

Abstract
There is extensive evidence that the mitogen-activated protein kinase (MAPK) signaling cascade mediates programmed cell death in neurons. However, current evidence that the mixed linage kinases (MLKs), upstream in this cascade, mediate cell death is based, in the in vivo context, entirely on pharmacological approaches. The compounds used in these studies have neither complete specificity nor selectivity among these kinases. Therefore, to better address the molecular specificity of the MLKs in mediating neuron death, we used dominant-negative constructs delivered by AAV (adenoassociated virus) vector transfer. We assessed effects in a neurotoxin model of parkinsonism, in which neuroprotection by pharmacologic MLK inhibition has been reported. We find that two dominant-negative forms of dual leucine zipper kinase (DLK) inhibit apoptosis and enhance long-term survival of dopamine neurons, but a dominant negative of MLK3 does not. Interestingly, the kinase-dead form of DLK not only blocks apoptosis but also has trophic effects on dopamine neurons. Although the MAPK cascade activates a number of downstream cell death mediators, we find that inhibition of DLK correlates closely with blockade of phosphorylation of c-jun and prevention of cell death. We conclude that DLK acts primarily through c-jun phosphorylation to mediate cell death in this model.
Keywords: apoptosis, programmed cell death, Parkinson's disease, substantia nigra, mixed lineage kinases, dopamine
Introduction
The mixed lineage kinases (MLKs) are upstream mediators of programmed cell death (PCD) in neurons, acting through the mitogen-activated protein kinase (MAPK) cascade to phosphorylate and activate the transcription factor c-jun (for review, see Gallo and Johnson, 2002; Wang et al., 2004; Silva et al., 2005a). Recently, there has developed a particular interest in the MLKs in relation to Parkinson's disease (PD), based on the discovery of disease-causing mutations in LRRK2 (leucine-rich repeat kinase) (Paisan-Ruiz et al., 2004; Zimprich et al., 2004), which bears homology to members of the MLK class (Mata et al., 2006). Its activity appears to be required for cellular toxicity (Greggio et al., 2006; Smith et al., 2006), and, interestingly, some of the disease-causing mutations result in increased kinase activity (West et al., 2005; Gloeckner et al., 2006; Greggio et al., 2006).
The concept that the MLKs may play a role in PCD, and thereby in the progression of PD, has been put to a clinical test in an investigation of 3,9-bis[(ethylthio)methyl]-K-252a (CEP1347), an MLK inhibitor (Waldmeier et al., 2006). Unfortunately, the trial failed to show efficacy to slow disease progression. However, this disappointing result cannot be taken as evidence that the overall strategy is a failure (for review, see Burke, 2007). It is unknown, for example, whether MLK inhibition in brain was actually achieved.
If we are to further consider inhibition of the MLKs as a potential neuroprotective approach, we must advance our understanding of their neurobiology as mediators of PCD in neurons. Although the role of the MLKs in mediating PCD in neurons has been supported by both pharmacologic and molecular approaches in the in vitro context (for review, see Silva et al., 2005a), this is not the case for in vivo studies. All of these studies have been based on pharmacologic inhibition of the MLKs with either CEP1347 or its analog bis-isopropylthiomethyl-K-252a (CEP11004) (Murakata et al., 2002) (for review, see Saporito et al., 2002). These compounds are not specific for the MLKs. Roux et al. (2002) have shown that CEP1347 also activates the survival signaling kinase Akt through an MLK-independent pathway. We showed that activation of Akt signaling in dopamine (DA) neurons of the substantia nigra (SN) has marked neurotrophic and antiapoptotic effects in vivo (Ries et al., 2006). Furthermore, these drugs are broadly inhibitory among kinases within the MLK class (Maroney et al., 2001; Murakata et al., 2002) and they do not permit identification of a selective role for any one of them.
To address these issues of the specificity and selectivity of the MLKs in PCD in SN DA neurons in vivo, we used an adenoassociated virus (AAV) vector approach to deliver dominant-negative (DN) forms of MLK3 and dual leucine zipper kinase (DLK), to assess their ability to suppress apoptosis in DA neurons of the SN induced by the neurotoxin 6-hydroxydopamine (6OHDA). We find that two dominant-negative forms of DLK, but not MLK3, are effective in suppressing apoptosis. Unexpectedly, we find that the “kinase-dead” dominant-negative form of DLK has trophic effects on SN DA neurons.
