

Abstract
A sequential three-component synthesis of functionalized benzisothiazoline-3-acetic acid 1,1-dioxides utilizing a domino Heck-aza-Michael pathway is reported. This one-pot procedure rapidly assembles functionalized benzylsulfonamides, which undergo a palladium-catalyzed, domino, Heck-aza-Michael transformation in an experimentally straightforward manner. This attractive protocol has been utilized to synthesize three combinatorial sub-libraries (I-III) comprising a total of 95 compounds in high purities (≥95% for 75 compounds), yield and quantities.
Keywords: Domino, Heck, Aza-Michael, Sultam
Introduction
Advances in high-throughput screening and the need for new pharmaceutical leads have prompted the development of new protocols to generate diverse libraries of drug-like compounds. In recent years, a number of facilitated protocols that utilize both solid-phase and solution-phase chemistry have emerged to meet this challenge. Despite success in this area, there are limited examples of protocols that take advantage of cross-reaction functionality to allow domino/tandem processes to occur in a multi-component one-pot procedure.i
Recently, we reported a one-pot, sequential three-component approach towards the synthesis of 1,2-benzisothiazoline-3-acetic acid 1,1-dioxides (Scheme 1).ii The key step in this protocol was the utilization of a domino Heck-aza-Michael (HaM) reaction for both the mode of cyclization and as a pathway for the incorporation of an additional point of diversification.
Scheme 1.

Sequential three-component domino Heck-aza-Michael
Utilizing the (HaM) protocol, a small proof of concept demonstrative library of 1,2-benzisothiazoline-3-acetic acid 1,1-dioxides and subsequent derivatives was reported.ii Building on this work, the application of a domino, HaM protocol in the synthesis of three combinatorial sub-libraries (I-III) utilizing a variety of reaction platforms (Figure 1) is herein reported. Additionally a wider range of coupling partners was utilized to ultimately afford structural diversity around the central core. In order to maximize their potential drug-like properties, the subsequent libraries feature a multi-faceted design scheme as well as in-silico screening against Lipinski's rule of five criteria. The in-silico data ranges include a molecular weight range under 500 g/mol, no more than 5 hydrogen bond donors and/or 10 hydrogen bond acceptors, and a partition coefficient log P (clogP) less than 5.0.iii Overall, the application of a domino Heck-aza-Michael (HaM) allows for rapid incorporation of functionality via the manipulation of the three individual components, allowing for the design of a library of diverse drug-like small molecules.
Figure 1.

Overview of the prepared libraries
Sultams (cyclic sulfonamide analogues) have emerged as important targets in drug discovery due to their extensive chemical and biological profiles.iv Though not found in nature, a number of benzofused sultams have recently surfaced in the literature, which display potent activity across a variety of biological targets. Such reports include inhibition of a variety of enzymes, including COX-2 (Ampiroxicam),v,vi HCV NS5b RNA-dependent RNA-polymerase,vii HIV integrase,viii cysteine proteases involved in the progression of maleriaix and lipoxygenases.x In addition, sultams have also shown antimycobacterial activity against M. Tuberculosis,xi and inhibition of melanin-concentrating hormone [MCH] (Figure 2).xii
Figure 2.

Representative biologically active sultams
This aforementioned biological profile is augmented by a number of inherent chemical properties possessed by both sultams and their sulfonamide precursors, including facile coupling/allylation pathways for sulfonamide and sultam formation, hydrolytic stability, polarity and their crystalline nature. Traditionally, sultams have been synthesized utilizing a variety of classical cyclization protocols such as Friedel-Craft,xiii dianion,xiv [3+2] cycloadditions,xv, Diels-Alder reaction,xvi and recently the application of an oxa/aza-Michael reaction.xvii However, recently there have been a number of transition metal-catalyzed protocols reported utilizing ring-closing methathesis (RCM),xviii Heck,xix as well as Au-,xx Cu-xxi and Rh-catalyzedxxii cyclization protocols for the generation of diverse sultams.
Results and Discussion
A 56-member library I was initially designed to expand on the prototype library previously reported,ii demonstrating the capability of the HaM protocol in a library format. The method allows for the generation of sultams with three points of diversification starting from commercially available α-bromobenzenesulfonyl chlorides coupled with a range of aromatic, cyclic and alkyl amines (Figure 3).xxiii,xix
Figure 3.

