

Abstract
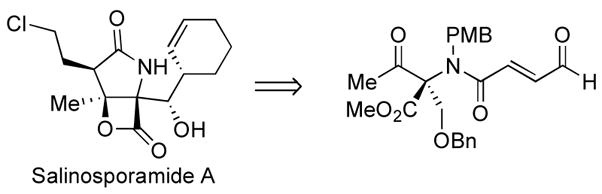
An N-heterocyclic carbene (NHC) catalyzed intramolecular lactonization to prepare densely functionalized bicyclic γ-lactam-γ-lactone adducts from enals is reported. This method has been applied to the formal synthesis of salinosporamide A, a potent 20S proteasome inhibitor and anti-cancer therapeutic.
1. Introduction
Over the past decade, significant advances have been achieved in reactions catalyzed by N-heterocyclic carbenes (NHCs).i Conceptually new reaction pathways have been identified, making possible the production of stereochemically rich heterocycles from simple, readily available starting materials under exceptionally mild reaction conditions. An intriguing feature of these reactions is the often exquisite control over competing reaction pathways. For example, α,β-unsaturated enals can react as either homoenolate or ester enolate equivalents (Figure 1). In intermolecular annulation reactions, we have been successful in achieving complete control over these reaction pathways through careful choice of catalysts and reaction conditions.2
Figure 1.

Reactive intermediates formed between NHCs and enals.
To further explore the ability of catalyst design to dictate the reaction outcome, we wished to examine the more challenging case of selective enolate versus homoenolate reactivity in a densly functionalized substrate that would lead to synthetically valuable bicyclic products. In this article, we document our studies on asymmetric intramolecular cyclizations catalyzed by N-heterocyclic carbenes and its application to a formal synthesis of salinosporamide A.
These studies originate from our recent discovery that α,β-unsaturated aldehydes undergo reactions with N-heterocyclic carbenes, leading to the catalytic generation of homoenolates or enolates via internal redox processes.3 We have applied the generation of homoenolates to the formation of γ-lactones2a and γ-lactams.4 We have also disclosed that enals and α-chloroaldehydes serve as precursors to ester enolate equivalents for highly enantioselective inverse electron demand Diels-Alder reactions.5 Building on these studies, Scheidt has reported intramolecular variants of both the homoenolate and enolate pathways and has alluded to competition between these two reaction manifolds in cyclization reactions.6
We recognized that an intramolecular cyclization via the homoenolate pathway would provide a concise entry into the salinosporamide class of natural products (Figure 2). Salinosporamide A7 is a secondary metabolite of the marine actinomycete bacteria of Salinospora strain CNB-392. It is a potent inhibitor of the 20S proteasome and has attracted much attention8 because of its impressive in vitro cytotoxic activity against many tumor cell lines. In order to execute this synthesis, however, we needed to identify catalysts and reaction conditions that would react selectively via the desired homoenolate pathway.
Figure 2.
2. Results and Discussion
2.1. Development of an NHC-catalyzed intramolecular cyclization-lactonization
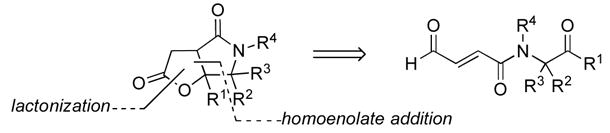
We sought to develop a general strategy toward the formation of cis-fused γ-lactam-γ-lactone adducts via an NHC-promoted intramolecular homoenolate addition of an enal to a tethered ketone followed by lactonization of the resulting alkoxide on the subsequently formed activated carboxylate (Figure 3).
Figure 3.
Retrosynthetic strategy toward γ-lactam-γ-lactone adducts.
Toward this end, aldehyde 1 was synthesized by the coupling of 29 and acid 3 (Scheme 1). However, when 1 was allowed to react with commercially available NHC precatalysts IMesCl or RMesCl10 in THF at 40 °C in the presence of DBU, the desired product was not observed but rather the N-substituted succinimide 4 was obtained exclusively via NHC-catalyzed redox generation of the activated carboxylate (Scheme 1).
Scheme 1.

Initially attempted NHC-catalyzed intramolecular cyclization. (HBTU=O-(benzotriazol-1 -yl)- N,N,N′,N′-tetramethyluronium hexafluorophosphate, DIPEA=N,N-diisopropylethylamine, DBU=1,8-diazabicyclo-[5.4.0]undec-7-ene).
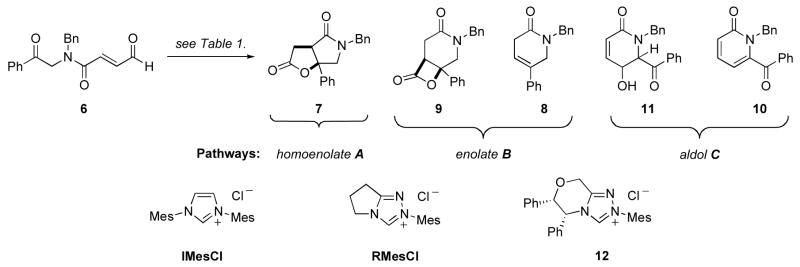
At this point we recognized the need to employ a protecting group on the amide prior to cyclization. The benzyl protecting group was chosen based on ease of the preparation of the amine coupling partner as well as the potential for its facile removal by catalytic hydrogenation. N-benzyl protected phenyl ketone 5 was synthesized from 2-bromoacetophenone and subsequently coupled with acid 3 using EDC. Deprotection of the acetal using aqueous hydrochloric acid afforded the aldehyde 6 (Scheme 2). We subjected the aldehyde to NHC-precatalyst RMesCl with DBU in THF at 40 °C and were pleased to find the desired product, albeit in low yield and as a complex mixture.
Scheme 2.

Synthesis of benzyl protected substrate 6. (Bn=benzyl, EDC= N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride, DMAP=4-(dimethylamino)pyridine).
Careful analysis of the reaction mixture revealed that in addition to the desired γ-lactam-γ-lactone product 7, 6-membered lactam 8 was also formed. A plausible catalytic cycle is presented in Figure 4. Deprotonation of the precatalyst provides the active NHC catalyst that undergoes nucleophilc attack on the enal. A proton transfer event leads to the Breslow intermediate, the fate of which is intimately related to the strength of base and reaction conditions employed. A resonance structure of the Breslow intermediate is the homoenolate equivalent; this can participate in an intramolecular nucleophilic addition to the ketone followed by lactonization via the resultant activated carboxylate to turn the catalyst over and produce the desired lactam 7 (pathway A). Protonation of the homoenolate, however, renders an NHC-enolate that can undergo an intramolecular keto-aldol addition. Lactonization would then result in expulsion of the catalyst and formation β-lactone 9, which readily undergoes decarboxylation11 to δ-lactam 8 (pathway B). From the reaction mixture, we also isolated 10, which we believe arises from a base-catalyzed olefin isomerization, followed by an intramolecular aldol addition (aldol pathway C). We corroborated pathway C by subjecting aldehyde 6 to DBU in THF at 40 °C; complete conversion to the α-pyridone 10 was observed (Scheme 3).
Figure 4.

Postulated catalytic cycles for NHC-promoted pathways A and B. (Mes=2,4,6-trimethylphenyl)
Scheme 3.

Base catalyzed intramolecular aldol pathway C.
Optimization was carried out in an attempt to bias the formation of the desired γ-lactam-γ-lactone, which arises from pathway A. The most pertinent results are summarized in Table 1. When imidazolium catalyst IMesCl was used in conjunction with strong tertiary amine base DBU, the intramolecular aldol pathway C predominated (entry 1). However, when a weaker tertiary amine base was employed, pathway C was completely suppressed but the formation of products arising from the enolate pathway B increased (entry 3). We attribute this to the facile protonation of the homoenolate by the conjugate acid of triethylamine. Use of the triazolium catalyst RMesCl yielded products of all three pathways with pathway B dominating when DBU was used (entry 2), and pathway A and B dominating when the weaker amine base triethylamine was used (entry 4). We further discovered that of the chiral triazolium NHC-precatalysts tested, pathway C could be suppressed when catalyst 12, reported by Scheidt and co-workers,12 was used along with either a strong base under dilution (entry 5), a bulky tertiary amine base in a chlorinated solvent (entry 6) or strong base in tBuOH (entry 7). However, we could never bias the product distribution to favor the desired γ-lactam-γ-lactone product in greater than 50% yield.
Table 1.
NHC-catalyzed cyclization-lactonization of substrate 6.
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| entry | Catalyst (mol%) | Base (mol%) | Solvent (conc.) | Temp. (°C) | A | : | B | : | Ca |
| 1 | IMesCl (20%) | DBU(15%) | THF (0.1M) | 40 | 1 | : | – | : | 10 |
| 2 | RMesCl (20%) | DBU(15%) | THF (0.1M) | 40 | 1 | : | 1 | : | 2 |
| 3 | IMesCl (20%) | NEt3(15%) | THF (0.1M) | 40 | 2.5 | : | 2 | : | – |
| 4 | RMesCl (20%) | NEt3(15%) | THF (0.1M) | 40 | 2.5 | : | 2.5 | : | 1 |
| 5 | 12(15%) | DBU(10%) | 10:1 THF:tBuOH (0.02 M) | 40 | 1 | : | 1 | : | – |
| 6 | 12(15%) | DIPEA(100%) | CH2Cl2 (0.1M) | 40 | 1 | : | 1 | : | – |
| 7 | 12(15%) | DBU(10%) | tBuOH (0.1M) | 40 | 6.2 | : | 5.3 | : | 1 |
| 8 | 12(15%) | DBU(10%) | tBuOH (0.1M) | 60 | 1.4 | : | 1 | : | – |
| 9 | 12(15%) | NEt3(10%) | tBuOH (0.1M) | 60 | 1.6 | : | 1 | : | – |
| 10 | 12(15%) | DBU(10%) | tBuOH (0.1M) | 80 | 1.4 | : | 1 | : | – |
All reactions listed proceeded with 100% conversion; isolated yields were not determined.