Materials and Methods
Generation of recombinant AAV.
Plasmids encoding human DN-DLK(K152A) and DN-MLK3(K144R) were kindly provided by Dr. Lloyd Greene (Columbia University, New York, NY). The dominant-negative activity of these kinase-dead constructs has previously been demonstrated in vitro (Xu et al., 2001). The DN-DLK(K152A) and DN-MLK3(K144R) sequences were modified by overlapping PCR to incorporate a FLAG-encoding sequence at the 3′ end and inserted into an AAV packaging construct, which incorporates the cytomegalovirus enhancer/chicken β-actin promoter and a woodchuck hepatitis virus postregulatory element, as previously described (Olson et al., 2006). The sequence encoding the leucine zipper (LZ) domain of human DLK was obtained by PCR from the human DLK clone using primers for the nucleotide sequence encoding amino acids 372–487. The LZ domain of DLK has been previously shown to have dominant-negative activity specific for DLK (Nihalani et al., 2000). The forward primer, with 5′ extension for FLAG-tag, was 5′-GGATCCACCATGGACTACAAGGACGACGATGACAAGCTCTCCACACCCC-3′; and the reverse primer was 5′-GAATTCACTCCATTGTGTTTCCATGCAGGA-3′. The PCR product was cloned in the AAV packaging construct as above. All nucleotide sequences in the AAV packaging constructs were confirmed before AAV production.
Recombinant AAV was packaged and purified as described previously (Olson et al., 2006). The genomic titer of each virus was determined using real-time PCR. The final titers were as follows: 2.4 × 1012 for AAV-MLK3(K144R); 3.4 × 1013 for AAV-DLK(K152A); and 5.1 × 1013 for AAV-DLK (LZ) [viral genomes per milliliter (vg/ml)]. EGFP was subcloned into the same AAV packaging construct, and viral stocks were used at a titer of 4.6 × 1012 (vg/ml). In separate experiments, this titer of AAV-GFP was found to be without effect on SN DA neurons, quantified by tyrosine hydroxylase (TH) immunohistochemistry and stereologic counts, at 28 d after injection.
Experimental animals.
Adult (8 week) male C57BL/6 mice weighing ∼25 g were obtained from Charles River Laboratories (Wilmington, MA). All injection procedures, described below, were approved by the Columbia University Animal Care and Use Committee.
Virus injection.
Mice were anesthetized with ketamine/xylazine solution and placed in a stereotaxic frame (Kopf Instruments, Tujunga, CA) with a mouse adapter. The tip of 5.0 μl syringe (Agilent, Santa Clara, CA) needle (26S) was inserted to the following stereotaxic coordinates: anteroposterior (AP), −0.35 cm; mediolateral (ML), +0.11 cm; dorsoventral (DV), −0.37 cm relative to bregma. These coordinates place the needle tip dorsal to the posterior SN. Viral vector suspension in a volume of 2.0 μl was injected at 0.1 μl/min over 20 min. After a wait of 5 min, the needle was slowly withdrawn. Successful transduction of DA neurons of the SN was confirmed histologically by double immunolabeling for FLAG and TH.
6OHDA lesion.
Adult mice received a unilateral intrastriatal injection of 6OHDA as described previously (Silva et al., 2005b). Briefly, mice were pretreated with desipramine, anesthetized with ketamine/xylazine solution, and placed in a stereotaxic frame. A solution of 6OHDA (5.0 μg/μl in 0.9% NaCl/0.02% ascorbate) was injected by microliter syringe at a rate of 0.5 μl/min by pump for a total dose of 15.0 μg/3 μl. Injection was performed into the left striatum at the following coordinates: AP, +0.09 cm; ML, +0.22 cm; DV, −0.25 cm relative to bregma. After a wait of 2 min, the needle was slowly withdrawn.
Immunohistochemistry.