Representative coupling partners for Heck-aza-Michael
Specific combinations of the three-components were chosen to evaluate the robustness of this protocol to peripheral functionality. In addition, Lipinski's rule of five also guided in-silico efforts in the selection of combinations. Based on previous work, it was anticipated that these substrates would be well tolerated under the reaction conditions, carrying out the preparation of library I in 1-dram vials on an aluminum reaction block.xxiv To this effect, α-bromobenzylsulfonyl chlorides (S1-3) were coupled with amines (A1-9) and stirred for 2 hours at room temperature. After such time, Et3N, Bu4NCl, Pd2(dba)3·CHCl3 and the corresponding Michael acceptors (M1-4) were added to the reaction mixture, which was heated to 110 °C and subjected to workup after 14 hours. Workup consisted of removal of DMF, suspension of the crude reaction mixture in EtOAc and filtration through a SiO2 SPE to remove inorganic salts and spent palladium. The crude material was analyzed by HPLC (UV 214 nm) and submitted to purification by mass-directed fractionation (MDF) to yield the anticipated products in modest to good yield and high purity (Table 1).xxv
Table 1.
Summary of Library I results
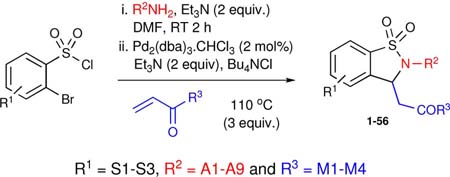
| entry | R1 | R2NH2 | R3 | yield (%) | purity (%) | product |
|---|---|---|---|---|---|---|
| 1 | S1 | A1 | M1 | 44 | 100 | 1 |
| 2 | S1 | A6 | M1 | 48 | 97 | 2 |
| 3 | S1 | A7 | M1 | 53 | 98 | 3 |
| 4 | S1 | A5 | M2 | 22 | 98 | 4 |
| 5 | S1 | A6 | M2 | 22 | 95 | 5 |
| 6 | S1 | A7 | M2 | 74 | 100 | 6 |
| 7 | S1 | A3 | M3 | 57 | 100 | 7 |
| 8 | S1 | A4 | M3 | 64 | 100 | 8 |
| 9 | S1 | A5 | M3 | 35 | 100 | 9 |
| 10 | S1 | A6 | M3 | 34 | 95 | 10 |
| 11 | S1 | A7 | M3 | 60 | 98 | 11 |
| 12 | S1 | A3 | M4 | 42 | 100 | 12 |
| 13 | S1 | A4 | M4 | 29 | 92 | 13 |
| 14 | S1 | A5 | M4 | 40 | 99 | 14 |
| 15 | S1 | A6 | M4 | 57 | 90 | 15 |
| 16 | S2 | A1 | M1 | 12 | 99 | 16 |
| 17 | S2 | A2 | M1 | 30 | 99 | 17 |
| 18 | S2 | A5 | M1 | 40 | 90 | 18 |
| 19 | S2 | A7 | M1 | 25 | 96 | 19 |
| 20 | S2 | A8 | M1 | 75 | 93 | 20 |
| 21 | S2 | A9 | M1 | 69 | 90 | 21 |
| 22 | S2 | A1 | M2 | 28 | 100 | 22 |
| 23 | S2 | A2 | M2 | 46 | 96 | 23 |
| 24 | S2 | A3 | M2 | 87 | 94 | 24 |
| 25 | S2 | A4 | M2 | 57 | 95 | 25 |
| 26 | S2 | A5 | M2 | 56 | 98 | 26 |
| 27 | S2 | A6 | M2 | 40 | 94 | 27 |
| 28 | S2 | A7 | M2 | 69 | 97 | 28 |
| 29 | S2 | A8 | M2 | 72 | 97 | 29 |
| 30 | S2 | A9 | M2 | 54 | 96 | 30 |
| 31 | S2 | A3 | M3 | 58 | 100 | 31 |
| 32 | S2 | A4 | M3 | 64 | 97 | 32 |
| 33 | S2 | A5 | M3 | 13 | 93 | 33 |
| 34 | S2 | A7 | M3 | 54 | 100 | 34 |
| 35 | S2 | A8 | M3 | 50 | 99 | 35 |
| 36 | S2 | A9 | M3 | 45 | 100 | 36 |
| 37 | S3 | A1 | M1 | 25 | 96 | 37 |
| 38 | S3 | A2 | M1 | 21 | 97 | 38 |
| 39 | S3 | A3 | M1 | 53 | 99 | 39 |
| 40 | S3 | A4 | M1 | 51 | 100 | 40 |
| 41 | S3 | A7 | M1 | 52 | 99 | 41 |
