

Abstract
This study aimed to examine the applicability of polyethyleneimine (PEI)-modified magnetic nanoparticles (GPEI) as a potential vascular drug/gene carrier to brain tumors. In vitro, GPEI exhibited high cell association and low cell toxicity – properties which are highly desirable for intracellular drug/gene delivery. In addition, a high saturation magnetization of 93 emu/g Fe was expected to facilitate magnetic targeting of GPEI to brain tumor lesions. However, following intravenous administration, GPEI could not be magnetically accumulated in tumors of rats harboring orthotopic 9L-gliosarcomas due to its poor pharmacokinetic properties, reflected by a negligibly low plasma AUC of 12 ± 3 μg Fe/ml*min. To improve “passive” GPEI presentation to brain tumor vasculature for subsequent “active” magnetic capture, we examined the intra-carotid route as an alternative for nanoparticle administration. Intra-carotid administration in conjunction with magnetic targeting resulted in 30-fold (p = 0.002) increase in tumor entrapment of GPEI compared to that seen with intravenous administration. In addition, magnetic accumulation of cationic GPEI (ζ-potential = + 37.2 mV) in tumor lesions was 5.2-fold higher (p = 0.004) than that achieved with slightly anionic G100 (ζ-potential = −12 mV) following intra-carotid administration, while no significant accumulation difference was detected between the two types of nanoparticles in the contra-lateral brain (p = 0.187). These promising results warrant further investigation of GPEI as a potential cell-permeable, magnetically-responsive platform for brain tumor delivery of drugs and genes.
1. Introduction
Malignant brain tumors are one of the most lethal types of cancer, which has defied all currently available treatment modalities including surgery, radiotherapy and chemotherapy. Despite three decades of research, the median survival for patients with malignant glioma remains only 48 weeks from diagnosis[1]. Although many potent cytotoxic drugs and genes are now available, their delivery to brain tumor lesions faces formidable challenges[2, 3]. Several routes of administration, including craniotomy, intracerebral injection and the vascular route have been explored for delivery of pharmacological agents to the brain tumor site[4]. Craniotomy and intracerebral injections involve direct brain intervention and may be associated with a risk of neurological and neurocognitive sequelae, due to possible damage to the surrounding healthy brain parenchyma[5, 6]. The vascular route presents a safer alternative, especially for repeated drug administration. Unfortunately, low accumulation of blood-borne agents in glioma lesions diminishes the therapeutic benefit of many potent drugs[7]. To improve intravascular delivery of drugs and genes, extensive research efforts have been dedicated to the development of colloidal drug/gene carriers (e.g. liposomes, nanoparticles) which could increase drug accumulation in brain tumor lesions[8].
Magnetic nanoparticles, composed of an iron oxide core and polymeric shell, present a particularly promising carrier for enhanced tumor delivery of therapeutic agents[9]. High magnetic susceptibility of the iron oxide core enables non-invasive manipulation of magnetic nanoparticles by magnetic fields. Localized magnetically-assisted capture of blood-borne magnetic nanoparticles, termed “magnetic targeting”, has been shown to enhance delivery of drugs to subcutaneous tumor lesions in both preclinical and clinical settings[10–12]. We have recently demonstrated the plausibility of the magnetic targeting approach for brain tumor delivery of starch-coated iron-oxide nanoparticles in 9L-glioma bearing rats[13].
While the iron oxide core of the nanoparticles is responsible for magnetic retention, the nanoparticle surface properties determine the particle interaction with both the payload and the physiological milieu. Surface charge appears to be especially important with regards to these interactions. In particular, cationic nanoparticle surface was shown to present several drug/gene delivery advantages over its anionic and electroneutral counterparts. For example, magnetic nanoparticles functionalized with a polycationic polyethyleneimine (PEI) were demonstrated to bind DNA and act as efficient transfection agents in vitro[14]. In addition, a positive surface charge was reported to enhance nanoparticle cellular uptake[15] and confer strong avidity towards the anionic proteoglycans in tumor vasculature[16, 17]. However, albeit its advantages, the cationic surface also inflicts a negative effect on nanoparticle pharmacokinetics, as the positively charged nanoparticles are known to be cleared from plasma extremely rapidly. For example, a plasma half-life of only 1–2 minutes was reported for polylysine-bearing cationic nanoparticles[18].
Short plasma half-life poses a major limitation for use of cationic nanoparticles as intravascular drug/gene carriers. The delivery of the nanoparticles to tumor vasculature is an important pre-requisite for their magnetic entrapment within the glioma lesion. It has been previously demonstrated that the amount of agent passively delivered to the tumor over time is directly proportional to its area under the concentration-time curve (AUC) in the arterial blood[19]. Thus, rapid systemic clearance and low AUC of cationic nanoparticles suggest their low passive presentation at the glioma site. One strategy to overcome this shortcoming is through administration of the nanoparticles via the carotid artery. Carotid artery is a major artery supplying the brain and administration of chemotherapeutic drugs via this artery has been widely explored as a means to improve brain tumor delivery[20, 21]. An advantage accrued with the arterial administration versus the intravenous route is due to higher exposure of the tumor vasculature to the drug on its first pass through circulation[19]. A higher first pass exposure could be utilized to substantially improve magnetic capture of cationic nanocarriers within glioma lesions.
This study aimed to examine the applicability of cationic magnetic nanoparticles as potential magnetically-responsive carriers for drugs/genes to brain tumors via the vascular route, utilizing intra-carotid administration to overcome the pharmacokinetic drawback. To address this goal, we first developed a facile method for surface modification of commercially-available magnetic nanoparticles with cationic PEI moieties and evaluated their physical properties and interaction with 9L-glioma cells in vitro. We then proceeded to explore the in vivo benefit of intra-carotid versus intravenous administration in magnetically-assisted delivery of these nanoparticles to tumors of 9L-gliosarcoma bearing rats.
2. Materials and methods
2.1 Materials
Iron oxide nanoparticles, coated with starch or gum arabic polysaccharide matrix, were generously contributed by Chemicell® (Berlin, Germany). These particles are referred to as G100 and Gara, respectively, throughout this report. Low molecular weight polyethyleneimine (PEI, MW ~ 1200) was purchased from Sigma-Aldrich. 1-Ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride (EDC) and N-hydroxysulfosuccinimide (sulfo-NHS) were obtained from Pierce (Rockford, IL, USA).
2.2 Preparation and characterization of PEI-modified nanoparticles
Surface modification of carboxyl-bearing Gara with primary amine-containing PEI was carried out using EDC coupling chemistry[22]. Briefly, Gara nanoparticles were mixed with PEI, EDC and sulfo-NHS at a molar ratio of 1(Fe):1:2:2 (pH = 6). This reaction mixture was incubated at RT for 48 hours. Mixtures of nanoparticles and PEI, without activating reagents, were employed as controls. The modified nanoparticles were purified on a magnetic separator, with deionized water, until the presence of free amines in the supernatant could no longer be detected with the ninhydrin assay, described below. The resulting nanoparticles, bearing pendant PEI chains, were termed GPEI.
