

Abstract
Selective binding by metalloproteins to their cognate metal ions is essential to cellular survival. How proteins originally acquired the ability to selectively bind metals and evolved a diverse array of metal-centered functions despite the availability of only a few metal-coordinating functionalities remains an open question. Using a rational design approach (Metal-Templated Interface Redesign), we describe the transformation of a monomeric electron transfer protein, cytochrome cb562, into a tetrameric assembly (C96RIDC-1) that stably and selectively binds Zn2+, and displays a metal-dependent conformational change reminiscent of a signaling protein. A thorough analysis of the metal binding properties of C96RIDC-14 reveals that it can also stably harbor other divalent metals with affinities that rival (Ni2+) or even exceed (Cu2+) those of Zn2+ on a per site basis. Nevertheless, this analysis suggests that our templating strategy also introduces an increased bias towards binding a higher number of Zn2+ ions (4 high affinity sites) versus Cu2+ or Ni2+ (2 high affinity sites), ultimately leading to the exclusive selectivity of C96RIDC-14 for Zn2 over those ions. More generally, our results indicate that an initial metal-driven nucleation event followed by the formation of a stable protein architecture around the metal provides a straightforward path for generating structural and functional diversity.
Introduction
The incorporation of metal ions into correct cellular targets is a formidable chemical task. Modern-day organisms use a number of strategies to ensure that a metal ion associates with the right target (frequently a protein), including the control of absolute and relative ambient metal concentrations,1 active delivery via chaperones,2 and compartmentalization.3 Nevertheless, these strategies still require the target protein to possess an intrinsic affinity and selectivity for the desired metal ion. This is achieved within the 3-D framework of proteins despite the availability of only a handful of metal-coordinating side chain functionalities.
An outstanding question is how protein structures have evolved to stably and selectively bind metal ions and developed metal-dependent functions, such as signal transduction (which necessitates conformational flexibility) and electron transfer or catalysis (which generally require rigid architectures). Some contemporary metalloproteins likely were based on pre-existing protein folds that acquired the ability to bind metals through random genetic events and subsequently attained their current structures and functions in the course of natural selection.4 In an alternative pathway, metal ions could first have templated the formation of a protein/peptide aggregate, followed by the evolution of the protein structure around the metal ion.5 Although this pathway would not have the benefit of re-using existing genetic material and a preformed scaffold, it may lead to greater flexibility towards generating bioinorganic diversity. We have developed a rational design approach (Metal-Templated Interface Redesign, MeTIR), which follows the time course of this latter hypothetical pathway (Scheme 1).6 Using MeTIR, we describe here the construction of an oligomeric protein architecture– C96RIDC-14–that stably and selectively binds Zn2+, and displays a large, metal-dependent conformational change akin to signaling/regulatory proteins.
Scheme 1.

Results and Discussion
Design Strategy and Initial Characterization of C96RIDC-14
We have previously shown that variants (termed MBPC’s) of a four-helix-bundle heme protein, cytochrome cb562, with appropriately installed metal-binding motifs on their surfaces (represented by Species 2 in Scheme 1) can adopt discrete supramolecular architectures upon metal coordination.7–9 Because the parent cyt cb562 (1) is a monomeric protein whose surface carries no bias towards oligomerization, its supramolecular assembly is largely dictated by metal coordination. Accordingly, all MBPC assemblies initially formed through metal binding (3) feature interfacial metal ions with saturated coordination spheres and ligand arrangements that obey the preferred stereochemistry of the metal ions: Zn2+ – tetrahedral, Cu2+ – tetragonal, Ni2+ – octahedral.9 If the protein assembly surrounding a particular metal ion can be stabilized without changing the overall supramolecular architecture, one would expect the binding affinity and specificity to increase for that metal ion. We decided to put this “template-and-stabilize” strategy to the test using a tetrameric Zn-mediated assembly, Zn4:MBPC-14 (Figure 1).
Figure 1.
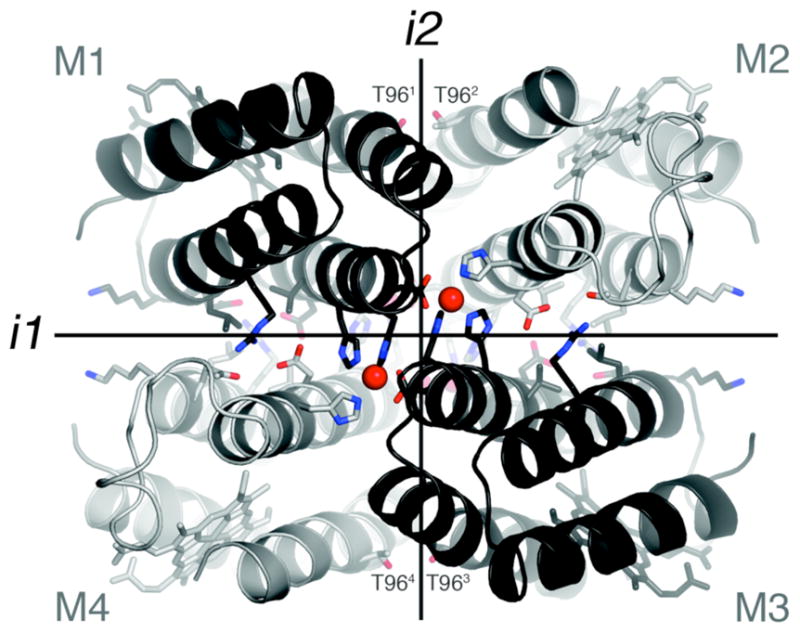
Interprotomeric interfaces in the D2-symmetric Zn4:MBPC-14 complex. Residues subjected to redesign as well as those that coordinate Zn2+ ions are shown as sticks.