Product pathway ratios determined from 1H NMR analysis of unpurified reaction mixtures.
From these results we hypothesized that there may exist a rotational barrier for the two rotamers of tertiary amide. As such, one rotamer might undergo intramolecular cyclization by the homoenolate faster than the other. The rate of this rotation might be in competition with the rate of protonation of the homoenolate that leads to the enolate which cyclizes to form the products of pathway B. To test this hypothesis, we subjected 6 to our optimized conditions at 60 °C and indeed a more favorable product distribution was achieved (entries 8 and 9). However, further increase in temperature did not improve the ratio of desired product (entry 10).
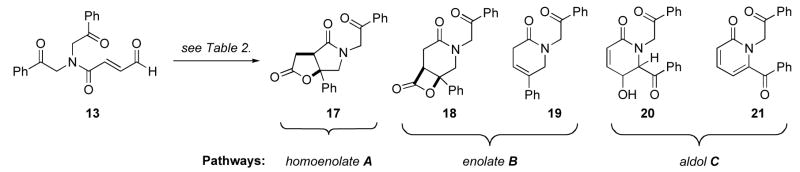
Similarly, we reasoned that a symmetrical tertiary amide would obviate the need for elevated temperatures to bias the homoenolate pathway A. Aldehyde 13 was synthesized to further probe our initial hypothesis (Scheme 4). The symmetrical bisketone 15 was synthesized in 2-steps from 2-bromoacetophenone and subsequently coupled with acid 3 using EDC. Deprotection of the diethyl acetal with aqueous hydrochloric acid furnished the symmetrical α,β-unsaturated aldehyde 13.
Scheme 4.

Synthesis of symmetrical substrate enal 13. (Bn=benzyl, EDC=N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride, DIPEA=N,N-diisopropylethylamine, DMAP=4-(dimethylamino)pyridine).
Initially triazolium catalysts were identified as superior to imidazolium catalysts, such as IMesCl, for obtaining the desired γ-lactam-γ-lactone product 17 (Table 2, entries 1–3). Further optimization identified 10:1 THF:tBuOH as the most suitable solvent system and RMesCl the best precatalyst for biasing pathway A (entries 4–13). It should be noted that strong bases DBU and tBuOK (entries 4, 5 and 7) as well as the weaker but bulkier tertiary amine base DIPEA (entry 6) helped to suppress pathway C. Lowering the temperature initially suppressed pathway C (entries 4 and 5) but further cooling suppressed pathway B while enhancing pathway C (entry 8). Dilution favored pathway A (entries 9–13), whereas raising the amount of base tended to favor pathway C for the strong base DBU (entry 11) but pathway B for the weaker base DIPEA (entry 12). The optimum conditions identified employed DBU at lower loading than the catalyst, moderate heating (40 °C), and dilute (0.01M), resulting in a 3:1 ratio of A:B (entry 13). Although we proved that the symmetrical substrate did provide a better ratio of the desired product, we could not easily purify 17 from 19.
Table 2.
NHC-catalyzed cyclization-lactonization of substrate 13.
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | Base (mol%) | Solvent (conc.) | Temp. (°C) | A | : | B | : | Ca |
| 1 | RMesCl (15%) | DBU(10%) | tBuOH (0.1M) | 60 | 3 | : | 1 | : | 2 |
| 2 | IMesCl (15%) | DBU(10%) | tBuOH (0.1M) | 60 | 1 | : | 1.5 | : | 8 |
| 3 | 12 (15%) | DBU(10%) | tBuOH (0.1M) | 60 | 4 | : | 1 | : | 1 |
| 4 | RMesCl (15%) | DBU(10%) | 10:1 THF:tBuOH (0.10 M) | 60 | 1 | : | 1.5 | : | - |
| 5 | RMesCl (15%) | DBU(10%) | 10:1 THF:tBuOH (0.10 M) | 40 | 1 | : | 1.3 | : | - |
| 6 | RMesCl (15%) | DIPEA(10%) | 10:1 THF:tBuOH (0.10 M) | 40 | 1 | : | 1.2 | : | - |
| 7 | RMesCl (15%) | tBuOK(10%) | 10:1 THF:tBuOH (0.10 M) | 40 | 1 | : | 1 | : | 1 |
| 8 | RMesCl (15%) | DBU(10%) | 10:1 THF:tBuOH (0.10 M) | 20 | 1 | : | - | : | 6.7 |
| 9 | RMesCl (15%) | DBU(10%) | 10:1 THF:tBuOH (0.05 M) | 40 | 4 | : | 1 | : | 1 |
| 10 | RMesCl (15%) | DIPEA(10%) | 10:1 THF:tBuOH (0.05 M) | 40 | 1 | : | 1 | : | trace |
| 11 | RMesCl (15%) | DBU (50%) | 10:1 THF:tBuOH (0.05 M) | 40 | 1 | : | - | : | 10 |
| 12 | RMesCl (15%) | DIPEA (50%) | 10:1 THF:tBuOH (0.05 M) | 40 | 1 | : | 1 | : | - |
| 13 | RMesCl (15%) | DBU (10%) | 10:1 THF:tBuOH (0.01 M) | 40 | 3 | : | 1 | : | trace |
All reactions listed proceeded with 100% conversion; isolated yields were not determined.
Product pathway ratios determined from 1H NMR analysis of umpurified reaction mixtures.
Of the two competing undesired pathways (B and C), pathway C was the easier to address. By blocking the α-position of the ketone, we could effectively eliminate pathway C since no enolizable hydrogens would exist. Also, there would be no concern for the use of either a strong base or an excess (compared to the precatalyst) of base. For this purpose, the geminal dimethyl substrate 22 was designed and synthesized (Scheme 5). Starting from Cbz-AIB, the acid was converted to the Weinreb amide 23 using DCC. N-Alkylation was carried out in DMF with NaH and benyl bromide providing 24 in quantitative yield. Formation of methyl ketone 25 was achieved using MeMgBr in Et2O, and the benzyl carbamate was removed by catalytic hydrogenation in acidic media to give 26. Amide formation with acid 3 proved most efficient via the isobutyl mixed anhydride at elevated temperature; standard coupling reagents such as HBTU, HATU, EDC were ineffective. Finally, deprotection of the diethyl acetal 27 afforded the desired aldehyde 22 in moderate yield due to its sensitivity to silica gel purification.
Scheme 5.

Synthesis of gem-dimethyl substrate 22. (Cbz= benzyl carboxy, Bn= benzyl, DCC= N,N′-dicyclohexylcarbodiimide, DMAP= 4-(dimethylamino) pyridine, DIPEA= N,N-diisopropylethylamine, NMM= N-methylmorpholine).
When aldehyde 22 was allowed to react in the presence of 15 mol% of RMesCl, 10 mol% of DBU at 0.05 M in 10:1 THF:tBuOH for 20 h, we identified only the desired γ-lactam-γ-lactone 28. We further identified that IMesCl was an even more effective catalyst for the desired transformation, providing the desired lactam in 79% isolated yield (Table 3, entry 1). We briefly screened several chiral precatalysts. Our triazolium N-mesityl aminoindanol-derived precatalyst 29 provided 28 in 75% yield but only 5% ee (entry 2). Our structurally related imidazolium N-mesityl aminoindanol-derived precatalyst 30 provided 28 in a reduced 50% yield and 9% ee (entry 3). Two chiral N-mesityl triazolium precatalysts, 12 and 31, developed by Scheidt12,13 proved just as effective with a slight increase in enantiomeric excess (entries 4 and 5). By employing precatalyst 31 and reducing the temperature to −20 °C, we obtained the γ-lactam-γ-lactone 28 with an enatiomeric excess of 40%.
Table 3.
NHC-catalyzed cyclization-lactonization of substrate 22.
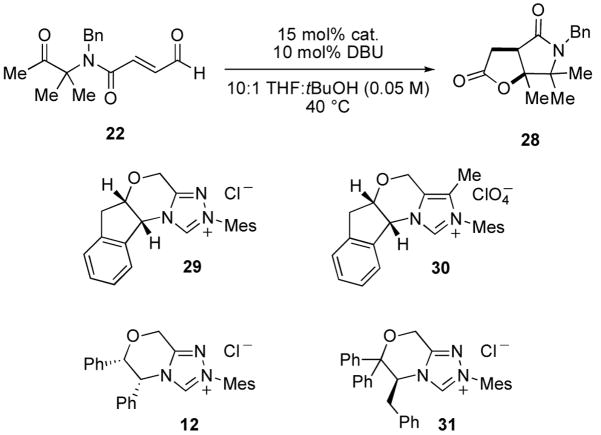
All reactions listed proceeded with 100% conversion.
Determined by chiral SFC analysis.
Performed at −20 °C.
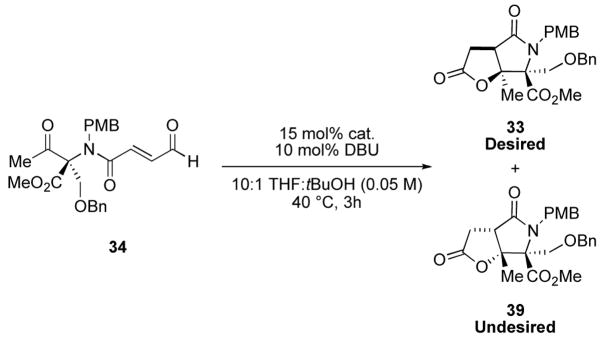
Encouraged by our results we sought to apply our methodology to streamlining the previously reported synthesis of salinosporamide A. During the course of our investigation, Lam and coworkers reported a formal synthesis of salinosporamide A from 32 via a nickel-catalyzed reductive aldol cyclization-lactonization strategy to construct 33.8g In parallel studies, we had identified intermediate 33 as a target for our NHC-promoted intramolecular cyclization-lactonization strategy (Figure 5) for a formal synthesis of salinosporamide A. Our retrosynthetic plan is outlined in Figure 6. Lactam 33 would be obtained from the NHC catalyzed cyclization-lactonization of aldehyde 34 which in turn would be synthesized in an analogous manner to our previous aldehyde substrates via amide bond formation between acid 3 and amine 36 followed by oxidation. The synthesis of 36 from L-threonine has previously been reported by Corey in his synthesis of salinosporamide A.8a
Figure 5.