Quantification of primary and secondary amines on the nanoparticle surface was carried out by ninhydrin colorimetric assay[23]. The ninhydrin reagent (500 μL of 0.2% w/v in 0.1M buffer phosphate, pH 9) was added to 200 μL of nanoparticle sample and the mixture heated in a boiling water bath for 15 minutes. Samples were cooled to room temperature and placed on a magnetic separator to remove the nanoparticles. The absorbance of supernatants was measured at 570 nm on a microplate reader (Power-Wave 340, Bio-Tek Instruments, Winooski, VT) and the amine content quantified using ethanolamine standard curves.
Zeta potential and hydrodynamic diameter of purified GPEI and Gara were measured on a PSS Nicomp 380/ZLS Zeta Potential and Submicron Particle Size Analyzer (Nicomp, Santa Barbara, CA). Magnetization measurements of aqueous nanoparticle preparations were performed at 293 K using a MPMS-XL Superconducting Quantum Interference Device (SQUID) magnetometer (Quantum Design Inc. San Diego, CA). Iron concentrations of nanoparticle preparations were determined by Inductively Coupled Plasma – Optical Emission Spectroscopy (ICP-OES) using an Optima 2000 DV spectrometer (Perkin-Elmer, Boston MA) as previously described[13].
2.3 Cell uptake assay
Rat 9L-glioma cells (Brain Tumor Research Center, University of California, San Francisco) were seeded in a 6-well plate. Cells (1.5 × 105 cells/well) in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 100 IU/mL penicillin, 100 μg/mL streptomycin and 0.29 mg of L-glutamine (complete medium, 2 mL) were allowed to attach overnight at 37°C in a humidified atmosphere of 5% CO2. The medium was then carefully aspirated and substituted with 2 ml of serum-free DMEM (control) or serum-free DMEM containing G100 or GPEI nanoparticles at a concentration of 45 μg Fe/mL (test). Cells were incubated at 37°C for 1 hour, washed three times with serum-free DMEM to remove unbound nanoparticles and further incubated overnight with complete medium. For qualitative analysis, the medium was replaced with PBS and images of the cells were acquired with a digital camera using phase contrast microscopy. For quantitative analysis, the cells were harvested with 0.5 mL trypsin-EDTA (0.25% trypsin, 1 mM EDTA) and complete medium added to inhibit trypsin. The cells were counted in a hemacytometer and washed by five cycles of dispersion in PBS, centrifugation and supernatant removal. After the last centrifugation and aspiration of the supernatant, cells were resuspended in 100μL of PBS, transferred to ESR (electron spin resonance) tubes and kept at −80°C until analysis.
2.4 Assay for nanoparticle cytotoxicity
The cytotoxicity of nanoparticles to 9L-glioma cells was assessed with the MTT (3-[4,5-dimethyl-thiazol-2-yl]-2,5-diphenyltetrazolium bromide) cell viability assay. The assay was performed essentially as described elsewhere with minor modifications[24]. Briefly, 9L cells were seeded in a 96-well plate at a density of 2 × 103 cells/well in 100 μL DMEM complete medium and allowed to adhere overnight at 37°C in a humidified atmosphere of 5% CO2. The medium was then replaced with 100 μL of either serum-free DMEM (control) or serum-free DMEM containing G100 or GPEI nanoparticles at a concentration of 45 μg Fe/mL (test). Cells were incubated for 3 hours at 37°C, then washed with fresh DMEM and further incubated overnight with complete medium. Since phenol red was found to interfere with the MTT assay, DMEM not containing phenol red was used for the overnight incubation step (Invitrogen, 21063–029). MTT solution (20 μL of 6 mg/mL in PBS) was added to each well and cells were incubated for 2 hours at 37°C and 5% CO2. The medium was then carefully replaced with 100 μL DMSO and the plates were incubated for 30 minutes at RT to dissolve formazan crystals. The absorbance was measured at 550 nm using a microplate reader (PowerWave 340, Bio-Tek instruments, Winooski, VT). To calculate the number of cells from the measured absorbance values, a calibration curve was constructed with known 9L cell concentrations, counted with a hemacytometer. The calibration curve was found to be linear within a range of 2 × 103–6 × 104 9L cells.
2.5 In vivo studies
All animal experiments were conducted according to protocols approved by the University of Michigan Committee on Use and Care of Animals (UCUCA).
2.5.1. Pharmacokinetic analysis
The pharmacokinetics of G100 and GPEI magnetic nanoparticles was studied in male Fisher 344 rats weighting 200–250 g. The nanoparticles were administered intravenously via tail vein and the blood was sampled through the cannulated carotid artery. The animals were anaesthetized by intraperitoneal injection of ketamine/xylazine mixture (87/13 mg/kg body weight). The left carotid artery of the animals was exposed by blunt dissection and ligated rostrally to occlude the flow. PE-10 tubing was then inserted caudally via a small incision in the arterial wall and secured in place by ligation. The intracarotid catheter was flushed with Heparin flush solution (Hepflush-10, 10 USP Units/ml, Abraxis Pharmaceutical Products, IL) and clamped. Tail veins of the animals were cannulated with a 26-gauge angiocatheter (Angiocath™, Becton Dickinson, Sandy, UT). The nanoparticle suspension in PBS was administered to rats via the tail vein catheter at a dose of 12 mg Fe/kg body weight. Blood samples of 100 μL were collected from the cannulated carotid artery in 0.5 mL Eppendorf tubes spiked with 10 μL of Heparin solution (5,000 USP Units/ml). Samples were acquired before and serially after nanoparticle administration at pre-set time intervals over 60 minute duration. Plasma fractions were immediately separated by centrifugation (3 minutes at 7000×g) and stored at −80°C until analysis by ESR, detailed below.
2.5.2 Induction of brain tumors
Intracerebral 9L tumors were induced in male Fisher 344 rats weighing 125–150 g according to a previously described procedure[25]. Briefly, rat 9L-glioma cells (Brain Tumor Research Center, University of California, San Francisco) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum, 100 IU/mL penicillin, 100 μg/mL streptomycin and 0.29 mg of L-glutamine at 37°C in a humidified atmosphere of 5% CO2. Prior to implantation, cells were grown to confluency in 100 mm culture dishes, harvested and resuspended in serum free DMEM at a concentration of ~105 cells/μL. The cell suspension (10 μL) was implanted in the right forebrain of the animals at a depth of 3 mm beneath the skull through a 1-mm-diameter burr hole. The surgical field was cleaned with 70% ethanol and the burr hole was filled with bone wax (Ethicon Inc., Summerfield, NJ) to prevent extracerebral extension of the tumor. The tumor volume of the animals was monitored with MRI beginning on day 10 after cell implantation to select tumors between 70 and 90 μL for magnetic targeting experiments.