MBPC-1 (2) is a cyt cb562 variant with two i, i+4 bis-His metal-binding motifs (H59/H63 and H73/H77) installed on its Helix3. Upon Zn coordination MBPC-1 forms a D2 (222) symmetrical tetramer, Zn4:MBPC-14, which is held together by four identical Zn ions coordinated to one bis-His motif (73/77) from one protomer, His63 from a second, and Asp74 from a third.7 Owing to its twofold dihedral symmetry, Zn4:MBPC-14 presents three pairs of C2-symmetric interfaces (i1, i2, i3) between its four protomeric constituents. Of these interfaces, only i1 presents an extensive surface (>1000 Å2) with close protein-protein contacts. We therefore undertook computationally-guided redesign of i1 to examine if a favorable set of interactions can be built into i1 to stabilize the entire Zn-driven assembly (Step b in Scheme 1).
A construct, RIDC-1, which features six mostly hydrophobic mutations (R34A/L38A/Q41W/K42S/D66W/V69I) in i1, indeed was found to form a considerably stabilized tetrameric assembly (Zn4:RIDC-14, 4) with an identical supramolecular geometry to the parent tetramer.6 Despite this stabilization, Zn4:RIDC-14 (like Zn4:MBPC-14) remains a dynamically exchanging assembly that does not stay intact, for instance, upon passage through a size-exclusion column. Consequently, it has not been possible to uncouple protein oligomerization from Zn binding and directly assess whether interface redesign has led to improvements in Zn affinity and selectivity. In order to obtain a stable tetrameric complex that would also form in the absence of Zn, we sought to further engineer RIDC-1.
As the dihedral symmetry of the Zn-tetramer dictates, the concurrent stabilization of any two of the three interfaces could in principle lead to the formation of a persistent tetrameric assembly. While interface i2 is not as tightly packed as i1 and therefore is less amenable to redesign, it presents position 96 from two protomers within distance for disulfide (SS) crosslinking (Figure 1). Thus, we combined the six mutations in i1 with the T96C mutation in i2 to generate C96RIDC-1. Under oxidizing conditions C96RIDC-1 readily forms an SS-crosslinked dimer as shown by SDS-PAGE electrophoresis (Figure S1). Sedimentation velocity (SV) experiments indicate that the predominant oligomeric form of C96RIDC-1 in solution is a tetramer (C96RIDC-14, 6), even in the absence of metals (Figure 2a). The tetramer-dimer dissociation constant (Kd(4mer-2mer)) for C96RIDC-14 has been determined by sedimentation equilibrium (SE) measurements to be <100 nM (Figure 2b), which to our knowledge makes it one of the most stable engineered protein complexes.
Figure 2.

(a) Sedimentation coefficient distributions for various cyt cb562 constructs at 5 μM monomeric concentration and equimolar Zn2+ where indicated. For MBPC-1 and RIDC-1, the Zn-induced tetrameric species are fully populated at >1 mM and >20 μM protein and Zn, respectively. At 5 μM protein and Zn, MBPC-1 is still predominantly monomeric (Smax= 1.8), while RIDC-1 is a mixture of dimeric (Smax= 2.8) and tetrameric (Smax= 4.5) forms. (b) Sedimentation equilibrium profile for 2.5 μM C96RIDC-12 obtained at 20000 rpm (see Figure S2 for other concentrations and speeds). The sedimentation data are equally well described by a dimer-tetramer equilibrium (Kd = 52 nM) or a tetramer-only model, which suggests the tetramer dissociation constant to be <100 nM.
Zn-dependent changes in C96RIDC-14 conformation
C96RIDC-14 remains tetrameric upon Zn binding. SV population distributions indicate that the resulting tetramer (5) forms at significantly lower protein and Zn concentrations and therefore is more stable than its progenitors Zn4:MBPC-14 and Zn4:RIDC-14 (Figure 2a). SV measurements also suggest that C96RIDC-14 undergoes a Zn-induced rearrangement, evidenced by a shift in its sedimentation coefficient from 4.25 S to 4.5 S. To elucidate this conformational change, we solved the crystal structures of C96RIDC-14 and its Zn adduct at 2.1 and 2.4-Å resolution, respectively (PDB ID’s 3IQ5 and 3IQ6).
As illustrated in Figure 3, these structures reveal a remarkable double-clothespin motion of the four protomers upon Zn-coordination, measuring ~16 Å at the N-terminus of Helix3. One of the keys to the simultaneous stability and conformational plasticity of C96RIDC-14 lies with the redesigned interface i1. Each of two equivalent i1 interfaces in C96RIDC-14 is formed between two protomers oriented in an antiparallel fashion. These interfaces feature an extensive network of hydrophobic interactions (~1300 Å2 buried surface), the main contributors being the two pairs of engineered Trp (41 and 66) and His (59 and 77) residues (Figure S3). Not surprisingly, this hydrophobic interface was predicted and found to be rather fluid.6 Upon binding Zn, the fluidity of the engineered hydrophobic interactions allows the four protomers to pivot around i1 and undergo the double-clothespin motion. The resulting architecture of Zn4:C96RIDC-14 features a well-packed hydrophobic core in i1 (~1400 Å2 buried surface) built around the engineered residues originally proposed by computation. Zn4:C96RIDC-14 is superimposable onto both Zn4:MBPC-14 and Zn4:RIDC-14 structures with respective root-mean-square deviations of 0.59 and 0.63 Å over 424 Cα’s (Figure S4). Importantly, the four equivalent Zn coordination environments remain unchanged as intended by the template-and-stabilize strategy.
Figure 3.

Crystal structures of C96RIDC-14 (left) and Zn4:C96RIDC-14 (right). Redesigned residues in i1 and i2 are shown as sticks. Interfacial SS-bond configurations are shown below each structure along with corresponding Fo – Fc omit difference maps (cyan mesh − 4.5 σ (apo), 3 σ (Zn); purple mesh − 11 σ (apo), 5 σ (Zn)). See Figure S4 for detailed views of interfacial residues.