Immunostaining for TH was performed as described previously (Kholodilov et al., 2004). Mice were perfused intracardially with 0.9% NaCl followed by 4.0% paraformaldehyde in 0.1 m phosphate buffer (PB), pH 7.1. The brain was carefully removed and blocked into midbrain and forebrain regions. The region containing the midbrain was postfixed for 1 week, cryoprotected in 20% sucrose overnight, and then rapidly frozen by immersion in isopentane on dry ice. A complete set of serial sections was then cut through the SN at 30 μm. Beginning with a random section between 1 and 4, every fourth section was processed, in keeping with the fractionator method of sampling (see below). Sections were processed free-floating. The primary antibody was rabbit anti-TH (Calbiochem, La Jolla, CA) at 1:750. Sections were then treated with biotinylated protein A and avidin-biotinylated horseradish peroxidase complexes (ABC; Vector Labs, Burlingame, CA). After immunoperoxidase staining, sections were thionin counterstained. The forebrain region containing the striatum was postfixed for 48 h, and then frozen without cryoprotection, and processed as described previously (Kholodilov et al., 2004). The optical density of striatal TH immunostaining was determined with an Imaging Research (St. Catherines, Ontario) Analytical Imaging Station.
For anti-FLAG immunostaining, mice were perfused with 4% paraformaldehyde, and the brains were postfixed for 24 h. After cryoprotection overnight, brains were frozen and sectioned through the SN at 30 μm. Sections were initially treated with Mouse-on-Mouse Blocking Reagent (Vector Labs) and then processed free-floating with a mouse monoclonal anti-FLAG antibody (Sigma, St. Louis, MO) at 1:1000. Sections were then incubated with biotinylated anti-mouse IgG (Vector Labs), followed by ABC.
To perform immunohistochemistry for phosphorylated c-jun, mice were anesthetized with ketamine/xylazine solution and hypothermia, and provided with supplemental oxygen at the rate of 4 L/min for 6 min. Mice were then perfused immediately with 4% paraformaldehyde in 0.1 m PB containing 0.2 mm Na-orthovanadate at a rate of 5 ml/min for 10 min. The brains were removed, postfixed for 24 h, and then cryoprotected with 20% sucrose in 0.1 m PB overnight at 4°C. SN sections were cut and processed according to the methods described above. Primary antibody was rabbit polyclonal anti-phospho-c-jun (Ser73) (Cell Signaling, Beverly, MA) at a dilution of 1: 50.
Immunostaining for NeuN was performed with a mouse monoclonal antibody (Chemicon, Temecula, CA) at 1:100.
Double immunofluorescence staining.
For TH and FLAG double staining, mice were perfused and their brains were removed and postfixed for 24 h. After cryoprotection overnight, brains were frozen and sectioned through the SN at 30 μm. Sections were blocked with Mouse-on-Mouse Blocking Reagent (Vector Labs) for 1 h at 4°C, and then incubated with the primary antibodies, mouse monoclonal anti-FLAG antibody (Sigma, St. Louis, MO) at 1:1000 and rabbit polyclonal anti-TH (Calbiochem) at 1:1000 in PBS containing Mouse-on-Mouse Diluent Working Solution (Vector Labs) and normal goat serum for 24 h at 4°C. Control sections were prepared either in the absence of primary antibody or with anti-FLAG or anti-TH alone. Sections were then incubated in a mixture of biotinylated anti-mouse IgG reagent and goat anti-rabbit Texas Red (Vector Labs) for 15 min at room temperature, followed by incubation in fluorescein avidin DCS for 1 h at a dilution of 1:100. After PBS wash, the sections were mounted and then coverslipped with DakoCytomation (Carpinteria, CA) anti-fade medium. Sections were viewed with a Nikon (Tokyo, Japan) Eclipse 80i fluorescence microscope.
For phospho-c-jun and FLAG double staining, mice were anesthetized by hypothermia with supplemental oxygen as described above and perfused. The brains were postfixed and cryoprotected. SN sections were cut and processed according to the methods described above. Primary anti-phospho-c-jun was diluted at 1:50. Anti-FLAG was diluted at 1:1000.
Determination of SN DA neuron numbers by stereologic analysis.
