in the latter compared to the liquid-ordered GUVs was indicated by the right shift of the distribution. As expected the distributions could well be described by Eq. 1, and yielded a diffusion coefficient of D = 2.2 ± 0.02 μm2/s for DOPC compared to GUVs comprising DPPC with D = 0.56 ± 0.02 μm2/s.
CLSM imaging of TAT peptide-membrane interaction using neutral GUVs
To examine the TAT peptide interaction with the model membranes, we incubated pre-formed GUVs with R-TAT or AF-TAT contained in the glucose solution. Interaction of the TAT peptides with neutral GUVs was monitored as a function of time by confocal laser scanning microscopy (Fig. S1 C). Over a time period of 30 min the fluorescence at the GUV surface increased on incubation with 2 μM of R-TAT. However, we did not detect any fluorescence within the GUVs. Even in a special case, where incidentally a GUV contained several internal membranes, which were connected to the outer membrane as could be seen in the DIC image, no labeling of the internal membranes was observed (Fig. S1 C, arrow, DIC image at 30 min).
Lateral mobility of single TAT on neutral GUV surfaces
To study the interaction of TAT peptides with the model membrane surface we incubated the neutral GUVs with 0.25 nM AF-TAT for 30 min. The concentration of the peptide was so low that single diffraction-limited spots of AF-TAT fluorescence became visible at the GUV surface in the single-molecule microscope, which could be tracked by high speed imaging yielding trajectories of TAT peptides on the GUV surface. The fluorescently labeled TAT peptides had a higher mobility than the lipid tracers, so the imaging rate had to be increased from 30 to 100 Hz to catch the fast motions without blurring the single molecule signals (see OSM, Movie S2). A respective jump distance analysis showed clearly a significantly higher mobility of AF-TAT compared to membrane-embedded tracer molecules (Table 2). The mobility of the AF-TAT peptide on the GUV surface was virtually identical for DOPC- and DPPC-based GUVs within the precision of the measurements (data not shown). All jump distance histograms could well be fitted using a single diffusion component. The mobility on the neutral GUV surfaces was DTAT = 5.3 ± 0.3 μm2/s for GUVs prepared form DPPC/cholesterol and DTAT = 5.2 ± 0.1 μm2/s for DOPC/ cholesterol containing GUVs. This confirmed our previous result that TAT peptide motion was independent of the internal lipid bilayer structure (25).
Table 2.
AF-TAT peptide mobility on GUVs
| Sample (cholesterol 20 mol %) | D1,SPT [μm2/s] | Fraction 1 | D2,SPT [μm2/s] | Fraction 2 |
|---|---|---|---|---|
| DPPC | 5.3 | 1 | — | — |
| DOPC | 5.2 | 1 | — | — |
| DOPC /DPPS = 80 − x /x | ||||
| X = 20 | 4.0 | 1 | — | — |
| X = 30 | 3.9 | 1 | — | — |
| X = 40 | 4.1 | 1 | — | — |
| DOPC /DPPE = 80 − x /x | ||||
| X = 10 | 5.2 | 0.96 | 0.3∗ | 0.04 |
| X = 20 | 5.3 | 0.67 | 0.4 | 0.33 |
| X = 30 | 3.6 | 0.36 | 0.3 | 0.64 |
DOPC, 1,2-dioleoyl-sn-glycero-3-phosphocholine; DPPC, 1,2-dipalmitoyl-sn-glycero-3-phosphocholine; DPPE, 1,2-dipalmitoleoyl-sn-glycero-3-phosphoethanolamine; DPPS, 1,2-dipalmitoyl-sn-glycero-3-phospho-L-serine; GUVs, giant unilamellar vesicles.
For this fit, the diffusion value was fixed to 0.3 to reduce fitting parameter correlation Error DSPT = ±0.1 μm2/s, ErrorFraction = ±0.05.
Interaction of TAT peptides with anionic model membranes
Electrostatics presumably plays a key role not only in interaction of TAT with membranes but probably also for the penetration process (23). Therefore, we studied the association of peptides with vesicles prepared from anionic and zwitterionic lipids in dependence on the surface charge density. Anionic GUVs were prepared from mixtures of DOPC/DOPS/cholesterol or DPPC/DPPS/cholesterol with a gradually increasing PS content to follow the effect of the anionic charge on TAT mobility and possibly translocation.
Imaging and tracking of single lipid analogs within anionic GUVs
After the analysis of lipid tracer mobility in neutral GUVs comparable measurements were carried out for anionic GUVs containing 40 mol % PS and 20 mol % cholesterol. We imaged TR-DHPE within anionic liquid-ordered and liquid-disordered GUVs containing DPPC/DPPS and DOPC/DOPS, respectively. As above, the mobility of the tracer molecules within the bilayers was quantified by a jump distance analysis. The histogram data could well be fitted by a single diffusion component (Fig. S2). The presence of the charged lipid did neither change the phase behavior nor the lipid tracer mobility considerably.