Proposed formal synthesis of salinosporamide A from aldehyde 34 employing our intramolecular NHC-cyclization.
Figure 6.

Retrosynthetic analysis for intermediate γ-lactam 33.
Attempted amide formation between 3 and 36 did not appreciably proceed in the presence of coupling reagents HBTU, HATU, PyBop or EDC. Instead, either a trace amount of the desired amide, decomposition of the amine, deprotection of the silyl group, or no reaction was observed under a variety of conditions. We attempted the amide bond formation via the mixed anhydride of acid 3. Neither the isobutyl, ispropenyl, nor 2,4,6-trichlorophenyl mixed anhydride provided any of the desired amide. Formation of the amide via the 2,3,4,5,6-pentafluorophenyl ester derivative of 3 also proved unsuccessful. Attempts to acylate amine 36 via an acid chloride formed by means of the Vilsmeier reagent and acid 3 yielded trace amounts of the desired product in certain cases, but often returned starting material and deprotected alcohol accompanied by decomposition of the acid chloride. After numerous attempts to couple amine 36 and acid 3 had failed, we revised our synthesis of aldehyde 34.
We instead accessed aldehyde 34 from amine 36 by first using a 3-step sequence of N-acylation with sorbyl chloride, silyl ether cleavage under aqueous acidic conditions, and Dess-Martin oxidation14 to afford ketone 37 in 60% overall yield. Next we performed a regioselective Sharpless dihydroxylation15 at the γ,δ-position of 37 using a procedure similar to one reported by Zhang and O’Doherty16 to access diol 38 as a single diastereomer in moderate yield. Finally diol cleavage was accomplished with sodium periodate in quantitative yield (Scheme 6).
Scheme 6.

Preparation of aldehyde 34 for NHC-catalyzed intramolecular cyclization-lactonization. (DIPEA=N,N-diisopropylethylamine, Dess-Martin= 1,1,1-triacetoxy-1,1-dihydro-1,2-benziodoxol-3 (1 H)-one, PMB=p-methoxybenzyl).
Having successfully synthesized key intermediate 34, we subjected the enal to our previously optimized conditions for cyclization of substrate 22. When 34 was allowed to react with 15 mol% IMesCl and 10 mol% DBU in 10:1 THF:tBuOH at 40 °C a complete conversion to the γ-lactam-γ-lactone products 33 and 39 was observed (Table 4, entry 1). Although the isolated yield was good (75%), the diastereomeric ratio was only 3:1 in favor of the undesired diastereomer. We therefore sought to bias the stereochemical outcome through the use of chiral catalyst. We employed our N-mesityl aminoindanol derived chiral triazolium catalyst 29 (entry 2); the diastereomeric ratio decreased to 1.2:1 still in favor of the undesired diastereomer but with increased yield (84%). Interestingly, the use of ent-29 provided nearly an identical outcome (entry 3). Triazolium salts, 12 and 31 which had previously led to higher enantiomeric ratios with substrate 22 gave slightly higher diastereomeric ratios: 1.7:1 and 1.5:1, respectively, but again in favor of the undesired diastereomer (entry 4–5). Therefore, choice of the appropriate catalyst, ent-29, did indeed influence the strong substrate control over the diastereoslective homoenolate addition to the ketone whereby a 1:1.1 ratio of desired:undersired lactam is our best result (see entry 3). Our NHC-catalyzed intramolecular cyclization-lactonization of enal 34 provided lactams 33 and 39 in excellent yield and represents a formal synthesis of salinospoaramide A.
Table 4.
NHC-catalyzed cyclization-lactonization of substrate 34.
 | |||
|---|---|---|---|
| Entry | Catalyst | Yielda | d.r. (desired:undesired) |
| 1 | IMesCl | 75% | 1:3 |
| 2 | 29 | 84% | 1:1.2 |
| 3 | ent-29 | 88% | 1:1.1 |
| 4 | 12 | 93% | 1:1.7 |
| 5 | 31 | 77% | 1:1.5 |
All reactions listed proceeded with 100% conversion.
Combined isolated yield of both diastereomers.
3. Conclusion
We have presented an NHC-catalyzed intramolecular cyclization-lactonization of enals to ketones tethered by an amide bond, producing densely functionalized γ-lactam-γ-lactone adducts. To demonstrate the utility of this method, we accomplished the formal synthesis of salinosporamide A, a potent 20S proteasome inhibitor, via intermediates reported by Corey and by Lam. The attraction of our synthesis is the use of a NHC-promoted intramolecular cylization-lactonization strategy to construct the carbocyclic core of salinosporamide A in a single high-yielding step.
4. Supporting Information
4.1. General Methods
All reactions utilizing air- or moisture-sensitive reagents were performed in dried glassware under an atmosphere of dry nitrogen. Dichloromethane was distilled over CaH2. Diethyl ether, THF and tert-butanol were distilled from Na0. Toluene and DMF were dried by passage over activated alumina under Ar atmosphere. All other reagents were used without further purification. Thin-layer chromatography (TLC) was performed on Merck precoated plates (silica gel 60 F254, Art 5715, 0.25 mm) and were visualized by fluorescence quenching under UV light or by staining with phosphomolybdic acid, cerium sulfate or potassium permanganate solutions. Silica-gel preparative thin-layer chromatography (PTLC) was performed using plates prepared from Merck Kieselgel 60 PF254 (Art 7747). Column chromatography was performed on E. Merck 13 Silica Gel 60 (230–400 Mesh) using a forced of 0.1–0.5 bar. *H and 13C NMR were measured on Bruker Avance II NMR at 500 and 125 MHz, respectively. Chemical shifts are expressed in parts per million (ppm) downfield from residual solvent peaks and coupling constants are reported in hertz (Hz). Splitting patterns are indicated as follows: br, broad; s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet. Amide rotamers, are marked by an asterisks (*). Infrared (IR) spectra were recorded on a JASCO FT:IR-430 spectrophotometer and are reported as wavenumbers (cm−1).
4.1.1. General Procedure for Catalytic Reactions
A vial was charged with the NHC precatalyst, the substrate, and purged with N2(g). Next, the solvent was added followed by the base under an atmosphere of nitrogen. The vial was sealed and stirred at the designated temperature until the starting material was consumed as indicated by TLC analysis. Finally, the solvent was removed under reduced pressure and the crude reaction mixture purified by silica gel chromatography.
4.1.2. (E)-ethyl 4,4-diethoxybut-2-enoate
To a 1.0 M solution of (E)-ethyl 4-oxobut-2-enoate (12.0 mL, 100 mmol, 1.00 equiv) in 200 proof EtOH cooled to 0 °C was added triethylorthoformate (12.8 mL, 120 mmol, 1.2 equiv) and conc. aq HCl (0.100 mL, 1.16 mmol, 0.0116 eqiuv). The solution was stirred at 0 °C for 4 h, allowed to warm to rt, and stirred until the disappearence of the starting material by TLC analysis (ca 4 h). The solution was concentrated under reduced pressure and dissolved in EtOAc (150 mL). This solution was washed with H2O (150 mL), and the aqueous layer back-extracted with EtOAc (150 mL). The combined organic fractions were washed with brine (200 mL), dried over Na2SO4, filtered and concentrated under reduced pressure to afford the title compound as a clear liquid (19.5 g, 88%). 1H NMR (CDCl3) δ 6.80 (dd, 1H, J = 15.8, 4.2 Hz), 6.13 (dd, 1H, J = 15.8, 1.3 Hz), 5.04 (dd, 1H, J = 4.2, 1.3 Hz), 4.20 (q, 2H, J = 7.1 Hz), 3.65 (dq, 2H, J = 7.1, 2.3 Hz), 3.52 (dq, 2H, J = 7.1, 2.4 Hz), 1.29 (t, 3H, J = 7.1 Hz), 1.22 (t, 6H, J = 7.1 Hz); 13C NMR (CDCl3) δ 166.3, 143.6, 124.2, 99.2, 61.4, 60.7, 15.3, 14.3; IR (thin film) ν 2981, 2927, 2878, 1722, 1446, 1373, 1304, 1269, 1181, 1058 cm−1; HRESI+/TOF-MS calcd for C10H18O4 [M]+ 202.1205, found 225.1106 [M+Na]+.
4.1.3. (E)-4,4-diethoxybut-2-enoic acid (3)
A 1N aq solution of NaOH (20 mL, 20 mmol, 1.0 equiv) was added slowly to a chilled (0 °C) 1 M solution of (E)-ethyl 4,4-diethoxybut-2-enoate (4.0 g, 20 mmol, 1.0 equiv) in THF (20 mL). The solution was allowed to warm to rt and stirred until the consumption of starting material was observed by TLC (ca 5 h). The solution was diluted with CH2Cl2 (100 mL) and the pH of the aqueous phase adjusted with 1N aq HCl until pH~2 was achieved. The organic layer was separated and the aqueous layer extracted with CH2Cl2 (100 mL). The organic layers were combined, dried over Na2SO4, filtered and concentrated under reduced pressure to afford the title compound as a yellow liquid (3.5 g, quant.). 1H NMR (CDCl3) δ 6.92 (dd, 1H, J = 15.8, 4.0 Hz), 6.16 (dd, 1H. J = 15.8, 1.4 Hz), 5.09 (dd, 1H, J = 4.0, 1.3 Hz), 3.66 (dq, 2H, J = 7.1, 2.4 Hz), 3.54 (dq, 2H, J = 7.1, 2.4 Hz), 1.23 (t, 6H, J = 7.1 Hz); 13C NMR (CDCl3) δ 170.7, 150.5, 125.0, 103.3, 66.2, 15.1; IR (thin film) ν 3104, 2981, 2937, 2898, 1796, 1761, 1377, 1348, 1136, 1013, 929, 890, 817 cm−1; HRESI−/TOF-MS calcd for C8H14O4 [M]+ 174.0892, found 173.0803 [M–H]−.