Stereologic analysis was performed under blinded conditions on coded slides. For each animal, the SN on both sides of the brain was analyzed. For each section, the entire SN was identified as the region of interest. Using StereoInvestigator software (MicroBrightField, Williston, VT), a fractionator probe was established for each section. The number of TH-positive neurons in each counting frame was then determined by focusing down through the section, using 100× objective under oil, as required by the optical disector method. Our criterion for counting an individual TH-positive neuron was the presence of its nucleus either within the counting frame, or touching the right or top frame lines (green), but not touching the left or bottom lines (red). The total number of TH-positive neurons for each side of the SN was then determined by the StereoInvestigator program.
The number of NeuN-positive neuronal nuclei was determined in a similar manner, except that the region of interest was confined to the SN pars compacta (SNpc).
The cell size of TH neurons in the SN was also determined by use of the StereoInvestigator program. For each brain, five representative sections (one caudal, two middle, and two rostral) were chosen, and five neurons were selected at five random sites from each section. For each group, 100 TH neurons were outlined and measured under a 100× oil-immersion objective.
Determination of the number of apoptotic profiles within the SN.
Apoptotic profiles were identified in the SN on TH-immunoperoxidase-stained and thionin-counterstained sections as described previously (Silva et al., 2005b).
Measurement of SN and striatal DA and metabolites.
For determination of SN and striatal levels of DA and its metabolites, each brain was placed in a mouse brain matrix and 2.0 mm coronal slices through the forebrain and mesencephalon were taken for dissection of the striatum and SN, respectively. The slices were placed anterior surface up on a glass plate, and the striatum punched out on each side with a 2.0 mm punch. For the SN, ventral mesencephalon was separated from dorsal by a horizontal cut just dorsal to the SN. Tissues were immediately frozen on dry ice. DA, 3,4-dihydroxyphenylacetic acid (DOPAC), and homovanillic acid (HVA) were determined by HPLC by Bioanalytical Systems and expressed as nanograms per sample as described previously (Kholodilov et al., 2004).
TH activity assay.
For measurement of TH activity, striata and SN tissues were dissected as described above, rapidly frozen on dry ice, and stored at −80°C. TH activity was assayed by a coupled decarboxylation assay, as described by Waymire et al. (1971), modified by Tank et al. (1986). All procedures were performed on ice or in a cold room unless otherwise stated. Frozen tissue was homogenized in 150 μl of 30 mm potassium phosphate, pH 6.8, 10 mm EDTA, and 50 mm NaF, and the homogenate was centrifuged at 13,000 × g for 15 min. The resulting supernatant was assayed for TH activity using saturating 4 mm 6-MPH4 (6-methy-5,6,7,8-tetrahydropterine) as cofactor and 0.1 mm [14C]tyrosine as substrate, which are essentially Vmax conditions. Reactions were performed for 10 min at 30°C. Protein was measured using bovine serum albumin as standard (Bradford, 1976). Results were expressed as picomoles of 14CO2 formed per minute per milligram of protein.
Statistical methods.
The difference between two groups was analyzed by the t test. Multiple comparisons among groups were performed by one-way ANOVA and Tukey's post hoc analysis. All statistical analyses were performed using SigmaStat software (SPSS Science, Chicago, IL).
Results
DN-DLK(K152A) has trophic effects on DA neurons of the SN
The ability of each AAV vector to successfully transduce DA neurons of the SNpc was assessed by performing immunoperoxidase staining and immunofluorescence double labeling for the FLAG epitope tag and TH as a marker for the dopaminergic phenotype in SNpc at 28 d after injection. These studies demonstrated extensive transduction of neurons within the SNpc, and confirmed expression of the dominant-negative constructs within DA neurons (Fig. 1).
Figure 1.