CLSM imaging of TAT peptide-membrane interaction using anionic GUVs
CLSM experiments were carried out in DOPC/DOPS/cholesterol or DPPC/DPPS/cholesterol lipid mixtures containing 20, 30, and 40 mol % of PS (Fig. 1, A–C). A further increase in the mol % of PS resulted in failure of GUVs by electroformation. Anionic GUVs were incubated with 2 μM AF-TAT containing glucose solution. A distinct labeling of the GUVs was already clearly visible after 5 min for 20% PS and 2 min for 30% and 40% PS. Generally, the labeling of the anionic GUVs was significantly stronger than that of neutral GUVs. At 40 mol % PS TAT peptide fluorescence was observed within GUVs indicating peptide translocation across the bilayer (Fig. 1 C, upper panel). To control the overall membrane integrity, this experiment was carried out in the presence of a 40 kDa fluorescein-labeled dextran, which was imaged in a separate fluorescence channel (Fig. 1, A–C, lower panel). This dextran has a Stokes radius of 4.3 nm (39,40) and remained completely excluded from the GUVs at AF-TAT concentrations of 2 μM. After 20 to 30 min several vesicles with 40% PS began to deform, collapse, and release their inner content (see Fig. 1, the lower GUV and Fig. S3 in the OSM).
Figure 1.
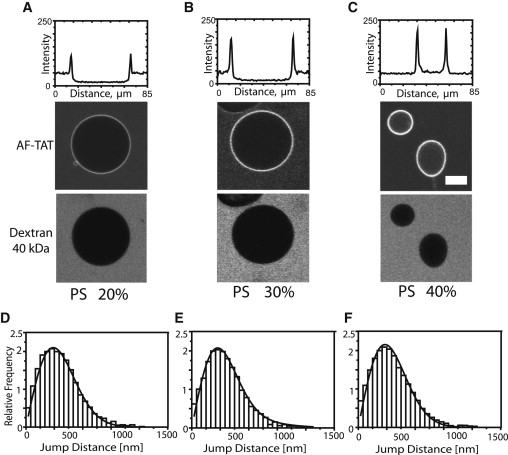
Interaction of TAT peptides with anionic model membranes. Confocal imaging of TAT peptide labeled with AlexaFluor 647 interaction with anionic GUV after 30 min of incubation. (A) Membrane with 20 mol % PS. (B) Membrane with 30 mol % PS. (C) Membrane with 40 mol % PS. The upper plots quantify the intensity of fluorescent TAT peptides along a central line across the GUVs. The internalization of TAT peptide can be seen in C. All experiments were done in the presence of 40 kDa FITC-dextran (lower panel). Scale bar: 20 μm. The histograms show the analysis of AF-TAT diffusion on anionic GUVs with (D) 20% PS, (E) 30%PS, and (F) 40% PS.
The CLSM images suggested an increasing interaction of the charged peptides with the anionic bilayers with increasing surface charges. Hence, we examined whether this would have an impact on the TAT mobility on the GUV surface. For SPT experiments, the GUVs comprising 20, 30, and 40 mol % PS were incubated with 0.25 nM AF-TAT for 30 min. In all cases the diffusion coefficient was D = 4.0 ± 0.2 μm2/s without a detectable mobility difference in the absence or presence of translocation (Fig. 1, D–F). A distinct difference, however, was observed for TAT peptide mobility on neutral versus anionic GUVs (Table 2). The mobility of TAT on the neutral GUV surfaces was faster than on the anionic GUVs. The average diffusion coefficient on the neutral GUVs was DSPT = 5.3 ± 0.2 μm2/s, whereas it amounted only to DSPT = 4.0 ± 0.2 μm2/s on anionic GUVs indicating an increased association strength of the cationic peptides to the anionic GUVs.
To gain deeper insight into the membrane translocation of TAT peptide in GUVs with 40 mol % PS, we incorporated different fluorescent tracer molecules with increasing molecular weight (Alexa647-maleimide, 1.3 kDa, Alexa546-dextran 3 kDa; Alexa546-dextran 70 kDa) inside the GUVs (41). After peptide addition the tracer fluorescence inside the GUVs was followed as a function of time to uncover a putative membrane leakage (Fig. 2). Indeed, these experiments showed the complete release of Alexa647 within 20 min, often followed by GUV rupture. For the 3 kDa dextran, a certain efflux of the tracer was observed. In contrast, the 70 kDa dextran molecules remained included in the GUVs. In absence of any TAT the GUVs were stable even after 24 h, and no fluorescent content was released. To verify that the observed fluorescence decay was not due to photobleaching of the tracers we carried out control experiments in similar conditions but without peptides. There was no change in fluorescence intensity for a similar number of images in these experiments (see OSM, Fig. S4). We concluded that the previous release of the tracer molecules from the vesicles was due to TAT peptide induced pores in the GUV membranes.
Figure 2.