4.1.4. (E)-4,4-diethoxy-N-(2-oxo-2-phenylethyl)but-2-enamide
N,N-Diiosopropylethylamine (0.52 mL, 3.0 mmol, 3.0 equiv) was added to a solution of 3 (0.35 g, 2.0 mmol, 2.0 equiv) and HBTU (0.76 g, 2.0 mmol, 2.0 equiv) in CH2Cl2 (10 mL). After 10 min of stirring, 2-oxo-2-phenylethanaminium chloride (0.17 g, 1.0 mmol, 1.0 equiv) was added in a single portion and the solution stirred at rt for 20 h. The solution was diluted with CH2Cl2 (15 mL) and washed with sat aq NaHCO3 (20 mL). The aqueous layer was extracted with CH2Cl2 (20 mL). The combined organic extracts were washed with sat aq NH4Cl (20 mL), dried over Na2SO4, filtered and concentrated under reduced pressure. Purification by column chromatography afforded the title compound as a white solid (0.195 g, 67%). 1H NMR (CDCl3) δ 8.00–7.98 (m, 2H), 7.63 (t, 1H, J = 7.4 Hz), 7.51 (t, 1H, J = 7.8 Hz), 6.78 (dd, 1H, J = 15.5, 3.8 Hz), 6.70 (bs, 1H), 6.28 (dd, 1H, J = 15.5, 1.4 Hz), 5.10 (dd, 1H, J = 3.8, 1.4 Hz), 4.84 (d, 2H, J = 4.3 Hz), 3.66 (dq, 2H, J = 7.1, 2.4 Hz), 3.53 (dq, 2H, J = 7.1, 2.4 Hz), 1.23 (t, 6H, J = 7.1 Hz); 13C NMR (CDCl3) δ 194.1, 165.2, 140.5, 134.4, 129.1, 128.1, 125.9, 99.2, 61.3, 46.7, 15.4; IR (thin film) ν 3311, 2976, 2873, 1678, 1638, 1377, 1058, 1003, 979 cm−1; HRESI+/TOF-MS calcd for C16H21NO4 [M]+ 291.1471, found 314.1356 [M+Na]+.
4.1.5. (E)-4-oxo-N-(2-oxo-2-phenylethyl)but-2-enamide (1)
A solution of (E)-4,4-diethoxy-N-(2-oxo-2-phenylethyl)but-2-enamide (0.10 g, 0.34 mmol, 1.0 equiv) in THF (4.0 mL) was treated with 2N aq HCl (1.2 mL, 2.4 mmol, 7.0 equiv) at rt and stirred until the consumption of starting material was observed by TLC (ca 5 min). The solution was diluted with H2O (15 mL) and EtOAc (15 mL); the organic layer was separated, washed with brine (15 mL), dried over Na2SO4, filtered and concentrated under reduced pressure affording the title compound as a light tan solid (0.074 g, quant.). 1H NMR (CDCl3) δ 9.79 (d, 1H, J = 7.4 Hz), 8.00 (dd, 2H, J = 8.4, 1.2 Hz), 7.66 (dt, 1H, J = 7.4, 1.2 Hz), 7.53 (t, 2H, J = 7.5 Hz), 7.02 (dd, 1H, J = 15.6, 7.4 Hz), 6.89 (d, 1H, J = 15.6 Hz), 4.89 (d, 1H, J = 4.3 Hz); 13C NMR (CDCl3) δ 193.5, 192.4, 163.4, 141.4, 138.3, 134.7, 129.2, 129.1, 128.2, 46.8; IR (thin film) ν 3316, 3060, 2922, 2848, 1695, 1675, 1670, 1540, 1358, 1230, 984 cm−1; HRESI+/TOF-MS calcd for C12H11NO3 [M]+ 217.0739, found 240.0622 [M+Na]+.
4.1.6. 1-(2-oxo-2-phenylethyl)pyrrolidine-2,5-dione (4)
An oven dried vial was charged with IMesCl (0.0075 g, 0.022 mmol, 0.20 equiv) and 3 (0.023 g, 0.11 mmol, 1.0 equiv) and purged with N2(g). To this mixture was added THF (1.0 mL) and DBU (3.3 μL, 0.022 mmol, 0.20 equiv), the vial sealed, and the reaction stirred at 40 °C for 12 h. The solution was concentrated under reduced pressure and purified by PTLC (2:1 hexanes:acetone). 1H NMR (CDCl3) δ 7.97 (dd, 2H, J = 8.4, 1.2 Hz), 7.63 (dt, 1H, J = 7.4, 1.2 Hz), 7.52–7.49 (m, 2H), 4.95 (s, 2H), 2.87 (s, 4H); 13C NMR (CDCl3) δ 190.3, 176.8, 134.3, 129.0, 128.3, 44.9, 28.5; HRESI+/TOF-MS calcd for C12H11NO3 [M]+ 217.0739, found 240.0627 [M+Na]+.
4.1.7. N-benzyl-2-oxo-2-phenylethanaminium chloride (5)17
A 1.0 M solution of 2-bromo-1-phenylethanone (10.0 g, 50.0 mmol, 1.00 equiv) in Et2O (50.0 mL) was added to a chilled (0 °C) 2.0 M solution of benzylamine (10.9 mL, 100 mmol, 2.00 equiv) in Et2O over a 10 minute period and stirred at 0 °C for 16 h. The precipitate was filtered and washed with Et2O. The filtrate was cooled to 0 °C and conc. HCl (4.0 mL) was slowly added. The resultant orange precipitate was filtered and washed with Et2O. The precipitate was then sonicated in 60 mL of 1:1 Et2O:EtOH (200 proof) until the solid appeared white. The precipitate was collected by filtration and washed with Et2O affording 2.80 g (crop 1, 21.4%) of the title compound as a white crystalline solid. The filtrate was then concentrated and the resulting solid sonicated in a 1:1 Et2O:EtOH (200 proof) solution until the solid appeared white. The precipitate was collected by filtration and washed with Et2O affording an additional 3.70 g (crop 2, 28.2%) of the title compound as a white powder. 1H NMR (dmso-d6) δ 9.45 (bs, 2H), 8.00–7.98 (m, 2H), 7.93–7.91 (m, 1H), 7.77–7.74 (m 1H), 7.63–7.60 (m, 2H), 7.56–7.54 (m, 2H), 7.48–7.44 (m, 2H), 4.82 (s, 2H), 4.21 (s, 2H); 13C NMR (dmso-d6) δ 192.2, 134.8, 133.7, 131.8, 130.3, 129.2, 128.8, 128.2, 128.1, 51.9, 50.2; IR (KBr) ν 2977, 2912, 2785, 2730, 2608, 2435, 1712, 1579, 1461, 1225, 742 cm−1; HRESI+/TOF-MS calcd for C15H16ClNO+ [M]+ 226.1226, found 226.1223 [M]+.
4.1.8. (E)-N-benzyl-4-oxo-N-(2-oxo-2-phenylethyl)but-2-enamide (6)
To a solution of 3 (1.50 g, 8.61 mmol, 2.00 equiv) in CH2Cl2 (50.0 mL) were sequentially added 5 (1.12 g, 4.28 mmol, 1.00 equiv), EDC (1.65 g, 8.60 mmol, 2.00 equiv), 4-dimethylaminopyridine (0.525 g, 4.30 mmol, 1.00 equiv), and triethylamine (3.00 mL, 21.5 mmol, 5.02 equiv) and the resulting brown solution stirred for 24 h. The solution was diluted with CH2Cl2 (100 mL), washed with 1N aq HCl (2 × 75 mL) followed by sat aq NaHCO3 (100 mL), dried over Na2SO4, filtered and concentrated under reduced pressure affording a brown foam. The foam was dissolved in THF (13 mL) and treated with 6N aq HCl (13 mL, 26 mmol, 6.0 equiv) for 1 h. After diluting the solution with CH2Cl2 (~70 mL) and H2O (~50 mL) the organic phase was separated. The aqueous phase was further extracted with CH2Cl2 (2 × 50 mL). The organic phases were combined, dried over Na2SO4, filtered and concentrated under reduced pressure to afford the crude aldehyde as a brown solid. Sonication of the crude solid in Et2O/EtOAc precipitated the pure aldehyde as a tan solid and as a ~3:1 mixture of amide rotamers that was collected by vacuum filtration and washed with Et2O (0.600 g, 46% over 2 steps). 1H NMR (CDCl3) δ 9.73 (d, 1H, J = 7.5 Hz), 9.64* (d, 1H, J = 5.2 Hz), 7.93 (d, 2H, J = 7.5 Hz), 7.85* (d, 2H, J = 7.5 Hz), 7.60–7.22 (m, 9H), 7.65–7.22* (m, 9H), 7.03 (dd, 1H, J = 15.5, 7.5 Hz), 7.03* (dd, 1H, J = 15.5, 7.5 Hz), 6.96 (d, 1H, J = 5.5 Hz), 6.96* (d, 1H, J = 5.5 Hz), 4.87 (s, 2H), 4.78 (s, 2H), 4.76* (s, 2H), 4.72* (s, 2H); 13C NMR (CDCl3) δ 193.4, 192.5, 165.8, 139.9, 139.7, 139.4, 138.9, 135.7, 135.2, 134.7, 134.2, 129.5, 129.4, 129.1, 129.1, 128.9, 128.6, 128.2, 128.1, 126.9, 52.8, 52.2, 50.6; IR (thin film) ν 3065, 3035, 2932, 1697, 1647, 1451, 1358, 1230, 1117, 1077, 994 cm−1; HRESI+/TOF-MS calcd for C19H17NO3 [M]+ 307.1208, found 308.1284 [M+H]+.