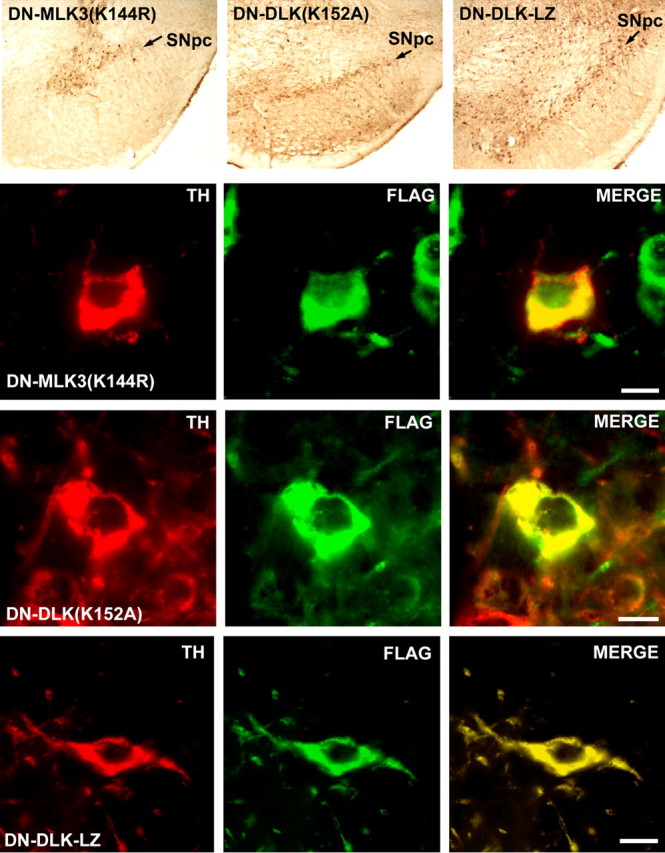
Transduction of DA neurons of the SN with AAV vectors expressing dominant-negative forms of MLKs. The top row of panels illustrates immunoperoxidase labeling (brown) for the FLAG epitope of each transgene in the SN. Positive immunoperoxidase labeling in the SNpc (arrows) is shown for each of the dominant-negative constructs. The following rows of panels demonstrate successful transduction of DA neurons within SNpc for SN-MLK3(K144R), DN-DL (K152A), and DN-DLK-LZ, respectively. The dopaminergic phenotype is demonstrated by immunofluorescent staining for TH (red); transgene expression is demonstrated by staining for the FLAG epitope (green); coexpression is demonstrated by yellow in the merged images in the right-hand panels. Scale bars: 10 μm.
Given the evidence that high-titer preparations of AAV can be associated with toxicity in DA neurons of the SNpc in vivo (Klein et al., 2006), we assessed stereologic counts of TH-immunostained neurons in SN at 28 d after intranigral vector injection. Neither AAV DN-MLK3(K144R) (Fig. 2A) nor AAV DN-DLK-LZ (data not shown) had any effect on numbers of SN DA neurons, but, unexpectedly, AAV DN-DLK(K152A) induced a modest (19%) but highly significant (p < 0.001) increase in their apparent number (Fig. 2A). This increase in cell number was accompanied by a 34% increase in neuronal size compared with contralateral control (p < 0.001) (Fig. 2B). To determine whether this increase in neuron number was attributable to an actual increase in the neuronal population of the SNpc, or rather to an increased ability to detect DA neurons based on augmented expression of the phenotype marker TH, we performed both immunostaining for a general neuronal marker (NeuN) and enzyme activity assays for TH. Stereologic counts of NeuN revealed no effect of AAV DN-DLK(K152A) (Fig. 2C). Assay of TH activity, however, revealed a modest (16%), but significant (p = 0.03) induction after AAV DN-DLK(K152A) (Fig. 2D). Changes in TH activity measured under Vmax conditions represent changes in TH protein levels (Tank et al., 1986). Not surprisingly, this modest induction was not associated with a change in DA or its metabolites in SN (Table 1). Nevertheless, we conclude that the apparent increase in the number of SNpc DA neurons after AAV DN-DLK(K152A) was not attributable to an increase in their actual number, but rather to increased TH expression and immunodetection, a phenomenon we had previously observed after injection of a constitutively active form of the neurotrophic signaling kinase Akt (Ries et al., 2006).
Figure 2.