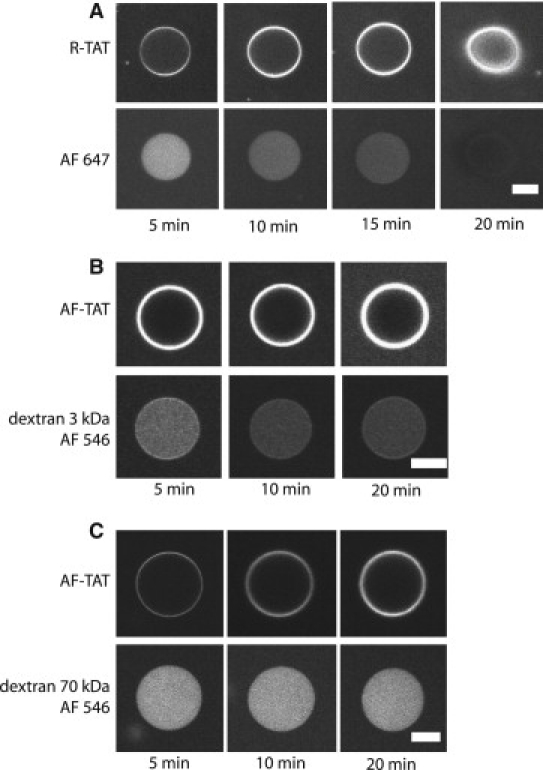
Pore formation by TAT peptides in GUVs with 40 mol % PS. (A) The upper panel shows the binding and translocation of 2 μM RTAT, and the lower panel the leakage of AlexaFluor 647, and final GUV disruption. (B) Upper panel, binding and translocation of AF-TAT peptide, lower panel shows the release of 3 kDa dextran labeled with AlexaFluor 546. (C) Upper panel, AF-TAT, lower panel 70 kDa dextran labeled with AlexaFluor 546. Scale bar, 20 μm.
Effect of counterions in the external solution
It can be assumed that the interaction between cationic TAT peptides and GUVs is dependent on the presence of ions in the solution. To examine this situation we carried out a set of experiments with GUVs that were transferred not into glucose solution, but into PBS containing glucose. In PBS visible TAT association with neutral and anionic GUVs was completely abrogated. Single TAT peptides were only rarely and for one or two images maximally observable on GUV surfaces, which prevented a quantitative mobility analysis. TAT peptide translocation, however, and tracer efflux (Alexa 488) occurred although either no PS or only 20% anionic lipids were present in the membrane (OSM Fig. S5). Hence, in physiological buffer there was an increased trend to pore formation by TAT peptides.
Interaction of TAT peptides with membranes containing lipids inducing a negative curvature
PE contains a primary amine group and PC a choline group. This results in a small headgroup and a negative intrinsic curvature for PE unlike to the zero intrinsic curvature of cylinder-shaped PC. PE is often found concentrated in the inner leaflet of membranes and plays an important role in the membrane fusion mechanism and vesicle formation (42). For that reason, PE is associated with a wide variety of biological functions including cell division, growth, reproduction, and motility (43,44). Because the previous experiments suggested that TAT interacts with lipid headgroups and not with the hydrophobic core, we investigated how the differences between PC and PE could induce changes in peptide mobility. It was not possible to generate vesicles by electroformation from pure PE. Therefore, we prepared mixtures of PC and PE. The prepared GUVs contained 10, 20, or 30 mol % PE and 20 mol % of cholesterol (Table 1).
Diffusion of single lipid analogs in model membranes containing PE
The analysis of lipid tracer mobility in PE containing GUVs was done similar to the experiments presented above. We examined two different lipid compositions, DPPC/DPPE/cholesterol and DOPC/DOPE/cholesterol, both with 30% of PE. Despite the fact that the hydrophobic core was identical to the GUVs containing PC and PS examined above, now we observed significantly lower diffusion coefficients, namely 0.41 ± 0.02 and 0.63 ± 0.02 μm2/s, respectively (Fig. 3). Obviously, the negative curvature of the phosphoethanolamine caused a distortion of the membrane topology resulting in a significant obstruction of lipid mobility.
Figure 3.
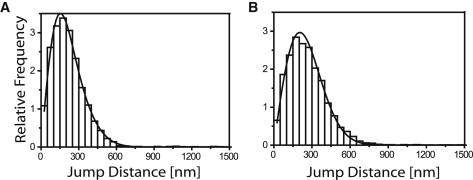
Mobility of lipid tracers on model membranes containing PE Jump distance analysis for TR-DHPE in GUV membranes containing 30 mol % PE: (A) DPPC/DPPE; (B) DOPC/DOPE.
Confocal imaging of TAT peptide-GUV interaction
The DOPC/DOPE/cholesterol respectively the DPPC/DPPE/cholesterol GUVs were incubated with 2 μM AF-TAT and fluorescein labeled 40 kDa dextran containing glucose solution. GUVs with molar fractions of PE from 10 mol % to 30 mol % were examined. For 10% of PE we observed no visible peptide translocation, just accumulation on the membrane. For 20 and 30% PE a clear TAT peptide translocation was observed after 30 min, which was related to the amount of PE contained in the membrane (Fig. 4, A–C). It appeared in the data shown in the upper panel as if the peptide reached a higher concentration within the GUV than outside, what is obviously against thermodynamic principles. Control experiments showed that this was due to the different sugar inside the GUVs than outside. The presence of sucrose instead of glucose in the medium results in an 80% increase in fluorescence intensity of rhodamine and AF-647. The sugar-dependent fluorescence of dyes such as FITC or TRITC can even be used to quantify glucose concentrations (45).
Figure 4.