| 42 | S3 | A8 | M1 | 31 | 91 | 42 |
| 43 | S3 | A9 | M1 | 42 | 96 | 43 |
| 44 | S3 | A1 | M2 | 50 | 95 | 44 |
| 45 d | S3 | A3 | M2 | 5 | 100 | 45 |
| 46 d | S3 | A4 | M2 | 7 | 100 | 46 |
| 47 d | S3 | A6 | M2 | 5 | 87 | 47 |
| 48 d | S3 | A7 | M2 | 6 | 97 | 48 |
| 49 d | S3 | A8 | M2 | 9 | 96 | 49 |
| 50 d | S3 | A9 | M2 | 9 | 93 | 50 |
| 51 | S3 | A2 | M3 | 27 | 97 | 51 |
| 52 | S3 | A3 | M3 | 42 | 93 | 52 |
| 53 | S3 | A4 | M3 | 46 | 95 | 53 |
| 54 | S3 | A7 | M3 | 35 | 92 | 54 |
| 55 | S3 | A8 | M3 | 44 | 92 | 55 |
| 56 | S3 | A9 | M3 | 40 | 99 | 56 |
aReaction conditions: 1 (0.273 mmol), Pd2(dba)3.CHCl3 (2 mol %), methyl acrylate (0.82 mmol), Bu4NCl (0.273 mmol) in DMF at 110 °C for 14 h.
bPurified by an automated preparative reverse phase HPLC (detected by mass spectroscopy).
cPurity was determined by HPLC with peak area (UV) at 214 nm.
When repeated in single reaction formation, good yields (50-80%) were achieved as predicted.
Overall, a total of 56 reactions afforded product in variable yield and purity, validating the scope and economy of the HaM strategy. Specifically, out of the 56 reactions carried out, 44 had a final purity of 95% or greater with reactions yielding good overall mass recovery.xxvi Having established the viability of the HaM protocol in the generation of libraries, we set out to design a library (Library II) of 1,2-benzisothiazoline-3-acetic acid 1,1-dioxides utilizing the Bohdan MiniBlock® platform. Under this premise, a library of compounds was prepared in parallel using a 26-member Bohdan MiniBlock. In an attempt to design more drug-like molecules a new collection of amines (A10-15) was employed (Table 2). In addition, reactions were carried out using stock solutions to streamline the process thereby granting it the potential for future automation. As with sub-library I, crude compounds were analyzed by HPLC (UV 214 nm) and submitted to purification by MDF. Overall, with the exception of compounds 60 and 81, all reactions worked with good yield and high purity. Notably, 20 out of the 24 reactions had a final purity of 95% or greater. Having validated the methodology, the protocol was implemented for the generation of 1,2-benzisothiazoline-3-acetic acid 1,1-dioxides 84-95 bearing both saturated and unsaturated side chains as previously reported (Table 3). The addition of one more point of diversification was accomplished by utilizing commercially available 2,5-dibromosulfonyl chloride. In this regard, 12 parallel reactions were carried out in a Radley Carousel®, utilizing stock solutions for the quick generation of the desired products in a sequential three-component coupling. Crude material was analyzed via HPLC (UV 214 nm) and submitted to purification by MDF. As expected, all reactions resulted in good yields and high purities yielding the desired compounds with good mass recovery.
Table 2.