Trophic effects of kinase-dead DN-DLK(K152A) on DA neurons of the SN. A, Stereological analysis of the number of TH-positive neurons in the SN at 28 d after AAV injection reveals a 19% increase in comparison with the contralateral noninjected control side (CON) (p < 0.001, ANOVA) (n = 8). No difference in number between the two sides was noted for mice injected with AAV DN-MLK3(K144R) (n = 8) or AAV DN-DLK-LZ (data not shown). B, The mean cross-sectional area of SN DA neurons was increased by 34% in comparison with contralateral controls at 28 d after injection in mice receiving AAV DN-DLK(K152A) (p < 0.001). No significant effect was observed in mice injected with AAV GFP. A total of 50 neurons (25 each in 2 mice) was examined for each condition. C, No effect was observed on the number of NeuN-stained neurons in the SNpc at 28 d after injection of either AAV GFP (n = 6) or AAV DN-DLK(K152A) (n = 5). This result suggests that the increased apparent number of TH-positive neurons in A is attributable to an increased expression of TH, rather than an actual increase in the number of neurons. Error bars indicate SEM. D, A modest (16%) but significant (p < 0.03, paired t test) increase in TH activity (picomoles of 14CO2 formed per minute per milligram of protein) is observed in SN on the side of AAV DN-DLK(K153A) compared with the contralateral control at 28 d after injection (n = 8). A nonsignificant trend (p = 0.3) is observed in the AAV GFP-injected mice (n = 8).
Table 1.
The Effect of DN DLK(K152A) on SN and striatal levels of dopamine and its metabolites
| Condition | n | Substantia nigra |
Striatum |
||||||
|---|---|---|---|---|---|---|---|---|---|
| DA | DOPAC | HVA | HVA/DA | DA | DOPAC | HVA | HVA/DA | ||
| GFP Con | 8 | 2.9 ± 0.1 | 1.1 ± 0.06 | 1.7 ± 0.08 | 0.6 ± 0.02 | 44.3 ± 1.6 | 5.1 ± 0.8 | 5.9 ± 0.4 | 0.13 ± 0.009 |
| GFP Exp | 8 | 2.9 ± 0.2 | 1.1 ± 0.04 | 1.8 ± 0.07 | 0.6 ± 0.02 | 54.6 ± 2.2 | 4.3 ± 0.4 | 6.3 ± 0.3 | 0.12 ± 0.009 |
| DLK(K152A) Con | 7 | 2.6 ± 0.1 | 1.0 ± 0.04 | 1.8 ± 0.06 | 0.7 ± 0.02 | 44.7 ± 0.8 | 5.3 ± 0.5 | 6.7 ± 0.4 | 0.15 ± 0.007 |
| DLK(K152A) Exp | 7 | 2.7 ± 0.1 | 1.0 ± 0.07 | 1.8 ± 0.09 | 0.7 ± 0.03 | 47.3 ± 2.3 | 5.7 ± 0.6 | 7.1 ± 0.4 | 0.14 ± 0.007 |
Although DN DLK(K152A) increased TH activity within the SN, the modest effect did not result in a change in levels of DA or its metabolites within either the SN or the striatum.
The trophic effects of AAV DN-DLK(K152A) did not extend to the axonal projections of nigrostriatal dopaminergic neurons. No effect was observed on the density of striatal immunoperoxidase staining (data not shown), or on the amount of striatal DA, its metabolites, or turnover (Table 1).
Dominant-negative forms of DLK, but not MLK3, block apoptosis in DA neurons in the 6OHDA model
To examine the relative roles of MLK3 and DLK in apoptosis in SNpc DA neurons in the 6OHDA neurotoxin model, we injected the toxin at 3 weeks after intranigral viral injection, and assessed apoptosis at 6 d after lesion. DN-DLK(K152A) and DN-DLK-LZ suppressed apoptosis, to 12 and 18%, respectively, of levels observed in AAV GFP-injected controls (Fig. 3A,B). Injection of AAV DN-MLK3(K144R), however, did not suppress apoptosis; there was, on the contrary, a trend for an increased number of profiles that did not achieve significance (Fig. 3C). Because production of AAV DN-MLK3(K144R) was achieved only to a titer of 2.4E12 vg/ml, this result could not be directly compared with those obtained for the DN-DLKs, which were produced in the E13 vg/ml range. We therefore diluted preparations of AAV DN-DLK(K152A) to 2.4E12 vg/ml. At this titer, DN-DLK(K152A) still suppressed apoptosis to 51% of levels observed in AAV GFP-injected controls, a significant effect (p = 0.01) (Fig. 3D). We therefore conclude that, at the viral titers examined, dominant-negative forms of DLK suppress apoptosis, but the kinase-dead dominant-negative form of MLK3 does not.