The interfacial SS bonds incorporated into i2 are the second key component for the bistability of C96RIDC-14. The electron density maps for C96RIDC-14 and Zn4:C96RIDC-14 clearly outline the C96-C96′ linkages, which are found in distinct conformations to accommodate the two different supramolecular arrangements (Figure 3). Using the SS-bonds as “loose hinges”, the pairs of protomers that share i2 undergo a significant translational and rotational motion relative to one another, while maintaining the ideal bond distance (2.05 Å) and the stereochemical requirements for an SS bond. Operating together, the fluid non-covalent interactions in i1 and the adaptable SS bonds in i2 allow the formation of two stable and interconvertible tetrameric architectures in the absence or presence of Zn.
Zn binding by C96RIDC-14
Having thus uncoupled protein oligomerization from metal binding, we examined if our interface templating strategy leads to increased Zn affinity. Because Zn is spectroscopically silent, its binding to C96RIDC-14 was assessed through two different indirect methods. In the first method modeled after a procedure used for determining uranyl binding to NikR,10 we used nitrilotriacetic acid (NTA) as a competing Zn ligand (Kd(Zn-NTA) = 18.2 nM at 22°C and μ = 0.15 M) and 4-(2-pyridylazo)resorcinol (PAR) as an indicator, which exhibits an increase in absorbance at 500 nm upon binding Zn.11 Briefly, increasing amounts of NTA were added to samples of nearly equimolar Zn and C96RIDC-14. The resulting mixtures were subjected to centrifugation in 10-kDa-cutoff protein concentrators, which allowed the separation of the Zn fraction bound to C96RIDC-14 from that bound to NTA at each NTA concentration. This, in turn, permitted the calculation of the extent of Zn binding to C96RIDC-14 versus free Zn concentration, which was inferred from the Zn-NTA binding equilibrium. The NTA titrations are reasonably well described by an n-equivalent binding sites model, and indicate that each C96RIDC-14 complex binds four Zn equivalents with an apparent dissociation constant of 3.3 nM (Figure 4a, see Figure S9 for fits to alternative binding models).
Figure 4.

(a) Zn-binding isotherm of C96RIDC-14 determined using NTA as a competing ligand. (b) Zn-binding isotherm for Fura-2–C96RIDC-14 competition experiments; corresponding changes in the Fura-2 absorbance spectrum are shown in the inset. The data are corrected for dilution and background absorbance by the protein. The sample contained 7.5 μM C96RIDC-14 and 11 μM Fura-2. The tick marks shown on the top x-axis correspond to theoretical endpoints for titration if C96RIDC-14 bound to one, two, three or four equivalents of Zn. The fits obtained using DynaFit are shown for the following different models: solid line, four consecutive Zn binding equilibria (1+1+1+1); dashed line, two consecutive binding equilibria (2+2); dotted line; single binding equilibrium (4×1). Equilibrium constants obtained with these different models are listed in Table 1.
In a second, “higher-resolution” method that obviates the separation step and the secondary indicator, we used the chromophore Fura-2 (Kd, Zn = 5.7 nM)12 as a competing Zn ligand/indicator in a similar fashion to previously published procedures.13 In these experiments, increasing amounts of Zn were added to samples that contained C96RIDC-14 and Fura-2, and the Zn-dependent changes in the Fura-2 spectrum were directly monitored (Figure 4b). These titrations reveal that Zn binding to C96RIDC-14 is best described by four individual, consecutive binding equilibria or nearly equally well by two consecutive binding events by pairs of Zn2+ ions, with corresponding dissociation constants that range from 0.5 nM to 60 nM in the case of the 1+1+1+1 model and from 0.5 to 40 nM in the case of the 2+2 model (Table 1).14
Table 1.
Association constants for various metal binding equilibrium models for C96RIDC-14 determined through competitive Fura-2 titrations (pH 7, 295 K). The total free energies for metal binding correspond to the free energy sums of individual equilibria (times their multiplicity) for every model. Corresponding titrations and fits are shown in Figure 4b (Zn2+), Figure 7 (Cu2+ and Ni2+) and Figure S11 (Co2+). Numbers in parentheses correspond to standard deviation in the last reported significant figure, were obtained through DynaFit, and do not include any experimental errors.
| Total metal equivalents | Number of Consecutive Binding Equilibria | Kd1 (M) | Kd2 (M) | Kd3 (M) | Kd4 (M) | Total -ΔG for metal binding (kJ mol−1) |
|---|---|---|---|---|---|---|
| 4 Zn2+ | 2 | 5.2(4) × 10−10 | 4.3(2) × 10−8 | 189 | ||
| 4 | 1.3(3) × 10−9 | 5.3(7) × 10−10 | 3.3(8) × 10−8 | 5.8(8) × 10−8 | 186 | |
| 2 Cu2+ | 1 | 1.0(1) × 10−12 | 136 | |||
| 2 | 2.5(3) × 10−13 | 1.4(1) × 10−12 | 138 | |||
| 2 Ni2+ | 1 | 8.0(9) × 10−9 | 92 | |||
| 2 | 9.0(1) × 10−10 | 4.9(5) × 10−9 | 93 | |||
| 1 Co2+ | 1 | 9(4) × 10−7 | 34 |
Owing primarily to its flexibility, C96RIDC-14 apparently can accommodate Zn binding through several different modes; in other words, the intermediate Zn bound states may and most likely do utilize different ligand sets from one another. In any case, given that i, i+4 bis-His motifs on α-helices display Zn dissociation constants in the low μM range,15 the Zn binding titrations suggest that the pre-formation of a tetrameric, templated acceptor complex results in a ≥1000-fold increase in Zn-binding affinity relative to the monomeric parent species, MBPC-1.
Zn binding selectivity of C96RIDC-14 over other divalent metal ions
To elucidate if C96RIDC-14 also displays increased selectivity for Zn binding, its interactions with several other divalent metal ions (M2+) were examined, including the neighboring Co2+, Ni2+ and Cu2+, which typically are effective competitors for Zn binding sites. To this end, C96RIDC-14 was incubated with the metal ion of interest in a noncoordinating buffer solution (20 mM 3-(N-morpholino)propanesulfonic acid, MOPS), followed by the separation of the C96RIDC-14-metal complex via gel filtration and subsequent metal analysis by ICP-OES. These experiments show that C96RIDC-14 retains ~1 equivalent of Co2+, ~2 equivalents of Ni2+ and ~4 equivalents of Cu2+ (Figure S5a).
