

Abstract
Engagement of toll-like receptors serve to link innate immune responses with adaptive immunity and can be exploited as powerful vaccine adjuvants for eliciting both primary and anamnestic immune responses. TLR7 agonists are highly immunostimulatory without inducing dominant proinflammatory cytokine responses. A structure-activity study was conducted on the TLR7-agonistic imidazoquinolines, starting with 1-(4-amino-2-((ethylamino)methyl)-1H-imidazo[4,5-c]quinolin-1-yl)-2-methylpropan-2-ol as a lead. Modifications of the secondary amine of the C2 ethylaminomethylene sidechain are poorly tolerated. The 4-amino group must be retained for activity. Replacement of the imidazole ring of the scaffold with triazole or cyclic urea led to complete loss of activity. A systematic exploration of N1-benzyl-C2-alkyl substituents showed a very distinct relationship between alkyl length and TLR7-agonistic potency with the optimal compound bearing a C2-n-butyl group. Transposition of the N1 and C2 substituents led to the identification of an extremely active TLR7-agonistic compound with an EC50 value of 8.6 nM. The relative potencies in human TLR7-based primary reporter gene assays were paralleled by interferon-α induction activities in whole human blood models.
Keywords: Toll-like receptor, TLR7, Imidazoquinoline, Vaccine adjuvants, Interferon, Innate immunity
Introduction
Toll-like receptors (TLRs) are pattern recognition receptors that recognize specific molecular patterns present in molecules that are broadly shared by pathogens, but are structurally distinct from host molecules.1;2 There are 10 TLRs in the human genome.2 The ligands for these receptors are highly conserved microbial molecules such as lipopolysaccharides (LPS) (recognized by TLR4), lipopeptides (TLR2 in combination with TLR1 or TLR6), flagellin (TLR5), single stranded RNA (TLR7 and TLR8), double stranded RNA (TLR3), CpG motif-containing DNA (recognized by TLR9), and profilin present on uropathogenic bacteria (TLR 11).3;4 TLR1, -2, -4, -5, and -6 respond to extracellular stimuli, while TLR3, -7, -8 and -9 respond to intracytoplasmic PAMPs, being associated with the endolysosomal compartment.2
The activation of TLRs by their cognate ligands leads to activation of innate immune effector mechanisms, including the production of pro-inflammatory cytokines, and up-regulation of MHC molecules and co-stimulatory signals in antigen-presenting cells as well as activating natural killer (NK) cells. The consequence of activation of the innate immune system mobilizes and amplifies specific adaptive immune responses involving both T- and B-cell effector functions.5-7 Thus, TLR stimuli serve to link innate and adaptive immunity5 and can therefore be exploited as powerful adjuvants in eliciting both primary and anamnestic immune responses.
We have recently examined representative members of virtually the entire compendium of known TLR agonists in a series of hierarchical assays including primary TLR-reporter assays, secondary indices of immune activation such as cytokine induction and activation of lymphocytic subsets in whole human blood, and tertiary screens characterizing transcriptomal activation patterns with a view to identifying optimal immunostimulatory chemotypes.8 Of all the innate immune stimuli examined, we found that TLR7 agonists were extraordinarily immunostimulatory, stimulating virtually all subsets of lymphocytes, and yet without inducing dominant proinflammatory cytokine responses (unlike TLR4 -5 or -8 agonists, which were proinflammatory and therefore may exert systemic toxicity).8 We therefore became especially desirous of evaluating TLR7-active compounds as potential vaccine adjuvants.
Long before endosomal TLR7 was discovered to serve as the primary sensor for short, single-stranded, GU-rich RNA sequences (ssRNA), mainly of viral origin,9-11 a number of small molecules were synthesized and evaluated in the 1970s and '80s for antiviral activities owing to their pronounced Type I interferon (IFN-α and -β) inducing properties.12-16 Although the mechanisms of innate immune stimulation of several of these compounds (such as tilorone14 and bromopirone16) remain yet to be formally elucidated, members of the 1H-imidazo[4,5-c]quinolines were found to be good Type I IFN inducers in human cell-derived assays,17 and FDA approval was obtained in 1997 for Imiquimod (1) for the treatment of basal cell carcinoma and actinic keratosis.18 It was not until 2002, however, that the mechanistic basis of IFN induction by the imidazoquinolines was found to be a consequence of TLR7 engagement and activation.19 Other than the original landmark studies performed by investigators at 3M Pharmaceuticals,17 structure-activity relationships of the imidazoquinoline chemotype remains poorly explored, perhaps attributable in part to recent interest in the 8-hydroxy-adenine compounds as alternate TLR7-agonists,20-23 which appear to lack emetic side-effects observed in ferrets upon oral administration.22
An aspect of our recent work in this area8;24;25 focuses on developing adjuvants for transcutaneous vaccines (needle-free vaccine patches).26-28 Given that imiquimod is already approved for topical use, it was of particular interest not only to explore chemical space around the imidazoquinolines, but also to carefully ‘immunophenotype’ such compounds in human TLR7-based assays to verify that they did not have any TLR8-driven proinflammatory properties. One of our goals was also to learn from such structure-activity studies optimal positions on the scaffold that would tolerate the introduction of electrophilic or photoactivable labels for purposes of developing self-adjuvanting vaccine constructs29;30 by covalently modifying protein antigens. We report here the synthesis and evaluation of a focused library of variously substituted 1H-imidazo[4,5-c]quinolines, and the identification of a novel, pure TLR7 agonist whose potency is 250 times that of imiquimod.
Results and Discussion
As alluded to earlier, long before the fact that imidazoquinolines specifically engaged and activated TLR7 was known, it was recognized that these compounds were potently adjuvantic31-34 in addition to displaying anti-viral effects via the induction of IFN-α/β.35;36 In human blood stimulated ex vivo with TLR7 agonists, upregulation of the surface expression of costimulatory molecules such as CD40, CD80, and CD86 occurred in both CD11c- plasmacytoid DCs and CD11c+ myeloid DCs; furthermore, the Th1 stimulatory ability of both DC subsets was enhanced in response to TLR7 ligands.37 Because TLR7 agonists transmit a T-helper-like signal to antibody-producing B cells, it is a highly effective adjuvant even for synthetic peptides that lack T-cell epitopes, possibly even replacing the function of T-helper cells, and providing a potential T-cell-independent vaccination strategy.33
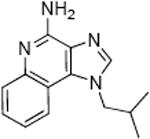
Our initial interest in exploring TLR7 agonists as vaccine adjuvants has been greatly reinforced by our recent observations that pure TLR7 agonists, unlike other TLR ligands, are potently immunostimulatory without activating inflammatory programs in human whole blood model systems.8 We elected to choose 1-(4-amino-2-((ethylamino)methyl)-1H-imidazo[4,5-c]quinolin-1-yl)-2-methylpropan-2-ol, 2 as our point of departure in examining structure-activity relationships in the imidazoquinolines, rather than imiquimod because we had earlier established that this analogue was more active than the parent compound, and was also verified to be a pure TLR7 agonist in human primary cells.8;24 We have recently reported the synthesis of 2.24 Furthermore, the only report on a detailed examination of SAR on the imidazoquinolines by the 3M group17 had used indirect bioassays (indirect assay for IFN-α using inhibition of virus-induced cytopathic effect) which predated the discovery and development of TLR7-specific assays. In the studies reported below, all compounds have been evaluated in human TLR7 and TLR8 reporter gene assays; highly active compounds were further examined for IFN-α induction in whole human blood using analyte-specific immunoassays.
Recent studies show that the TLR7 agonistic imidazoquinolines are indeed potently adjuvantic in vaccine constructs;38;39 however, the route of administration appears to be crucial since intradermal, but not subcutaneous administration of the vaccine adjuvanted with 2 is immunogenic and protective.38 Epidermal Langerhans cells as well as dermal dendritic cells (myeloid and plasmacytoid subsets)40 function as specialized professional antigen presenting cells in the skin.41;42 The plasmacytoid dendritic cell subset appears to be the target cell-type for TLR7 agonists.43 We hypothesized that the C2 ethylaminomethylene substituent of 2 renders the molecule sufficiently polar as to hinder transcutaneous penetration to effectively access and stimulate plasmacytoid dendritic cells. It was therefore desirable to evaluate more hydrophobic analogues of 2 as potential candidate transcutaneous vaccine adjuvants.
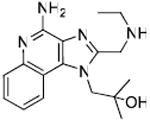
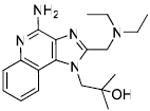
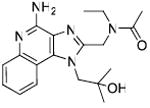
Unlike TLR2, TLR3, and TLR4 for which crystal structures are available as complexes with their cognate ligands,44 a detailed structural characterization of the mode of binding of the imidazoquinolines to TLR7 is not yet available. Therefore, our strategy in examining structure-activity relationships was focused rather on exploring chemical space systematically around the validated lead compound 2, toward realizing the two major objectives mentioned earlier. We began with derivatization of the secondary amine of the C2 ethylaminomethylene sidechain in an effort to examine whether this could be a potential site for introducing photoactivable or electrophilic functionalities. We found that modifications on this substituent are poorly tolerated, with only the N-ethyl analogue 3 (Scheme 1) retaining partial activity, and acyl (4, 5) and guanidine45 (6) derivatives being bereft of activity (Table 1). The loss of activity appeared not to be a consequence of altered basicity of the amine; surmising that it may, in part, be due either to the hydrophobicity of the substituent, or the conformal constraint imposed by the amide or guanidine, we asked if a polar spacer group, such as triethyleneglycol (11) may mitigate loss of activity, but we were proven wrong because 11 was also found to be completely inactive. Concurrent with the syntheses of the above compounds, we also synthesized the N-propyleneisothiocyanate derivative (14) via the N-propyleneamino (13) and N-ethylenenitrile (12) precursors. Compound 14 was not tested in reporter gene assays because we anticipated spurious results on account of its reactivity; the reaction of 14 with the spermine derivative (used as a model amine) afforded the expected thiourea adduct (16) which, disappointingly, was also inactive. These results conclusively showed that the secondary amine on the C2 substituent was not amenable to modifications. Replacement of the terminal ethyl group on the C2 substituent with an n-undecanyl group in 22 resulted in complete loss of activity (Table 1), signaling that the length of the C2 substituent was crucial; this was to become dramatically apparent when 32, with a pentyl substituent on C2 was evaluated later (see below).
Scheme 1.

Syntheses of 2-imidazolyl sidechain-modified analogues of 2.
Table 1.
EC50 values (μM) of compounds in human TLR7-specific reporter gene assay.
| Structure | Compound Number | TLR7-Agonistic Activity (μM) |
|---|---|---|
 |
1 | 2.12 |
 |
2 | 2.0 |
 |
3 | 7.71 |
 |
4 | ND |
 |
5 | ND |
 |
6 | ND |
 |
11 | ND |
 |
12 | 8.39 |
 |
13 | ND |
 |
16 | ND |
 |
22 | ND |
 |
28 | 0.955 |
 |
29 | 0.472 |
 |
30 | 0.647 |
 |
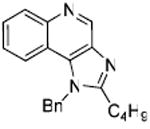
31 | 0.0591 |
 |
32 | 0.0915 |
 |
33 | 0.479 |
 |
34 | 0.822 |
 |
35 | 3.35 |
 |
36 | ND |
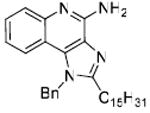 |
37 | ND |
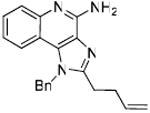 |
38 | 0.262 |
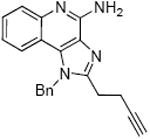 |
43 | 0.209 |
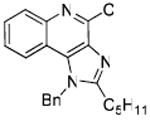 |
44 | ND |
 |
46 | ND |
 |
47 | ND |
 |
48 | 0.723 |
 |
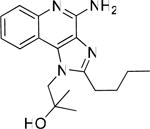
49 | 0.873 |
 |
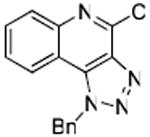
50 | ND |
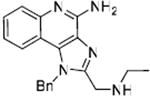 |
52 | 0.385 |
 |
54 | 0.00858 |
 |
55 | ND |
 |
56 | ND |
 |
57 | ND |
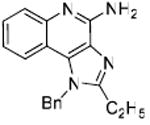
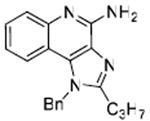
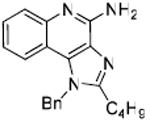
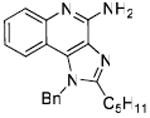
Desiring more lipophilic analogues which are expected to permeate the dermal barrier better, we sought to examine N1-benzyl analogues (Scheme 3) noting that similar substitutions have resulted in augmented activity in the 8-hydroxy-adenine series.20-23 Compound 32 was the first to be synthesized in this series which, to our pleasant surprise showed an activity twenty-fold greater than 2. Coupled with our observation that 22 was completely inactive, we sought to systematically explore SAR of the N1-benzyl C2 alkyl substituents. As depicted in Fig. 2, a very distinct relationship between alkyl length and TLR7-agonistic potency was observed, with the optimal compound being 31 (C2-n-butyl), with an EC50 of 59 nM (Table 1). n-Butylene (38) and n-butylyne (43) analogues showed decrease in potency. It is noteworthy that a C2-n-butyl, N1-phenyl analogue synthesized by the 3M group17 was not the most active; our results indicate that an N1-benzyl substituent is preferred.