Figure 3.

Dominant-negative forms of DLK inhibit apoptosis in SN DA neurons after intrastriatal 6OHDA. A, Adult male mice were injected with either AAV GFP or AAV DN-DLK(K152A) and then with 6OHDA 3 weeks later. At 6 d after lesion, animals were killed, and the number of apoptotic profiles in the SNpc was determined. AAV DN-DLK(K152A) reduced the number of apoptotic profiles to 12% of the number observed in AAV GFP controls [p < 0.001, t test; n = 8 GFP controls; n = 7 DN-DLK(K152A)]. B, In a similar experiment, AAV DN-DLK-LZ (n = 7) reduced the number of apoptotic profiles to 18% of that observed in AAV GFP controls (n = 7) (p = 0.02, t test). C, AAV DN-MLK3(K144R) had no effect on the number of apoptotic profiles after 6OHDA. D, To directly compare kinase-dead DN-DLK(K152A) to DN-MLK3(K154R), the viral stocks of AAV DN-DLK(K152A) were diluted to a titer equal to that of AAV DN-MLK3(K144R) (2.4E12 vg/ml). Even at this diluted titer, AAV DN-DLK3(K152A) retained the ability to suppress apoptosis (p = 0.01, t test; n = 11). Error bars indicate SEM.
Previous studies have shown that an acute suppression of apoptosis in neurotoxin models does not necessarily provide a lasting protection of SN DA neurons (Silva et al., 2005b), because such an apparent decrease could be attributable to an altered time course of apoptotic cell death, or the rate of clearance of apoptotic profiles. We therefore examined by TH immunostaining and stereological analysis the number of SN dopaminergic neurons remaining at 28 d after lesion in mice injected with the dominant-negative forms of the DLKs compared with AAV GFP controls. Both DN-DLK(K152A) and DN-DLK-LZ provided significant protection at 28 d after lesion. AAV DN-DLK(K152A)-injected mice had 55% survival of DA neurons, compared with only 31% in controls (p = 0.005) (Fig. 4A). AAV DN-DLK-LZ mice showed an even greater protective effect, with a 75% survival rate, compared with 31% in a second AAV GFP control group, processed in parallel (Fig. 4B). When the number of surviving DA neurons is expressed as a percentage of the contralateral, noninjected control side, the protective effect of AAV DN-DLK-LZ (75 ± 3%) was significantly greater than that of AAV DN-DLK(K152A) (55 ± 7%) (p = 0.01, t test). In addition to protecting the survival of SN DA neurons, AAV DLK-LZ also protected them from the atrophy, which occurs after 6OHDA lesion. Whereas AAV GFP injected mice lost 24% of the cross-sectional area of SN DA neurons, AAV DLK-LZ mice showed only a 7% (not significant) loss (Fig. 4D). Despite their inhibition of apoptosis in SNpc, and their lasting protection of dopamine neurons, neither dominant-negative form of DLK protected dopaminergic axon terminals in the striatum (Fig. 4E).
Figure 4.