In competition studies, the initial C96RIDC-14/metal mixture was additionally incubated with Zn at various metal/Zn ratios prior to gel filtration and metal analysis. Each competition experiment was also carried out in reverse order - incubation with Zn followed by addition of other metals - to ascertain the formation of thermodynamic products (Figure S5b). These experiments reveal that C96RIDC-14 displays significant Zn selectivity over all ions except Cu2+, especially relative to its parent structure, MBPC-1 (Figures 5, S5b and S7). Previous studies have shown the affinity of the i, i+4 bis-His motif for Zn2+ to be comparable to that for Ni2+ and 5–10 fold higher than that for Co2+,15,16 following the Irving-Williams(IW) series.17 In contrast, Zn2+ completely outcompetes Co2+ for C96RIDC-14 binding at all ratios measured (up to 100 Co:1 Zn), and has an effective affinity roughly 100-fold higher than Ni2+.
Figure 5.

Extent of divalent metal ion binding to C96RIDC-14 in competition experiments.
Cu2+ presents a special case in terms of Zn selectivity. Due to a combination of its d9 configuration and high Lewis acidity, Cu2+ is situated at the top of the IW series, leading to its higher affinity for most ligand platforms designed for specific Zn binding13,18 and even natural Zn enzymes.19 The results shown in Figure 5 initially indicated that neither Cu2+ nor Zn2+ outcompete each other for C96RIDC-14 binding; rather, each tetrameric C96RIDC-14 unit appeared to bind ~3 equivalents of each ion in the non-coordinating MOPS buffer solution. To ascertain whether this apparent oversaturation of C96RIDC-14 is due to the binding of Cu2+ or Zn2+ ions to the C96RIDC-14 surface (in addition to the core binding sites) or due to the formation of an actual Cu-Zn heterometallic core species, we obtained crystals of C96RIDC-14 grown in the presence of equimolar amounts of both ions. The resulting 2.1-Å resolution diffraction data reveal a structure (PDB ID: 3M79) identical to that of Zn4:C96RIDC-14. To identify the four observed interfacial metal ions in this complex, we collected full data sets at the Zn and Cu K edges (1.28 Å and 1.38 Å), respectively. The corresponding anomalous difference maps clearly show that the interfacial ions are Zn2+, and that there are no detectable Cu2+ ions associated with the core or the surface of the tetramer (Figures 6a and b), despite the fact the crystal clearly contained Cu as indicated by an X-ray fluorescence excitation scan (Figure 6c). These observations suggest that Zn2+ outcompetes Cu2+ completely for binding to the core sites. This is further supported by the finding that when the Zn-Cu competition experiments are carried out in a weakly coordinating buffer solution (20 mM Tris(hydroxymethyl)aminomethane, TRIS), the amount of Cu2+ associated with C96RIDC-14 is significantly diminished, whereas the amount of bound Zn2+ stays constant (see the last two rightmost bars in Figure 5).
Figure 6.

Anomalous difference maps (4σ) for the C96RIDC-14 structures grown in the presence of equimolar Cu2+ and Zn2+ obtained at the Zn (a) or Cu (b) K edges. As expected, the heme Fe centers show anomalous signals at both wavelengths, whereas the core metal sites do the same only at the higher energy Zn edge, unambiguously identifying them as Zn ions. (c) X-ray fluorescence excitation scans of the same crystal – which was thoroughly washed with non-metal containing solutions – indicate the presence of both Zn and Cu.
To describe the Zn selectivity of C96RIDC-14 in a more quantitative fashion, we examined its affinity for Co2+, Ni2+ and Cu2+, again using Fura-2 as a competing ligand (Kd, Fura-Co = 8.6 nM,20 Kd, Fura-Ni = 6.9 nM,12 Kd, Fura-Cu = 0.3 pM12). The titrations indicate that C96RIDC-14 has one weak Co2+ binding site that barely competes with Fura-2 (Figure S11 and Table 1), whereas it can accommodate two equivalents of Ni2+ and Cu2+ with comparable affinities to Fura-2 (Figure 7 and Table 1).21 Both Ni2+ and Cu2+ binding curves are well described by two consecutive binding equilibria. Although on a per site basis the derived affinities are either similar to those for Zn2+ (in the case of Ni2+) or considerably higher (in the case of Cu2+) in line with the IW series, the higher multiplicity for Zn2+ binding apparently results in a more favorable overall free energy (Table 1), and ultimately in the Zn selectivity of C96RIDC-14 over these ions.21 Thus, templated interface engineering leads to increased bias not only towards Zn coordination geometry but also towards Zn binding multiplicity, which, to our knowledge, is a rare, and perhaps unique, case in designed/synthetic systems. Studies are currently underway to elucidate the coordination modes/environments of Co2+, Ni2+ and Cu2+ to C96RIDC-14 (which we expect to be different from each other and from Zn2+) and associated changes in protein structure.
Figure 7.

(a) Cu2+ and (b) Ni2+ binding isotherms for Fura-2–C96RIDC-14 competition experiments; corresponding changes in the Fura-2 absorbance spectrum are shown in the inset. The samples contained 7.5 μM C96RIDC-14 and 11 μM Fura-2. The tick marks shown on the top x-axis correspond to theoretical endpoints for titration if C96RIDC-14 bound to one, two, three or four equivalents of metal. The fits obtained using DynaFit are shown for the following different models: solid line, two consecutive binding equilibria (1+1); dashed line, single binding equilibrium (2). Equilibrium constants obtained with these different models are listed in Table 1.