Scheme 3.

Syntheses of 1-benzyl-2-(alkyl)-1H-imidazo[4,5-c]quinolin-4-amines.
Figure 2.

Alkyl chain length dependence of TLR7-agonistic activity profiles of the N1-benzyl-C2-alkyl compounds (right panel). The activity of 2 (reference compound) is shown on the left. Error bars represent standard deviations obtained on quadruplicate samples.
In recognition that bioactivity readouts using cell-culture systems may not always reflect with fidelity in vivo behavior owing to differential plasma protein binding (that we ourselves have observed and characterized),46 it was important to verify that the activity profiles observed in TLR7-specific reporter gene assays (Table 1) was also seen in whole blood with its full complement of plasma proteins. It was gratifying that we observed the expected 31≫2∼34 activity profile in whole blood IFN-α induction experiments (Fig. 3).
Figure 3.

Dose-dependent induction of IFN-α in whole human blood by 2, 31, and 34. Plasma IFN-α was assayed in triplicate by ELISA.
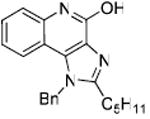
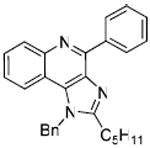
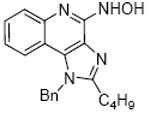
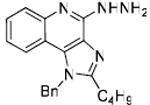
Bioisosteric replacements of the C4-NH2 group with -NMe, -OH and -SMe groups on the imiquimod scaffold have been explored earlier,17 and it was of interest to examine if the previously-described SAR would hold true in our analogues, and also to test whether the introduction of phenyl, -NHNH2 and -NHOH groups would alter TLR7 activity (Scheme 4). The 4-Cl precursor (44) and the desamino analogue 50, as well as the 4-OH (46) and 4-Ph (47) analogues were completely inactive, while the 4-NHOH (48) and 4-NHNH2 (49) compounds were substantially weaker (Table 1) than their 4-NH2 counterparts (31, 32). These results indicate the importance of the preservation of the NH2 group on C4 for maximal activity.
Scheme 4.

Syntheses of 4-substituted-1-benzyl-2-(alkyl)-1H-imidazo[4,5-c]quinolines.
Up to this point in our SAR studies, the most active and chemically distinct species were compound 2, and the N1-benzyl-C2-n-butyl analogue 31. We were curious to examine whether transposing the N1 and C2 substituents on these molecules would result in enhanced activity. Fortuitously, both the ‘hybrids’ (52, 54; Scheme 5) were very active. 54 was found to be extraordinarily potent with an EC50 of 8.6 nM (Fig. 4, Table 1).
Scheme 5.

Syntheses of C-2/N-1 substituent-swapped 31/2 hybrids.
Figure 4.

TLR7-agonistic activities of active compounds in a human TLR7-specific reporter gene assay. Error bars represent standard deviations obtained on quadruplicate samples.
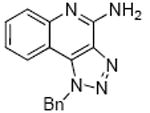
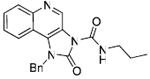
Finally, we attempted to modify the imidazole ring of the scaffold. The triazole compounds 55 and 56, as well as the cyclic urea 57 were completely inactive, which emphasizes the role of the recognition of the imidazole ring system by TLR7. Mention must also be made that these studies have been useful in delineating possible sites for introducing labeling functionalities. Preliminary studies (data not shown) show that the N1-benzyl substituent may be appropriate for introducing arylazido functional groups, while the quinoline ring may be amenable to introducing electrophilic groups. These results will be communicated in a future publication.
All of the C2-alkyl compounds reported herein were tested for TLR8 activity using the thiazoloquinoline CL075 (3M002) as a reference TLR8 agonist,47;48 and none had any appreciable activity in these assays. It should also be pointed out that while potencies of compounds are conventionally represented as EC50 (or IC50) values, that metric alone appears to be inadequate in rank-ordering the activities of the active compounds tested. An inspection of Fig. 4 indicates, for instance, that while the EC50 values of both imiquimod and 2 are comparable (∼ 2 μM), compound 2 induces a more robust response than imiquimod. Similarly, the area-under-curve estimates of 54 (analogous to combined ‘avidity’ and affinity measures of antigen-antibody interactions)49;50 indicate that consideration of the EC50 values alone may underestimate its potency.
Given that TLR7 is sequestered in an acidic endolysosomal compartment, and these compounds are weak bases and are therefore expected to accumulate in that acidic organelle, we wondered if the observed SAR could simply be a consequence of differential hydrophobicity of the compounds, modulating uptake into the cell and entry into the endolysosomal compartment activity, rather than governing specific molecular interactions with TLR7. A comparison of observed retention times on C18-reverse-phase mass-chromatograms (a measure of hydrophobicity)51;52 with the potencies of the active compounds indicate a distinct deviation from the trend of 54, suggesting specific and idiosyncratic effects, rather than bulk-effects (Fig. 5).
Figure 5.

Correlation of TLR7-agonistic potency and hydrophobicity (measured as retention time on C18 reverse-phase chromatography).
These structure-activity studies have been instructive and rewarding in that they not only complement the seminal work at 3M, but also have led to the discovery of highly-potent, lipophilic, human TLR7 agonist, 54. We note, not without irony, that this highly potent compound differs from its parent molecule by only a replacement of a secondary amine by a methylene unit. The oral bioavailability of 54 is predicted to be good, and it is possible that the systemic IFN-α inducing effects of this imidazoquinoline may find additional uses in the treatment of conditions such as hepatitis,53 chronic myelogenous54 and hairy cell leukemias.55 However, for purposes of developing dermal vaccine adjuvants, 31 may be more efficacious for reasons discussed earlier. Compounds 54 and 31 are being exhaustively immunoprofiled and characterized in a variety of assays.
Experimental Section
Chemistry
All of the solvents and reagents used were obtained commercially and used as such unless noted otherwise. Moisture- or air-sensitive reactions were conducted under nitrogen atmosphere in oven-dried (120 °C) glass apparatus. The solvents were removed under reduced pressure using standard rotary evaporators. Flash column chromatography was carried out using RediSep Rf ‘Gold’ high performance silica columns on CombiFlash Rf instrument unless otherwise mentioned, while thin-layer chromatography was carried out on silica gel CCM pre-coated aluminum sheets. Purity for all final compounds was confirmed to be greater than 97% by LC-MS using a Zorbax Eclipse Plus 4.6 mm × 150 mm, 5 μm analytical reverse phase C18 column with H2O-isopropanol or H2O-CH3CN gradients and an Agilent ESI-TOF mass spectrometer (mass accuracy of 3 ppm) operating in the positive ion (or negative ion, as appropriate) acquisition mode.
Synthesis of Compound 3: 1-(4-Amino-2-((diethylamino)methyl)-1H-imidazo[4,5-c]quinolin-1-yl)-2-methylpropan-2-ol
To a solution of trifluoroacetate salt of 2 (75 mg, 0.14 mmol) in anhydrous MeOH, were added acetaldehyde (6.6 mg, 0.15 mmol), 12-14 drops of acetic acid and sodium cyanoborohydride (10 mg, 0.15 mmol). After 4 hours the solvent was removed using vacuum and the residue was purified using column chromatography (7% MeOH/dichloromethane) to obtain the compound 3 (16 mg, 34%). 1H NMR (500 MHz, MeOD) δ 8.36 (d, J = 7.8 Hz, 1H), 7.73 (dd, J = 8.4, 1.0 Hz, 1H), 7.69 – 7.64 (m, 1H), 7.52 (ddd, J = 8.4, 7.2, 1.2 Hz, 1H), 4.83 (s, 2H), 4.75 – 4.63 (m, 2H), 3.42 (dd, J = 14.2, 7.0 Hz, 4H), 1.39 – 1.29 (m, 6H), 1.21 (dd, J = 13.5, 6.2 Hz, 6H). 13C NMR (126 MHz, MeOD) δ 150.85, 150.16, 138.41, 136.09, 131.52, 126.42, 126.18, 123.69, 120.11, 114.65, 72.54, 56.68, 49.70, 49.56, 27.65, 9.45. MS (ESI) calculated for C19H27N5O, m/z 341.22, found 342.24 (M + H)+.
Synthesis of Compound 4: N-((4-Amino-1-(2-hydroxy-2-methylpropyl)-1H-imidazo[4,5-c]quinolin-2-yl)methyl)-N-ethylacetamide
To a solution of trifluoroacetate salt of 2 (40 mg, 0.074 mmol) in anhydrous THF, were added triethlyamine (23 mg, 0.22 mmol) and acetyl chloride (6 mg, 0.08 mmol). After 30 minutes, the solvent was removed under vacuum. The residue was dissolved in EtOAc, washed with water and dried over Na2SO4. The EtOAc fraction was then concentrated under vacuum and the residue was purified using column chromatography (6% MeOH/dichloromethane) to obtain the compound 4 (22 mg, 85%). 1H NMR (400 MHz, MeOD) δ 8.34 (d, J = 8.3 Hz, 1H), 7.71 (d, J = 8.3 Hz, 1H), 7.53 (t, J = 7.7 Hz, 1H), 7.38 (t, J = 7.7 Hz, 1H), 5.23 (s, 2H), 4.81 (s, 2H), 3.63 (q, J = 7.1 Hz, 2H), 2.23 (s, 3H), 1.34 – 1.13 (m, 9H). 13C NMR (101 MHz, MeOD) δ 172.18, 154.62, 136.88, 134.07, 129.68, 129.48, 124.60, 122.64, 122.31, 118.21, 113.67, 70.85, 65.49, 55.12, 43.70, 41.79, 19.68, 14.03, 12.30. MS (ESI) calculated for C19H25N5O2, m/z 355.20, found 356.21 (M + H)+.
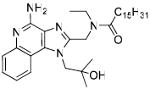
Synthesis of Compound 5: N-((4-Amino-1-(2-hydroxy-2-methylpropyl)-1H-imidazo[4,5-c]quinolin-2-yl)methyl)-N-ethylpalmitamide
To a solution of trifluoroacetate salt of 2 (20 mg, 0.05 mmol) in anhydrous THF, were added triethylamine (15 mg, 0.15 mmol) and palmitoyl chloride (16 mg, 0.06 mmol). After 30 minutes the solvent was removed under vacuum, residue was dissolved in EtOAc and washed with water. The EtOAc fraction was then dried under vacuum and the residue was purified using column chromatography (5% MeOH/dichloromethane) to obtain the compound 5 (10 mg, 30%). 1H NMR (400 MHz, CDCl3) δ 8.12 (d, J = 8.1 Hz, 1H), 7.88 (d, J = 8.3 Hz, 1H), 7.54 (t, J = 7.6 Hz, 1H), 7.38 (t, J = 7.8 Hz, 1H), 4.75 (s, 2H), 3.56 (q, J = 6.8 Hz, 2H), 2.45 – 2.34 (m, 2H), 1.76 – 1.57 (m, 2H), 1.58 – 1.11 (m, 35H), 0.90 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 192.82, 173.95, 152.03, 150.36, 135.45, 130.90, 128.29, 124.10, 123.24, 120.62, 114.81, 71.96, 55.58, 42.81, 42.30, 33.06, 31.92, 29.68, 29.65, 29.52, 29.47, 29.42, 29.35, 25.37, 22.69, 14.12, 13.70, 5.61. MS (ESI) calculated for C33H53N5O2, m/z 551.42, found 552.41 (M + H)+.
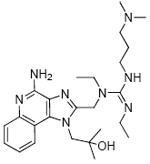
Synthesis of Compound 6: 1-((4-Amino-1-(2-hydroxy-2-methylpropyl)-1H-imidazo[4,5-c]quinolin-2-yl)methyl)-3-(3-(dimethylamino)propyl)-1,2-diethylguanidine
To a solution of trifluoroacetate salt of 2 (50mg, 0.092 mmol) in anhydrous DMF, were added triethylamine (51 mg, 0.51 mmol), EDCI.HCl (53 mg, 0.28 mmol) and a catalytic amount of DMAP. The reaction was stirred for 24-30 hours. The solvent was then removed under vacuum, and the residue was purified using C18 reverse-phase column chromatography to obtain the compound 6 (9 mg, 24%). 1H NMR (400 MHz, MeOD) δ 8.48 (d, J = 8.4 Hz, 1H), 7.84 (d, J = 7.4 Hz, 1H), 7.76 (t, J = 7.7 Hz, 1H), 7.63 (t, J = 7.2 Hz, 1H), 5.18 (s, 2H), 4.85 – 4.71 (m, 2H), 3.58 (q, J = 7.3 Hz, 2H), 3.54 – 3.41 (m, 4H), 3.27 – 3.18 (m, 2H), 2.90 (d, J = 3.7 Hz, 6H), 2.27 – 2.00 (m, 2H), 1.30 (dt, J = 14.4, 7.2 Hz, 12H). 13C NMR (101 MHz, MeOD) δ 160.10, 153.79, 137.08, 134.45, 129.86, 124.93, 122.19, 118.63, 113.38, 71.07, 55.30, 54.69, 46.25, 45.16, 42.06, 41.41, 39.78, 24.50, 13.79, 11.77. MS (ESI) calculated for C25H40N8O, m/z 468.33, found 469.33 (M + H)+ and 235.17 (M + 2H)2+.