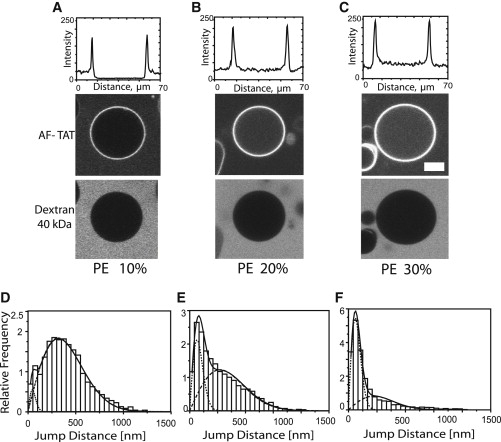
Interaction of TAT peptides with model membranes containing PE Confocal imaging of AF-TAT peptide interaction with GUV membranes comprising (A) 10 mol % PE, (B) 20 mol % PE, and (C) 30 mol % PE. The upper plots show the intensity of TAT peptide fluorescence along the central line of the images below. Internalization of TAT peptides can clearly be seen in B and C. All experiments were done in the presence of 40 kDa FITC-dextran outside the GUVs (lower panel). Observation time was 30 min after peptide addition. The lower histograms show the analysis of AF-TAT diffusion on the respective GUVs with (D) 10% PE, (E) 20%PE, and (F) 30% PS. Scale bar, 10 μm.
To gain a deeper insight of the translocation mode, similar leakage experiments as in the case of GUVs containing PS were carried out (Fig. 5). On incubation with 2 μM AF-TAT AlexaFluor 647 dye molecules were completely released from GUVs containing 30 mol % PE within 20 min, however, both fluorescent dextrans, 3 kDa and 70 kDa, did not pass the respective GUV membranes. Thus we confirmed the existence of TAT-induced pores in GUVs. In addition, we found that TAT peptides induce differently sized pores in PS and PE containing membranes, because the fluorescent 3 kDa dextran was released from PS containing but not from PE containing GUVs.
Figure 5.
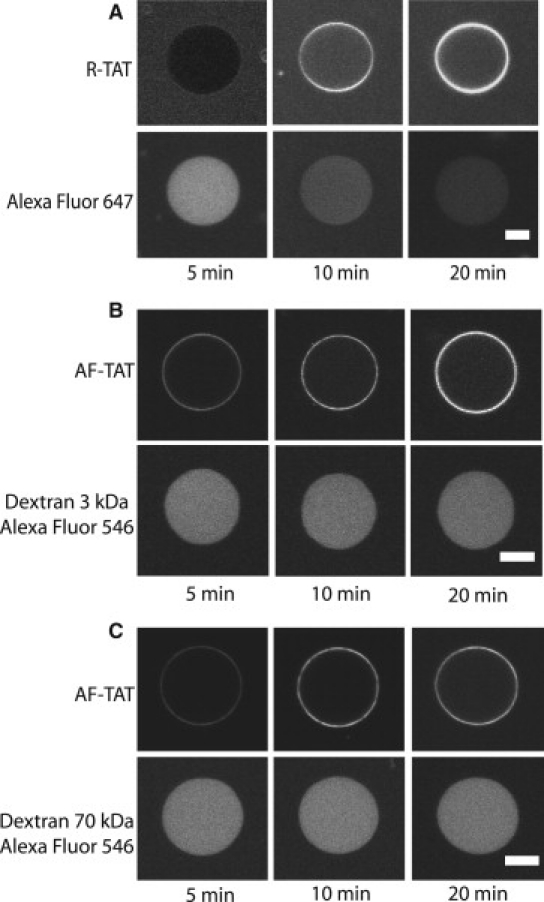
Pore formation of TAT peptides in membranes containing 30 mol % PE. (A) Upper panel shows the internalization of R-TAT; lower panel shows the rapid efflux of AlexaFluor 647 from GUVs comprising 30 mol % PE. (B) Upper panel, internalization of R-TAT; lower panel, no efflux of AlexaFluor 546 labeled 3 kDa dextran molecules was detected. (C) Upper panel, internalization of R-TAT; lower panel, no efflux of AlexaFluor 546 labeled 70 kDa dextran molecules was detected. Scale bar, 20 μm.
Lateral mobility of single TATs on the GUV surface
As above SPT experiments of fluorescently labeled single TAT peptides were carried out on GUVs containing up to 30 mol % PE after an incubation with 0.25 nM AF-TAT in glucose for 30 min. Single AF-TAT trajectories were recorded, and the mobility characteristics were quantified by means of the jump distance histograms (Fig. 4, D–F). For 10% PE, where no peptide translocation occurred, the mobility was similar to that on neutral GUVs, suggesting alike to this case, that the peptide was not incorporated into but rather floating on the membrane. Notably, for higher PE molar ratios, where a distinct TAT translocation was observed by confocal microscopy, the jump distance histograms were clearly bimodal, and two diffusion coefficients were required for a satisfactory fit of the data. The higher coefficient, D1 = 5.2 ± 0.1 μm2/s, was similar to the values, which were determined for TAT peptides floating on the membranes. Suspiciously, the second coefficient, D2 = 0.3 ± 0.1 μm2/s, was even lower than the lipid mobility (Table 2). For GUVs with 30% PE, the diffusion coefficients were even more reduced. The occurrence of two distinct mobility coefficients indicated that TAT peptides first attach to the membrane in relatively loose manner as observed above, presumably due to the partially negative charge of PE, and then form particular, large molecular aggregates on or within the membranes, which comprise several lipid molecules.