Representative components and products synthesized using Bohdan MiniBlock platform
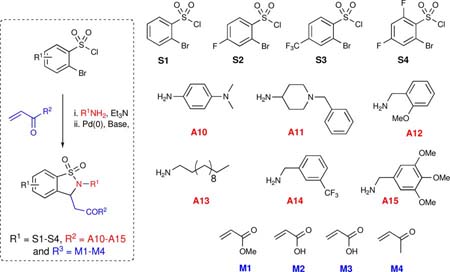
| entry | R1 | R2NH2 | R3 | yield (%) | purity (%) | product |
|---|---|---|---|---|---|---|
| 1 | S1 | A10 | M1 | 92 | 98 | 57 |
| 2 | S1 | A11 | M4 | 12 | 98 | 58 |
| 3 | S1 | A12 | M3 | 44 | 100 | 59 |
| 4 | S1 | A13 | M4 | 6 | 100 | 60 |
| 5 | S1 | A14 | M2 | 56 | 100 | 61 |
| 6 | S1 | A15 | M2 | 68 | 100 | 62 |
| 7 | S2 | A10 | M1 | 93 | 99 | 63 |
| 8 | S2 | A10 | M4 | 82 | 99 | 64 |
| 9 | S2 | A11 | M3 | 16 | 100 | 65 |
| 10 | S2 | A12 | M1 | 84 | 90 | 66 |
| 11 | S2 | A12 | M3 | 16 | 97 | 67 |
| 12 | S2 | A15 | M2 | 82 | 100 | 68 |
| 13 | S3 | A10 | M1 | 40 | 100 | 69 |
| 14 | S3 | A10 | M3 | 32 | 95 | 70 |
| 15 | S3 | A10 | M4 | 52 | 100 | 71 |
| 16 | S3 | A11 | M3 | 18 | 91 | 72 |
| 17 | S3 | A12 | M1 | 36 | 92 | 73 |
| 18 | S3 | A12 | M4 | 20 | 100 | 74 |
| 19 | S3 | A13 | M1 | 30 | 100 | 75 |
| 20 | S3 | A13 | M3 | 34 | 93 | 76 |
| 21 | S3 | A13 | M4 | 16 | 100 | 77 |
| 22 | S4 | A10 | M1 | 32 | 100 | 78 |
| 23 | S4 | A11 | M1 | 30 | 99 | 79 |
| 24 | S4 | A11 | M3 | 24 | 97 | 80 |
| 22 | S4 | A12 | M3 | 8 | 100 | 81 |
| 23 | S4 | A13 | M1 | 22 | 100 | 82 |
| 24 | S4 | A13 | M4 | 16 | 100 | 83 |
a Reaction conditions: 1 (0.136 mmol), Pd2(dba)3.CHCl3 (2 mol %), Michael acceptor (0.40 mmol), Bu4NCl (0.136 mmol) in DMF at 110 °C for 14 h. b Purified by an automated preparative reverse phase HPLC (detected by mass spectroscopy). c Purity was determined by HPLC with peak area (UV) at 214 nm.
Table 3.
Representative components and products synthesized using the Radley Carousel® platform
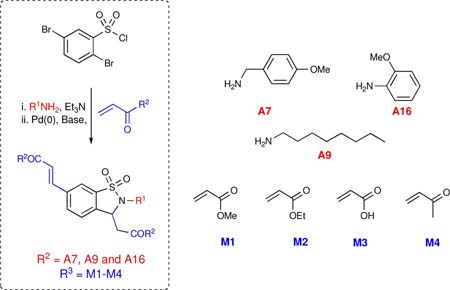
| entry | R2NH2 | R3 | yield (%) | purity (%) | compound |
|---|---|---|---|---|---|
| 1 | A7 | M1 | 18 | 100 | 84 |
| 2 | A7 | M2 | 45 | 93 | 85 |
| 3 | A7 | M3 | 62 | 90 | 86 |
| 4 | A7 | M4 | 86 | 100 | 87 |
| 5 | A9 | M1 | 69 | 100 | 88 |
| 6 | A9 | M2 | 90 | 100 | 89 |
| 7 | A9 | M3 | 86 | 99 | 90 |
| 8 | A9 | M4 | 59 | 100 | 91 |
| 9 | A16 | M1 | 78 | 100 | 92 |
| 10 | A16 | M2 | 81 | 100 | 93 |
| 11 | A16 | M3 | 60 | 100 | 94 |
| 12 | A16 | M4 | 90 | 90 | 95 |
a Reaction conditions: 1 (0.136 mmol), Pd2(dba)3.CHCl3 (2 mol %), methyl acrylate (0.40 mmol), Bu4NCl (0.136 mmol) in DMF at 110 °C for 14 h. b Purified by an automated preparative reverse phase HPLC (detected by mass spectroscopy). c Purity was determined by HPLC with peak area (UV) at 214 nm.