Synthesis of Compound 9: tert-Butyl 2-(2-(2-hydroxyethoxy)ethoxy)ethylcarbamate
To a solution of 7 (500 mg, 2.1 mmol) in anhydrous dichloromethane, cooled to 0 °C, were added triethylamine (420 mg, 4.2 mmol), tosyl chloride (609 mg, 3.54 mmol) and a catalytic amount of DMAP. The reaction was stirred for 16 hours and then the mixture was washed with water and dried over Na2SO4. The solvent was then removed under vacuum. This residue O-tosylated compound was dissolved in anhydrous DMF, to which sodium azide (273 mg, 4.2 mmol) was added and the reaction was heated at 70 °C for 1 hour. The solvent was then removed under vacuum and the residue was dissolved in EtOAc, washed with water and dried over Na2SO4. The EtOAc fraction was concentrated under vacuum to obtain the compound 8 (600mg). This 8 was then dissolved in MeOH and di-tert-butyl dicarbonate (687 mg, 3.5 mmol) was added to it. The solution was subjected to catalytic hydrogenolysis using Pd(OH)2/C at 60 psi hydrogen pressure for 4 hours. The solution was then filtered through celite and filtrate was evaporated under reduced pressure to afford the residue, which was purified using column chromatography (3% MeOH/dichloromethane) to obtain the compound 9 (320 mg, 62%). 1H NMR (400 MHz, CDCl3) δ 5.13 (s, 1H), 3.77 (d, J = 3.5 Hz, 2H), 3.70 – 3.61 (m, 6H), 3.58 (t, J = 5.2 Hz, 2H), 3.34 (d, J = 4.6 Hz, 2H), 2.50 (s, 1H), 1.46 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 159.62, 79.34, 72.55, 70.43, 70.29, 61.78, 40.34, 28.41. MS (ESI) calculated for C11H23NO5, m/z 249.16, found 272.15 (M + Na+).
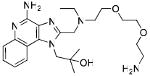
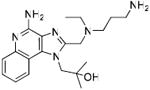
Synthesis of Compound 11: 1-(4-Amino-2-(((2-(2-(2-aminoethoxy)ethoxy)ethyl) (ethyl)amino)methyl)-1H-imidazo[4,5-c]quinolin-1-yl)-2-methylpropan-2-ol
To a solution of 9 (200mg, 0.8 mmol) in 4 ml of 0.3M Dess Martin periodinane solution in dichloromethane, 10-12 drops of acetic acid were added and the reaction was stirred for 4-5 hours. The solvent was then removed under vacuum and the residue was purified using column chromatography (40-50% EtOAc/dichloromethane) to obtain the intermediate aldehyde derivative 10 (71 mg). To a solution of trifluoroacetate salt of 2 (150 mg, 0.29 mmol) in anhydrous MeOH, were added 10 (71 mg, 0.29 mmol), 6-8 drops of acetic acid, and sodium cyanoborohydride (22 mg, 0.35 mmol). The solution was stirred for 14-15 hours and the solvent was removed under vacuum to obtain the residue, which was purified using column chromatography (10% MeOH/dichloromethane) to obtain the intermediate N-Boc derivative. This was dissolved in 5 ml of trifluoroacetic acid and stirred for 30 minutes, followed by removal of the solvent by purging nitrogen and drying under vacuum to obtain the trifluoroacetate salt of the compound 11 (110 mg, 60%). 1H NMR (400 MHz, MeOD) δ 8.49 (d, J = 8.4 Hz, 1H), 7.84 (d, J = 7.4 Hz, 1H), 7.76 (t, J = 7.3 Hz, 1H), 7.63 (t, J = 7.2 Hz, 1H), 4.82 (s, 4H), 3.88 (t, J = 5.1 Hz, 2H), 3.70 (d, J = 6.5 Hz, 6H), 3.50 (s, 2H), 3.38 (s, 2H), 3.17 – 3.09 (m, 2H), 1.55 – 1.10 (m, 9H). 13C NMR (101 MHz, MeOD) δ 149.53, 148.21, 136.86, 134.61, 130.12, 122.23, 118.64, 113.15, 71.15, 69.99, 69.93, 66.50, 64.83, 55.36, 52.60, 49.74, 48.69, 46.46, 39.10, 26.24, 7.96. MS (ESI) calculated for C23H36N6O3, m/z 444.29, found 467.27 (M + Na+) and 223.15 (M + 2H)2+.
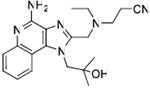
Synthesis of Compound 12: 3-(((4-Amino-1-(2-hydroxy-2-methylpropyl)-1H-imidazo[4,5-c]quinolin-2-yl)methyl)(ethyl)amino)propanenitrile
To a solution of 2 (250 mg, 0.46 mmol) in anhydrous MeOH, were added triethylamine (117 mg, 1.16 mmol) and acrylonitrile (61 mg, 1.16 mmol). The reaction was stirred for 30 hours, followed by removal of the solvent under vacuum. The residue was dissolved in EtOAc, washed with water and dried over Na2SO4. The EtOAc fraction was evaporated under vacuum and the residue was purified using column chromatography (11% MeOH/dichloromethane) to obtain the compound 12 (145 mg, 86%). 1H NMR (400 MHz, CDCl3) δ 8.06 (d, J = 8.4 Hz, 1H), 7.87 (d, J = 8.3 Hz, 1H), 7.55 (t, J = 7.6 Hz, 1H), 7.36 (t, J = 7.7 Hz, 1H), 6.04 (s, 2H), 4.88 (s, 2H), 4.12 (s, 2H), 2.94 (t, J = 6.7 Hz, 2H), 2.75 (q, J = 7.0 Hz, 2H), 2.54 (t, J = 6.7 Hz, 2H), 1.35 (s, 6H), 1.19 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 150.98, 150.41, 135.44, 127.81, 126.16, 122.48, 122.40, 122.18, 120.20, 118.64, 115.30, 71.49, 56.48, 50.85, 47.92, 47.64, 27.99, 15.33, 11.12. MS (ESI) calculated for C20H26N6O, m/z 366.22, found 367.22 (M + H)+.
Synthesis of Compound 13: 1-(4-Amino-2-(((3-aminopropyl)(ethyl)amino)methyl)-1H-imidazo[4,5-c]quinolin-1-yl)-2-methylpropan-2-ol
To a solution of 12 (160 mg, 0.44 mmol) in glacial acetic acid, a catalytic amount of Pd(OH)2/C was added and the reaction mixture was subjected to hydrogenation at 55 psi hydrogen pressure for 4 hours. The solution was then filtered. The filtrate was concentrated to obtain the acetate salt of the compound 13 (55 mg). This residue (20 mg, 0.05 mmol) was then dissolved in MeOH and di-tert-butyl dicarbonate (18 mg, 0.08 mmol) was added. After 30 minutes, the solvent was removed under vacuum and the residue was purified using column chromatography (15% MeOH/dichloromethane) to obtain the intermediate N-Boc derivative. This was then dissolved in 2 ml of HCl solution in dioxane and stirred for 2 hours. The solvent was then removed under vacuum to obtain the hydrochloride salt of the compound 13 (15 mg, 74%). 1H NMR (400 MHz, MeOD) δ 8.51 (d, J = 8.2 Hz, 1H), 7.85 (d, J = 8.3 Hz, 1H), 7.78 (t, J = 7.9 Hz, 1H), 7.65 (t, J = 7.7 Hz, 1H), 5.09 (s, 2H), 3.74 – 3.50 (m, 6H), 3.12 (d, J = 7.0 Hz, 2H), 2.32 (s, 2H), 1.51 (t, J = 6.9 Hz, 3H), 1.35 (s, 6H). 13C NMR (126 MHz, MeOD) δ 156.18, 151.90, 140.30, 137.56, 129.79, 126.47, 124.63, 123.13, 123.00, 72.46, 56.49, 53.02, 52.27, 48.77, 40.46, 27.84, 24.89, 11.74. MS (ESI) calculated for C20H30N6O, m/z 370.25, found 371.26 (M + H)+.
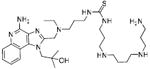
Synthesis of Compound 16: 1-(3-(((4-amino-1-(2-hydroxy-2-methylpropyl)-1H-imidazo[4,5-c]quinolin-2-yl)methyl)(ethyl)amino)propyl)-3-(3-(4-(3-aminopropylamino)butylamino)propyl) thiourea
To a solution of 13 (35 mg, 0.07 mmol) in anhydrous ethanol, were added triethylamine (22 mg, 0.21 mmol) and CS2 (54 mg, 0.71 mmol). The reaction mixture was stirred for 30 minutes and then cooled to 0 °C. Di-tert-butyl dicarbonate (15 mg, 0.07 mmol) and a catalytic amount of DMAP dissolved in ethanol were added to the reaction mixture. After 30 minutes, the solvent was removed under vacuum and the residue was purified using column chromatography to obtain the compound 15. To a solution of 15 (10mg, 0.02 mmol) in anhydrous dichloromethane, N1,N5,N10-tris boc spermine (37 mg, 0.073 mmol) was added. The reaction mixture was stirred for 3 hours. The solvent was then removed under vacuum and the residue was purified using column chromatography (6% MeOH/dichloromethane) to obtain the intermediate N-Boc derivative. This was dissolved in 5 ml of trifluoroacetic acid and stirred for 30 minutes, followed by removal of the solvent by purging nitrogen and drying under vacuum to obtain the trifluoroacetate salt of the compound 16 (5 mg, 20%). 1H NMR (500 MHz, MeOD) δ 8.45 (d, J = 8.5 Hz, 1H), 7.83 (d, J = 8.4 Hz, 1H), 7.76 (t, J = 7.8 Hz, 1H), 7.67 – 7.56 (m, 1H), 4.96 (d, J = 6.9 Hz, 2H), 4.81 – 4.70 (m, 2H), 3.77 – 3.38 (m, 8H), 3.13 (dd, J = 18.0, 10.2 Hz, 2H), 3.10 – 2.96 (m, 8H), 2.16 – 2.04 (m, 4H), 1.97 – 1.88 (m, 2H), 1.84 – 1.74 (m, 4H), 1.44 (t, J = 7.2 Hz, 3H), 1.29 (s, 6H). 13C NMR (126 MHz, MeOD) δ 150.81, 138.54, 138.41, 135.99, 131.60, 126.50, 123.71, 120.06, 114.65, 72.55, 56.76, 52.24, 50.71, 49.86, 49.68, 48.26, 48.06, 46.25, 45.87, 37.80, 27.48, 25.39, 24.29, 9.47. MS (ESI) calculated for C31H54N10OS, m/z 614.42, found 615.43 (M + H)+ and 308.22 (M + 2H)2+.
Synthesis of Compound 19: 2-(tert-Butoxycarbonyl(undecyl)amino)acetic acid.24
To a solution of 17 (200 mg, 1.6 mmol) in anhydrous MeOH, were added undecanal (218 mg, 1.28 mmol), 5-6 drops of acetic acid and sodium cyanoborohydride (111 mg, 1.76 mmol). The reaction was stirred for 3 hours and then the solvent was removed under vacuum. The residue was dissolved in dichloromethane and washed with water, brine and dried over Na2SO4. The dichloromethane fraction was then concentrated to obtain the residue 18, which was dissolved in MeOH, followed by the addition of triethylamine (202 mg, 2 mmol) and di-tert-butyl dicarbonate (436 mg, 2 mmol). The reaction was stirred for 4 hours and the solvent was removed under vacuum. The residue was then dissolved in a solvent mixture of THF:MeOH (3:1) and lithium hydroxide that was dissolved in water was added until the pH went above 12. The reaction was stirred for 24 hours, followed by removal of the solvent under vacuum. The residue was dissolved in water and the solution was acidified using 10% HCl until pH was less than 2. The water fraction was then extracted with EtOAc, followed by washing the EtOAc fraction with water and drying over Na2SO4. The EtOAc fraction was then concentrated under vacuum to obtain the compound 19 (320 mg, 61%). MS (ESI) calculated for C18H35NO4, m/z 329.26, found 328.25 (M - H)-.