4.1.9. N-benzyl-2-oxo-N-(2-oxo-2-phenylethyl)-2-phenylethanaminium bromide (14)
Prepared by modification of a published procedure:18 A 2.0 M solution of benzylamine (5.5 mL, 50 mmol, 1.0 equiv) in benzene (25 mL) was added to a 1.0 M solution of 2-bromo-1-phenylethanone (10 g, 50 mmol, 2.0 equiv) in benzene (50 mL). The resultant suspension was then diluted by the addition of an additional 30 mL benzene. The suspension was heated under reflux at 100 °C for 2 days. After allowing the reaction to cool to rt, the precipitate was collected by vacuum filtration and washed with benzene (75 mL). The precipitate was suspended in MeOH (75 mL) and sonicated for 30 min. The precipitate was collected by vacuum filtration and washed with MeOH (25 mL). Residual solvent was removed under reduced pressure affording the title compound as a white powder (8.0 g, 75%). 1H NMR (dmso-d6) δ 7.88 (d, 4H, J = 7.3 Hz), 7.71 (t, 2H, J = 7.4 Hz), 7.60–7.55 (m, 6H), 7.34–7.32 (m, 3H), 5.05 (bs, 4H), 4.51 (bs, 2H); 13C NMR (dmso-d6) δ 191.4, 134.9, 132.0, 129.9, 129.2, 128.8, 128.4, 128.1, 60.2, 60.0; IR (KBr) ν 3011, 2977, 2922, 1692, 1604, 1451, 1254, 895 cm−1; HRESI+/TOF-MS calcd for C16H22NO2+ [M]+ 344.1645, found 344.1647 [M]+.
4.1.10. Bis(2-oxo-2-phenylethyl)ammonium bromide (15)
A sclenck flask was charged with 10% Pd/C (0.172 g, 0.162 mmol, 0.010 equiv of Pd) was evacuated and backfilled with H2(g) 10 times. MeOH (75.0 mL) was added followed by 14 (6.88 g, 16.2 mmol, 1.0 equiv). H2(g) was bubbled through the suspension until all of 14 had dissolved (ca. 8 h). The solution was filtered through celite to remove the Pd/C, the Celite cake was washed with MeOH, and the filtrate concentrated under reduced pressure. The crude solid was recrystalized from MeOH to afford the title compound as a white solid (4.6 g, 85%). 1H NMR (dmso-d6) δ 9.54 (bs, 2H), 8.00 (d, 4H, J = 7.6 Hz), 7.76 (t, 2H, J = 7.4 Hz), 7.62 (t, 4H, J = 7.7 Hz), 4.84 (s, 4H); 13C NMR (dmso-d6) δ 192.2, 134.9, 133.6, 129.3, 128.3, 52.3; IR (KBr) ν 3001, 2912, 2814, 2716, 2588, 1687, 1599, 1554, 1368, 1259, 969 cm−1; HRESI+/TOF-MS calcd for C15H16NO+ [M]+ 226.1226, found 226.1232 [M]+.
4.1.11. (E)-4,4-diethoxy-N,N-bis(2-oxo-2-phenylethyl)but-2-enamide (16)
In a single portion, 15 (4.0 g, 12 mmol, 1.0 equiv) was added to a solution of 3 (4.2 g, 24 mmol, 2.0 equiv) in CH2Cl2 (120 mL). Next, EDC (4.6 g, 24 mmol, 2.0 equiv), 4-dimethylaminopyridine (1.5 g, 12 mmol, 1.0 equiv) and N,N′-diisopropylethylamine (10.5 mL, 60 mmol, 5.0 equiv) were sequentially added. The brown solution was stirred for 24 h at rt. After concentration of the solution under reduced pressure, the crude material was dissolved in EtOAc (250 mL) and sequentially washed with an aq 10% citric acid solution (2 × 125 mL), H2O (100 mL), sat aq NaHCO3 (2 × 125 mL), H2O (1 × 100 mL), and brine (1 × 100 mL). The EtOAc solution was then dried over Na2SO4, filtered and concentrated under reduced pressure. Purification by flash column chromatography (2:1 hexanes: EtOAc) afforded the title compound as a yellow foam (3.1 g, 63%). 1H NMR (CDCl3) δ 7.95 (m, 4H), 7.63–7.55 (m, 2H), 7.50–7.43 (m, 4H), 6.74 (dd, 1H, J = 15.3, 3.8 Hz), 6.38 (d, 1H, 15.3 Hz), 5.01 (s, 4H), 4.99 (d, 1H, J = 3.8 Hz), 3.57–3.53 (m, 2H), 3.46–3.42 (m, 2H), 1.10 (t, 6H, J = 7.1 Hz); 13C NMR (CDCl3) δ 194.9, 194.0, 167.2, 142.1, 135.1, 134.6, 134.3, 133.9, 129.1, 128.9, 128.2, 128.0, 122.4, 99.2, 61.2, 54.9, 52.9, 15.2; IR (thin film) ν 3063, 2976, 2927, 1697, 1658, 1449, 1346, 1229, 1117, 1059, 1004, 756 cm−1; HRESI+/TOF-MS calcd for C24H27NO5 [M]+ 409.1889, found 432.1788 [M+Na]+.
4.1.12. (E)-4-oxo-N,N-bis(2-oxo-2-phenylethyl)but-2-enamide (13)
A 0.33 M solution of 16 (3.1 g, 7.6 mmol, 1.0 equiv) in THF (23 mL) was treated with 2N aq HCl (23 mL, 46 mmol, 6.0 equiv) and stirred vigorously for 1 h at rt. The solution was diluted with EtOAc (125 mL), washed with brine (100 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to afford the title compound as a yellow foam (2.5 g, quant). 1H NMR (CDCl3) δ 9.66 (d, 1H, J = 7.1 Hz), 7.99–7.95 (m, 3H), 7.66–7.60 (m, 2H), 7.54–7.46 (m, 4H), 7.02 (d, 1H, J = 15.7 Hz), 6.94 (dd, 1H, J = 15.7, 7.1 Hz), 5.05 (s, 4H); 13C NMR (CDCl3) δ 194.1, 193.3, 192.4, 166.0, 139.0, 136.1, 134.8, 134.7, 134.2, 134.1, 129.2, 129.0, 128.1, 128.1, 55.0, 53.1; IR (thin film) ν 3068, 2932, 2834, 2742, 1697, 1654, 1600, 1449, 1351, 1229, 1112, 917, 756 cm−1; HRESI+/TOF-MS calcd for C20H17NO4 [M]+ 335.1158, found 358.1031 [M+Na]+.
4.1.13. Benzyl 1-(methoxy(methyl)amino)-2-methyl-1-oxopropan-2-ylcarbamate (23)
To stirring solution of Cbz-AIB19 (23.0 g, 96.9 mmol, 1.00 equiv) in CH2Cl2 (400 mL) was sequentially added N,O-dimethyl hydroxylamine hydrochloride (11.3 g, 116 mmol, 1.20 equiv), 4-dimethylaminopyridine (14.2 g, 116 mmol, 1.20 equiv), N,N′-diisopropylethylamine (20.0 mL, 116 mmol, 1.2 equiv), and N,N′-dicyclohexylcarbodiimide (11.3 g, 116 mmol, 1.20 equiv). The resultant suspension was stirred for 5 days. The precipitate was filtered and the filtrate diluted with CH2Cl2 (400 mL). This solution was sequentially washed with an aq 10% citric acid solution (2 × 400 mL), sat aq NaHCO3 (2 × 400 mL), and H2O (2 × 100 mL), dried over Na2SO4, filtered and concentrated under reduced pressure. Purification by flash column chromatography (3:2 hexanes:EtOAc) afforded the title compound as white solid (18.0 g, 67 %). 1H NMR (CDCl3) δ 7.35–7.29 (m, 5H), 5.77 (bs, 1H), 5.08 (s, 2H), 3.60 (s, 3H), 3.19 (s, 3H) 1.60 (s, 6H); 13C NMR (CDCl3) δ 174.5, 154.7, 136.8, 128.6, 128.2, 66.4, 60.8, 57.3, 34.0, 24.2; IR (thin film) ν 3327, 3034, 2985, 2937, 1717, 1649, 1527, 1454, 1386, 1367, 1258, 1073, 995, 746, 698 cm−1; HRESI+/TOF-MS calcd for C14H20N2O4 [M]+ 280.1432, found 303.1311 [M+Na]+.
4.1.14. Benzyl benzyl(1-(methoxy(methyl)amino)-2-methyl-1-oxopropan-2-yl)carbamate (24)
To a stirring solution of 23 (9.16 g, 32.7 mmol, 1.00 equiv) in DMF (165 mL) cooled to 0 °C was added NaH (60% mineral oil dispersion, 1.57 g, 39.2 mmol, 1.20 equiv). After effervescence had ceased (ca 10–15 min), benzyl bromide (4.30 mL, 36.0 mmol, 1.10 equiv) was slowly added. The solution was allowed to warm to rt and stirred for 24 h. The solution was poured into a seperatory funnel containing a solution of 1:1 sat aq NH4Cl:brine (500 mL) and extracted with EtOAc (2 × 300 mL). The combined organic fractions were then washed with brine (500 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. Purification by flash column chromatography (gradient, hexanes:EtOAc 2:1 → 4:3 → 1:1) afforded the title compound as a white solid (12.1 g, quant). 1H NMR (CDCl3) δ 7.30–7.24 (m, 10H), 5.22 (s, 3H), 4.68 (s, 2H), 3.38 (bs, 3H), 3.11 (bs, 3H), 1.43 (s, 6H); 13C NMR (CDCl3) δ 175.2, 139.3, 136.5, 128.6, 128.4, 128.2, 127.5, 127.2, 67.5, 62.3, 60.0, 47.7, 33.8, 25.1; IR (thin film) ν 3033, 2990, 2939, 1697, 1665, 1401, 1356, 1228, 1095, 998 699 cm−1; HRESI+/TOF-MS calcd for C21H26N2O4 [M]+ 370.1893, found 393.1798 [M+Na]+.