Conclusions
Metal-templated synthesis has been a powerful approach to construct ligands with enforced coordination geometries that provide stable and specific metal binding.22–24 We have shown here that such templating strategies used for smaller systems can also be applied to proteins, which, owing to their extensive surfaces rich in chemical functionality, allow the formation of an extensive network of covalent and non-covalent interactions around the template. Importantly, the templating of protein interfaces around the target metal ion, Zn2+, has led to the evolution of a flexible, multi-14 stable protein complex that not only presents an increased bias towards the tetrahedral Zn2+ coordination geometry, but also towards the Zn2+ binding multiplicity of four, ultimately resulting in significantly increased selectivity over other divalent ions including Cu2+. It is well established that Cu2+ can readily outcompete Zn2+ (or any other physiologically important metal ion) for binding even rigid, tetrahedral coordination environments that favor Zn2+.19 Such thermodynamic dominance by the Cu2+ ion indeed necessitates various cellular strategies – often operating under kinetic control – to be employed for the incorporation of other metal ions into their intended protein targets.25 Our findings suggest that control of metal binding multiplicity may be a viable thermodynamic strategy alongside the design and restraint of the inner-sphere coordination environment to enhance metal selectivity in natural or synthetic systems.
Our study further demonstrates that cyt cb562 – a monomeric, putative electron transfer protein – can be transformed into a Zn-responsive complex through a minimal number of mutations that amount to less than 10% of its amino acid sequence. It is conceptually straightforward to envision how the Zn4:C96RIDC-14 architecture can be further rigidified and modified around the Zn centers to promote metal-based functions such as Lewis acid catalysis. While the role of metal templating in the evolution of metalloproteins can only be postulated, MeTIR clearly provides a practical route to generating structural and functional diversity. It remains to be seen if this approach can also be extended to non-metallic substrates, which could have served as nucleants in the early emergence of protein folds.26,27
Experimental Section
Site-Directed Mutagenesis and Protein Expression/Purification
The T96C mutation was introduced into pET-ridc16 using QuikChange (Stratagene) site-directed mutagenesis and primers obtained from Integrated DNA technologies, yielding the expression vector pET-C96ridc1. pET-C96ridc1 was transformed into XL-1 blue E. coli cells, purified using the QIAprep Spin Miniprep Kit (QIAGEN) and sequenced by Retrogen. pET-C96ridc1 was transformed into BL21(DE3) E. coli cells along with the ccm heme maturation gene cassette plasmid, pEC86.28 Cells were plated on LB agar containing 100 μg/ml ampicillin and 34 μg/ml chloramphenicol and grown overnight. 3 mL starter cultures were inoculated from the resulting colonies, grown to an Abs600 of 0.6 and used to inoculate 1 L of LB medium. 1-L cultures were then incubated for 16 hours with rotary shaking at 250 rpm. No induction was necessary.
Protein was obtained by sonicating cells in the presence of lysozyme, bringing the lysate to pH 5 with HCl and isolating the soluble fraction by centrifugation at 16,000 g, 4° C, for 1 hr. Initial purification was by cation-exchange chromatography on a CM-Sepharose matrix (Amersham Biosciences) using an NaCl step gradient in sodium acetate (pH 5). After dialysis into sodium phosphate (pH 8), the protein was further purified by anion exchange on an Uno-Q (BioRad) column using a DuoFlow chromatography workstation (BioRad) and a linear NaCl gradient. A protein sample was then exchanged into water, mixed 1:1 with sinapinic acid matrix (Agilent Technologies) and subjected to MALDI mass spectrometry to verify the T96C mutation (expected mass 12305 amu, observed 12299 amu).
Dimeric C96RIDC-12 was separated from monomeric protein on a preparative scale size exclusion chromatography column (GE Healthcare) packed with Superdex 75 (GE Healthcare) resin equilibrated in 20 mM Tris HCl (pH 7) and 150 mM NaCl. Separation of dimer (which effectively is a tetramer at concentrations > 1 μM) from monomer was verified by non-reducing SDS-PAGE gel electrophoresis using a 15% acrylamide gel. Typical protein preparations yielded 8–10 mg of protein per liter of culture based on an extinction coefficient of 148,000 M−1cm−1 at 415 nm. After purification, dimeric C96RIDC-12 was concentrated to ~2 mM using an Amicon stirred cell (Millipore), flash frozen in liquid nitrogen and stored at −80° C.
Analytical Ultracentrifugation
SV and SE samples were prepared in 20 mM Tris (pH 7) and 150 mM NaCl using appropriate volumes of 5 mM metal stock solutions or 50 mM EDTA. After incubating in the presence of M2+ or EDTA for 1 hour, SV measurements were carried out at 25° C on a Beckman XL-I Analytical Ultracentrifuge using an An-60 Ti rotor at 41,000 rpm for a total of 250 scans per sample. The following wavelengths were used to monitor C96RIDC-12 sedimentation at different protein concentrations: 415 nm (5 μM), 440 nm (30 μM). All data were processed using SEDFIT29 software with the following fixed parameters: buffer density (r) = 1.0049 g/ml; bufferviscosity = 0.010214 poise; Vbar = 0.73084.