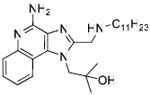
Synthesis of Compound 22: 1-(4-Amino-2-((undecylamino)methyl)-1H-imidazo[4,5-c]quinolin-1-yl)-2-methylpropan-2-ol
To a solution of 19 (171 mg, 0.52 mmol) in anhydrous DMF, were added HATU (217 mg, 0.57 mmol), triethylamine (58 mg, 0.57 mmol), a catalytic amount of DMAP and 20 (120 mg, 0.52 mmol). The reaction mixture was stirred for 3-4 hours. The solvent was evaporated under vacuum. The residue was dissolved in EtOAc, washed with water and dried over Na2SO4 to obtain the residue. This was dissolved in 10 mL of ethanol and a solution of NaOH(105 mg, 2.62 mmol) dissolved in 1 mL of water was added to it. The reaction mixture was refluxed for 5-6 hours, followed by evaporation under reduced pressure. The residue was purified by column chromatography (5% MeOH/dichloromethane) to obtain the compound 21 (165 mg). To a solution of 21 in a solvent mixture of MeOH:dichloromethane:chloroform (0.1:1:1), 3-chloroperoxybenzoic acid (135 mg, 0.79 mmol) was added and the solution was refluxed at 45-50 °C for 40 minutes. The solvent was then removed and the residue was purified using column chromatography (7% MeOH/dichloromethane) to obtain the N-oxide derivative (75 mg). This was then dissolved in anhydrous dichloromethane, followed by the addition of benzoyl isocyanate (37 mg, 0.25 mmol) and heated at 45 °C for 15 minutes. The solvent was then removed under vacuum and the residue was dissolved in anhydrous MeOH, followed by the addition of excess sodium methoxide and heating at 80 °C for 1 hour. The solvent was then removed under vacuum and the residue was purified using column chromatography (8% MeOH/dichloromethane) to obtain the intermediate N-Boc derivative (27 mg). This was dissolved in 5 ml of trifluoroacetic acid and stirred for 30 minutes, followed by removal of the solvent by purging nitrogen and drying under vacuum to obtain the trifluoroacetate salt of the compound 22 (33 mg, 12%). 1H NMR (400 MHz, CDCl3) δ 14.65 (s, 1H), 10.11 (s, 2H), 8.06 (s, 1H), 7.99 (d, J = 8.3 Hz, 1H), 7.44 (t, J = 7.6 Hz, 1H), 7.37 (t, J = 7.8 Hz, 1H), 7.20 (d, J = 8.6 Hz, 1H), 4.76 (s, 2H), 4.61 (s, 2H), 3.23 (s, 2H), 1.93 (s, 2H), 1.50 – 1.11 (m, 22H), 0.91 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 149.04, 148.91, 136.14, 134.18, 129.98, 124.99, 124.25, 120.76, 119.24, 112.18, 72.07, 49.07, 44.01, 31.89, 29.55, 29.49, 29.34, 29.31, 28.98, 26.58, 25.46, 22.67, 14.10, 5.61. MS (ESI) calculated for C26H41N5O, m/z 439.33, found 440.33 (M + H)+ and 220.67 (M + 2H)2+.
Synthesis of Compound 24: 3-Nitroquinoline-2,4-diol
Compound 23 (5 g, 31 mmol) was dissolved in 30 mL of nitric acid and stirred at room temperature for 10 minutes, followed by heating the reaction mixture at 75 °C for another 15 minutes. The reaction mixture was then allowed to cool down to room temperature and was added to ice-water mixture to precipitate the product. The solid, yellow precipitate was filtered and dried to obtain the compound 24 (5.9 g, 92%). 1H NMR (400 MHz, DMSO) δ 12.00 (s, 1H), 8.04 (d, J = 7.9 Hz, 1H), 7.66 (t, J = 7.5 Hz, 1H), 7.34 (d, J = 8.2 Hz, 1H), 7.28 (t, J = 7.7 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 156.69, 156.23, 138.56, 133.61, 127.71, 124.92, 122.81, 116.32, 114.47. MS (ESI) calculated for C9H6N2O4, m/z 206.03, found 205.04 (M - H)-.
Synthesis of Compound 25: 2,4-Dichloro-3-nitroquinoline
Compound 24 (2 g, 9.7 mmol) was dissolved in 20 mL of phenylphosphonyl dichloride and heated at 135 °C for 3 hours. The reaction mixture was poured into ice-water and stirred vigorously to obtain the precipitate, which was filtered and dried to afford the compound 25 (2.24 g, 74%). 1H NMR (400 MHz, CDCl3) δ 8.30 (ddd, J = 8.4, 1.4, 0.6 Hz, 1H), 8.14 (ddd, J = 8.5, 1.2, 0.6 Hz, 1H), 7.97 (ddd, J = 8.5, 7.0, 1.4 Hz, 1H), 7.84 (ddd, J = 8.3, 7.0, 1.2 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 146.64, 139.73, 135.62, 133.44, 129.76, 129.37, 125.22, 124.46.
Synthesis of Compound 26: N-Benzyl-2-chloro-3-nitroquinolin-4-amine
To a solution of 25 (2.24 g, 9.26 mmol) in 30 mL of anhydrous dichloromethane, were added triethylamine (1.4 g, 13.9 mmol) and benzylamine (1.19 g, 11.1 mmol). The reaction mixture was refluxed at 45 °C for 30 minutes. The solvent was then evaporated under vacuum and water was added to the residue to obtain the precipitate. This was filtered, washed several times with water and dried to obtain the compound 26 (2.1 g, 73%). 1H NMR (400 MHz, DMSO) δ 8.54 (dd, J = 12.9, 7.3 Hz, 2H), 7.89 – 7.81 (m, 2H), 7.69 (td, J = 8.6, 5.0 Hz, 1H), 7.39 – 7.21 (m, 5H), 4.46 (d, J = 6.3 Hz, 2H). 13C NMR (101 MHz, DMSO) δ 145.79, 144.82, 141.51, 138.27, 132.70, 129.08, 128.94, 127.89, 127.40, 127.15, 126.96, 123.52, 120.12, 47.24. MS (ESI) calculated for C16H12ClN3O2, m/z 313.06, found 312.06 (M - H)-.
Synthesis of Compound 27: N4-Benzyl-2-chloroquinoline-3,4-diamine
To a solution of 26 (2.21g, 7.06 mmol) in 20 mL of EtOAc, were added a catalytic amount of Pt/C and Na2SO4. The reaction mixture was subjected to hydrogenation at 55 psi hydrogen pressure for 3 hours. The reaction mixture was then filtered through celite and the filtrate was evaporated under vacuum to obtain the compound 27 (1.8 g, 90%). 1H NMR (400 MHz, MeOD) δ 7.90 (d, J = 8.2 Hz, 1H), 7.70 (d, J = 8.4 Hz, 1H), 7.43 (t, J = 7.6 Hz, 1H), 7.38 – 7.31 (m, 3H), 7.25 (t, J = 7.5 Hz, 2H), 7.20 (d, J = 7.0 Hz, 1H), 4.51 (s, 2H). 13C NMR (101 MHz, MeOD) δ 141.54, 141.28, 139.93, 137.86, 128.45, 128.04, 127.44, 126.96, 126.80, 126.22, 125.31, 123.22, 121.40, 49.56. MS (ESI) calculated for C16H14ClN3, m/z 283.09, found 284.10 (M + H)+.
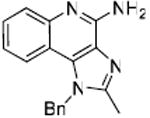
General Procedure for Synthesis of C-2 alkyl imidazoquioline compounds (28-38). Synthesis of Compound 32: 1-Benzyl-2-pentyl-1H-imidazo[4,5-c]quinolin-4-amine
To a solution of 27 (50 mg, 0.18 mmol) in anhydrous THF, were added triethylamine (26 mg, 0.19 mmol) and hexanoyl chloride. The reaction mixture was stirred at room temperature for 6 hours. The solvent was then removed under vacuum, and the residue was dissolved in ethyl acetate and washed with water. The ethyl acetate fraction was dried using Na2SO4 and evaporated under vacuum to obtain the intermediate amide compound. This was dissolved in 0.1 ml of 2M solution of ammonia in MeOH. The sealed reaction vessel was heated in microwave at 150 °C for 1 hour. The solvent was then removed under vacuum and the residue was purified using column chromatography (7% MeOH/dichloromethane) to obtain the compound 32 (8 mg; unoptimized yields). 1H NMR (500 MHz, CDCl3) δ 7.81 (d, J = 8.3 Hz, 1H), 7.72 (dd, J = 8.3, 1.0 Hz, 1H), 7.44 (qd, J = 7.0, 3.5 Hz, 1H), 7.38 – 7.29 (m, 3H), 7.18 – 7.10 (m, 1H), 7.06 (d, J = 6.9 Hz, 2H), 5.74 (s, 2H), 5.60 (s, 2H), 2.95 – 2.79 (m, 2H), 1.80 (dt, J = 15.6, 7.6 Hz, 2H), 1.42 – 1.27 (m, 4H), 0.87 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 153.50, 150.28, 134.61, 133.41, 128.61, 127.40, 126.51, 125.94, 125.85, 124.85, 121.69, 119.07, 114.42, 48.20, 30.85, 26.97, 26.77, 21.65, 13.23. MS (ESI) calculated for C22H24N4, m/z 344.20, found 345.21 (M + H)+. 2D 1H COSY and NOESY experiments (included in Supporting Information) provided unambiguous assignment of 1H resonances.
28: 1-Benzyl-2-methyl-1H-imidazo[4,5-c]quinolin-4-amine
1H NMR (500 MHz, CDCl3) δ 7.81 (dd, J = 8.4, 0.8 Hz, 1H), 7.75 (dd, J = 8.3, 1.0 Hz, 1H), 7.45 (ddd, J = 8.4, 7.1, 1.4 Hz, 1H), 7.38 – 7.28 (m, 3H), 7.15 (ddd, J = 8.3, 7.1, 1.2 Hz, 1H), 7.10 – 7.05 (m, 2H), 5.73 (s, 2H), 5.64 (s, 2H), 2.63 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 149.17, 148.91, 142.26, 133.27, 132.64, 127.74, 126.56, 125.74, 124.90, 124.82, 123.96, 120.90, 117.99, 113.37, 47.59, 12.20. MS (ESI) calculated for C18H16N4, m/z 288.14, found 289.15 (M + H)+.
29: 1-Benzyl-2-ethyl-1H-imidazo[4,5-c]quinolin-4-amine
1H NMR (500 MHz, CDCl3) δ 7.80 (dd, J = 8.4, 0.8 Hz, 1H), 7.73 (dd, J = 8.3, 1.0 Hz, 1H), 7.44 (qd, J = 7.2, 3.7 Hz, 1H), 7.37 – 7.28 (m, 3H), 7.13 (ddd, J = 8.3, 7.0, 1.3 Hz, 1H), 7.09 – 7.01 (m, 2H), 5.73 (s, 2H), 5.59 (s, 2H), 2.97 – 2.86 (m, 2H), 1.41 (t, J = 7.5 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 153.83, 149.91, 143.24, 134.14, 133.05, 128.19, 126.96, 126.04, 125.64, 125.52, 124.43, 121.20, 118.62, 114.05, 47.69, 19.72, 10.98. MS (ESI) calculated for C19H18N4, m/z 302.15, found 303.16 (M + H)+.
30: 1-Benzyl-2-propyl-1H-imidazo[4,5-c]quinolin-4-amine
1H NMR (500 MHz, CDCl3) δ 7.80 (dd, J = 8.4, 0.8 Hz, 1H), 7.71 (dd, J = 8.3, 1.0 Hz, 1H), 7.49 – 7.42 (m, 1H), 7.40 – 7.30 (m, 3H), 7.18 – 7.10 (m, 1H), 7.10 – 7.05 (m, 2H), 5.74 (s, 2H), 5.66 (s, 2H), 2.92 – 2.80 (m, 2H), 1.91 – 1.85 (m, 2H), 1.03 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 153.50, 150.46, 143.54, 134.77, 133.58, 128.82, 127.60, 126.73, 126.12, 126.05, 125.01, 121.90, 119.30, 114.59, 48.36, 28.88, 20.92, 13.52. MS (ESI) calculated for C20H20N4, m/z 316.17, found 317.17 (M + H)+.
31: 1-Benzyl-2-butyl-1H-imidazo[4,5-c]quinolin-4-amine
1H NMR (500 MHz, CDCl3) δ 7.80 (dd, J = 8.4, 0.8 Hz, 1H), 7.72 (dd, J = 8.3, 1.0 Hz, 1H), 7.43 (ddd, J = 8.4, 7.0, 1.4 Hz, 1H), 7.36 – 7.28 (m, 3H), 7.12 (ddd, J = 8.3, 7.1, 1.2 Hz, 1H), 7.08 – 7.04 (m, 2H), 5.73 (s, 2H), 5.56 (s, 2H), 2.91 – 2.83 (m, 2H), 1.84 – 1.74 (m, 2H), 1.43 (dq, J = 14.8, 7.4 Hz, 2H), 0.92 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 152.56, 149.54, 142.97, 133.87, 132.54, 127.81, 126.58, 125.61, 125.34, 125.21, 124.06, 120.78, 118.25, 113.71, 47.38, 28.54, 25.71, 21.04, 12.28. MS (ESI) calculated for C21H22N4, m/z 330.18, found 331.19 (M + H)+.