4.1.15. Benzyl benzyl(2-methyl-3-oxobutan-2-yl)carbamate (25)
Methyl magnesium bromide (3.0 M in Et2O, 55.0 mL, 165 mmol, 5.00 equiv) was added slowly to a chilled solution (0 °C) of 24 (12.1 g, 32.7 mmol, 1.00 equiv) in Et2O (165 mL). The suspension was allowed to warm to rt and stirred for 20 h. The suspension was cooled again to 0 °C and ice was carefully and slowly added over a 2 h period until the addition of more ice no longer caused gas evolution. The solution was decanted from the precipitate into a seperatory funnel and diluted with EtOAc (300 mL) and H2O (300 mL). The aqueous phase was removed; the organic phase was washed with brine (250 mL), dried over Na2SO4, filtered and concentrated under reduced pressure. The crude material was sonicated in Et2O and the title compound precipitated as a white solid that was collected by vacuum filtration (crop 1: 5.5 g, 59%). The filtrate was concentrated and then sonicated in a 3:2 solution of hexanes:Et2O. Again the title compound precipitated as a white solid and was collected by vacuum filtration (crop 2: 2.0 g, 19%). 1H NMR (CDCl3) δ 7.30–7.24 (m, 10H), 5.16 (s, 2H), 4.64 (s, 2H), 2.05 (bs, 3H), 1.26 (s, 6H); 13C NMR (CDCl3) δ 207.5, 156.6, 139.4, 136.2, 128.7, 128.6, 128.3, 127.3, 127.1, 67.9, 66.9, 47.5, 34.0, 23.6, 23.1; IR (thin film) ν 3063, 2985, 2941, 1722, 1693, 1458, 1405, 1351, 1253, 1220, 1088, 741 cm−1; HRESI+/TOF-MS calcd for C20H23NO3 [M]+ 325.1678, found 348.1579 [M+Na]+.
4.1.16. N-Benzyl-2-methyl-3-oxobutan-2-aminium chloride (26)
A 2-neck flask was purged with N2(g) and then charged with 10% Pd/C (1.14 g, 1.08 mmol, 0.0500 equiv of Pd). The reaction vessel was evacuated and back-filled with H2(g) three times before EtOH (200 proof, 100 mL) was added. Next, 25 (7.00 g, 21.5 mmol, 1.00 equiv) was added followed by conc. HCl (3.70 mL, 43.0 mmol, 2.00 equiv). The heterogeneous solution was stirred at rt under an atmosphere of H2(g) until the starting material was consumed as indicated by TLC analysis. Upon completion of the reaction, the solids were removed by filtration through a pad of Celite and the filter cake washed with MeOH. The filtrate was concentrated under reduced pressure to provide a crude white solid. The crude material was suspended in CH2Cl2 (200 mL), treated with sat aq NaHCO3 and stirred at rt for 2 h. The biphasic mixture was transferred to a seperatory funnel, the organic phase removed and the aqueous phase extracted with CH2Cl2 (100 mL). The combined organic phases were washed with brine (150 mL), dried over Na2SO4, filtered and concentrated under reduced pressure providing a crude oil. The oil was subjected to high vacuum to remove any 3-amino-3-methylbutan-2-one that resulted from over hydrogenation. The remaining crude material was dissolved in Et2O (100 mL), cooled to 0 °C, treated with anhydrous HCl (4.0 M in dioxane, 5.40 mL, 21.6 mmol), and stirred for 30 min. The resultant precipitate was collected by vacuum filtration to afford the title compound as a white solid (4.31 g, 88%). 1H NMR (CDCl3) δ 9.89 (bs, 2H), 7.64 (d, 2H, J = 7.0 Hz), 7.35–7.30 (m, 3H), 3.86 (t, 2H, J = 5.9 Hz), 2.18 (s, 3H), 1.54 (s, 6H); 13C NMR (CDCl3) δ 204.7, 131.4, 130.5, 129.5, 128.9, 67.4, 48.0, 24.5, 21.6; IR (thin film) ν 3424, 3034, 2917, 2855, 1720, 1547, 1453, 1133, 757, 702 cm−1; HRESI+/TOF-MS calcd for C12H18NO3 [M]+ 192.1383, found 192.1390 [M]+.
4.1.17. (E)-N-benzyl-4,4-diethoxy-N-(2-methyl-3-oxobutan-2-yl)but-2-enamide (27)
Isobutyl chloroformate (0.52 mL, 4.0 mmol, 1.0 equiv) was dropwise added to a solution of 3 (0.70 g, 4.0 mmol, 1.0 equiv) and N-methylmorpholine (2.2 mL, 20 mmol, 5.0 equiv) in THF (20 mL) at −10 °C. The resultant suspension was stirred for 20 min at −10 °C before the addition of 26 (1.1 g, 4.8 mmol, 1.2 equiv). The reaction vessel was equipped with a water-jacketed condenser and heated at 50 °C for 20 h. After cooling to rt, the suspension was poured onto an aqueous 10 % citric solution (50 mL) and extracted with EtOAc (75 mL). The organic extract was sequentially washed with aq 10 % citric acid solution (50 mL), water (30 mL), sat aq NaHCO3 (2 × 50 mL), water (30 mL), and brine (30 mL). The solution was dried over Na2SO4, filtered and concentrated under reduced pressure. Purification by flash column chromatography (3:2 hexanes:EtOAc doped with 0.5%/vol NEt3) provided the title compound (1.1 g, 78 %). 1H NMR (CDCl3) δ 7.40–7.35 (m, 4H), 7.31–7.28 (t, 1H, J = 6.8 Hz), 6.82 (dd, 1H, J = 15.2, 4.0 Hz), 6.45 (d, 1H, J = 15.2 Hz), 4.96 (d, 1H, J = 4.0 Hz), 4.67 (s, 2H), 3.59–3.53 (m, 2H), 3.45–3.39 (m, 2H), 2.18 (s, 3H), 1.30 (s, 6H), 1.12 (t, 6H, J = 7.0 Hz); 13C NMR (CDCl3) δ 206.1, 167.2, 142.9, 138.3, 129.1, 127.7, 126.3, 123.7 99.5, 66.4, 61.5, 47.8, 24.4, 22.9, 15.2; IR (thin film) ν 3034, 2980, 2931, 2883, 1717, 1663, 1619, 1410, 1351, 1121, 1053, 737 cm−1; HRESI+/TOF-MS calcd for C20H29NO4 [M]+ 347.2097, found 348.2163 [M+H]+.
4.1.18. (E)-N-benzyl-N-(2-methyl-3-oxobutan-2-yl)-4-oxobut-2-enamide (22)
A chilled (0 °C) solution of 27 (0.25 g, 0.72 mmol, 1.0 equiv) in THF (3.6 mL) was treated with 2N aq HCl (2.1 mL, 4.3 mmol, 6.0 equiv). The solution was slowly allowed to warm to rt. Upon completion, as indicated by TLC analysis, the solution was diluted with EtOAc (25 mL), washed with brine (20 mL), dried over Na2SO4, filtered and concentrated under reduced pressure. Purification by flash column chromatography (3:2 hexanes:EtOAc doped with 0.5%/vol NEt3) provided the title compound as a white solid (0.13 g, 64%). 1H NMR (CDCl3) δ; 9.62 (d, 1H, J = 7.0 Hz), 7.46–7.33 (m, 5H), 7.06–6.97 (m, 2H), 4.74 (s, 2H), 2.23 (s, 3H), 1.37 (s, 6H); 13C NMR (CDCl3) δ 206.0, 192.3, 165.8, 140.1, 137.4, 129.4, 128.2, 126.1, 66.8, 48.0, 24.7, 23.0; IR (thin film) ν 3034, 2985, 2931, 2849, 2736, 1712, 1693, 1644, 1419, 1351, 1254, 1117, 1029, 976 cm−1; HRESI+/TOF-MS calcd for C16H19NO3 [M]+ 273.1365, found 274.1440 [M+H]+.
4.1.19. (3aR,6aR)-5-benzyl-6,6,6a-trimethyltetrahydro-2H-furo[2,3-c]pyrrole-2,4(5H)-dione (28)
Prepared according to the general procedure. The following procedure is representative: An oven-dried vial was charged with IMesCl (13.4 mg, 0.0393 mmol, 0.15 equiv) and 22 (72.0 mg, 0.263 mmol, 1.00 equiv). The vial was purged with N2(g) and charged with 10:1 THF:tBuOH (5.2 mL) and DBU (0.25 M solution in 10:1 THF:tBuOH, 0.10 mL, 0.025 mmol, 0.10 equiv). The vial was capped and the reaction stirred at 40 °C for 4.5 h. The solvent was removed under reduced pressure and the crude solid purified by flash column chromatography (gradient, hexanes:EtOAc:iPrOH, 49:49:2→47.5:47.5:5) affording the title compound as a white foam (57.0 mg, 79%). 1H NMR (CDCl3) δ 7.31–7.28 (m, 2H), 7.26–7.21 (m, 3H), 4.52 (d, 1H, J = 15.5 Hz), 4.41 (d, 1H, J = 15.5 Hz), 3.08 (dd, 1H, J = 18.0, 0.7 Hz), 3.03 (d, 1H, J = 9.0 Hz), 2.89 (dd, 1H, J = 18.0, 9.0 Hz), 1.45 (s, 3H), 1.26 (s, 3H), 1.07 (s, 3H); 13C NMR (CDCl3) δ 174.0, 171.9, 137.9, 128.9, 127.6, 127.5, 90.2, 65.9, 46.5, 43.5, 31.7, 24.7, 20.2, 18.9; IR (thin film) ν 2976, 2937, 1780, 1693, 1410, 1268, 1229, 1200, 1122, 1083, 946 cm−1; HRESI+/TOF-MS calcd for C16H19NO3 [M]+ 273.1365, found 296.1249 [M+Na]+.