2.5.3 Magnetic Resonance Imaging (MRI) studies
MRI experiments were performed on an 18-cm horizontal-bore, 7 Tesla Varian Unity Inova imaging system (Varian, Palo Alto, CA). Animals were anesthetized with 1.5 % isoflurane/air mixture and imaged using a 35-mm-diameter quadrature RF head coil (USA Instruments Inc, OH). Animals were maintained at 37°C inside the magnet using a thermostated circulating water bath. To visualize the tumor localization within the rat brain, 13 axial sections of the brain were acquired with a T2-weighted fast spin echo sequence using the following parameters: repetition time (TR) = 4 s, echo time (TE) = 60 ms, field of view = 30 × 30 over 128 × 128 matrix, slice thickness = 1 mm, slice separation = 2 mm, four signal averages per phase encoding step. To determine nanoparticle distribution in the brain, 13 gradient echo (GE) axial slices of the brain were collected before the nanoparticle administration (baseline scans) and immediately following magnetic targeting. GE images were acquired with the following parameters: TR = 20 ms, TE = 5 ms, field of view = 30 × 30 over 128 × 128 matrix, slice thickness = 1 mm.
2.5.4 Magnetic targeting
Magnetic targeting studies were carried out in tumor-bearing rats using either the intravenous or intra-arterial route for nanoparticle administration. For intravenous administration, the tail vein was cannulated with a 26-gauge angiocatheter (Angiocath™, Becton Dickinson, Sandy, UT). For intra-arterial administration, the right carotid artery was cannulated according to a previously described method [26].
Our magnetic setup was optimized to achieve sharp gradient of magnetic flux density at the target location. Briefly, a small cylindrical neodymium-iron-boron magnet (NdFeB, 9 mm in diameter) was attached to the pole of a dipole electromagnet (GMW associates, Model 3470). The animals were placed supinely on a platform with their heads positioned directly on the pole face of the small magnet. The magnetic field density at the pole face of the small magnet was adjusted to 350 mT. Animals were then injected with nanoparticle suspension at a dose of 12 mg Fe/kg through either an intravenous or intra-carotid catheter, and retained in the magnetic field for 30 min. The rats were imaged with MRI before the administration of nanoparticles and after the magnetic targeting as described in Section 2.5.3. Immediately following MRI, the animals were sacrificed, dissected and the isolated brain divided into right and left hemispheres. The tumor was carefully separated from the normal tissue of the right hemisphere. The left hemisphere and tumor tissues were frozen and kept at −80°C until analysis by ESR.
2.6 Ex vivo analysis of tissue and plasma samples by Electron Spin Resonance (ESR) spectroscopy
Nanoparticle concentrations were determined by ESR spectroscopy by a previously described method [27]. Briefly, ESR spectra of the samples were acquired using an EMX ESR spectrometer (Bruker Instruments Inc., Billerica, MA) equipped with a liquid nitrogen cryostat. The acquisition parameters were: resonant frequency: ~9.2 GHz, microwave power: 20 mW, temperature: 145 K, modulation amplitude: 5 G and receiver gain of 5 × 104 and 5 × 103 for tissue and plasma samples, respectively. Due to the 100 kHz modulation of the magnetic field, the measured signal was the derivative dP/dH of the absorbed microwave power with respect to the external static field H. The integral ∫P(H)dH is known to be proportional to the amount of resonating electronic spins present in the sample. Therefore, the double integral of the ESR spectra of samples was calculated to quantify the nanoparticles. Calibration curves were constructed with nanoparticle solutions of known iron concentrations. The data were corrected for background tissue absorption using control tissue samples from animals not exposed to nanoparticles or control plasma samples collected prior to nanoparticle injection.
2.7 Quantitative data analysis
The tumor delivery advantage, Rd, resulting from arterial nanoparticle administration as compared to the intravenous route was calculated by a previously described equation [19]:
| [1] |
Where D is the dose, F the blood flow through the infused artery, and CO the cardiac output; Ira and Irv are the arterial AUCs for the systemic recirculation of the substance after arterial and venous administration, respectively.
The AUC was estimated numerically by a linear trapezoidal integration method; integration was performed on the interval of 0–30 minutes after nanoparticle administration, corresponding to the duration of magnetic targeting.
2.8 Statistical analysis
Data are presented as mean ± SE, unless indicated otherwise.
SPSS 15.0 statistical software package (SPSS Inc., Chicago, IL) was used for data analysis. Group means were compared using an unpaired two-tailed t-test (experiments comparing two groups of cells/animals) or one-way ANOVA (experiment comparing 3 groups of cells). The assumption of the homogeneity of variances was assessed using Levene’s test. Statistical difference within data sets (two groups) that were found to violate the assumption of the homogeneity of variances was inferred by Welch-Satterthwaite t-test not assuming equal variances. A p-value of <0.05 was considered statistically significant.
3. Results
3.1 GPEI formation and characterization
GPEI nanoparticles were prepared by covalent grafting of low molecular weight branched PEI chains onto the surface of Gara as depicted in Figure 1. Since primary and secondary amines constitute about 75% of all PEI amino groups [14], the success of PEI surface grafting can be readily assessed with the ninhydrin assay [28]. Ninhydrin reacts with immobilized amines to generate a soluble Ruhemann’s purple chromophore, which can be detected in the supernatant [23]. The absorbance of the Ruhemann’s complex at 570 nm is proportional to the surface amine density. Figure 2 demonstrates that, following thorough purification, covalently modified GPEI nanoparticles exhibited 14-fold higher absorbance at 570 nm than the physical mixture of Gara and PEI at the same nanoparticle concentration. Since no loss of amine groups was detected with additional purification of GPEI, the high amine content of the GPEI surface can be attributed to covalent PEI grafting and not to physical surface adsorption.
Figure 1.
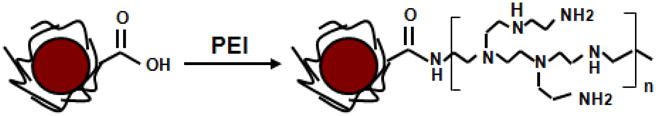
Surface modification of carboxyl-bearing Gara nanoparticles with PEI
Figure 2.