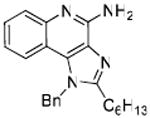
33: 1-Benzyl-2-hexyl-1H-imidazo[4,5-c]quinolin-4-amine
1H NMR (400 MHz, CDCl3) δ 7.82 (d, J = 8.4 Hz, 1H), 7.72 (d, J = 8.2 Hz, 1H), 7.45 (t, J = 7.7 Hz, 1H), 7.38 – 7.29 (m, 3H), 7.13 (t, J = 7.6 Hz, 1H), 7.07 (d, J = 7.4 Hz, 2H), 5.73 (s, 2H), 5.63 (s, 2H), 2.93 – 2.83 (m, 2H), 1.87 – 1.73 (m, 2H), 1.40 (dd, J = 14.3, 6.7 Hz, 2H), 1.34 – 1.24 (m, 4H), 0.88 (t, J = 6.5 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 154.01, 151.10, 144.59, 135.38, 133.98, 129.25, 128.02, 127.02, 126.87, 126.69, 125.54, 122.17, 119.70, 115.20, 48.84, 31.44, 29.05, 27.90, 27.46, 22.46, 14.01. MS (ESI) calculated for C23H26N4, m/z 358.22, found 359.22 (M + H)+.
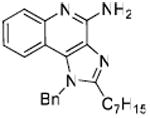
34: 1-Benzyl-2-heptyl-1H-imidazo[4,5-c]quinolin-4-amine
1H NMR (500 MHz, CDCl3) δ 7.81 (dd, J = 8.4, 0.8 Hz, 1H), 7.71 (dd, J = 8.3, 1.0 Hz, 1H), 7.44 (ddd, J = 8.4, 7.1, 1.3 Hz, 1H), 7.35 – 7.29 (m, 3H), 7.12 (ddd, J = 8.2, 7.1, 1.2 Hz, 1H), 7.05 (d, J = 6.9 Hz, 2H), 5.72 (s, 4H), 2.86 (dd, J = 18.2, 10.2 Hz, 2H), 1.79 (dt, J = 15.5, 7.7 Hz, 2H), 1.39 (dt, J = 14.9, 7.0 Hz, 2H), 1.34 – 1.18 (m, 6H), 0.86 (t, J = 7.0 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 152.52, 149.22, 142.11, 133.55, 132.39, 127.57, 126.36, 125.49, 124.86, 124.69, 123.80, 120.69, 118.05, 113.31, 47.15, 29.89, 27.60, 27.19, 26.21, 25.75, 20.85, 12.34. MS (ESI) calculated for C24H28N4, m/z 372.23, found 373.24 (M + H)+.
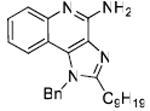
35: 1-Benzyl-2-nonyl-1H-imidazo[4,5-c]quinolin-4-amine
1H NMR (500 MHz, CDCl3) δ 7.85 – 7.79 (m, 1H), 7.71 (dd, J = 8.3, 0.9 Hz, 1H), 7.44 (qd, J = 6.9, 3.4 Hz, 1H), 7.38 – 7.29 (m, 3H), 7.13 (ddd, J = 8.2, 7.1, 1.2 Hz, 1H), 7.05 (d, J = 6.9 Hz, 2H), 5.85 (s, 2H), 5.72 (s, 2H), 2.90 – 2.84 (m, 2H), 1.87 – 1.71 (m, 2H), 1.44 – 1.33 (m, 2H), 1.33 – 1.18 (m, 10H), 0.87 (t, J = 7.0 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 152.31, 148.72, 141.13, 133.07, 132.10, 127.20, 126.00, 125.25, 124.35, 123.92, 123.40, 120.47, 117.71, 112.77, 46.79, 29.72, 27.59, 27.26, 27.24, 27.13, 27.12, 25.78, 25.35, 20.53, 12.00. MS (ESI) calculated for C26H32N4, m/z 400.26, found 401.27 (M + H)+.
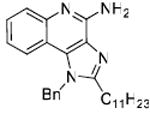
36: 1-Benzyl-2-undecyl-1H-imidazo[4,5-c]quinolin-4-amine
1H NMR (500 MHz, CDCl3) δ 7.81 (d, J = 8.2 Hz, 1H), 7.71 (d, J = 7.7 Hz, 1H), 7.47 – 7.42 (m, 1H), 7.36 – 7.28 (m, 3H), 7.15 – 7.11 (m, 1H), 7.06 (d, J = 7.0 Hz, 2H), 5.73 (s, 2H), 5.70 (s, 2H), 3.02 – 2.71 (m, 2H), 1.79 (dt, J = 15.5, 7.8 Hz, 2H), 1.44 – 1.34 (m, 2H), 1.34 – 1.18 (m, 14H), 0.88 (t, J = 7.0 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 152.56, 149.23, 142.11, 133.58, 132.43, 127.60, 126.39, 125.53, 124.89, 124.73, 123.83, 120.73, 118.08, 113.34, 47.19, 30.20, 27.88, 27.73, 27.67, 27.62, 27.56, 26.24, 25.78, 20.98, 12.43. MS (ESI) calculated for C28H36N4, m/z 428.29, found 429.30 (M + H)+.
37: 1-Benzyl-2-pentadecyl-1H-imidazo[4,5-c]quinolin-4-amine
1H NMR (500 MHz, CDCl3) δ 7.81 (dd, J = 8.4, 0.8 Hz, 1H), 7.72 (dd, J = 8.3, 1.0 Hz, 1H), 7.44 (ddd, J = 8.4, 7.1, 1.3 Hz, 1H), 7.36 – 7.27 (m, 3H), 7.13 (ddd, J = 8.3, 7.1, 1.2 Hz, 1H), 7.09 – 7.02 (m, 2H), 5.73 (s, 2H), 5.69 (s, 2H), 2.91 – 2.83 (m, 2H), 1.79 (dt, J = 15.5, 7.7 Hz, 2H), 1.59 – 1.33 (m, 2H), 1.33 – 1.16 (m, 22H), 0.88 (t, J = 7.0 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 151.99, 148.66, 141.59, 133.01, 131.85, 127.03, 125.82, 124.96, 124.32, 124.17, 123.26, 120.16, 117.50, 112.78, 46.62, 29.66, 27.43, 27.40, 27.39, 27.37, 27.32, 27.17, 27.11, 27.10, 26.99, 25.68, 25.21, 20.43, 11.86. MS (ESI) calculated for C32H44N4, m/z 484.36, found 485.36 (M + H)+.
38: 1-Benzyl-2-(but-3-enyl)-1H-imidazo[4,5-c]quinolin-4-amine
1H NMR (500 MHz, CDCl3) δ 7.80 (dd, J = 8.4, 0.8 Hz, 1H), 7.73 (dd, J = 8.3, 1.0 Hz, 1H), 7.46 – 7.42 (m, 1H), 7.36 – 7.29 (m, 3H), 7.13 (ddd, J = 8.2, 7.1, 1.2 Hz, 1H), 7.08 – 7.05 (m, 2H), 5.96 – 5.84 (m, 1H), 5.74 (s, 2H), 5.58 (s, 2H), 5.11 – 4.97 (m, 2H), 3.03 – 2.93 (m, 2H), 2.64 – 2.54 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 150.99, 148.87, 134.50, 133.06, 131.93, 127.16, 125.97, 125.02, 124.64, 124.58, 123.43, 120.17, 117.57, 113.92, 46.72, 29.59, 24.77. MS (ESI) calculated for C21H20N4, m/z 328.17, found 329.18 (M + H)+.
Synthesis of Compound 40: N-Benzyl-3-nitroquinolin-4-amine
To a solution of 39 (2.03 mg, 9.76 mmol) in 20 mL of anhydrous dichloromethane, were added triethylamine (1.48 g, 14.64 mmol) and benzylamine (1.36 g, 12.69 mmol). The reaction mixture was refluxed at 45 °C for 30 minutes. The solvent was then evaporated under vacuum and water was added to the residue to obtain the precipitate This was filtered, washed several times with water and dried to obtain the compound 40 (2.6 g, 95%). 1H NMR (400 MHz, CDCl3) δ 9.89 (s, 1H), 9.42 (s, 1H), 8.31 (d, J = 8.6 Hz, 1H), 8.04 (d, J = 8.4 Hz, 1H), 7.79 (t, J = 7.6 Hz, 1H), 7.56 – 7.35 (m, 6H), 5.13 (d, J = 5.7 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 151.01, 150.64, 147.37, 136.84, 132.73, 130.51, 129.36, 128.54, 127.16, 126.84, 126.33, 125.58, 119.14, 53.06. MS (ESI) calculated for C16H13N3O2, m/z 279.10, found 280.11 (M + H)+.
Synthesis of Compound 41: N4-Benzylquinoline-3,4-diamine
To a solution of 40 (2.6 g, 7.06 mmol) in 10 mL of EtOAc, were added a catalytic amount of Pt/C and Na2SO4. The reaction mixture was subjected to hydrogenation at 55 psi hydrogen pressure for 4 hours. The reaction mixture was then filtered through celite and the filtrate was evaporated under vacuum to obtain the compound 41 (2.3 g, 96%). 1H NMR (400 MHz, CDCl3) δ 8.53 (s, 1H), 8.00 (d, J = 8.3 Hz, 1H), 7.75 (d, J = 8.2 Hz, 1H), 7.56 – 7.29 (m, 7H), 4.43 (s, 2H), 4.05 (s, 1H), 3.78 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 143.59, 143.48, 141.10, 139.77, 129.89, 129.84, 128.81, 127.77, 127.60, 126.05, 125.76, 120.06, 99.98, 50.98. MS (ESI) calculated for C16H15N3, m/z 249.13, found 250.13 (M + H)+.
Synthesis of Compound 43: 1-Benzyl-2-(but-3-ynyl)-1H-imidazo[4,5-c]quinolin-4-amine
To a solution of pentynoic acid (37 mg, 0.38 mmol) in anhydrous DMF, were added HBTU (142 mg, 0.38 mmol), triethylamine (52 mg, 0.51 mmol), a catalytic amount of DMAP and 41 (85 mg, 0.34 mmol). The reaction mixture was then heated to 90 °C for 1 hour. The solvent was then removed under vacuum. The residue was dissolved in EtOAc, washed with water, dried over Na2SO4 and then concentrated to obtain the residue. This was purified using column chromatography (7% MeOH/dichloromethane) to obtain the compound 42 (60 mg). To a solution of 42 in a solvent mixture of MeOH:dichloromethane:chloroform (0.1:1:1), was added 3-chloroperoxybenzoic acid (83 mg, 0.48 mmol), and the solution was refluxed at 45-50 °C for 40 minutes. The solvent was then removed under vacuum and the residue was purified using column chromatography (8% MeOH/dichloromethane) to obtain the N-oxide derivative (30 mg). This was then dissolved in anhydrous dichloromethane, followed by the addition of benzoyl isocyanate (20 mg, 0.14 mmol) and heated at 45 °C for 15 minutes. The solvent was then removed under vacuum and the residue was dissolved in anhydrous MeOH, followed by the addition of excess sodium methoxide. The reaction mixture was then heated at 80 °C for 1 hour. The solvent was removed under vacuum and the residue was purified using column chromatography (6% MeOH/dichloromethane) to obtain the compound 43 (15 mg, 12%). 1H NMR (500 MHz, CDCl3) δ 7.81 (dd, J = 8.4, 0.8 Hz, 1H), 7.74 (dd, J = 8.3, 1.0 Hz, 1H), 7.46 (ddd, J = 8.4, 5.7, 1.3 Hz, 1H), 7.37 – 7.30 (m, 3H), 7.15 (ddd, J = 8.3, 7.1, 1.2 Hz, 1H), 7.09 – 7.05 (m, 2H), 5.79 (s, 2H), 5.68 (s, 2H), 3.11 (dd, J = 8.8, 6.4 Hz, 2H), 2.87 – 2.71 (m, 2H), 1.99 (t, J = 2.6 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 150.49, 149.42, 133.45, 132.82, 127.91, 126.75, 126.01, 125.19, 124.98, 124.08, 121.13, 118.33, 113.52, 81.16, 68.15, 47.46, 25.06, 15.63. MS (ESI) calculated for C21H18N4, m/z 326.15, found 327.16 (M + H)+.