In conclusion, the successful demonstration of a sequential three-component synthesis of functionalized benzisothiazoline-3-acetic acid 1,1-dioxides utilizing a domino Heck-aza-Michael pathway has been accomplished. This one-pot, three step procedure, rapidly assembles functionalized benzylsulfonamides, which undergo a palladium-catalyzed domino Heck-aza-Michael transformation in an experimentally straightforward manner. This protocol was demonstrated on a variety of platforms (Reaction blocks, Bohdan MiniBlock® and Radley Carousel®) producing overall three combinatorial sub-libraries (I-III) comprising a total of 95 pure compounds in high purities (≥95% for 75 compounds) and good quantities. The evaluation of biological activities of the compounds reported herein in high-throughput screens is currently underway. Future efforts will continue to focus on the development of new methodology for the synthesis of diverse, functionalized libraries and their biological evaluation.
Experimental Section
General procedures: All air and moisture sensitive reactions were carried out in flame- or oven-dried glassware under argon atmosphere using standard gas tight syringes, cannula, and septa. Stirring was achieved with oven-dried, magnetic stir bars. CH3CN was purified by passage through the Solv-Tek purification system employing activated Al2O3 (Grubbs, R. H.; Rosen, R. K.; Timmers, F. J. Organometallics 1996, 15, 1518-1520). Et3N was purified by passage over basic alumina and stored over KOH. Flash column chromatography was performed with SiO2 from Sorbent Technology (30930M-25, Silica Gel 60A, 40-63 um). Thin layer chromatography was performed on silica gel 60F254 plates (EM-5717, Merck). Deuterated solvents were purchased from Cambridge Isotope laboratories. 1H and 13C NMR spectra were recorded on a Bruker DRX-400 NMR spectrometer operating at 400 MHz and 100 MHz respectively; or a Bruker Avance operating at 500 MHz and 125 MHz respectively. High-resolution mass spectrometry (HRMS) and FAB spectra were obtained in one of two manners: (i) on a VG Instrument ZAB double-focusing mass spectrometer and (ii) on a LCT Premier Spectrometer (Micromass UK Limited) operating on ESI (MeOH). All library syntheses using block technology were performed using a 24-position Mettler-Toledo Bohdan MiniBlock XT under an argon atmosphere in oven-dried Autochem 17 × 100 mm round bottom tubes. Parallel evaporations were performed using a GeneVac EZ-2 plus evaporator. Automated preparative reverse-phase HPLC purification was performed using a Waters 2767 Mass-Directed Fractionation system (2767 sample manager, 2525 Binary Pump, 515 Make-up pump) with a Waters ZQ quadrapole spectrometer and detected by UV (270 nm, Waters Xterra MS C-18 column, 19×150 mm, elution with the appropriate gradient of CH3CN in pH 9.8 buffered aqueous ammonium formate at 18 mL min-1 flow rate). Purity was determined by reverse-phase HPLC with peak area (UV) at 214 nm using a Waters Alliance 2795 system (Waters Xterra MS C-18 column, 4.6×150 mm, elution with a linear gradient of 5% CH3CN in pH 9.8 buffered aqueous ammonium formate to 100% CH3CN at 1.0 mL/min flow rate).
General procedure (A) for the synthesis of Library I (1-56) on Reaction Blocks in 1 dram vials
Into a 1-dram vial was added amine (0.237 mmol), Et3N (0.546 mmol) and dry DMF (0.60 mL) and the reaction was stirred at RT for 15 minutes. After such time, α-bromobenzenesulfonyl chlorides (0.237 mmol) were added and the reaction was stirred for 2 hrs. To the reaction vial was added Et3N (0.546 mmol), Bu4NCl (0.237 mmol), Pd2(dba)3•CHCl3 (2 mol%) and dry DMF (1.4 mL). After stirring for 5 min at RT, Michael acceptor (0.819 mmol) was added and the reaction vial was placed immediately into a preheated reaction block. The reaction was stirred at 110 °C for 14 hrs after which time the reaction was cooled and concentrated under reduced pressure. The crude was suspended in EtOAc, filtered through a SiO2 SPE and analyzed by HPLC (UV 214 nm). Crude material with purity below 90% was submitted to purification by MDF.