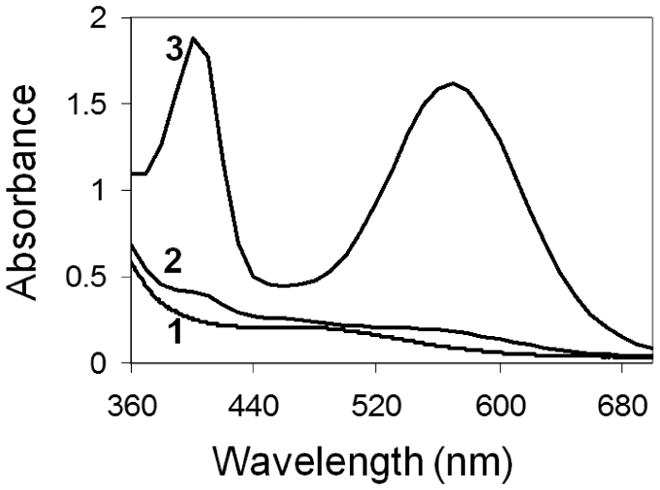
Analysis of surface amine content of (1) Gara, (2) purified Gara/PEI mixture, and (3) GPEI nanoparticles using ninhydrin colorimetric assay – absorbance of the Ruhemann’s purple chromophore at 570 nm is proportional to the amine content
Grafting of PEI to the surface of amine-bearing magnetic nanoparticles was previously accomplished with a two-step procedure, requiring glutaraldehyde as a crosslinker [14]. In our study, PEI chains were attached directly to carboxyl-bearing nanoparticle surface via a one-step EDC coupling chemistry. A previously reported procedure resulted in nitrogen content of 0.635 μmol per mg of nanoparticles, corresponding to about 15.5 nmol PEI (MW ~ 1800 Da) per mg of nanoparticles. In our study, the amine density on the nanoparticle surface was estimated to be 0.437 μmol/mg nanoparticles. This amine content corresponds to about 15.6 nmol PEI (MW ~ 1200 Da) per mg of nanoparticles. Thus, in spite of the simplified procedure employed, our PEI grafting efficiency was comparable to that achieved in the previous study.
Further physical characterization of GPEI nanoparticles included evaluation of ζ-potential, size distribution and magnetic properties. Measurement of ζ-potential served to further corroborate the success of surface PEI grafting. At a pH of 5.5, the ζ–potential of Gara was measured to be −36.1 mV. This negative ζ-potential can be ascribed to the high content of glucoronic acid found in Gara surface coating as reported by the manufacturer. In contrast, GPEI exhibited a positive ζ-potential of +37.2 mV at the same pH. The shift in ζ-potential (Figure 3A) can clearly be attributed to the PEI graft, which provided GPEI surface with a high density of protonable amine groups. Mean hydrodynamic diameter of the nanoparticles was found to increase slightly from 189 nm for Gara to 225 nm for GPEI due to PEI grafting (Figure 3B). Yet, GPEI still exhibited monomodal size distribution with a polydispersity index (PDI) of 0.147, comparable to that of the starting material Gara (PDI = 0.136). Measurement of induced magnetization by SQUID magnetometry showed no significant alteration in nanoparticle magnetic properties due to surface PEI grafting (Figure 3C). Magnetization curves exhibited no hysteresis and no remanent magnetization, consistent with superparamagnetic behavior. Saturation magnetization of GPEI was found to be 93 emu/g Fe, suggesting that GPEI should be amenable to magnetic targeting.
Figure 3.

Characterization of GPEI nanoparticles as compared to the starting material Gara. A.ζ-potential (at pH=5.5) measured by electrophoretic light scattering, B. particle size distribution determined by dynamic light scattering, C. induced magnetization measured with MPMS-XL SQUID magnetometer
The in vitro and the in vivo behavior of the GPEI nanoparticles were assessed and compared to that of commercially available starch-coated magnetic nanoparticles (G100). G100 nanoparticles have been extensively studied in magnetic targeting applications. The physical properties of G100 nanoparticles are summarized in Table 1.
Table 1.
Physical properties of G100 magnetic nanoparticles. Data expressed as MEAN ± SD
3.2 Cell uptake and cytotoxicity
Many therapeutic agents exert their pharmacological action inside the cell. Therefore, the ability of a nanocarrier to internalize into cells can augment its drug delivery performance. The interaction of GPEI and G100 nanoparticles with 9L-glioma cells was assessed qualitatively with optical microscopy and quantitatively with ESR spectroscopy. Optical micrographs, obtained after nanoparticle incubation with 9L-glioma cells and extensive washing, revealed appearance of dark pigmentation in the GPEI-exposed but not the G100-exposed cells (Figure 4A). This pigmentation qualitatively indicated enhanced cellular uptake of GPEI compared to G100. Quantitative analysis further demonstrated that the extent of cellular uptake was 138–fold higher (p = 0.013, Figure 4B) for GPEI (58 ± 17 pg Fe/cell) than for the G100 (0.42 ± 0.05 pg Fe/cell). Despite the significantly enhanced cellular uptake, the polycationic nanoparticle surface did not increase cell toxicity compared to the slightly anionic G100, as MTT cell viability test revealed no significant difference (p = 0.712) between the GPEI-treated, G100-treated and untreated control cells (Figure 4C).
Figure 4.
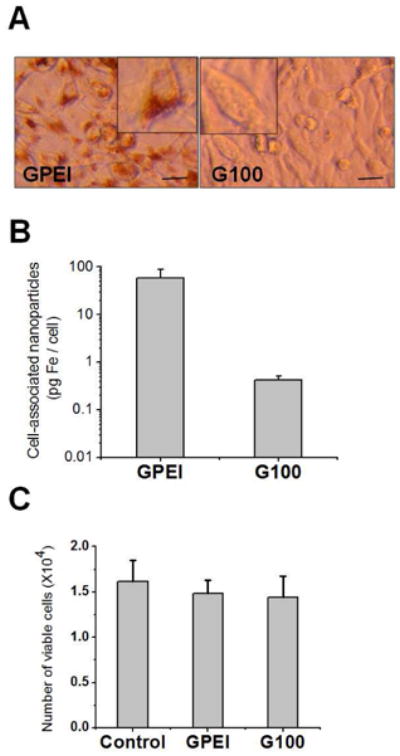
Interaction of G100 and GPEI nanoparticles with 9L-glioma cells. A. Optical micrographs of 9L-glioma cells incubated with GPEI and G100. Scale bar = 20 μm. [Inset: Digital magnification (2×) of representative cells demonstrates increased uptake of GPEI compared to G100] B. Uptake of GPEI and G100 nanoparticles by 9L-glioma cells quantified by EPR spectroscopy. C. Cytotoxicity assessment of GPEI and G100 nanoparticles to 9L-glioma cells using MTT cell viability assay.