Synthesis of Compound 44: 1-Benzyl-4-chloro-2-pentyl-1H-imidazo[4,5-c]quinoline
To a solution 27 (200 mg, 0.71 mmol) in anhydrous THF, were added triethylamine (179 mg, 1.77 mmol) and hexanoyl chloride (143 mg, 1.06 mmol). The reaction mixture was then stirred for 6-8 hours, followed by removal of the solvent under vacuum. The residue was dissolved in EtOAc, washed with water, brine and then dried over Na2SO4 to obtain the intermediate amide compound. This was dissolved in MeOH, followed by the addition of calcium oxide. The reaction mixture was then heated in microwave at 110 °C for 1 hour. The solvent was removed under vacuum and the residue was purified using column chromatography (3% MeOH/dichloromethane) to obtain the compound 44 (170 mg, 66%). 1H NMR (400 MHz, CDCl3) δ 8.16 (d, J = 8.4 Hz, 1H), 7.88 (d, J = 8.4 Hz, 1H), 7.60 (t, J = 7.7 Hz, 1H), 7.42 (t, J = 7.7 Hz, 1H), 7.38 – 7.30 (m, 3H), 7.05 (d, J = 7.0 Hz, 2H), 5.81 (s, 2H), 3.05 – 2.95 (m, 2H), 1.84 (dt, J = 15.6, 7.6 Hz, 2H), 1.48 – 1.24 (m, 4H), 0.88 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 156.40, 143.96, 143.60, 135.40, 134.74, 133.74, 129.93, 129.42, 128.30, 127.56, 126.58, 125.40, 119.91, 117.29, 49.19, 31.63, 27.91, 27.70, 22.29, 13.90. MS (ESI) calculated for C22H22ClN3, m/z 363.15, found 364.15 (M + H)+.
Synthesis of Compound 46: 1-Benzyl-2-pentyl-1H-imidazo[4,5-c]quinolin-4-ol
The compound 44 (50 mg, 0.14 mmol) was dissolved in 6M HCl and the solution was heated in microwave at 110 °C for 1 hour. The solvent was then removed under vacuum and the residue was purified using column chromatography (7% MeOH/dichloromethane) to obtain the compound 46 (38 mg, 80%). 1H NMR (400 MHz, DMSO) δ 11.59 (s, 1H), 7.74 (d, J = 8.2 Hz, 1H), 7.43 (d, J = 8.2 Hz, 1H), 7.39 – 7.30 (m, J = 8.3 Hz, 3H), 7.30 – 7.23 (m, J = 7.2 Hz, 1H), 7.08 – 7.00 (m, J = 7.5 Hz, 3H), 5.83 (s, 2H), 2.83 (t, J = 7.5 Hz, 2H), 1.79 – 1.65 (m, 2H), 1.40 – 1.16 (m, 4H), 0.82 (t, J = 7.0 Hz, 3H). 13C NMR (101 MHz, DMSO) δ 157.78, 154.36, 137.04, 136.73, 134.44, 131.09, 129.43, 128.08, 127.95, 125.94, 121.98, 121.27, 116.69, 112.04, 48.44, 31.22, 27.11, 26.69, 22.30, 14.26. MS (ESI) calculated for C22H23N3, m/z 345.18, found 346.19 (M + H)+.
Synthesis of Compound 47: 1-Benzyl-2-pentyl-4-phenyl-1H-imidazo[4,5-c]quinoline
Suzuki coupling of the compound 44 with phenyl boronic acid was performed. To a solution of 44 (70 mg, 0.19 mmol) in THF, were added polystyrene-bound PPh3-Pd (9.5 mg, 0.00095 mmol), K2CO3 (40 mg, 0.29 mmol) dissolved in water and phenyl boronic acid (28 mg, 0.23 mmol). The reaction was heated at 80 °C for 10-12 hours. The solution was filtered to remove the solid resin and the filtrate was evaporated under vacuum to obtain the residue. This was purified using column chromatography (30% EtOAc/hexane) to obtain the compound 47 (25 mg, 33%). 1H NMR (400 MHz, CDCl3) δ 8.81 – 8.77 (m, 1H), 8.33 (dd, J = 8.5, 0.8 Hz, 1H), 7.93 (dd, J = 8.4, 0.8 Hz, 1H), 7.64 – 7.58 (m, 3H), 7.55 – 7.49 (m, 1H), 7.36 (dddd, J = 9.8, 6.9, 6.1, 2.2 Hz, 4H), 7.12 – 7.07 (m, 2H), 5.84 (s, 2H), 3.03 – 2.85 (m, 2H), 1.91 (dt, J = 15.5, 7.6 Hz, 2H), 1.42 (ddt, J = 36.4, 21.9, 7.4 Hz, 4H), 0.92 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 155.17, 151.02, 144.50, 138.18, 135.43, 135.10, 135.01, 130.96, 130.01, 129.31, 129.27, 128.37, 128.05, 126.74, 125.76, 125.52, 119.61, 117.17, 48.92, 31.58, 27.56, 27.48, 22.37, 13.97. MS (ESI) calculated for C28H27N3, m/z 405.22, found 406.23 (M + H)+.
Synthesis of Compound 48: N-(1-Benzyl-2-butyl-1H-imidazo[4,5-c]quinolin-4-yl)hydroxylamine
To a solution of 27 (100 mg, 0.35 mmol) in anhydrous THF, were added triethylamine (72 mg, 0.71 mmol) and valeryl chloride (51 mg, 0.42 mmol). The reaction mixture was then stirred for 6-8 hours, followed by removal of the solvent under vacuum. The residue was dissolved in EtOAc, washed with water, brine and then dried over Na2SO4 to obtain the residue. This was dissolved in MeOH, followed by the addition of calcium oxide. The reaction mixture was then heated in microwave at 110 °C for 1 hour. The solvent was removed under vacuum and the residue was purified using column chromatography (3% MeOH/dichloromethane) to obtain 45 (75 mg, 62%). To a solution of 45 (25 mg, 0.072 mmol) in anhydrous MeOH, were added triethylamine (72 mg, 0.72 mmol) and hydroxylamine hydrochloride (76 mg, 1.08 mmol). The reaction mixture was heated at 50 °C for 10-12 hours. The solvent was then removed under vacuum and the residue was dissolved in EtOAc, washed with water and dried over Na2SO4. The EtOAc fraction was then evaporated under vacuum and the residue was purified using basic-alumina (pH>7) column chromatography (30% MeOH/dichloromethane) to obtain the compound 48 (10 mg, 40%). 1H NMR (500 MHz, CDCl3) δ 7.41 (d, J = 8.1 Hz, 1H), 7.32 (dq, J = 25.0, 7.2 Hz, 4H), 7.21 (s, 1H), 7.06 (d, J = 7.1 Hz, 2H), 6.88 (dd, J = 10.6, 5.7 Hz, 1H), 5.61 (s, 2H), 2.84 – 2.77 (m, 2H), 1.78 (dt, J = 15.5, 7.7 Hz, 2H), 1.40 (dt, J = 15.0, 7.4 Hz, 2H), 0.90 – 0.86 (m, 3H). 13C NMR (126 MHz, CDCl3) δ 151.78, 133.18, 127.13, 125.87, 125.13, 123.30, 119.10, 117.94, 46.50, 27.68, 24.81, 20.34, 11.58. MS (ESI) calculated for C21H22N4O, m/z 346.18, found 347.19 (M + H)+.
Synthesis of Compound 49: 1-Benzyl-2-butyl-4-hydrazinyl-1H-imidazo[4,5-c]quinoline
To a solution of 45 (60 mg, 0.17 mmol) in anhydrous MeOH, was added tert-butyl carbazate (57 mg, 0.43 mmol). After 15 hours, the solvent was removed under vacuum and the residue was purified using column chromatography (4% MeOH/dichloromethane) to obtain the intermediate N-Boc derivative. This was then dissolved in 5 ml of trifluoroacetic acid and stirred for 30 minutes, followed by removal of the solvent by purging nitrogen and drying under vacuum to obtain the trifluoroacetate salt of the compound 49 (55 mg, 57%). 1H NMR (500 MHz, MeOD) δ 7.82 (d, J = 8.4 Hz, 1H), 7.79 (d, J = 8.3 Hz, 1H), 7.45 – 7.40 (m, 1H), 7.28 (dt, J = 25.5, 7.2 Hz, 3H), 7.15 – 7.10 (m, 1H), 7.03 (d, J = 7.4 Hz, 2H), 5.82 (s, 2H), 2.94 – 2.90 (m, 2H), 1.79 – 1.69 (m, 2H), 1.40 (dq, J = 14.8, 7.4 Hz, 2H), 0.90 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, MeOD) δ 154.65, 151.25, 143.19, 135.64, 133.54, 128.81, 128.68, 127.41, 126.96, 125.31, 125.10, 124.68, 122.23, 120.02, 114.47, 48.23, 29.28, 26.26, 21.84, 12.51. MS (ESI) calculated for C21H23N5, m/z 345.20, found 346.20 (M + H)+.
Synthesis of Compound 50: 1-Benzyl-2-butyl-1H-imidazo[4,5-c]quinoline
To a solution of 41 (300 mg, 1.221 mmol) in anhydrous THF, were added triethylamine (183 mg, 1.82 mmol) and valeryl chloride (174 mg, 1.45 mmol). The reaction mixture was then stirred for 6-8 hours, followed by removal of the solvent under vacuum. The residue was dissolved in EtOAc, washed with water, brine and then dried over Na2SO4 to obtain the intermediate amide compound. This was dissolved in MeOH, followed by the addition of calcium oxide. The reaction mixture was then heated in microwave at 110 °C for an hour. The solvent was removed under vacuum and the residue was purified using column chromatography (9% MeOH/dichloromethane) to obtain the compound 50 (360 mg, 94%). 1H NMR (400 MHz, CDCl3) δ 9.38 (s, 1H), 8.25 (d, J = 8.1 Hz, 1H), 7.93 (d, J = 8.5 Hz, 1H), 7.66 – 7.57 (m, 1H), 7.42 (dd, J = 11.2, 4.1 Hz, 1H), 7.40 – 7.31 (m, 3H), 7.06 (d, J = 6.6 Hz, 2H), 5.80 (s, 2H), 3.02 – 2.85 (m, 2H), 1.90 (dt, J = 15.4, 7.7 Hz, 2H), 1.57 – 1.39 (m, 2H), 0.96 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 155.81, 144.98, 144.75, 136.53, 135.14, 134.10, 130.83, 129.35, 128.15, 126.72, 126.29, 125.47, 119.88, 117.58, 48.94, 29.57, 27.18, 22.53, 13.77. MS (ESI) calculated for C21H21N3, m/z 315.17, found 316.18 (M + H)+.
Synthesis of Compound 52: 1-Benzyl-2-((ethylamino)methyl)-1H-imidazo[4,5-c]quinolin-4-amine
To a solution of 2-(tert-butoxycarbonyl(ethyl)amino)acetic acid (90 mg, 0.44 mmol) in anhydrous DMF, were added HBTU (167 mg, 0.44 mmol), triethylamine (53 mg, 0.52 mmol), a catalytic amount of DMAP and 41 (100 mg, 0.4 mmol). The reaction mixture was then heated to 70 °C for 18 hours. The solvent was then removed under vacuum. The residue was dissolved in EtOAc, washed with water, dried using Na2SO4 and then concentrated to obtain the residue, which was purified using column chromatography (6% MeOH/dichloromethane) to obtain the compound 51 (133 mg). To a solution of 51 in a solvent mixture of MeOH:dichloromethane:chloroform (0.1:1:1), 3-chloroperoxybenzoic acid (138 mg, 0.8 mmol) was added and the solution was refluxed at 45-50 °C for 40 minutes. The solvent was then removed under vacuum and the residue was purified using column chromatography (10% MeOH/dichloromethane) to obtain the N-oxide derivative (31 mg). This was then dissolved in anhydrous dichloromethane, followed by the addition of benzoyl isocyanate (16 mg, 0.11 mmol) and heated at 45 °C for 15 minutes. The solvent was then removed under vacuum and the residue was dissolved in anhydrous MeOH, followed by the addition of excess sodium methoxide and heated at 80 °C for 1 hour. The solvent was then removed under vacuum and the residue was purified using column chromatography (8% MeOH/dichloromethane) to obtain the intermediate N-Boc derivative. This was dissolved in 5 ml of trifluoroacetic acid and stirred for 30 minutes, followed by removal of the solvent by purging nitrogen and drying under vacuum to obtain the trifluoroacetate salt of the compound 52 (26 mg, 20%). 1H NMR (400 MHz, MeOD) δ 8.02 (d, J = 8.4 Hz, 1H), 7.82 (d, J = 8.4 Hz, 1H), 7.69 (t, J = 7.8 Hz, 1H), 7.49 – 7.32 (m, 4H), 7.15 (d, J = 7.6 Hz, 2H), 6.00 (s, 2H), 4.71 (s, 2H), 3.38 (q, J = 7.3 Hz, 2H), 1.45 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, MeOD) δ 149.52, 148.45, 136.86, 134.46, 134.03, 130.18, 129.15, 128.12, 125.29, 125.15, 124.97, 121.76, 118.34, 112.47, 48.94, 43.17, 42.36, 10.06. MS (ESI) calculated for C20H21N5, m/z 331.18, found 332.19 (M + H)+.