General procedure (B) for the synthesis of Library II (57-83) in a Bohdan MiniBlock
Into a MiniBlock reaction tube, was added a stock solution of amine (0.136 mmol) in dry DMF (0.10 mL) followed by Et3N (0.273 mmol) in dry DMF (0.10 mL) and reaction was stirred at RT for 15 minutes. A stock solution of α-bromobenzenesulfonyl chloride (0.136 mmol) in dry DMF (0.10 mL) was added and the reaction was stirred for 2 hrs. After such time, a stock solution of Et3N (0.273 mmol), Bu4NCl (0.136 mmol) and Pd2(dba)3•CHCl3 (2 mol%) in dry DMF (0.7 mL) was added to the reaction mixture. The MiniBlock was then heated to 110 °C and the Michael acceptor (0.410 mmol) was added. After stirring at 110 °C for 14 hrs, the crude reaction was cooled to RT and concentrated under reduced pressure. The crude was suspended in EtOAc, filtered through a SiO2 SPE and analyzed by HPLC (UV 214 nm). Crude material with purity below 90% was submitted to purification by MDF.
General procedure (C) for the synthesis of Library III (84-95) in a Radleys Carousel
Into a reaction tube contained within a 12-port Radley Carousel®, was added a stock solution of amine (0.136 mmol) in dry DMF (0.10 mL) followed by Et3N (0.273 mmol) in dry DMF (0.10 mL) and the reaction was stirred at RT for 15 minutes. A stock solution of α-bromobenzenesulfonyl chloride (0.136 mmol) in dry DMF (0.10 mL) was added and the reaction was stirred for 2 hrs. After such time, a stock solution of Et3N (0.273 mmol), Bu4NCl (0.136 mmol) and Pd2(dba)3•CHCl3 (2 mol%) in dry DMF (0.7 mL) was added to the reaction mixture. The MiniBlock was then heated to 110 °C after which time, the Michael acceptor (0.410 mmol) was added. After stirring at 110 °C for 14 hrs, the crude reaction was cooled to RT and concentrated under reduced pressure. The crude was suspended in EtOAc, filtered through a SiO2 SPE and analyzed by HPLC (UV 214 nm). Crude material with purity below 90% was submitted to purification by mass-directed fractionation (MDF).
Supplementary Material
Acknowledgment
This work was generously supported by funds provided by the National Institutes of General Medical Sciences (KU Chemical Methodologies and Library Development Center, P50 GM069663), NIGMS Pilot Scale Libraries Program (NIH P41GM076302) and NIH COBRE award P20 RR015563 with additional funds from the State of Kansas. Undergraduate funding was provided by the NIH K-INBRE award and KU Center of Research (K.Y. and K.V).