3.3 Magnetic targeting of GPEI with intravenous administration
The promising in vitro results prompted us to examine the in vivo feasibility of delivering GPEI to tumors of 9L-glioma bearing rats via magnetic targeting. We first attempted to deliver GPEI via intravenous administration. Magnetic resonance imaging (MRI) was used to validate nanoparticle delivery to the tumor tissue. The tumor lesion is visible as a hyperintense region on the T2-weighted MRI baseline scans of the brain (Figure 5: T2 baseline). The presence of iron oxide nanoparticles at a particular spatial location is reflected by a pronounced hypointensity (negative contrast) on gradient echo (GE) MRI scans. Axial GE brain scan, obtained after magnetic targeting of G100 nanoparticles, shows a hypointense region corresponding to the tumor location (Figure 5A). The loss of signal indicates G100 accumulation within the tumor lesion. In contrast, with GPEI administration no signal reduction is observed on the post-targeting GE scan compared to the baseline (Figure 5B). The lack of signal reduction indicates that GPEI nanoparticles were not accumulated in the tumor lesion.
Figure 5.
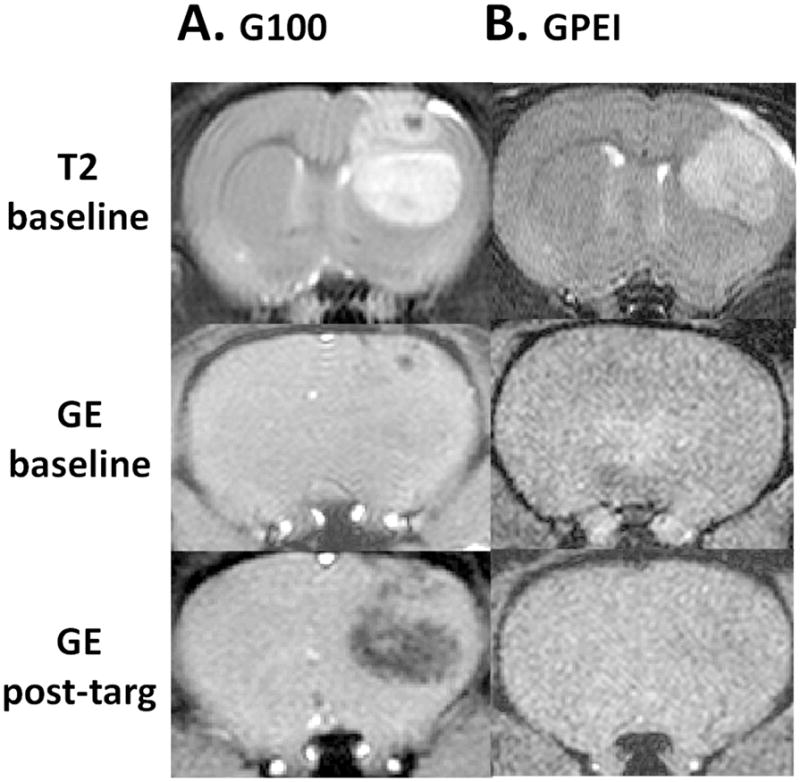
Representative subsets of axial MRI head-scans of 9L-glioma bearing rats before (baseline) intravenous administration of (A) G100 or (B) GPEI and after magnetic targeting (post-targ).
Our previous work established that magnetic nanoparticles have to be passively delivered to the brain tumor vasculature in order to be subjected to magnetic capture [13]. To assess the efficiency of GPEI passive delivery, we determined GPEI concentrations in arterial plasma over time following intravenous administration. The carotid artery ipsilateral to the tumor was used for blood collection to accurately reflect the arterial input of nanoparticles to the tumor microvasculature. Figure 6A presents the resulting pharmacokinetic profile of GPEI compared to that of G100 after intravenous administration of both types of nanoparticles at the same dose of 12 mg Fe/kg. It can be seen that the positive surface charge has induced substantial deterioration in nanoparticle pharmacokinetic profile, as the GPEI plasma AUC (12 ± 3 μg Fe/ml*min, Figure 6B) was found to be 78–fold lower (p = 0.002) than that of G100 (934 ± 44 μg Fe/ml*min).
Figure 6.
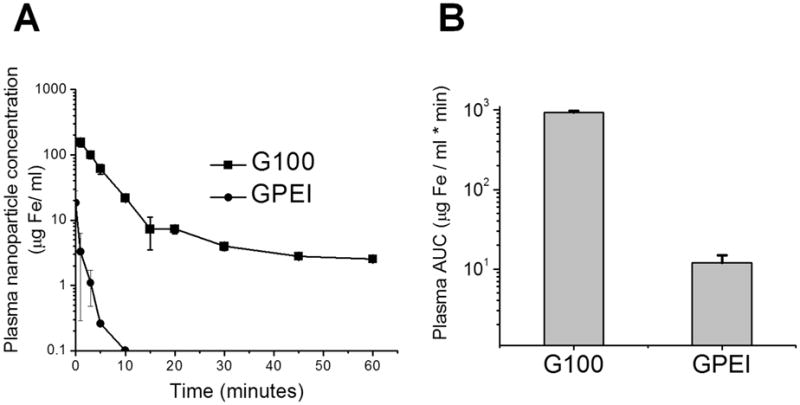
A. Plasma concentrations of magnetic nanoparticles after intravenous administration of G100 and GPEI at a dose of 12 mg Fe/kg. B. Plasma AUC calculated for the concentration-time profiles of G100 and GPEI
The low systemic AUC of GPEI nanoparticles prompted us to examine administration of GPEI via the intra-carotid route to enhance nanoparticle presentation to tumor vasculature. Passive tumor delivery advantage resulting from administration of an agent via intra-arterial versus intravenous route, Rd, can be estimated using Equation [1] (Section 2.7). Assuming that the fraction of total dose lost during first passage through the tumor vasculature is small; the systemic recirculation AUC for intra-arterial administration Ira is approximately equal to Irv. Based on this assumption and using literature values for the carotid blood flow and cardiac output measured in Fisher rats (see Table 2), the value of Rd was estimated to be 27, clearly warranting intra-carotid administration.
Table 2.
Parameters used for estimation of the advantage of intra-carotid GPEI nanoparticle administration, Rd
3.4 Magnetic targeting of GPEI with intra-carotid administration
An experimental advantage of intra-carotid administration with respect to the GPEI magnetic capture is clearly visible on the representative MRI images of Figure 7. In animals which were administered with GPEI via carotid artery, the post-targeting GE MRI scan of the brain showed a pronouncedly hypointense region (Figure 7A). This loss of signal within the region of the image corresponding to the tumor location reveals accumulation of nanoparticles in the tumor lesion. In contrast, in animals administered with GPEI via the intravenous route, no difference could be visually discerned between the post-targeting and the baseline GE scans (Figure 7B). Quantitative determination of nanoparticle concentrations in excised tumor tissues further revealed that intra-carotid administration of GPEI resulted in a 30-fold higher glioma accumulation of nanoparticles (222.8 ± 66.7 nmol Fe/g tissue, p = 0.002) compared to that achieved with intravenous administration (7.3 ± 0.8 nmol Fe/g tissue).
Figure 7.