Synthesis of Compound 54: 1-(4-Amino-2-butyl-1H-imidazo[4,5-c]quinolin-1-yl)-2-methylpropan-2-ol
To a solution of 20 (100 mg, 0.43 mmol) in anhydrous THF, were added triethylamine (66 mg, 0.65 mmol) and valeryl chloride (62 mg, 0.52 mmol). The reaction mixture was then stirred for 6-8 hours, followed by removal of the solvent under vacuum. The residue was dissolved in EtOAc, washed with water, brine and then dried over Na2SO4 to obtain the intermediate amide compound. This was dissolved in MeOH, followed by the addition of calcium oxide and was heated in microwave at 110 °C for 1 hour. The solvent was then removed and the residue was purified using column chromatography (9% MeOH/dichloromethane) to obtain the compound 53 (58 mg). To a solution of 53 in a solvent mixture of MeOH:dichloromethane:chloroform (0.1:1:1), was added 3-chloroperoxybenzoic acid (84 mg, 0.49 mmol) and the solution was refluxed at 45-50 °C for 40 minutes. The solvent was then removed and the residue was purified using column chromatography (20% MeOH/dichloromethane) to obtain the N-oxide derivative (55 mg). This was then dissolved in anhydrous dichloromethane, followed by the addition of benzoyl isocyanate (39 mg, 0.26 mmol) and heated at 45 °C for 15 minutes. The solvent was then removed under vacuum and the residue was dissolved in anhydrous MeOH, followed by the addition of excess sodium methoxide. The reaction mixture was then heated at 80 °C for an hour. The solvent was removed under vacuum and the residue was purified using column chromatography (11% MeOH/dichloromethane) to obtain the compound 54 (40 mg, 30%). 1H NMR (400 MHz, MeOD) δ 8.26 (d, J = 8.3 Hz, 1H), 7.69 (d, J = 8.4 Hz, 1H), 7.47 (t, J = 7.7 Hz, 1H), 7.31 (t, J = 7.7 Hz, 1H), 4.63 (s, 2H), 3.20 – 3.02 (m, 2H), 1.98 – 1.80 (m, 2H), 1.62 – 1.45 (m, 2H), 1.27 (s, 6H), 1.04 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, MeOD) δ 156.33, 151.22, 143.80, 134.53, 126.75, 125.41, 125.12, 121.49, 120.93, 115.31, 71.16, 54.80, 29.72, 27.12, 26.52, 22.24, 12.86. MS (ESI) calculated for C18H24N4O, m/z 312.20, found 313.20 (M + H)+.
Synthesis of Compound 55: 1-Benzyl-4-chloro-1H-[1,2,3]triazolo[4,5-c]quinoline
To a solution of 27 (60 mg, 0.21 mmol) in 1.5 ml of 1:1 acetic acid/water at 0 °C, was added 0.5N aqueous solution of sodium nitrite (0.56 ml, 0.28 mmol) and the reaction was stirred for 2 hours. The solvent was then removed under vacuum. The residue was dissolved in EtOAc, washed with water and dried over Na2SO4. The EtOAc fraction was then concentrated under vacuum to obtain the compound 55 (58 mg, 94%). 1H NMR (500 MHz, CDCl3) δ 8.12 (dd, J = 8.4, 0.7 Hz, 1H), 7.96 (dd, J = 8.2, 0.9 Hz, 1H), 7.70 (ddd, J = 8.4, 7.2, 1.4 Hz, 1H), 7.52 (ddd, J = 8.3, 7.2, 1.2 Hz, 1H), 7.30 – 7.21 (m, 3H), 7.12 – 7.09 (m, 2H), 6.19 (s, 2H). 13C NMR (126 MHz, CDCl3) δ 142.94, 141.16, 136.74, 132.71, 131.61, 128.30, 127.80, 127.24, 126.61, 125.80, 124.24, 124.20, 120.01, 112.71, 52.00. MS (ESI) calculated for C16H11ClN4, m/z 294.07, found 317.06 (M + Na+) and 612.11 (M + M + Na+).
Synthesis of Compound 56: 1-Benzyl-1H-[1,2,3]triazolo[4,5-c]quinolin-4-amine
Compound 55 (46 mg, 0.16 mmol) was dissolved in 0.1 ml of 2M ammonia solution in MeOH and the reaction was heated in microwave at 150 °C for 1 hour. The solvent was then removed under vacuum and the residue was purified using column chromatography (65% EtOAc/hexane) to obtain the compound 56 (35 mg, 82%). 1H NMR (500 MHz, TFA) δ 8.25 – 8.13 (m, 1H), 7.87 (d, J = 7.2 Hz, 1H), 7.79 (d, J = 8.0 Hz, 1H), 7.63 (t, J = 6.1 Hz, 1H), 7.39 (s, 3H), 7.24 (s, 2H), 6.38 (d, J = 23.6 Hz, 2H). 13C NMR (126 MHz, TFA) δ 134.33, 133.27, 131.52, 129.18, 129.06, 127.08, 125.83, 123.58, 118.48, 117.54, 115.29, 113.04, 110.78, 54.87. MS (ESI) calculated for C16H11N5, m/z 275.12, found 276.12 (M + H)+.
Synthesis of Compound 57: 1-Benzyl-2-oxo-N-propyl-1H-imidazo[4,5-c]quinoline-3(2H)-carboxamide
To a solution of 41 (30 mg, 0.12 mmol) in anhydrous THF, were added triethylamine (15 mg, 0.14 mmol) and n-propyl isocyanate (33 mg, 0.39 mmol); the n-propylamine released as a consequence of cyclization en route to the cyclic urea was found to consume n-propyl isocyanate, necessitating the use of additional equivalents of the reagent. The reaction was heated in microwave at 100 °C for 1 hour. The solvent was then removed under vacuum and the residue was purified using column chromatography (6% MeOH/dichloromethane) to obtain the compound 57 (26 mg, 61%). 1H NMR (500 MHz, CDCl3) δ 9.90 (s, 1H), 8.79 (s, J = 5.4 Hz, 1H), 8.15 (d, J = 8.5, 0.6 Hz, 1H), 7.91 (d, J = 8.6, 0.6 Hz, 1H), 7.60 (t, J = 8.4, 6.9, 1.3 Hz, 1H), 7.40 (t, J = 8.3, 6.9, 1.2 Hz, 1H), 7.37 – 7.32 (m, 2H), 7.31 – 7.28 (m, 1H), 7.26 (d, J = 6.7, 5.4 Hz, 2H), 5.60 (s, 2H), 3.49 – 3.45 (m, J = 7.1, 5.8 Hz, 2H), 1.78 – 1.68 (m, 2H), 1.04 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 151.08, 148.67, 143.44, 136.25, 132.69, 128.54, 127.08, 127.02, 125.87, 125.74, 124.61, 123.68, 118.43, 117.87, 112.78, 44.45, 39.68, 20.57, 9.22. MS (ESI) calculated for C21H20N4O2, m/z 360.16, found 361.17 (M + H)+.
NF-κB induction
The induction of NF-κB was quantified using HEK-Blue-7 cells and HEK-Blue-8 cells as previously described by us.8;24 HEK293 cells were stably transfected with human TLR7 (or human TLR8), MD2, and secreted alkaline phosphatase (sAP), and were maintained in HEK-Blue™ Selection medium containing zeocin and normocin. Stable expression of secreted alkaline phosphatase (sAP) under control of NF-κB/AP-1 promoters is inducible by the TLR7 (or TLR8) agonists, and extracellular sAP in the supernatant is proportional to NF-κB induction. HEK-Blue cells were incubated at a density of ∼105 cells/ml in a volume of 80 μl/well, in 384-well, flat-bottomed, cell culture-treated microtiter plates until confluency was achieved, and subsequently graded concentrations of stimuli. sAP was assayed spectrophotometrically using an alkaline phosphatase-specific chromogen (present in HEK-detection medium as supplied by the vendor) at 620 nm.
IFN-α induction in whole human blood
Aliquots (3 mL) of heparin-anticoagulated blood obtained from healthy human donors after informed consent and stimulated for 12 h with graded concentrations of test compounds. The plasma was isolated by centrifugation, diluted 1:20, and IFN-α was assayed in triplicate using a high-sensitivity human IFN-α-specific ELISA kit (PBL Interferon Source, Piscataway, NJ).
Supplementary Material
Figure 1.

Structures of Imiquimod and 2.
Scheme 2.

Syntheses of 2-imidazolyl sidechain-modified analogues of 2.
Scheme 6.

Syntheses of imidazole-modified analogues.
Acknowledgments
This work was supported by NIH/NIAID contract HHSN272200900033C.
Abbreviations
- AP-1
activator protein-1
- CD
cluster of differentiation
- DC
dendritic cells
- DMAP
4-dimethylaminopyridine
- DMF
N,N-dimethylformamide
- EC50
half-maximal effective concentration
- EDCI.HCl
1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride
- ELISA
enzyme linked immunosorbent assay
- ESI-TOF
electrospray ionization-time of flight
- HATU
2-(1H-7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl uronium hexafluorophosphate
- HBTU
O-benzotriazole-N,N,N′,N′-tetramethyl uronium hexafluorophosphate
- HEK
human embryonic kidney
- IFN
interferon
- LPS
lipopolysaccharide
- MHC
major histocompatibility complex
- NF-κB
nuclear factor-kappa B
- NK
natural killer
- PAMP
pathogen-associated molecular patterns
- sAP
secreted alkaline phophatase
- SAR
structure activity relationship
- ssRNA
single stranded RNA
- Th1
helper T–type 1
- THF
tetrahydrofuran
- TLR
Toll-like receptor
Footnotes
Supporting Information: Characterization of intermediates and final compounds (1H, 13C, mass spectra). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Kawai T, Akira S. TLR signaling. Semin Immunol. 2007;19:24–32. doi: 10.1016/j.smim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 2.Kumagai Y, Takeuchi O, Akira S. Pathogen recognition by innate receptors. J Infect Chemother. 2008;14:86–92. doi: 10.1007/s10156-008-0596-1. [DOI] [PubMed] [Google Scholar]
- 3.Akira S. Innate immunity to pathogens: diversity in receptors for microbial recognition. Immunol Rev. 2009;227:5–8. doi: 10.1111/j.1600-065X.2008.00739.x. [DOI] [PubMed] [Google Scholar]
- 4.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 5.Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nature Immunol. 2001;2:675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- 6.Cottalorda A, Verschelde C, Marcais A, Tomkowiak M, Musette P, Uematsu S, Akira S, Marvel J, Bonnefoy-Berard N. TLR2 engagement on CD8 T cells lowers the threshold for optimal antigen-induced T cell activation. Eur J Immunol. 2006;36:1684–1693. doi: 10.1002/eji.200636181. [DOI] [PubMed] [Google Scholar]
- 7.Kaisho T, Akira S. Toll-like receptors as adjuvant receptors. Biochim Biophys Acta. 2002;1589:1–13. doi: 10.1016/s0167-4889(01)00182-3. [DOI] [PubMed] [Google Scholar]
- 8.Hood JD, Warshakoon HJ, Kimbrell MR, Shukla NM, Malladi S, Wang X, David SA. Immunoprofiling toll-like receptor ligands: Comparison of immunostimulatory and proinflammatory profiles in ex vivo human blood models. Hum Vaccin. 2010;6:1–14. doi: 10.4161/hv.6.4.10866. [DOI] [PubMed] [Google Scholar]
- 9.Lee J, Chuang TH, Redecke V, She L, Pitha PM, Carson DA, Raz E, Cottam HB. Molecular basis for the immunostimulatory activity of guanine nucleoside analogs: activation of Toll-like receptor 7. Proc Natl Acad Sci U S A. 2003;100:6646–6651. doi: 10.1073/pnas.0631696100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crozat K, Beutler B. TLR7: A new sensor of viral infection. Proc Natl Acad Sci U S A. 2004;101:6835–6836. doi: 10.1073/pnas.0401347101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303:1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 12.Wierenga W, Skulnick HI, Stringfellow DA, Weed SD, Renis HE, Eidson EE. 5-substituted 2-amino-6-phenyl-4(3H)-pyrimidinones. Antiviral- and interferon-inducing agents. J Med Chem. 1980;23:237–239. doi: 10.1021/jm00177a004. [DOI] [PubMed] [Google Scholar]
- 13.Li LH, Wallace TL, Wierenga W, Skulnick HI, DeKoning TF. Antitumor activity of pyrimidinones, a class of small-molecule biological response modifiers. J Biol Response Mod. 1987;6:44–55. [PubMed] [Google Scholar]
- 14.Stringfellow DA, Glasgow LA. Tilorone hydrochloride: an oral interferon-inducing agent. Antimicrob Agents Chemother. 1972;2:73–78. doi: 10.1128/aac.2.2.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stringfellow DA. Comparation interferon- inducing and antiviral properties of 2-amino-5-bromo-6-methyl-4-pyrimidinol (U-25,166), tilorone hydrochloride, and polyinosinic-polycytidylic acid. Antimicrob Agents Chemother. 1977;11:984–992. doi: 10.1128/aac.11.6.984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hamilton RD, Wynalda MA, Fitzpatrick FA, Teagarden DL, Hamdy AH, Snider BG, Weed SD, Stringfellow DA. Comparison between circulating interferon and drug levels following administration of 2-amino-5-bromo-6-phenyl-4(3H)-pyrimidinone (ABPP) to different animal species. J Interferon Res. 1982;2:317–327. doi: 10.1089/jir.1982.2.317. [DOI] [PubMed] [Google Scholar]