Discussion
TAT peptides are of great interest due to their efficiency in cell penetration and cargo delivery and their potential as drug-delivery candidates (17,46). In this study we examined the interaction of TAT peptides with model membrane systems with defined charge and specific geometrical properties. We discovered that TAT peptides are not only capable of directly penetrating such membranes in a passive manner, but they can also form physical pores, which could be passed by tracer molecules.
The composition of mammalian cell membranes was taken as a reference for choosing the lipids, which were used to form the GUVs. Biological membranes are electrically charged, but the fraction of charged phospholipids is only ∼10 mol %. Phospholipids with an ethanolamine headgroup occur frequently in natural membranes with high mol % (47). Focusing on the effect of charged lipids and lipids with a PE headgroup we kept our model systems as simple as possible. Because of this simplification, we expect that our findings to show basic principles of peptide-membrane interaction.
To obtain a reference we first analyzed the mobility of a lipid tracer molecule, TR-DHPE, within the membranes of GUVs made from lipid mixtures based on PC, PS, and PE by high-speed and high-sensitivity single molecule tracking. We found that the mobility of the tracer in neutral membranes is 0.56 and 2.2 μm2/s for PC-based liquid-ordered and liquid-disordered phases, respectively. The diffusion coefficients did not differ in anionic membranes containing up to 30 mol % PS for the same hydrophobic chains verifying that the anionic charge of the headgroup does not significantly modify lipid mobility. In contrast, in membranes containing PE and PC the tracer molecules had lower diffusion coefficients, although the used lipids contained the same hydrophobic chain. For liquid-ordered membranes the mobility was as low as 0.41 μm2/s and for unordered membranes it was reduced to 0.63 μm2/s. Obviously, PE introduced severe distortions of the membrane structure (48). PE has a relatively small headgroup, which results in a significantly lower area per lipid leading to a high order of the hydrocarbon lipid tails and corresponding low lipid mobility. Furthermore, comparative studies of PE and PC using 2H-NMR showed that PE molecules tend to form inter- and intramolecular hydrogen bonds, including association with other lipid types (49). The amine group as a hydrogen-donor can strongly interact with other phosphate/carbonyl groups or water, which function as hydrogen-acceptors. Probably, these strong intermolecular interactions cause a further reduction of lipid mobility.
To analyze the general features of TAT peptide interaction with the chosen model membrane systems we incubated preformed GUVs with red-fluorescent TAT peptides in glucose solution. The distribution of TAT peptides outside, on the surface and potentially inside the GUVs was monitored by confocal laser scanning microscopy. Generally, 40 kDa green-fluorescent dextran molecules were used to check GUV integrity.
TAT peptide fluorescence increased with time on the surface of GUVs made from neutral lipids, but we did not detect any fluorescence within the vesicles. Hence, TAT peptides were not able to traverse the membrane at concentrations up to 2 μM under the experimental conditions used. Imaging of single TAT peptide molecules on the surface of the GUVs showed that the peptide mobility was significantly higher than that of the lipids and did not depend on the lipid phase confirming our previous result that TAT peptides are not inserted but rather floating on the membrane (25).
When the molar ratio of the anionic lipid PS in the model membrane was gradually increased TAT peptide association to the membrane became noticeably enhanced. At a mole fraction of 40% PS in the model membrane TAT peptides traversed the GUV membranes. Often, the GUVs became so unstable that they disrupted and released their content. If TAT peptides can induce pores into GUV membranes, then small molecules should be able to flow across the membranes. To test this possibility, we carried out efflux experiments with small fluorescent molecules. Indeed, GUVs comprising 40% PS in their membrane and containing the dye Alexa 647 released these 1.3 kDa tracer molecules within 20 min after peptide addition. A fluorescent 3 kDa dextran tracer was released from GUVs within 30 min of incubation with 2 μM TAT. In contrast to these small molecules, 70 kDa dextran molecules remained within the GUVs.
Cationic TAT peptides interacted strongly with anionic GUVs. Simultaneously, the mobility of the peptides was slowed down compared to neutral membranes. However, this effect was not dependent on the mole fraction of PS present in the membrane. Even at the highest anionic charge concentration examined—with ongoing peptide translocation—we did not observe a further reduction of TAT mobility. We concluded that the TAT peptides first accumulate on the membrane due to the electrostatic interactions. At a certain concentration pores were formed, which allowed peptides and tracers to pass. The life time of the pores must be extremely short, because they were not observable by single molecule tracking. These results for TAT peptides corroborated recent experiments and computer simulations carried out by Herce and Garcia (14) and Herce et al. (16) for nona-arginine and TAT. These researchers showed the flow of ions across and thus the presence of pores within a black lipid membrane system in the presence of the peptides.
To focus on the aspect of membrane charge the above experiments were carried out in solutions without ions. In physiological solution we observed a strong reduction of the association of TAT and GUV membranes, probably because the positive charges on the TAT peptide were shielded by counterions in the buffer. Interestingly, TAT peptide translocation and tracer efflux occurred although either no or only a small percentage of anionic lipids were present in the membrane. Therefore, in physiological buffer TAT peptides had a significantly increased capability to form pores in GUV membranes.