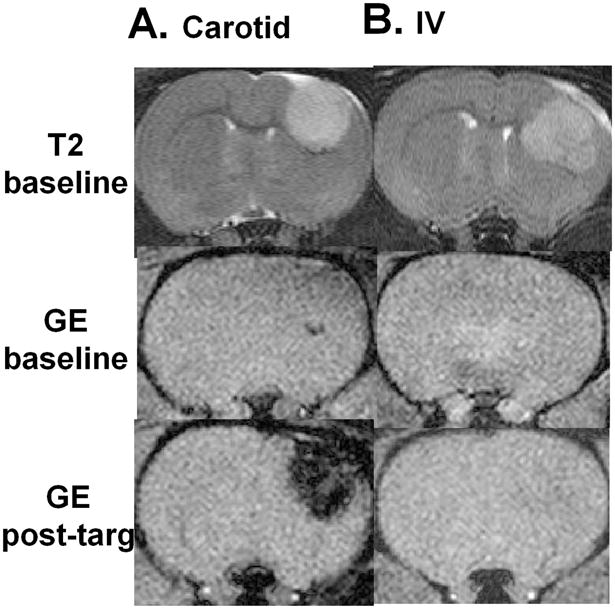
Representative subsets of axial MRI head-scans of 9L-glioma bearing rats before (baseline) (A) intra-carotid or (B) intravenous administration of GPEI and after magnetic targeting (post-targ).
We next aimed to assess the role of the positive surface charge in magnetic tumor targeting of nanoparticles following intra-carotid administration. To this regard, we compared the glioma magnetic capture of GPEI and G100, both administered at the same dose of 12 mg Fe/kg via the intra-carotid route. The results of quantitative nanoparticle analysis in excised tumor and contra-lateral brain tissues are presented in Figure 8. Tumor accumulation of GPEI nanoparticles (Figure 8) was found to be 5.2-fold higher (222.8 ± 66.7 nmol Fe/g tissue, p = 0.004) than that of the G100 nanoparticles (42.7 ± 19.5 nmol Fe/g tissue). In contrast to tumor tissue, no significant difference was found between concentrations of GPEI (8.3 ± 3.1 nmol Fe/g tissue) and G100 (4.6 ± 1.1 nmol Fe/g) in the contra-lateral brain tissues (p = 0.187).
Figure 8.

(A) Nanoparticle accumulation in tumor and contra-lateral brain tissues in magnetically targeted rats after intra-carotid administration of G100 or GPEI at a dose of 12 mg Fe/kg; (B) Target selectivity of GPEI and G100 nanoparticles for tumor versus contra-lateral brain tissue.
4. Discussion
Intravascular delivery of therapeutic agents to brain tumor lesions is one of the most challenging goals of chemotherapy. High local drug concentrations are required to eradicate the tumor mass. In addition, selectivity of brain tumor delivery is of utmost importance due to the high sensitivity of the normal brain to damage that can compromise functional capacity and exacerbate morbidity [2]. Our previous studies revealed that enhanced brain tumor accumulation of iron-oxide based nanoparticles, a potential carrier of therapeutic agents, could be achieved with magnetic targeting [13]. Nanocarriers bearing positive surface charge were previously demonstrated to avidly associate with tumor cells and tumor microvasculature [29, 30], and thus could potentially enhance the selectivity of tumor targeting. The present work investigated magnetically-targeted delivery of model cationic magnetic nanoparticles, GPEI, which combine the characteristics of the cationic surface with the magnetic properties of an iron oxide core.
GPEI were found to exhibit superparamagnetic behavior; i.e. accrual of a net magnetic moment only when subjected to an external magnetic field [31]. The magnetic field source employed in the present study was optimized to generate a short-range, sharp-gradient magnetic field topography to avoid non-specific nanoparticle aggregation away from the target site. Due to the rapid decay of the magnetic field density with distance from the tumor site, blood-borne nanoparticles have to be brought in close proximity to the region of peak magnetic flux in order to experience a force of magnetic attraction sufficient for capture. Therefore, the targeting procedure here employed essentially constitutes a combination of both passive and active targeting approaches. While the active targeting component is associated with magnetic interaction, the passive component is responsible for delivering the nanoparticles into the region of magnetic flux gradient via the vascular route.
Passive delivery to brain tumor lesion relies upon the pharmacokinetic properties of the nanocarriers. Pharmacokinetics of starch-coated G100 nanoparticles allowed successful presentation of these slightly negatively charged nanoparticles (ζ-potential = −12 ± 6 mV; Table 1) to the tumor vasculature, thus facilitating magnetic entrapment. In contrast, a positive surface charge (ζ-potential = +37.2 mV, Figure 2) was found to abolish GPEI nanoparticle delivery to brain tumor lesions following intravenous administration. Rapid opsonization with negatively charged plasma proteins, reported for other cationic nanoparticles[32], could provide a plausible explanation for the expedited plasma clearance of GPEI. Irrespective of the mechanism, a negligibly low AUC of GPEI (12 ± 3 μg Fe/ml*min) indicated that the failure of their magnetic accumulation within the tumor could be clearly attributed to their inability to passively reach the tumor site.
To improve the passive delivery of GPEI nanoparticles to brain tumor vasculature, we examined administration of nanoparticles via the carotid artery. Intra-carotid administration is a clinically-viable alternative to the intravenous route for brain tumor delivery of therapeutic agents [33, 34]. Previous theoretical work revealed that the extent of intra-arterial delivery advantage over the intravenous route, termed Rd, can be predicted based on physiological parameters and the pharmacokinetic profile of the agent after systemic bolus injection [19]. Theoretical estimate of Rd calculated for GPEI nanoparticles suggested 27-fold higher exposure of the tumor vasculature to GPEI following intra-carotid administration as compared with the intravenous route. Interestingly, experimental data revealed 30-fold higher magnetic accumulation of GPEI in tumor lesion with intra-carotid versus intravenous administration, consistent with theoretical predictions. These results underscore the remarkable benefit of intra-arterial administration in conjunction with magnetic targeting. Moreover, the Rd parameter may serve as a useful predictor of the intra-carotid administration advantage in magnetic brain tumor targeting.
The major finding of the present study was that surface modification with polycationic PEI resulted in 5.2-fold higher tumor accumulation of GPEI nanoparticles as compared with G100. Furthermore, the nanoparticle accumulation enhancement was found to be tumor specific and was not accompanied by a corresponding rise of nanoparticle concentration in the normal brain. Compromised blood-brain-barrier functions in gliomas, manifested by multiple structural abnormalities including interendothelial gaps and fenestrations, were shown to result in increased microvascular permeability to macromolecules and nanoparticles [35]. Cationic microspheres of about 1–2 μm in diameter, which are over 4-fold larger than GPEI nanoparticles employed in the present study, were previously found to localize in the interstitial space of an experimental brain tumor [36]. Thus, our nanoparticles are likely to extravasate upon their passage through the tumor vasculature and come into contact with tumor cells. The present study demonstrated that in vitro association of 9L-glioma cells with GPEI was over 100-fold higher than that with G100. It is therefore possible that following removal of the external magnetic field, a fraction of magnetically-retained G100 nanoparticles demagnetize, leak into vasculature and are washed away from the target site by blood flow, while demagnetized, cationic GPEI nanoparticles remain electrostatically bound to the tumor and endothelial cells, thus resulting in higher tumor accumulation and selectivity. However, additional studies are required to elucidate the mechanism of this phenomenon.