- 17.Gerster JF, Lindstrom KJ, Miller RL, Tomai MA, Birmachu W, Bomersine SN, Gibson SJ, Imbertson LM, Jacobson JR, Knafla RT, Maye PV, Nikolaides N, Oneyemi FY, Parkhurst GJ, Pecore SE, Reiter MJ, Scribner LS, Testerman TL, Thompson NJ, Wagner TL, Weeks CE, Andre JD, Lagain D, Bastard Y, Lupu M. Synthesis and structure-activity-relationships of 1H-imidazo[4,5-c]quinolines that induce interferon production. J Med Chem. 2005;48:3481–3491. doi: 10.1021/jm049211v. [DOI] [PubMed] [Google Scholar]
- 18.Miller RL, Gerster JF, Owens ML, Slade HB, Tomai MA. Imiquimod applied topically: a novel immune response modifier and new class of drug. Int J Immunopharmacol. 1999;21:1–14. doi: 10.1016/s0192-0561(98)00068-x. [DOI] [PubMed] [Google Scholar]
- 19.Hemmi H, Kaisho T, Takeuchi O, Sato S, Sanjo H, Hoshino K, Horiuchi T, Tomizawa H, Takeda K, Akira S. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat Immunol. 2002;3:196–200. doi: 10.1038/ni758. [DOI] [PubMed] [Google Scholar]
- 20.Weterings JJ, Khan S, van der Heden van Noort GJ, Melief CJ, Overkleeft HS, van der Burg SH, Ossendorp F, Van der Marel GA, Filippov DV. 2-Azidoalkoxy-7-hydro-8-oxoadenine derivatives as TLR7 agonists inducing dendritic cell maturation. Bioorg Med Chem Lett. 2009;19:2249–2251. doi: 10.1016/j.bmcl.2009.02.095. [DOI] [PubMed] [Google Scholar]
- 21.Hirota K, Kazaoka K, Niimoto I, Kumihara H, Sajiki H, Isobe Y, Takaku H, Tobe M, Ogita H, Ogino T, Ichii S, Kurimoto A, Kawakami H. Discovery of 8-hydroxyadenines as a novel type of interferon inducer. J Med Chem. 2002;45:5419–5422. doi: 10.1021/jm0203581. [DOI] [PubMed] [Google Scholar]
- 22.Isobe Y, Tobe M, Ogita H, Kurimoto A, Ogino T, Kawakami H, Takaku H, Sajiki H, Hirota K, Hayashi H. Synthesis and structure-activity relationships of 2-substituted-8-hydroxyadenine derivatives as orally available interferon inducers without emetic side effects. Bioorg Med Chem. 2003;11:3641–3647. doi: 10.1016/s0968-0896(03)00369-9. [DOI] [PubMed] [Google Scholar]
- 23.Kurimoto A, Ogino T, Ichii S, Isobe Y, Tobe M, Ogita H, Takaku H, Sajiki H, Hirota K, Kawakami H. Synthesis and evaluation of 2-substituted 8-hydroxyadenines as potent interferon inducers with improved oral bioavailabilities. Bioorg Med Chem. 2004;12:1091–1099. doi: 10.1016/j.bmc.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 24.Shukla NM, Kimbrell MR, Malladi SS, David SA. Regioisomerism-dependent TLR7 agonism and antagonism in an imidazoquinoline. Bioorg Med Chem Lett. 2009;19:2211–2214. doi: 10.1016/j.bmcl.2009.02.100. [DOI] [PubMed] [Google Scholar]
- 25.Warshakoon HJ, Hood JD, Kimbrell MR, Malladi S, Wu WY, Shukla NM, Agnihotri G, Sil D, David SA. Potential adjuvantic properties of innate immune stimuli. Hum Vaccin. 2009;5:381–394. doi: 10.4161/hv.5.6.8175. [DOI] [PubMed] [Google Scholar]
- 26.Nicolas JF, Guy B. Intradermal, epidermal and transcutaneous vaccination: from immunology to clinical practice. Expert Rev Vaccines. 2008;7:1201–1214. doi: 10.1586/14760584.7.8.1201. [DOI] [PubMed] [Google Scholar]
- 27.Skountzou I, Kang SM. Transcutaneous immunization with influenza vaccines. Curr Top Microbiol Immunol. 2009;333:347–68. doi: 10.1007/978-3-540-92165-3_17. [DOI] [PubMed] [Google Scholar]
- 28.Giudice EL, Campbell JD. Needle-free vaccine delivery. Adv Drug Deliv Rev. 2006;58:68–89. doi: 10.1016/j.addr.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 29.Goldstein G, Chicca JJ. A universal anti-HIV-1 Tat epitope vaccine that is fully synthetic and self-adjuvanting. Vaccine. 2010;28:1008–1014. doi: 10.1016/j.vaccine.2009.10.129. [DOI] [PubMed] [Google Scholar]
- 30.Fujita Y, Abdel-Aal AB, Wimmer N, Batzloff MR, Good MF, Toth I. Synthesis and immunological evaluation of self-adjuvanting glycolipopeptide vaccine candidates. Bioorg Med Chem. 2008;16:8907–8913. doi: 10.1016/j.bmc.2008.08.064. [DOI] [PubMed] [Google Scholar]
- 31.Gupta S, Vayuvegula B, Gollapudi S. Substituted guanine ribonucleosides as B cell activators. Clin Immunol Immunopathol. 1991;61:S21–S27. doi: 10.1016/s0090-1229(05)80034-0. [DOI] [PubMed] [Google Scholar]
- 32.Pope BL, Chourmouzis E, Sigindere J, MacIntyre JP, Capetola RJ, Lau CY. In vivo activation of natural killer cells and priming of IL-2 responsive cytolytic cells by loxoribine (7-allyl-8-oxoguanosine) Cell Immunol. 1993;147:302–312. doi: 10.1006/cimm.1993.1071. [DOI] [PubMed] [Google Scholar]
- 33.Goodman MG. A new approach to vaccine adjuvants. Immunopotentiation by intracellular T-helper-like signals transmitted by loxoribine. Pharm Biotechnol. 1995;6:581–609. [PubMed] [Google Scholar]
- 34.Reitz AB, Goodman MG, Pope BL, Argentieri DC, Bell SC, Burr LE, Chourmouzis E, Come J, Goodman JH, Klaubert DH. Small-molecule immunostimulants. Synthesis activity of 7,8-disubstituted guanosines structurally related compounds. J Med Chem. 1994;37:3561–3578. doi: 10.1021/jm00047a014. [DOI] [PubMed] [Google Scholar]
- 35.Durand V, Wong SY, Tough DF, Le Bon A. Shaping of adaptive immune responses to soluble proteins by TLR agonists: a role for IFN-alpha/beta. Immunol Cell Biol. 2004;82:596–602. doi: 10.1111/j.0818-9641.2004.01285.x. [DOI] [PubMed] [Google Scholar]
- 36.Heil F, Ahmad-Nejad P, Hemmi H, Hochrein H, Ampenberger F, Gellert T, Dietrich H, Lipford G, Takeda K, Akira S, Wagner H, Bauer S. The Toll-like receptor 7 (TLR7)-specific stimulus loxoribine uncovers a strong relationship within the TLR7, 8 and 9 subfamily. Eur J Immunol. 2003;33:2987–2997. doi: 10.1002/eji.200324238. [DOI] [PubMed] [Google Scholar]
- 37.Ito T, Amakawa R, Kaisho T, Hemmi H, Tajima K, Uehira K, Ozaki Y, Tomizawa H, Akira S, Fukuhara S. Interferon-alpha and interleukin-12 are induced differentially by Toll-like receptor 7 ligands in human blood dendritic cell subsets. J Exp Med. 2002;195:1507–1512. doi: 10.1084/jem.20020207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baldwin SL, Bertholet S, Kahn M, Zharkikh I, Ireton GC, Vedvick TS, Reed SG, Coler RN. Intradermal immunization improves protective efficacy of a novel TB vaccine candidate. Vaccine. 2009;27:3063–3071. doi: 10.1016/j.vaccine.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang WW, Matlashewski G. Immunization with a Toll-like receptor 7 and/or 8 agonist vaccine adjuvant increases protective immunity against Leishmania major in BALB/c mice. Infect Immun. 2008;76:3777–3783. doi: 10.1128/IAI.01527-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zaba LC, Krueger JG, Lowes MA. Resident and “inflammatory” dendritic cells in human skin. J Invest Dermatol. 2009;129:302–308. doi: 10.1038/jid.2008.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miller LS, Modlin RL. Toll-like receptors in the skin. Semin Immunopathol. 2007;29:15–26. doi: 10.1007/s00281-007-0061-8. [DOI] [PubMed] [Google Scholar]
- 42.Mathers AR, Larregina AT. Professional antigen-presenting cells of the skin. Immunol Res. 2006;36:127–136. doi: 10.1385/IR:36:1:127. [DOI] [PubMed] [Google Scholar]
- 43.Rajagopal D, Paturel C, Morel Y, Uematsu S, Akira S, Diebold SS. Plasmacytoid dendritic cell-derived type I interferon is crucial for the adjuvant activity of Toll-like receptor 7 agonists. Blood. 2010;115:1949–1957. doi: 10.1182/blood-2009-08-238543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jin MS, Lee JO. Structures of TLR-ligand complexes. Curr Opin Immunol. 2008;20:414–419. doi: 10.1016/j.coi.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 45.Adediran SA, Day TP, Sil D, Kimbrell MR, Warshakoon HJ, Malladi SS, David SA. Synthesis of a highly water-soluble derivative of amphotericin B with attenuated proinflammatory activity. Mol Pharm. 2009;6:1582–1590. doi: 10.1021/mp9001602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nguyen TB, Kumar EV, Sil D, Wood SJ, Miller KA, Warshakoon HJ, Datta A, David SA. Controlling plasma protein binding: structural correlates of interactions of hydrophobic polyamine endotoxin sequestrants with human serum albumin. Mol Pharm. 2008;5:1131–1137. doi: 10.1021/mp8001123. [DOI] [PubMed] [Google Scholar]
- 47.Gorden KB, Gorski KS, Gibson SJ, Kedl RM, Kieper WC, Qiu X, Tomai MA, Alkan SS, Vasilakos JP. Synthetic TLR agonists reveal functional differences between human TLR7 and TLR8. J Immunol. 2005;174:1259–1268. doi: 10.4049/jimmunol.174.3.1259. [DOI] [PubMed] [Google Scholar]
- 48.Gorski KS, Waller EL, Bjornton-Severson J, Hanten JA, Riter CL, Kieper WC, Gorden KB, Miller JS, Vasilakos JP, Tomai MA, Alkan SS. Distinct indirect pathways govern human NK-cell activation by TLR-7 and TLR-8 agonists. Int Immunol. 2006;18:1115–1126. doi: 10.1093/intimm/dxl046. [DOI] [PubMed] [Google Scholar]
- 49.Middlebrooks BL, Voss PG, Douglas WL, Toom PM. Procedure for selecting monoclonal antibodies for use in a ligand displacement assay of serum antibody levels. J Immunoassay. 1991;12:125–144. doi: 10.1080/01971529108055061. [DOI] [PubMed] [Google Scholar]
- 50.Simon F, Rahimy C, Krivine A, Levine M, Pepin JM, Lapierre D, Denamur E, Vernoux L, De Crepy A, Blot P. Antibody avidity measurement immune complex dissociation for serological diagnosis of vertically acquired HIV-1 infection. J Acquir Immune Defic Syndr. 1993;6:201–207. [PubMed] [Google Scholar]
- 51.Liu X, Hefesha H, Tanaka H, Scriba G, Fahr A. Lipophilicity measurement of drugs by reversed phase HPLC over wide pH range using an alkaline-resistant silica-based stationary phase, XBridge Shield RP(18) Chem Pharm Bull (Tokyo) 2008;56:1417–1422. doi: 10.1248/cpb.56.1417. [DOI] [PubMed] [Google Scholar]
- 52.Fuguet E, Rafols C, Bosch E, Roses M. Determination of the chromatographic hydrophobicity index for ionisable solutes. J Chromatogr A. 2007;1173:110–119. doi: 10.1016/j.chroma.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 53.Aghemo A, Rumi MG, Colombo M. Pegylated IFN-α2a and ribavirin in the treatment of hepatitis C. Expert Rev Anti Infect Ther. 2009;7:925–935. doi: 10.1586/eri.09.70. [DOI] [PubMed] [Google Scholar]
- 54.Guilhot F, Roy L, Saulnier PJ, Guilhot J. Interferon in chronic myeloid leukaemia: past and future. Best Pract Res Clin Haematol. 2009;22:315–329. doi: 10.1016/j.beha.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 55.Cannon T, Mobarek D, Wegge J, Tabbara IA. Hairy cell leukemia: current concepts. Cancer Invest. 2008;26:860–865. doi: 10.1080/07357900801965034. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.