SE measurements were carried out at 25° C using speeds between 10,000 and 20,000 rpm. Scans were taken at 14 and 16 hours and visually inspected to verify that sedimentation equilibrium was achieved. The following wavelengths were used to monitor C96RIDC-12 sedimentation at different protein concentrations: 415 nm (1 μM), 420 nm (2.5 μM), and 500 nm (12.5 μM). 16-hr scans were fit to a monomer-dimer (where monomer = C96RIDC-12) or a dimer-only model using SEDPHAT.30 The molecular mass of C96RIDC-12 (24610 Da) and the menisci were fixed while floating the association constant. Standard deviation for the resulting log10(K) value was determined through Monte-Carlo analysis within SEDPHAT.30
Crystallography
All crystals were grown by sitting drop vapor diffusion at room temperature (20–25° C) in drops consisting of 2 μL of protein and 1 μL of precipitant solution. For apo and Zn2+ crystals, a 2.1 mM protein stock solution in 20 mM Tris (pH 7) and 150 mM NaCl was used. The precipitant solution for apo-C96RIDC-14 crystals consisted of 100 mM Bis-Tris (pH 6.5) and 30% PEG 400. The precipitant solution for Zn4:C96RIDC-14 crystals was 100 mM Tris (pH 7.5), 20% PEG 2000 and 2.5 mM ZnCl2. Crystallization of C96RIDC-14 in the presence of Zn and Cu (Zn/CuC96RIDC-14) was performed by incubating 60 μM protein with one molar equivalent of Zn for one hour followed by addition of one equivalent of Cu and incubation overnight to ensure the formation of the thermodynamic product. The mixture was then concentrated to 2.6 mM using 4 mL Amicon Ultra (Millipore) centrifugal filters. Crystallization was performed as above with a precipitant solution consisting of 24% PEG 2000 and 100 mM Bis-Tris (pH 6.5). Crystals used for diffraction were exchanged stepwise into a solution containing 20% glycerol as a cryoprotectant and frozen in liquid nitrogen or directly in the cryostream.
Diffraction data were collected at the Stanford Synchrotron Radiation Laboratory (SSRL) Beamline 9-2 for C96RIDC-14 and Zn4:C96RIDC-14 or Beamline 7-1 for Zn/CuC96RIDC-14 at 100 K. 1.0-Å radiation datasets were collected for all crystals and additional datasets were collected using 1.28-Å and 1.38-Å radiation, corresponding to Zn and Cu K edges, respectively, for Zn/CuC96RIDC-14. The data were processed using MOSFLM and SCALA.31 The structures of C96RIDC-14, Zn4:C96RIDC-14 and Zn/CuC96RIDC-14 were determined at 2.05, 2.35 and 2.1 Å resolution, respectively, by molecular replacement with MOLREP,32 using the RIDC-1 monomeric structure (PDB ID: 3HNI) as the search model. Rigid-body, positional and thermal refinement with CNS33 or REFMAC,34 along with manual model rebuilding and water/ligand placement with XFIT35 or COOT36 produced the final models. For all structures, non-crystallographic symmetry (NCS) restraints (tight main-chain and medium side-chain restraints) were applied throughout the positional/thermal refinement process. The Ramachandran plots were calculated with PROCHECK.37 All figures were produced with PYMOL (www.pymol.org). Data collection and refinement statistics are summarized in Table S1.
Inductively-coupled plasma-optical emission (ICP-OES) spectroscopy
ICP-OES samples were in 20 mM MOPS (pH 7) and 150 mM NaCl or 20 mM Tris (pH 7) and 150 mM NaCl. To determine the stoichiometry of metal binding to C96RIDC-14, 60 μM protein was incubated with one or two molar equivalents of the desired metal ion. For competition studies, 60 μM protein was incubated with one molar equivalent of Zn2+ for one hour, after which, one, ten or one hundred molar equivalents of competing metal (M2+) were added, bringing the final volume to 1 ml. The same experiments were also done in reverse order. All samples were allowed to equilibrate overnight at room temperature and free/loosely bound metal was subsequently removed using a 10DG gel filtration column (Bio-Rad). Samples were prepared for ICP-OES by diluting 1.3 mL of protein solution collected off the 10DG column to a final volume of 2 ml and adding 90 μl of 69% reagent grade nitric acid (Fluka) to achieve a final concentration of 3%. Standards were prepared from 1000 ppm certified ICP-OES metal stock solutions (Ricca) by mixing equal volumes of all metal analytes and diluting to a final concentration of 200 ppm of each metal. A standard curve with eleven points between 0.05 and 10 ppm was then constructed by diluting appropriate volumes of the 200-ppm stock to 10 ml with 3% nitric acid in deionized water. Data were collected on a Perkin-Elmer Optima 3000 DV ICP-OES spectrometer located at the Analytical Facility of the Scripps Institute of Oceanography. Because each C96RIDC-1 monomer contains one Fe atom as part of the covalently linked heme molecule, the experimentally determined M(II):Fe ratios directly yielded the M2+:protein ratios. Wavelengths used for the detection of various metal ions were as follows: Mg (279.077, 280.271 and 285.213 nm), Ca (315.887 and 317.993), Fe (234.349, 238.204, 239.562, 259.939 and 273.055 nm), Co (228.616 and 238.892 nm), Ni (221.648 and 231.604 nm), Cu (224.7, 222.778, 221.459, 327.393 and 324.752 nm), and Zn (202.548, 206.2 and 213.857 nm). Values reported for each metal are averages of those for all wavelengths indicated.
Competitive binding assays using nitrilotriacetic acid (NTA)
The stability constant for Zn2+ binding to C96RIDC-14 was determined based on a previously published protocol for the determination of uranyl binding to NikR,10 with the exception that NTA was substituted as the competing ligand. Titrations were performed in 20 mM MOPS (pH 7) and 150 mM NaCl at 22° C. Trace metal was removed from all buffers by passage through a Chelex 100 (Bio-Rad) column.
For each titration point, 22 μM protein was mixed with 28 μM of Zn and variable amounts of NTA. Samples were allowed to equilibrate overnight at room temperature and subsequently centrifuged in 0.5 mL Amicon Ultra (Millipore) protein concentrators (10 KDa MW cutoff) for ten minutes at 2000 rpm to allow free Zn, free NTA and NTA:Zn to pass through the membrane. Zn concentrations were determined for the flow-through and protein chamber at each titration point using 4-(2-pyridylazo)resorcinol (PAR), which exhibits an increase in aborbance at 500 nm upon binding Zn (Δε500 = 59100 M−1cm−1 in buffer and 54800 M−1cm−1 in 5 M guanidine hydrochloride), essentially as previously described.11 1800 μL of 1 mM PAR was added to a 1-cm pathlength cuvette, and the initial absorbance spectrum was recorded. Subsequently, 100 ml of flow-through was added, allowed to stir for a minimum of five minutes to ensure that equilibrium had been reached, and absorbance at 500 nm was again recorded. Metal content in the protein chamber was determined similarly to flow-through samples, except that 1800 μl of 5 M buffered guanidine hydrochloride was used to denature protein and allow the complete release of bound metal for measurement. Absorbance at 500 nm was corrected for dilution and protein absorbance, and Zn concentrations determined using the Δε500 values listed above.