Because we could induce pores in GUV membranes by charging the membrane, we became interested in the effect of topologic distortions of the membrane bilayer. To this end we carried out analogous experiments with GUVs containing PE lipids, which induce locally a negative membrane curvature. Indeed, confocal microscopy showed a distinct association of TAT peptides with PE/PC membranes and a rapid TAT peptide translocation into the GUV interior at a mole fraction of 20 mol % PE. Further increasing the PE mole fraction to 30 mol % yielded a higher translocation rate. We did not, however, observe any GUV destruction. Again, small tracer molecules were released from the GUVs, but neither 3 nor 70 kDa dextrans.
Most interestingly, single molecule tracking of fluorescent TAT peptides on the surface of PE containing GUVs revealed two distinct mobile fractions. One fraction had a largely reduced mobility, 0.3 μm2/s, and the other mobile fraction showed the mobility of TAT floating on the membrane with D = 5.2 μm2/s. The portion of the slow mobility component increased by a factor of 3 when the PE mole fraction in the membrane was increased from 10 to 30%. Suspiciously, this slow fraction was even less mobile than the lipids themselves in these membranes (D = 0.4–0.6 μm2/s). This slow diffusion coefficient indicated the presence of large molecular complexes comprising TAT peptides and lipids. The PE headgroup is small and carries a partial charge, and presumably TAT peptide can associate tightly to these headgroups thus forming molecular aggregates, which certainly include several PE lipids. At higher peptide concentration these distortions might eventually lead to the formation of pores, as was observed in the efflux experiments. These experiments indicated that the pores had a diameter d with 1.3 nm ≤ d < 2 nm, because Alexa 647 but not the 3 kDa dextran could pass the pores.
We found that TAT peptides accumulated on highly anionic membranes and internalized very rapidly. In some cases TAT provoked membrane destabilization leading to vesicle disruption, an effect that might be assigned rather to a carpet-like lysis mechanism. Furthermore, we observed rapid translocation across membranes containing lipids, which induce a negative curvature. In both cases we revealed the presence of nanometer-sized pores because small, but not larger, tracer molecules could pass the membranes. We believe that TAT peptide translocation is clearly dependent on membrane charge and composition.
Supporting Material
Two movies and five figures are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(10)00438-8.
Supporting Material
Acknowledgments
This work was supported by the European Commission under the 6th Framework Programme through the Marie-Curie Action BIOCONTROL (contract MCRTN-33439) and the German Research Foundation through the Collaborative Research Center SFB 624 Templates-Functional Chemical Matrices.
References
- 1.Derossi D., Chassaing G., Prochiantz A. Trojan peptides: the penetratin system for intracellular delivery. Trends Cell Biol. 1998;8:84–87. [PubMed] [Google Scholar]
- 2.Lindgren M., Hällbrink M., Langel U. Cell-penetrating peptides. Trends Pharmacol. Sci. 2000;21:99–103. doi: 10.1016/s0165-6147(00)01447-4. [DOI] [PubMed] [Google Scholar]
- 3.Schwarze S.R., Hruska K.A., Dowdy S.F. Protein transduction: unrestricted delivery into all cells? Trends Cell Biol. 2000;10:290–295. doi: 10.1016/s0962-8924(00)01771-2. [DOI] [PubMed] [Google Scholar]
- 4.Fischer R., Fotin-Mleczek M., Brock R. Break on through to the other side-biophysics and cell biology shed light on cell-penetrating peptides. ChemBioChem. 2005;6:2126–2142. doi: 10.1002/cbic.200500044. [DOI] [PubMed] [Google Scholar]
- 5.Drin G., Cottin S., Temsamani J. Studies on the internalization mechanism of cationic cell-penetrating peptides. J. Biol. Chem. 2003;278:31192–31201. doi: 10.1074/jbc.M303938200. [DOI] [PubMed] [Google Scholar]
- 6.Fittipaldi A., Ferrari A., Giacca M. Cell membrane lipid rafts mediate caveolar endocytosis of HIV-1 Tat fusion proteins. J. Biol. Chem. 2003;278:34141–34149. doi: 10.1074/jbc.M303045200. [DOI] [PubMed] [Google Scholar]
- 7.Ferrari A., Pellegrini V., Beltram F. Caveolae-mediated internalization of extracellular HIV-1 tat fusion proteins visualized in real time. Mol. Ther. 2003;8:284–294. doi: 10.1016/s1525-0016(03)00122-9. [DOI] [PubMed] [Google Scholar]
- 8.Richard J.P., Melikov K., Lebleu B. Cell-penetrating peptides. A reevaluation of the mechanism of cellular uptake. J. Biol. Chem. 2003;278:585–590. doi: 10.1074/jbc.M209548200. [DOI] [PubMed] [Google Scholar]