Footnotes
Supporting Information Available: Experimental procedures, tabulated results for all libraries, and full characterization data for representative compounds. This material is available free of charge via the Internet at http://acs.pubs.org/
References and Notes
- (i).(a) Keppler A,F, Gariani RA, Lopes DG, Comasseto JV. Tetrahedron Lett. 2009;50:2181–2184. [Google Scholar]; (b) Chai DI, Lautens M. J. Org. Chem. 2009 doi: 10.1021/jo900053b. ASAP. [DOI] [PubMed] [Google Scholar]; (c) Spandl RJ, Rudyk H, Spring DR. Chem. Comm. 2008:3001–3002. doi: 10.1039/b807278g. [DOI] [PubMed] [Google Scholar]; (d) Fustero S, Jiménez D, Sánchez-Roselló M, del Pozo C. J. Am. Chem. Soc. 2007;129:6700–6701. doi: 10.1021/ja0709829. [DOI] [PubMed] [Google Scholar]; (e) Bi H-P, Liu X-Y, Gou F-R, Guo L-N, Duan X-H, Shu X-Z, Liang Y-M. Angew. Chem. Int. Ed. 2007;46:7068–7071. doi: 10.1002/anie.200702238. [DOI] [PubMed] [Google Scholar]; (d) Zeng Y, Reddy DS, Hirt E, Aubé J. Org. Lett. 2004;6:4993–4995. doi: 10.1021/ol047809r. [DOI] [PubMed] [Google Scholar]
- (ii).Rolfe A, Young K, Hanson PR. Eur. J. Org. Chem. 2008:5254–5262. doi: 10.1002/ejoc.200800651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (iii).(a) Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Adv Drug. Del Rev. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]; (b) Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Adv. Drug. Del. Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]; (c) Oprea TI, Davis AM, Teague SJ, Leeson PD. J. Chem. Inf. Comput. Sci. 2001;41:1308–1315. doi: 10.1021/ci010366a. [DOI] [PubMed] [Google Scholar]; (d) Ghose AK, Viswanadhan VN, Wendoloski JJ. J. Comb. Chem. 1999;1:55–68. doi: 10.1021/cc9800071. [DOI] [PubMed] [Google Scholar]
- (iv).(a) Drews J. 2000;287:1960–1964. doi: 10.1126/science.287.5460.1960. [DOI] [PubMed] [Google Scholar]; (b) Scozzafaa A, Owa T, Mastrolorenzo A, Supuran CT. Curr. Med. Chem. 2003;10:925–953. doi: 10.2174/0929867033457647. [DOI] [PubMed] [Google Scholar]
- (v).Levy L. Drugs Future. 1992;17:451–454. [Google Scholar]
- (vi).Rabasseda X, Hopkins SL. Drugs Today. 1994;30:557–563. [Google Scholar]
- (vii).Hendrick RT, Spencer SR, Blake JF, Fell JB, Fischer JP, Stengel PJ, Leveque VJP, LePogam S, Rajyaguru S, Najera I, Joesy JA, Swallow S. Bioorg. Med. Chem. Lett. 2009;19:410–414. doi: 10.1016/j.bmcl.2008.11.060. [DOI] [PubMed] [Google Scholar]
- (viii).Zhuang L, Wai JS, Embrey MW, Fisher TE, Egbertson MS, Payne LS, Guare JP, Jr., Vacca JP, Hazuda DJ, Felock PJ, Wolfe AL, Stillmock KA, Witmer MV, Moyer G, Schleif WA, Gabryelski LJ, Leonard YM, Lynch JJ, Jr., Michelson SR, Young SD. J. Med. Chem. 2003;46:453–456. doi: 10.1021/jm025553u. [DOI] [PubMed] [Google Scholar]
- (ix).Valente C, Guedes RC, Moreira R, Iley J, Gut J, Rosental PJ. Biorg. Med. Chem. Lett. 2006;16:4115–4119. doi: 10.1016/j.bmcl.2006.04.079. [DOI] [PubMed] [Google Scholar]
- (x).Misu Y, Togo H. Org. Biomol. Chem. 2003;1:1342–1346. doi: 10.1039/b301330h. [DOI] [PubMed] [Google Scholar]
- (xi).Güzel Ö, Salman A. Bioorg. Med. Chem. 2006;14:7804–7815. doi: 10.1016/j.bmc.2006.07.065. [DOI] [PubMed] [Google Scholar]
- (xii).Cavasotto CN, Orry AJW, Murgolo NJ, Czarniecki MF, Kocsi SA, Hawes BE, O'Neil KA, Hine H, Burton MS, Voigt JH, Abagyan RA, Bayne ML, Monsma F,J., Jr J. Med. Chem. 2008;51:581–588. doi: 10.1021/jm070759m. [DOI] [PubMed] [Google Scholar]