The fraction of protein bound to metal as a function of competing ligand concentration was determined by the following equation:
where [M]P.C. is the concentration of metal in the protein chamber, [M]F.T. is the concentration of metal in the flow-through and [P]T is the total concentration of protein measured after centrifugation. For calculating dissociation constants, the concentration of free metal was determined based on the concentration and dissociation constant of the competing ligand (NTA) and metal in the flow-through using the program MaxChelator (http://maxchelator.stanford.edu). The data were fit to the following models38 using Igor Pro v. 6.02a (Wavemetrics, Inc.).
| (1) |
| (2) |
| (3) |
where B is the molar equivalents of metal bound per C96RIDC-14, n is the number of binding sites, [M] is the concentration of free metal and K1–4 are stoichiometric association constants. Equation (1) assumes four identical binding sites with invariant affinities. Equation (2) assumes two stepwise binding events each of which involves binding of two Zn2+. Equation (3) assumes four stepwise binding events.
Binding affinity of Fura-2 for Ni2+, Cu2+ and Zn2+
1 mg of lyophilized Fura-2 (Invitrogen) was suspended in 1 mL of deionized water and its concentration was determined based on a published extinction coefficient of 27,000 M−1cm−1 at 362 nm.39 EGTA competition binding assays were used to determine dissociation constants for Ni2+, Cu2+ and Zn2+ using the following EGTA:M2+ logK values obtained from the online program MaxChelator (http://maxchelator.stanford.edu): Cu2+:EGTA − 13.2, Ni2+ − 9.0, Zn2+ − 8.1. These values are corrected for pH, temperature and ionic strength. In a 3 mL quartz cuvette, 11 μM Fura-2 was mixed with 100 μM EGTA in 20 mM MOPS (pH 7) and 150 mM NaCl to a final volume of 2 mL at 22° C. M2+ was titrated into the solution with an equilibration time of 10 minutes at ambient conditions between each addition, and absorbance spectra were recorded from 190–800 nm using a Hewlett Packard 8452A diode array spectrophotometer (Figure S10). For Cu2+, a separate titration into a solution of 100 μM buffered EGTA was conducted and subtracted from the Fura-2 titration points to account for absorbance from aqueous Cu2+ and the Cu2+:EGTA complex. Absorbance and M2+ concentrations were corrected for dilution and data was fit to a 1:1 M2+:Fura-2 model using DynaFit40 scripts (Figure S10) modeling the competition between EGTA and Fura-2 for M2+. The Zn2+:Fura-2 value determined by this method (logK = 8.2) compares favorably with a value previously determined at pH 7.15 (logK = 8.5).41 The logK values determined for the Fura-2 complexes of Cu2+ and Ni2+ complexes are 12.5 and 8.2, respectively.
Competitive binding assays using Fura-2
Competition assays using the Fura-2:M2+ dissociation constants determined above were used to measure C96RIDC-14:M2+ affinities. In a 3-mL quartz cuvette, a stock solution of C96RIDC-14 was diluted to 7.5 μM in 20 mM MOPS (pH 7) and 150 mM NaCl at 22° C and absorbance was recorded. Fura-2 was added to a final concentration of 10 μM and absorbance again recorded. Stock metal solutions were then titrated into the Fura-2/C96RIDC-14 mixture in ~2 μM steps, allowed to equilibrate with stirring for 10 minutes at ambient conditions and absorbance recorded. Protein and metal concentrations were corrected for dilution, protein absorbance was subtracted from total absorbance and plots of Abs373 versus total metal added were fit to various models using custom DynaFit scripts (see Supporting Information). Co titrations were performed similarly to Zn, Ni and Cu, but with the following modifications due to its lower affinity for C96RIDC-14: C96RIDC-14 concentration was increased to 21 μM and the Fura-2 concentration was decreased to 8 μM. Also, titrations were performed in a 0.5 cm pathlength cuvette to allow for the use of a higher protein concentration without saturating the detector.
Supplementary Material
Acknowledgments
We thank Prof. Thomas O’Halloran for helpful discussions. Support for this work was provided by NIH (training grants to J.D.B. and A.M.M.; predoctoral fellowship to E.N.S.), NSF (CHE-0908115, structural work), DOE (DE-FG02-10ER46677, competitive metal binding titrations) and the Arnold and Mabel Beckman Foundation (F.A.T.). Portions of this research were carried out at SSRL, operated by Stanford University on behalf of DOE.