- 9.Kaplan I.M., Wadia J.S., Dowdy S.F. Cationic TAT peptide transduction domain enters cells by macropinocytosis. J. Control. Release. 2005;102:247–253. doi: 10.1016/j.jconrel.2004.10.018. [DOI] [PubMed] [Google Scholar]
- 10.Khalil I.A., Kogure K., Harashima H. High density of octa arginine stimulates macropinocytosis leading to efficient intracellular trafficking for gene expression. J. Biol. Chem. 2006;281:3544–3551. doi: 10.1074/jbc.M503202200. [DOI] [PubMed] [Google Scholar]
- 11.Duchardt F., Fotin-Mleczek M., Brock R. A comprehensive model for the cellular uptake of cationic cell-penetrating peptides. Traffic. 2007;8:848–866. doi: 10.1111/j.1600-0854.2007.00572.x. [DOI] [PubMed] [Google Scholar]
- 12.Patel L.N., Zaro J.L., Shen W.C. Cell penetrating peptides: intracellular pathways and pharmaceutical perspectives. Pharm. Res. 2007;24:1977–1992. doi: 10.1007/s11095-007-9303-7. [DOI] [PubMed] [Google Scholar]
- 13.Prochiantz A. Messenger proteins: homeoproteins, TAT and others. Curr. Opin. Cell Biol. 2000;12:400–406. doi: 10.1016/s0955-0674(00)00108-3. [DOI] [PubMed] [Google Scholar]
- 14.Herce H.D., Garcia A.E. Molecular dynamics simulations suggest a mechanism for translocation of the HIV-1 TAT peptide across lipid membranes. Proc. Natl. Acad. Sci. USA. 2007;104:20805–20810. doi: 10.1073/pnas.0706574105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mishra A., Gordon V.D., Wong G.C. HIV TAT forms pores in membranes by inducing saddle-splay curvature: potential role of bidentate hydrogen bonding. Angew. Chem. Int. Ed. Engl. 2008;47:2986–2989. doi: 10.1002/anie.200704444. [DOI] [PubMed] [Google Scholar]
- 16.Herce H.D., Garcia A.E., Milesi V. Arginine-rich peptides destabilize the plasma membrane, consistent with a pore formation translocation mechanism of cell-penetrating peptides. Biophys. J. 2009;97:1917–1925. doi: 10.1016/j.bpj.2009.05.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tünnemann G., Martin R.M., Cardoso M.C. Cargo-dependent mode of uptake and bioavailability of TAT-containing proteins and peptides in living cells. FASEB J. 2006;20:1775–1784. doi: 10.1096/fj.05-5523com. [DOI] [PubMed] [Google Scholar]
- 18.Rothbard J.B., Kreider E., Wender P.A. Arginine-rich molecular transporters for drug delivery: role of backbone spacing in cellular uptake. J. Med. Chem. 2002;45:3612–3618. doi: 10.1021/jm0105676. [DOI] [PubMed] [Google Scholar]
- 19.Tünnemann G., Ter-Avetisyan G., Cardoso M.C. Live-cell analysis of cell penetration ability and toxicity of oligo-arginines. J. Pept. Sci. 2008;14:469–476. doi: 10.1002/psc.968. [DOI] [PubMed] [Google Scholar]
- 20.Torchilin V.P. Recent approaches to intracellular delivery of drugs and DNA and organelle targeting. Annu. Rev. Biomed. Eng. 2006;8:343–375. doi: 10.1146/annurev.bioeng.8.061505.095735. [DOI] [PubMed] [Google Scholar]
- 21.Berry C.C. Intracellular delivery of nanoparticles via the HIV-1 tat peptide. Nanomed. 2008;3:357–365. doi: 10.2217/17435889.3.3.357. [DOI] [PubMed] [Google Scholar]
- 22.Tiriveedhi V., Butko P. A fluorescence spectroscopy study on the interactions of the TAT-PTD peptide with model lipid membranes. Biochem. 2007;46:3888–3895. doi: 10.1021/bi602527t. [DOI] [PubMed] [Google Scholar]
- 23.Ziegler A., Blatter X.L., Seelig J. Protein transduction domains of HIV-1 and SIV TAT interact with charged lipid vesicles. Binding mechanism and thermodynamic analysis. Biochem. 2003;42:9185–9194. doi: 10.1021/bi0346805. [DOI] [PubMed] [Google Scholar]
- 24.Ziegler A., Seelig J. Binding and clustering of glycosaminoglycans: a common property of mono- and multivalent cell-penetrating compounds. Biophys. J. 2008;94:2142–2149. doi: 10.1529/biophysj.107.113472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ciobanasu C., Harms E., Kubitscheck U. Cell-penetrating HIV1 TAT peptides float on model lipid bilayers. Biochem. 2009;48:4728–4737. doi: 10.1021/bi900365s. [DOI] [PubMed] [Google Scholar]
- 26.Siebrasse J.P., Grünwald D., Kubitscheck U. Single-molecule tracking in eukaryotic cell nuclei. Anal. Bioanal. Chem. 2007;387:41–44. doi: 10.1007/s00216-006-0763-0. [DOI] [PubMed] [Google Scholar]
- 27.Saxton M.J., Jacobson K. Single-particle tracking: applications to membrane dynamics. Annu. Rev. Biophys. Biomol. Struct. 1997;26:373–399. doi: 10.1146/annurev.biophys.26.1.373. [DOI] [PubMed] [Google Scholar]
- 28.Schütz G.J., Sonnleitner M., Schindler H. Single molecule microscopy of biomembranes (review) Mol. Membr. Biol. 2000;17:17–29. doi: 10.1080/096876800294452. [DOI] [PubMed] [Google Scholar]