- (xiii).(a) Bravo RD, Canepa AA. Syn. Comm. 2002;32:3675–3680. [Google Scholar]; (b) Orazi OO, Corral RA, Bravo R. J. Het. Chem. 1986;23:1701–1708. [Google Scholar]; (b) Katritzky AR, Wu J, Rachwal S, Rachwal B, Macomber DW, Smith TP. Org. Prep. Proced. Int. 1992;24:463–467. [Google Scholar]
- (xiv).Lee J, Zhong Y-L, Reamer RA, Askin D. Org. Lett. 2003;5:4175–4177. doi: 10.1021/ol0356183. [DOI] [PubMed] [Google Scholar]
- (xv).Chiacchio U, Corsaro A, Rescifina A, Bkaithan M, Grassi G, Piperno A. Tetrahedron. 2001;57:3425–3433. [Google Scholar]
- (xvi).(a) Metz P, Seng D, Fröhlich R. Synlett. 1996:741–742. [Google Scholar]; (b) Plietker B, Seng D, Fröhlich R, Metz P. Tetrahedron. 2000;56:873–879. [Google Scholar]; (c) Rogatchov VO, Bernsmann H, Schwab P, Fröhlich R, Wibbeling B, Metz P. Tetrahedron Lett. 2002;43:4753–4756. [Google Scholar]; (d) Greig IR, Trozer MJ, Wright PT. Org. Lett. 2001;3:369–371. doi: 10.1021/ol006863e. [DOI] [PubMed] [Google Scholar]
- (xvii).(a) Zhou A, Hanson PR. Org. Lett. 2008;10:2951–2954. doi: 10.1021/ol8009072. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhou A, Rayabarapu KD, Hanson PR. Org. Lett. 2009:531–534. doi: 10.1021/ol802467f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (xviii).(a) McReynolds MD, Dougherty JM, Hanson PR. Chem. Rev. 2004;104:2239–2258. doi: 10.1021/cr020109k. [DOI] [PubMed] [Google Scholar]; (b) Jiménez-Hopkins M, Hanson PR. Org. Lett. 2008;10:2951–2954. doi: 10.1021/ol8009072. and references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Jeon KO, Rayabarapu D, Rolfe A, Volp K, Omar I, Hanson PR. Tetrahedron. 2009 doi: 10.1016/j.tet.2009.03.080. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (xix).(a) Merten S, Fröhlich R, Kataeva O. Adv. Synth. Catal. 2005;347:754–758. [Google Scholar]; (b) Vasudevan A, Tseng P-S, Djuric SW. Tetrahedron Lett. 2006;47:8591–8593. [Google Scholar]; (c) Rayabarapu DK, Zhou A, Jeon KO, Samarakoon T, Rolfe A, Siddiqui H, Hanson PR. Tetrahedron. 2009;65:3180–3188. doi: 10.1016/j.tet.2008.11.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (xx).Liu X-Y, Li C-H, Che C-M. Org. Lett. 2006;8:2707–2710. doi: 10.1021/ol060719x. [DOI] [PubMed] [Google Scholar]
- (xxi).Dauban H, Dodd RH. Org. Lett. 2000;2:2327–2329. doi: 10.1021/ol000130c. [DOI] [PubMed] [Google Scholar]
- (xxii).Liang J-L, Yuan S-X, Chan PWH, Che C-M. Org. Lett. 2002;4:4507–4510. doi: 10.1021/ol0270475. [DOI] [PubMed] [Google Scholar]
- (xxiii).For probing chemical space for broad biological activity, it is many times desirable to assay racemic products due to potential variable activity between enantiomers of the same molecule. However, if a desirable biological hit is produced from this collection of compounds, the synthesis of each enantiomer will be pursued using either asymmetric catalysts or selective hydrogenation of the saturated derivative. Colantoni D, Fioravanti S, Pellacani L, Tardella P. Org. Lett. 2004;6:197–200. doi: 10.1021/ol0361554.. Shibuguchi T, Mihara H, Kuramochi, Ohshima O, Shibasaki M. Chem. Asian J. 2007;2:794–801. doi: 10.1002/asia.200700070..
- (xxiv). http://www.chemglass.com/products_new/np-reactionblock.html. Accessed 7-23-08.
- (xxv).The standards for compound purity and quantity are based on those requested for the National Institutes of Health Molecular Libraries Small Molecule Repository (http://mlsmr.glgp.com/MLSMR_HomePage/submitcompounds.html). The compound purities were determined by reverse-phase HPLC with peak area (UV) at 214 nm.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.