4.1.20. (R)-methyl 2-(benzyloxymethyl)-2-((2E,4E)-N-(4-methoxybenzyl)hexa-2,4-dienamido)-3-oxobutanoate (37)
To a 0.50 M solution of 36 (0.357 g, 0.80 mmol, 1.0 equiv) in CH2Cl2 (1.6 mL) cooled to 0 °C was added N,N′-diisopropylethylamine (0.21 mL, 1.2 mmol, 1.5 equiv). Sorbyl chloride20 (0.125 g, 0.96 mmol, 1.2 equiv) was added dropwise and the solution stirred for 0.5 h at 0 °C before being allowed to warm to rt and stirred an additional 22 h. The solution was diluted with CH2Cl2 (20 mL), washed with brine (20 mL), dried over Na2SO4, filtered and concentrated under reduced pressure. The crude material was dissolved in THF (4.0 mL) and treated with 6N aq HCl(1.0 mL, 6.0 mmol, 7.5 equiv) at rt for 3 h. The solution was diluted with EtOAc (25 mL), washed with brine (20 mL), dried over Na2SO4, filtered and concentrated under reduced pressure. The crude orange oil was dissolved in CH2Cl2 (8.0 mL), Dess-Martin periodinane (0.41 g, 0.96 mmol, 1.2 equiv) was added and the suspension stirred overnight. The solvent was removed under reduced pressure and the crude material taken up in EtOAc (30 mL). This was washed with a 1:1 solution of sat aq NaHCO3:sat aq Na2S2O3 (20 mL), washed with brine (20 mL), dried over Na2SO4, filtered and concentrated under reduced pressure. Purification by flash column chromatography (4:1 hexanes:EtOAc doped with 0.1%/vol NEt3) provided the title compound as a light yellow foam (0.225 g, 60% over 3 steps). 1H NMR (CDCl3) δ 7.33–7.26 (m, 6H), 7.12–7.11 (m, 2H), 6.90 (d, 2H, J = 8.5 Hz), 6.11-6.05 (m, 2H), 6.02 (d, 1H, J = 14.8 Hz), 4.91 (d, 1H, J = 18.4 Hz), 4.76 (d, 1H, J = 18.4 Hz), 4.30 (d, 1H, J = 11.9 Hz), 4.26 (d, 1H, J = 11.9 Hz), 3.82 (s, 3H), 3.79–3.74 (m, 2H), 3.77 (s, 3H), 2.42 (s, 3H), 1.78 (d, 3H, J = 5.0 Hz); 13C NMR (CDCl3) δ 198.1, 169.9, 168.8, 158.8, 145.4, 139.1, 137.0, 130.9, 130.2, 128.5, 128.0, 127.6, 127.3, 117.9, 114.2, 73.8, 70.7, 55.4, 52.9, 48.9, 28.1, 18.7; IR (thin film) ν 3029, 3000, 2951, 2839, 1737, 1654, 1617, 1517, 1356, 1249, 1175, 1098, 1029, 1000 cm−1; HRESI+/TOF-MS calcd for C27H31NO6 [M]+ 465.2151, found 466.2240 [M+H]+.
4.1.21. (2R)-methyl 2-(benzyloxymethyl)-2-((E)-4,5-dihydroxy-N-(4-methoxybenzyl)hex-2-enamido)-3-oxobutanoate (38)
To a round-bottom flask charged with 37 (0.042 g, 0.090 mmol, 1.0 equiv) was sequentially added AD-mix α (0.126 g), (DHQ)2PHAL (0.0077 g, 0.0099 mmol, 0.11 equiv), NaHCO3 (0.0023 g, 0.27 mmol, 3.0 equiv), MeSO2NH2 (0.0086 g, 0.090 mmol, 1.0 equiv) and a solution of H2O:tBuOH (5:3, 1.5 mL). The mixture was cooled to 0 °C and OsO4 (4% wt in H2O, 0.0023 g, 0.0090 mmol, 0.10 equiv) added. The mixture was allowed to warm to rt and stirred vigorously for 24 h before being quenched with sat aq Na2SO3 (0.5 mL). The aqueous mixture was extracted with EtOAc (4×7 mL), the combined extracts washed with 2N aq KOH (5 mL) and brine (5 mL), dried over Na2SO4, filtered and concentrated under reduced pressure. Purification by flash column chromatography (15:1 CHCl3:MeOH) yielded the title compound as a light tan foam (0.020 g, 44%). 1H NMR (CDCl3) δ 7.37–7.26 (m, 5H), 7.12–7.11 (m, 2H), 6.89 (d, 2H, J = 8.6 Hz), 6.87-6.83 (m, 1H), 6.42 (d, J = 15.0 Hz), 4.91 (d, 1H, J = 18.4 Hz), 4.79 (d, 1H, J = 18.3 Hz), 4.29 (d, 1H, J = 11.9 Hz), 4.26 (d, 1H, J = 11.9 Hz), 3.93 (bd, 1H, J = 4.1 Hz), 3.81 (s, 3H), 3.78 (s, 3H), 3.74 (s, 2H), 3.62 (bs, 1H), 2.56 (d, 1H, J = 5.0 Hz), 2.41 (s, 3H), 1.70 (bs, 1H), 1.14 (d, 3H, J = 6.3 Hz); 13C NMR (CDCl3) δ 198.1, 169.2, 168.6, 158.9, 146.4, 136.8, 130.5, 128.5, 128.1, 127.7, 127.3, 121.6, 114.2, 77.5, 76.0, 73.9, 70.5, 70.2, 55.4, 53.0, 49.0, 28.2, 19.1; IR (thin film) ν 3420, 3029, 2932, 2878, 1737, 1712, 1658, 1615, 1517, 1409, 1361, 1244, 1092, 824 cm−1; HRESI+/TOF-MS calcd for C27H33NO8 [M]+ 499.2206, found 500.2278 [M+H]+.
4.1.22. (R,E)-methyl 2-(benzyloxymethyl)-2-(N-(4-methoxybenzyl)-4-oxobut-2-enamido)-3-oxobutanoate (34)
A solution of 38 (0.043 g, 0.086 mmol, 1.0 equiv) in CH2Cl2 (1.0 mL) was treated with sat aq NaHCO3 (0.10 mL) and NaIO4 at rt for 20 h. The heterogeneous mixture was filtered through a plug of Na2SO4 that was further washed with CH2Cl2 (15 mL). The filtrate was concentrated under reduced pressure to afford the title compound as a white foam (0.039 g, quant). 1H NMR (CDCl3) δ 9.56 (d, 1H, J = 7.1 Hz), 7.38–7.33 (m, 2H), 7.31–7.26 (m, 3H), 7.13–7.11 (m, 2H), 7.01–6.92 (m, 4H), 4.97 (d, 1H, J = 18.4 Hz), 4.81 (d, 1H, J = 18.6 Hz), 4.32 (d, 1H, J = 11.9 Hz), 4.28 (d, 1H, J = 11.7 Hz), 3.83 (s, 3H), 3.81 (s, 3H), 3.79 (s, 2H), 2.45 (s, 3H); 13C NMR (CDCl3) δ 197.8, 192.4, 168.0, 167.5, 159.2, 140.2, 139.6, 136.5, 129.7, 128.6, 128.2, 127.8, 127.2, 114.5, 77.9, 74.0, 70.3, 55.5, 53.2, 49.2, 28.1; IR (thin film) ν 3006, 2953, 2922, 2834, 1742, 1694, 1651, 1513, 1412, 1357, 1248, 1110 cm−1; HRESI+/TOF-MS calcd for C25H27NO7 [M]+ 453.1788, found 476.1665 [M+Na]+.
4.1.23. (3aR,6R,6aS)-methyl 6-(benzyloxymethyl)-5-(4-methoxybenzyl)-6a-methyl-2,4-dioxohexahydro-2H-furo[2,3-c]pyrrole-6-carboxylate (33) and (3aS,6R,6aR)-methyl 6-(benzyloxymethyl)-5-(4-methoxybenzyl)-6a-methyl-2,4-dioxohexahydro-2H-furo[2,3-c] pyrrole-6-carboxylate (39)
Prepared according to the general procedure. The following procedure is representative: An oven-dried vial containing 34 (12.4 mg, 0.027 mmol, 1.0 equiv) was charged with IMesCl (1.4 mg, 0.0040 mmol, 0.15 equiv). The vial was purged with N2(g) and then charged with 10:1 THF:tBuOH (0.55 mL) and DBU (0.50 μL, 0.0036 mmol, 0.10 equiv). The vial was capped and the reaction stirred at 40 °C for 6 h. The solvent was removed under reduced pressure and the crude solid purified by flash column chromatography (hexanes:acetone, 3:1) to afford the mixture of diastereomers as a white film (9.3 mg, 75%).