Footnotes
Supporting Information Available. Additional experimental details, figures and tables on protein preparation, crystallography, analytical ultracentrifugation and binding assays. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Outten CE, O’Halloran TV. Science. 2001;292:2488. doi: 10.1126/science.1060331. [DOI] [PubMed] [Google Scholar]
- 2.Rosenzweig AC. Acc Chem Res. 2001;34:119. doi: 10.1021/ar000012p. [DOI] [PubMed] [Google Scholar]
- 3.Tottey S, Waldron KJ, Firbank SJ, Reale B, Bessant C, Sato K, Cheek TR, Gray J, Banfield MJ, Dennison C, Robinson NJ. Nature. 2008;455:1138. doi: 10.1038/nature07340. [DOI] [PubMed] [Google Scholar]
- 4.Blundell TL. The evolution of metal-binding sites in proteins. Symposium Press London; University of Sussex: 1977. [Google Scholar]
- 5.Liu CL, Xu HB. J Inorg Biochem. 2002;88:77. doi: 10.1016/s0162-0134(01)00337-3. [DOI] [PubMed] [Google Scholar]
- 6.Salgado EN, Ambroggio XI, Brodin JD, Lewis RA, Kuhlman B, Tezcan FA. Proc Natl Acad Sci USA. 2010;107:1827. doi: 10.1073/pnas.0906852107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salgado EN, Faraone-Mennella J, Tezcan FA. J Am Chem Soc. 2007;129:13374. doi: 10.1021/ja075261o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salgado EN, Lewis RA, Faraone-Mennella J, Tezcan FA. J Am Chem Soc. 2008;130:6082. doi: 10.1021/ja8012177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Salgado EN, Lewis RA, Mossin S, Rheingold AL, Tezcan FA. Inorg Chem. 2009;48:2726. doi: 10.1021/ic9001237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wegner SV, Boyaci H, Chen H, Jensen MP, He C. Angew Chem Intl Ed Eng. 2009;48:2339. doi: 10.1002/anie.200805262. [DOI] [PubMed] [Google Scholar]
- 11.McCall KA, Fierke CA. Anal Biochem. 2000;284:307. doi: 10.1006/abio.2000.4706. [DOI] [PubMed] [Google Scholar]
- 12.The dissociation constants of Fura-2 complexes with Zn2+, Cu2+ and Ni2+ were determined separately using EGTA as a competing ligand. See Experimental Section for more details.
- 13.Walkup GK, Imperiali B. J Am Chem Soc. 1997;119:3443. [Google Scholar]
- 14.With the data in hand, we consider the 2+2 model to be more plausible than the 1+1+1+1 model, which would require a 3-Zn bound intermediate state that is not likely (though not impossible) to form within the D2 symmetry of the C96RIDC-14 scaffold.
- 15.Krantz BA, Sosnick TR. Nat Struct Biol. 2001;8:1042. doi: 10.1038/nsb723. [DOI] [PubMed] [Google Scholar]
- 16.Ghadiri MR, Choi C. J Am Chem Soc. 1990;112:1630. [Google Scholar]
- 17.Frausto da Silva JJR, Williams RJP. The biological chemistry of the elements. Oxford University Press; Oxford: 2001. [Google Scholar]
- 18.Nolan EM, Ryu JW, Jaworski J, Feazell RP, Sheng M, Lippard SJ. J Am Chem Soc. 2006;128:15517. doi: 10.1021/ja065759a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hunt JA, Ahmed M, Fierke CA. Biochemistry. 1999;38:9054. doi: 10.1021/bi9900166. [DOI] [PubMed] [Google Scholar]
- 20.Kwan CY, Putney JW. J Biol Chem. 1990;265:678. [PubMed] [Google Scholar]
- 21.As previously mentioned, ICP-OES results indicate two high-affinity binding sites for Ni and four for Cu on C96RIDC-14. The two Cu binding sites/modes not observed in the Fura-2 competition experiments may either be less tightly bound to the core or to the surface of C96RIDC-14. If they are bound to the core in a fashion that would compete with Zn binding, their average dissociation constants would have to be ~40 μM each in order to compensate for the free energy difference between the four-Zn-bound and two-Cu-bound species listed in Table 1.
- 22.Thompson MC, Busch DH. J Am Chem Soc. 1964;86:3651. [Google Scholar]
- 23.Creaser II, Geue RJ, Harrowfield JM, Herlt AJ, Sargeson AM, Snow MR, Springborg J. J Am Chem Soc. 1982;104:6016. [Google Scholar]
- 24.McMurry TJ, Raymond KN, Smith PH. Science. 1989;244:938. doi: 10.1126/science.2658057. [DOI] [PubMed] [Google Scholar]
- 25.Waldron KJ, Robinson NJ. Nature Rev Microbiol. 2009;7:25. doi: 10.1038/nrmicro2057. [DOI] [PubMed] [Google Scholar]
- 26.Pande VS, Grosberg AY, Tanaka T. Proc Natl Acad Sci USA. 1994;91:12976. doi: 10.1073/pnas.91.26.12976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saito S, Sasai M, Yomo T. Proc Natl Acad Sci USA. 1997;94:11324. doi: 10.1073/pnas.94.21.11324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Braun M, Thony-Meyer L. Proc Natl Acad Sci USA. 2004;101:12830. doi: 10.1073/pnas.0402435101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schuck P. Biophys Chem. 2004;108:187. doi: 10.1016/j.bpc.2003.10.016. [DOI] [PubMed] [Google Scholar]
- 30.Vistica J, Dam J, Balbo A, Yikilmaz E, Mariuzza RA, Rouault TA, Schuck P. Anal Biochem. 2004;326:234. doi: 10.1016/j.ab.2003.12.014. [DOI] [PubMed] [Google Scholar]
- 31.Collaborative Computational Project, Number 4. The CCP4 Suite: Programs for Protein Crystallography. Acta Cryst. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 32.Vagin A, Teplyakov A. J Appl Cryst. 1998;30:1022. [Google Scholar]
- 33.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Acta Crystallogr D. 1998;54:905. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 34.Murshudov G, Vagin A, Dodson E. Acta Cryst. 1996;D53:240. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 35.McRee DE. J Mol Graphics. 1992;10:44. [Google Scholar]
- 36.Emsley P, Cowtan K. Acta Cryst. 2004;D60:2126. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 37.Laskowski RA, Macarthur MW, Moss DS, Thornton JM. J Appl Crystallogr. 1993;26:283. [Google Scholar]
- 38.Klotz IM. Ligand-receptor energetics: a guide for the perplexed. John Wiley & Sons, Inc; New York: 1997. [Google Scholar]
- 39.Grynkiewicz G, Poenie M, Tsien RY. J Biol Chem. 1985;260:3440. [PubMed] [Google Scholar]
- 40.Kuzmic P. Anal Biochem. 1996;237:260. doi: 10.1006/abio.1996.0238. [DOI] [PubMed] [Google Scholar]
- 41.Atar D, Backx PH, Appel MM, Gao WD, Marban E. J Biol Chem. 1995;270:2473. doi: 10.1074/jbc.270.6.2473. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.