Although delivery of cationic magnetic microspheres to brain tumor lesions has been assessed before[36], brain tumor targeting of PEI-bearing magnetic nanoparticles has not been previously examined. The particular appeal of magnetic nanoparticles surface-grafted with PEI lays in the possibility of their utilization for intracellular delivery of drugs and genes. High intracellular uptake of GPEI demonstrated in this study can be potentially utilized to increase the intracellular load of therapeutic compounds and circumvent mechanisms of drug resistance[37]. Most interestingly, previous investigation showed that PEI-bearing magnetic nanoparticles with properties similar to those of GPEI exhibited high DNA binding capacity and high transfection efficiency in vitro[14]. Furthermore, many promising tumoricidal agents including suicide genes and apoptosis inducing genes have already been developed[2] and can be tailored to achieve high tumor specificity[38]. Yet, evolution of these agents into therapeutic modalities for brain tumor treatment awaits development of low-toxicity brain tumor carriers [2]. To this regard, our methodology for in vivo brain tumor delivery of low-toxicity cell-permeant GPEI may offer an attractive way to enhance the therapeutic potential of tumor-specific cytotoxic agents.
5. Conclusions
Results presented reveal that cationic magnetic nanoparticles GPEI, explored in this study, exhibit high cell penetration ability and low cell toxicity – properties which are highly desirable for intracellular drug/gene delivery. In addition, GPEI could be magnetically captured in glioma lesions following clinically-viable intra-carotid administration. Furthermore, the extent of GPEI accumulation was 5.2-fold higher than that of G100 in the tumor lesions, but not in the contra-lateral normal brain, revealing higher target selectivity of cationic nanoparticles. These results warrant further investigation of GPEI as a potential nanocarrier for drug/gene delivery to glioma lesions.
Acknowledgments
We thank Dr. Christian Bergemann from Chemicell (Germany) for the generous gift of iron oxide nanoparticles. We acknowledge the Center for Molecular Imaging at the University of Michigan for providing access to the MRI facility. This work was supported in part by NIH RO1 Grants CA114612 and NS066945, as well as a research grant from the Hartwell Foundation. This research was also partially sponsored by Grant R31-2008-000-10103-01 from the World Class University (WCU) project of the MEST and NRF of South Korea. Victor C. Yang is currently a Participating Faculty in the Department of Molecular Medicine and Biopharmaceutical Sciences, College of Medicine & College of Pharmacy, Seoul National University, South Korea. Beata Chertok was a recipient of the University of Michigan Rackham Graduate School Pre-doctoral Fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fisher PG, Buffler PA. Malignant gliomas in 2005: Where to go from here? JAMA. 2005;293(5):615–7. doi: 10.1001/jama.293.5.615. [DOI] [PubMed] [Google Scholar]
- 2.Lam P, Breakefield X. Potential of gene therapy for brain tumors. Hum Mol Genet. 2001;10(7):777–87. doi: 10.1093/hmg/10.7.777. [DOI] [PubMed] [Google Scholar]
- 3.Blasberg RG, Groothuis DR. Chemotherapy of brain tumors: Physiological and pharmacokinetic considerations. Semin Oncol. 1986;13(1):70–82. [PubMed] [Google Scholar]
- 4.Gutman RL, Peacock G, Lu DR. Targeted drug delivery for brain cancer treatment. J Control Release. 2000;65(1–2):31–41. doi: 10.1016/s0168-3659(99)00229-1. [DOI] [PubMed] [Google Scholar]
- 5.Packer R. Brain tumors in children. Arch Neurol. 1999;56(4):421–5. doi: 10.1001/archneur.56.4.421. [DOI] [PubMed] [Google Scholar]
- 6.Weitzner MA, Meyers CA. Cognitive functioning and quality of life in malignant glioma patients: A review of the literature. Psychooncology. 1997;6(3):169–77. doi: 10.1002/(SICI)1099-1611(199709)6:3<169::AID-PON269>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 7.Groothuis DR. The blood-brain and blood-tumor barriers: A review of strategies for increasing drug delivery. Neuro Oncol. 2000;2(1):45–59. doi: 10.1093/neuonc/2.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garcia-Garcia E, Andrieux K, Gil S, Couvreur P. Colloidal carriers and blood-brain barrier (bbb) translocation: A way to deliver drugs to the brain? Int J Pharm. 2005;298(2):274–92. doi: 10.1016/j.ijpharm.2005.03.031. [DOI] [PubMed] [Google Scholar]
- 9.Dobson J. Magnetic nanoparticles for drug delivery. Drug Dev Res. 2006;67(1):55–60. [Google Scholar]
- 10.Alexiou C, Arnold W, Klein RJ, Parak FG, Hulin P, Bergemann C, et al. Locoregional cancer treatment with magnetic drug targeting. Cancer Res. 2000;60(23):6641–8. [PubMed] [Google Scholar]
- 11.Lubbe AS, Bergemann C, Huhnt W, Fricke T, Riess H, Brock JW, et al. Preclinical experiences with magnetic drug targeting: Tolerance and efficacy. Cancer Res. 1996;56(20):4694–701. [PubMed] [Google Scholar]
- 12.Lubbe AS, Bergemann C, Riess H, Schriever F, Reichardt P, Possinger K, et al. Clinical experiences with magnetic drug targeting: A phase I study with 4′-epidoxorubicin in 14 patients with advanced solid tumors. Cancer Res. 1996;56(20):4686–93. [PubMed] [Google Scholar]
- 13.Chertok B, Moffat B, David A, Yu F, Bergemann C, Ross B, et al. Iron oxide nanoparticles as a drug delivery vehicle for MRI monitored magnetic targeting of brain tumors. Biomaterials. 2008;29(4):487–496. doi: 10.1016/j.biomaterials.2007.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McBain S, Yiu H, El Haj A, Dobson J. Polyethyleneimine functionalized iron oxide nanoparticles as agents for DNA delivery and transfection. J Mater Chem. 2007;17(24):2561–2565. [Google Scholar]
- 15.Koch A, Reynolds F, Kircher M, Merkle H, Weissleder R, Josephson L. Uptake and metabolism of a dual fluorochrome tat-nanoparticle in hela cells. Bioconjugate Chem. 2003;14(6):1115–1121. doi: 10.1021/bc034123v. [DOI] [PubMed] [Google Scholar]