Data for 3321
1H NMR (CDCl3) δ 7.33–7.26 (m, 5H), 7.09 (d, 2H, J = 6.8 Hz), 6.78 (d, 2H, J = 8.6 Hz), 5.04 (d, 1H, J = 15.2 Hz), 4.28 (d, 1H, J = 15.2 Hz), 3.91 (d, 1H, J = 11.5 Hz), 3.83–3.80 (m, 2H), 3.80 (s, 3H), 3.74 (s, 3H), 3.21 (d, 1H, J = 10.4 Hz), 3.04 (d, 1H, J = 9.3 Hz), 2.95 (d, 1H, J = 18.3 Hz), 2.80 (dd, 1H, J = 18.3, 9.6 Hz), 1.60 (s, 3H); 13C NMR (CDCl3) δ 173.7, 173.1, 167.1, 159.2, 136.9, 130.2, 129.6, 128.6, 128.1, 127.5, 113.8, 88.6, 76.0, 73.0, 67.9, 55.4, 53.0, 47.7, 45.3, 30.9, 19.6; IR (thin film) ν 2956, 2922, 2853, 1790, 1761, 1702, 1614, 1512, 1454, 1400, 1249, 1181, 1131, 737 cm−1; HRESI+/TOF-MS calcd for C25H27NO7 [M]+ 453.1788, found 476.1690 [M+H]+.
Data for 39
1H NMR (CDCl3) δ 7.36–7.29 (m, 3H), 7.20 (d, 2H, J = 6.8 Hz), 7.08 (d, 2H, J = 8.6 Hz), 6.78 (d, 2H, J = 8.6 Hz), 4.67 (d, 1H, J = 15.1 Hz), 4.37 (d, 1H, J = 15.1 Hz), 4.30 (d, 1H, J = 11.6 Hz), 4.25 (d, 1H, J = 11.6 Hz), 3.89 (d, 1H, J = 10.2 Hz), 3.78 (s, 3H), 3.71 (d, 1H, J = 10.0 Hz), 3.63 (s, 3H), 3.04–2.99 (m, 2H), 2.84 (dd, 1H, J = 18.4, 10.0 Hz), 1.47 (s, 3H); 13C NMR (CDCl3) δ 173.6, 173.4, 169.4, 160.0, 137.3, 129.4, 129.2, 128.5, 128.0, 127.9, 86.8, 75.5, 73.8, 68.9, 55.4, 53.0, 47.2, 45.3, 31.1, 21.6; IR (thin film) ν 2951, 2927, 2853, 1790, 1746, 1702, 1610, 1512, 1439, 1249, 1127, 737 cm−1; HRESI+/TOF-MS calcd for C25H27NO7 [M]+ 453.1788, found 476.1691 [M+H]+.
Supplementary Material
Acknowledgments
We are grateful to the NIH (GM-079339) and for generous gifts from Boehringer-Ingelheim, Bristol Myers Squibb, Eli Lilly, and Roche for support of this research program. J.W.B is a fellow of the Packard Foundation, the Beckman Foundation, the Sloan Foundation, and a Cottrell Scholar. J.R.S. would like to thank Dr. M. Elisa Juarez–Garcia, Dr. Hiroshi Ishida, and Dr. Michael Rommel for helpful discussions.
Footnotes
Supplementary Material
Copies of 1HNMR and 13CNMR spectra for all new compounds associated with this article can be found in the online version at doi:xx.xxxx/j.tet.xxxx.xx.xxx.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- i.For excellent reviews of NHC-catalyzed reactions, please see: Enders D, Balensiefer T. Acc Chem Res. 2004;37:534–541. doi: 10.1021/ar030050j.Christmann M. Angew Chem, Int Ed. 2005;44:2632–2634. doi: 10.1002/anie.200500761.Zeitler K. Angew Chem, Int Ed. 2005;117:7506–7510. doi: 10.1002/anie.200502617.Marion N, Diez-Gonzalez S, Nolan SP. Angew Chem, Int Ed. 2007;46:2988–3000. doi: 10.1002/anie.200603380.Enders D, Niemeier O, Henseler A. Chem Rev. 2007;107:5606–5655. doi: 10.1021/cr068372z.Nair F, Vellalath S, Babu BP. Chem Soc Rev. 2008;37:2691–2698. doi: 10.1039/b719083m.
- 2.For an example on of the effect of reaction conditions in NHC annulations see: Sohn S, Bode JW. Org Lett. 2005;7:3873–3876. doi: 10.1021/ol051269w.For an example related to catalyst effects see: Struble JR, Kaeobamrung J, Bode JW. Org Lett. 2008;10:957–960. doi: 10.1021/ol800006m.
- 3.Sohn SS, Rosen EL, Bode JW. J Am Chem Soc. 2004;126:14370–14371. doi: 10.1021/ja044714b. [DOI] [PubMed] [Google Scholar]
- 4.(a) He M, Bode JW. Org Lett. 2005;I:3131–3134. doi: 10.1021/ol051234w. [DOI] [PubMed] [Google Scholar]; (b) Rommel M, Fukuzumi T, Bode JW. J Am Chem Soc. 2008;130:17266–17267. doi: 10.1021/ja807937m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) He M, Struble JR, Bode JW. J Am Chem Soc. 2006:8148–8150. doi: 10.1021/ja062707c. [DOI] [PubMed] [Google Scholar]; (b) He M, Uc GJ, Bode JW. J Am Chem Soc. 2006;128:15088–15089. doi: 10.1021/ja066380r. [DOI] [PubMed] [Google Scholar]; (c) He M, Beahm BJ, Bode JW. Org Lett. 2008;10:3817–3820. doi: 10.1021/ol801502h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Phillips EM, Wadamoto M, Chan A, Scheidt KA. Angew Chem, Int Ed. 2007;46:3107–3110. doi: 10.1002/anie.200605235. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wadamoto M, Phillips EM, Reynolds TE, Scheidt KA. J Am Chem Soc. 2007;129:10098–10099. doi: 10.1021/ja073987e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Maki BE, Chan A, Scheidt KA. Synthesis. 2008;8:1306–1315. doi: 10.1055/s-2008-1072516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feling RH, Buchanan GO, Mincer TJ, Kauffman CA, Jensen PR, Fenical W. Angew Chem, Int Ed. 2003;42:355–357. doi: 10.1002/anie.200390115. [DOI] [PubMed] [Google Scholar]
- 8.Many total and formal syntheses have been reported: some notable examples are listed here: Reddy LR, Saravanan P, Corey EJ. J Am Chem Soc. 2004;126:6230–6231. doi: 10.1021/ja048613p.Reddy LR, Fournier JF, Reddy BVS, Corey EJ. Org Lett. 2005;7:2699–2701. doi: 10.1021/ol0508734.Ling T, Macherla VR, Manam RR, McArthur KA, Potts BCM. Org Lett. 2007;9:2289–2292. doi: 10.1021/ol0706051.Ma G, Nguyen H, Romo D. Org Lett. 2007;9:2143–2146. doi: 10.1021/ol070616u.Fukuda T, Sugiyama K, Arima S, Harigaya Y, Nagamitsu T, Omura S. Org Lett. 2008;10:4239–4242. doi: 10.1021/ol8016066.Mulholland NP, Pattenden G, Walters IAS. Org Biomol Chem. 2008;6:2782–2789. doi: 10.1039/b803818j.Margalef IV, Rupnicki L, Lam HW. Tetrahedron. 2008;64:7896–7901.
- 9.Abdalla GM, Sowell JW. J Heterocycl Chem. 1987;24:297–301. [Google Scholar]
- 10.First synthesized in our group, we conveniently term 2-Mesityl-2,5,6,7-tetrahydropyrrolo[2,1 -c] [1,2,4]triazol-4-ium chloride as RMesCl. This precatalyst is commercially available from Aldrich, cat. No. 688487.
- 11.For NHC-catalyzed annulations with concomitant decarboxylation of β–lactones see Nair V, Vellalath S, Poonoth M, Suresh E. J Am Chem Soc. 2006;128:8736–8737. doi: 10.1021/ja0625677.Chiang PC, Kaeobamrung J, Bode JW. J Am Chem Soc. 2007;129:3520–3521. doi: 10.1021/ja0705543.(c) ref. 6b.
- 12.Phillips EM, Wadamoto M, Chan A, Scheidt KA. Angew Chem, Int Ed. 2007;46:3107–3110. doi: 10.1002/anie.200605235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chan A, Scheidt KA. J Am Chem Soc. 2007;129:5334–5335. doi: 10.1021/ja0709167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dess DB, Martin JC. J Am Chem Soc. 1991;113:7277–7278. [Google Scholar]
- 15.Sharpless KB, Amberg W, Bennani YL, Crispino GA, Hartung J, Jeong K-S, Kwong H-L, Morikawa K, Wang Z-M, Xu D, Zhang X-L. J Org Chem. 1992;57:2768–2771. [Google Scholar]
- 16.Zhang Y, O’Doherty GA. Tetrahedron. 2005;61:6337– 6351. [Google Scholar]
- 17.Fourrey J-L, Beauhaire J, Yuan CW. J Chem Soc, Perkin Trans 1. 1987:1841–1843. [Google Scholar]
- 18.Arduengo AJ, III, Stewart CA, Davidson F, Dixon DA, Becker JY, Culley SA, Mizen MB. J Am Chem Soc. 1987;109:627–647. [Google Scholar]
- 19.Kiviranta PH, Leppänen J, Rinne VM, Suuronen T, Kyrylenko O, Kyrylenko S, Kuusisto E, Tervo AJ, Järvinen T, Salminen A, Poso A, Wallén EAA. Bioorg Med Chem Lett. 2007;17:2448–2451. doi: 10.1016/j.bmcl.2007.02.023. [DOI] [PubMed] [Google Scholar]
- 20.Davies SG, Haggitt JR, Ichihara O, Kelly RJ, Leech MA, Mortimer AJP, Roberts PM, Smith AD. Org Biomol Chem. 2004;2:2630–2649. doi: 10.1039/B404962D. [DOI] [PubMed] [Google Scholar]
- 21.Although the 1H NMR spectrum for 33 is an exact match to Lam’s spectrum, there exists a descrepancy in our reported data at resonance 6.78 ppm due to a typo in Lam’s report (δ 6.60 ppm); please see ref. 8g and the supporting information therein.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.