- 29.Dimitrov D.S., Angelova M.I. Lipid swelling and liposome formation mediated by electric fields. Bioelectrochem. Bioenerg. 1988;19:323–336. [Google Scholar]
- 30.Menger F.M., Keiper J.S. Chemistry and physics of giant vesicles as biomembrane models. Curr. Opin. Chem. Biol. 1998;2:726–732. doi: 10.1016/s1367-5931(98)80110-5. [DOI] [PubMed] [Google Scholar]
- 31.Abramoff M.D., Magelhaes P.J., Ram S.J. Image processing with ImageJ. Biophot. Int. 2004;11:36–42. [Google Scholar]
- 32.Crank J. Clarendon Press; Oxford: 1975. The Mathematics of Diffusion. [Google Scholar]
- 33.Hauser H.G.P. The Structure of Biological Membranes. In: Yeagle Ph. L., editor. CRC Press; Boca Raton: 2005. [Google Scholar]
- 34.Willumeit R., Kumpugdee M., Andrä J. Structural rearrangement of model membranes by the peptide antibiotic NK-2. Biochim. Biophys. Acta. 2005;1669:125–134. doi: 10.1016/j.bbamem.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 35.Scherfeld D., Kahya N., Schwille P. Lipid dynamics and domain formation in model membranes composed of ternary mixtures of unsaturated and saturated phosphatidylcholines and cholesterol. Biophys. J. 2003;85:3758–3768. doi: 10.1016/S0006-3495(03)74791-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schmidt T., Schütz G.J., Schindler H. Characterization of photophysics and mobility of single molecules in a fluid lipid membrane. J. Phys. Chem. 1995;99:17662–17668. [Google Scholar]
- 37.Murcia M.J., Garg S., Naumann C.A. Single-molecule fluorescence microscopy to determine phospholipid lateral diffusion. Methods Mol. Biol. 2007;400:277–294. doi: 10.1007/978-1-59745-519-0_19. [DOI] [PubMed] [Google Scholar]
- 38.Kues T., Dickmanns A., Kubitscheck U. High intranuclear mobility and dynamic clustering of the splicing factor U1 snRNP observed by single particle tracking. Proc. Natl. Acad. Sci. USA. 2001;98:12021–12026. doi: 10.1073/pnas.211250098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Persky B., Hendrix M.J. Artificial matrix barriers: a diffusion study utilizing dextrans and microspheres. Anat. Rec. 1990;228:15–22. doi: 10.1002/ar.1092280104. [DOI] [PubMed] [Google Scholar]
- 40.Yao Y., Lenhoff A.M. Pore size distributions of ion exchangers and relation to protein binding capacity. J. Chromatogr. 2006;1126:107–119. doi: 10.1016/j.chroma.2006.06.057. [DOI] [PubMed] [Google Scholar]
- 41.Ambroggio E.E., Separovic F., Bagatolli L.A. Direct visualization of membrane leakage induced by the antibiotic peptides: maculatin, citropin, and aurein. Biophys. J. 2005;89:1874–1881. doi: 10.1529/biophysj.105.066589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Birner R., Bürgermeister M., Daum G. Roles of phosphatidylethanolamine and of its several biosynthetic pathways in Saccharomyces cerevisiae. Mol. Biol. Cell. 2001;12:997–1007. doi: 10.1091/mbc.12.4.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Opekarová M., Robl I., Tanner W. Phosphatidyl ethanolamine is essential for targeting the arginine transporter Can1p to the plasma membrane of yeast. Biochim. Biophys. Acta. 2002;1564:9–13. doi: 10.1016/s0005-2736(02)00455-8. [DOI] [PubMed] [Google Scholar]
- 44.Kearns D.B., Robinson J., Shimkets L.J. Pseudomonas aeruginosa exhibits directed twitching motility up phosphatidylethanolamine gradients. J. Bacteriol. 2001;183:763–767. doi: 10.1128/JB.183.2.763-767.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chaudhary A., Raina M., Srivastava R. Evaluation of glucose sensitive affinity binding assay entrapped in fluorescent dissolved-core alginate microspheres. Biotechnol. Bioeng. 2009;104:1075–1085. doi: 10.1002/bit.22500. [DOI] [PubMed] [Google Scholar]
- 46.Stewart K.M., Horton K.L., Kelley S.O. Cell-penetrating peptides as delivery vehicles for biology and medicine. Org. Biomol. Chem. 2008;6:2242–2255. doi: 10.1039/b719950c. [DOI] [PubMed] [Google Scholar]
- 47.Alberts B., Johnson A., Walter P. Garland Science; New York: 2002. Molecular Biology of the Cell. [Google Scholar]
- 48.Leekumjorn S., Sum A.K. Molecular simulation study of structural and dynamic properties of mixed DPPC/DPPE bilayers. Biophys. J. 2006;90:3951–3965. doi: 10.1529/biophysj.105.076596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boggs J.M. Lipid intermolecular hydrogen bonding: influence on structural organization and membrane function. Biochim. Biophys. Acta. 1987;906:353–404. doi: 10.1016/0304-4157(87)90017-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.