- 16.Campbell R, Fukumura D, Brown E, Mazzola L, Izumi Y, Jain R, et al. Cationic charge determines the distribution of liposomes between the vascular and extravascular compartments of tumors. Cancer Res. 2002;62(23):6831–6836. [PubMed] [Google Scholar]
- 17.Vincent S, Depace D, Finkelstein S. Distribution of anionic sites on the capillary endothelium in an experimental brain-tumor model. Microcirc Endoth Lym. 1988;4(1):45–67. [PubMed] [Google Scholar]
- 18.Papisov M, Bogdanov A, Schaffer B, Nossiff N, Shen T, Weissleder R, et al. Colloidal magnetic-resonance contrast agents - effect of particle surface on biodistribution. J Magn Magn Mater. 1993;122(1–3):383–386. [Google Scholar]
- 19.Eckman WW, Patlak CS, Fenstermacher JD. A critical evaluation of the principles governing the advantages of intra-arterial infusions. J Pharmacokinet Biopharm. 1974;2(3):257–85. doi: 10.1007/BF01059765. [DOI] [PubMed] [Google Scholar]
- 20.Yamada K, Ushio Y, Hayakawa T, Arita N, Huang T, Nagatani M, et al. Distribution of radiolabeled 1-(4-amino-2-methyl-5-pyrimidinyl)methyl-3-(2-chloroethyl)-3-nitrosourea hydrochloride in rat-brain tumor - intraarterial versus intravenous administration. Cancer Res. 1987;47(8):2123–2128. [PubMed] [Google Scholar]
- 21.Chiras J, Dormont D, Fauchon F, Debussche C, Bories J. Intra-arterial chemotherapy of malignant gliomas. J Neuroradiol. 1988;15(1):31–48. [PubMed] [Google Scholar]
- 22.Holzapfel V, Lorenz M, Weiss CK, Schrezenmeier H, Landfester K, Mailander V. Synthesis and biomedical applications of functionalized fluorescent and magnetic dual reporter nanoparticles as obtained in the miniemulsion process. J Phys: Condens Matter. 2006;18:S2581–S2594. [Google Scholar]
- 23.Kaiser E, Colescott RL, Bossinger CD, Cook PI. Color test for detection of free terminal amino groups in the solid-phase synthesis of peptides. Anal Biochem. 1970;34(2):595–8. doi: 10.1016/0003-2697(70)90146-6. [DOI] [PubMed] [Google Scholar]
- 24.Mosmann T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(1–2):55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 25.Ross B, Zhao Y-J, Neal E, Stegman L, Ercolani M, Ben-Yoseph O, et al. Contributions of cell kill and posttreatment tumor growth rates to the repopulation of intracerebral 9L tumors after chemotherapy: An mri study. Proc Natl Acad Sci USA. 1998;95:7012–7017. doi: 10.1073/pnas.95.12.7012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rodriguez M, Barraso N. An improved method for carotid-artery infusion without vessel occlusion. Physiology & Behavior. 1992;52(6):1211–1213. doi: 10.1016/0031-9384(92)90485-k. [DOI] [PubMed] [Google Scholar]
- 27.Chertok B, Cole AJ, David AE, Yang VC. Comparison of electron spin resonance spectroscopy and inductively-coupled plasma optical emission spectroscopy for biodistribution analysis of iron-oxide nanoparticles. Molecular Pharmaceutics. 2010;7(2):375–385. doi: 10.1021/mp900161h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Friedman M. Applications of the ninhydrin reaction for analysis of amino acids, peptides, and proteins to agricultural and biomedical sciences. J Agric Food Chem. 2004;52(3):385–406. doi: 10.1021/jf030490p. [DOI] [PubMed] [Google Scholar]
- 29.Thurston G, McLean JW, Rizen M, Baluk P, Haskell A, Murphy TJ, et al. Cationic liposomes target angiogenic endothelial cells in tumors and chronic inflammation in mice. J Clin Invest. 1998;101(7):1401–13. doi: 10.1172/JCI965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dandamudi S, Campbell R. Development and characterization of magnetic cationic liposomes for targeting tumor microvasculature. Biochim Biophys Acta. 2007;1768(3):427–38. doi: 10.1016/j.bbamem.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 31.Wang YX, Hussain SM, Krestin GP. Superparamagnetic iron oxide contrast agents: Physicochemical characteristics and applications in mr imaging. Eur Radiol. 2001;11(11):2319–31. doi: 10.1007/s003300100908. [DOI] [PubMed] [Google Scholar]
- 32.Brigger I, Morizet J, Laudani L, Aubert G, Appel M, Velasco V, et al. Negative preclinical results with stealth((r)) nanospheres-encapsulated doxorubicin in an orthotopic murine brain tumor model. J Control Release. 2004;100(1):29–40. doi: 10.1016/j.jconrel.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 33.Joshi S, Emala CW, Pile-Spellman J. Intra-arterial drug delivery: A concise review. J Neurosurg Anesthesiol. 2007;19(2):111–9. doi: 10.1097/ANA.0b013e318033da41. [DOI] [PubMed] [Google Scholar]
- 34.Doolittle ND, Miner ME, Hall WA, Siegal T, Jerome E, Osztie E, et al. Safety and efficacy of a multicenter study using intraarterial chemotherapy in conjunction with osmotic opening of the blood-brain barrier for the treatment of patients with malignant brain tumors. Cancer. 2000;88(3):637–47. doi: 10.1002/(sici)1097-0142(20000201)88:3<637::aid-cncr22>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 35.Vajkoczy P, Menger MD. Vascular microenvironment in gliomas. J Neurooncol. 2000;50(1–2):99–108. doi: 10.1023/a:1006474832189. [DOI] [PubMed] [Google Scholar]
- 36.Pulfer S, Gallo J. Enhanced brain tumor selectivity of cationic magnetic polysaccharide microspheres. J Drug Target. 1998;6(3):215–227. doi: 10.3109/10611869808997896. [DOI] [PubMed] [Google Scholar]
- 37.Cuvier C, Roblot-Treupel L, Millot JM, Lizard G, Chevillard S, Manfait M, et al. Doxorubicin-loaded nanospheres bypass tumor cell multidrug resistance. Biochem Pharmacol. 1992;44(3):509–17. doi: 10.1016/0006-2952(92)90443-m. [DOI] [PubMed] [Google Scholar]
- 38.Anderson DG, Peng W, Akinc A, Hossain N, Kohn A, Padera R, et al. A polymer library approach to suicide gene therapy for cancer. Proc Natl Acad Sci U S A. 2004;101(45):16028–33. doi: 10.1073/pnas.0407218101. [DOI] [PMC free article] [PubMed] [Google Scholar]