Dominant-negative forms of DLK enhance the long-term survival of SN DA neurons after intrastriatal 6OHDA. A, At 3 weeks after intranigral injection of AAV, mice received intrastriatal 6OHDA. At 28 d after 6OHDA, mice were killed and processed for immunoperoxidase staining of TH within SN neurons and axons. At this postlesion time point, there is no suppression of TH phenotype, and SN DA neuron counts determined by TH staining are approximately equal to those determined by Fluorogold retrograde labeling (Sauer and Oertel, 1994). AAV DN-DLK(K152A) increased the number of surviving DA neurons to 55% of the contralateral control (CON) noninjected side, which represents an 80% increase over the number of surviving neurons in the AAV GFP group [p = 0.005, ANOVA; n = 8 and 7, AAV GFP and AAV DN-DLK(K152A), respectively]. B, In a similar experiment, AAV DLK-LZ increased the number of surviving DA neurons to 75% of the contralateral control, which represents a 153% increase over the number of surviving neurons in the AAV GFP group (p < 0.001, ANOVA; n = 6 and 8 AAV GFP and AAV DN-DLK-LZ, respectively). C, A representative set of sections stained for TH (brown) and Nissl-counterstained after injection of AAVs and unilateral intrastriatal 6OHDA. At 28 d after 6OHDA, a mouse given AAV GFP shows very few surviving DA neurons in the SNpc. A mouse given AAV DN-DLK-LZ shows substantial preservation of DA neurons. D, AAV DN-DLZ-LZ preserves DA neuron size after 6OHDA. In the AAV GFP control condition, there is a 24% loss of cross-sectional area (p < 0.001, ANOVA; n = 100 neurons), whereas in the AAV DN-DLK-LZ condition there is only a 7% trend (p = 0.4, NS). Error bars indicate SEM. E, Neither AAV DN-DLK(K152A) nor AAV DN-DLK-LZ protected dopaminergic axons. Representative TH-immunostained striatal sections demonstrate that, for both GFP (top) and AAV DLK-LZ (bottom), there is near-complete loss of striatal TH staining after intrastriatal 6OHDA. Similar results were observed with AAV DN-DLK(K152A) (data not shown).
The neuroprotection provided by AAV DN-DLK-LZ correlates with inhibition of c-jun phosphorylation
In these experiments, even the most effective of the two dominant-negative forms of DLK, DLK-LZ, still demonstrated a 25% loss of SN dopaminergic neurons after 6OHDA. We therefore investigated the reasons for this persistent cell death. Analysis of the efficiency of transduction at 28 d after intranigral injection of AAV DN-DLK-LZ revealed that 71 ± 6% were transduced (Fig. 5A). This percentage is not significantly different from the percentage of neurons that survive after transduction (75 ± 3%). We therefore conclude that the persistent neuron death in the 6OHDA model after AAV DN-DLK-LZ can be mainly, if not entirely, attributable to the level of transduction efficiency.
Figure 5.

Transduction with AAV DN-DLK-LZ blocks phosphorylation of c-jun. A, To determine the transduction efficiency of AAV DN-DLK-LZ, n = 4 mice were injected into the SN and killed 4 weeks later for double-labeling of TH and FLAG. In sections from each animal, n = 25 TH-positive profiles (n = 100 total) were assessed for FLAG staining; 71.1 ± 6% of TH profiles were FLAG-positive. B, C, The efficacy of AAV DN-DLK-LZ to suppress c-jun phosphorylation was assessed at the population level, by counting the number of PO4-c-jun-positive immunostained profiles at 6 d after 6OHDA, in mice which received either AAV GFP (n = 7) or AAV DN-DLK-LZ (n = 10). Absolute counts are shown in B; counts as a percentage of the mean among the AAV GFP mice is shown in C. AAV DN-DLK-LZ reduced the number of PO4-c-jun profiles to 38 ± 13% of the number observed in AAV GFP mice. Error bars indicate SEM. D, The relationship between transduction with AAV DN-DLK-LZ and phosphorylation of c-jun after 6OHDA was examined at the cellular level by double-labeling for FLAG and PO4-c-jun at 6 d after 6OHDA. Of 752 profiles examined in sections from three mice, 449 (59.7%) stained for PO4-c-jun alone, 300 (39.8%) stained for FLAG alone, and only 3 profiles (0.4%) stained for both. Thus, phosphorylation of c-jun in the presence of AAV DN-DLK-LZ was exceedingly rare. A representative field demonstrates the discordance between FLAG and PO4-c-jun staining at the cellular level. E, Transduction of SNpc neurons with AAV DN-MLK3(K144R) does not block phosphorylation of c-jun after 6OHDA. The abundance of PO4-c-jun-positive nuclear profiles is about the same after AAV DN-MLK3(K144R) as it is after AAV GFP, as shown in a representative low-power micrograph in E and confirmed by quantitative analysis (data not shown). Representative phospho-c-jun nuclear staining is shown at high magnification in the inset in E. Positive phospho-c-jun staining in the GFP and MLK3(K144R) conditions, on the side of the 6OHDA injection, is shown in the low-power micrographs encircled in red.
We next sought to determine to what degree prevention of cell death by successful transduction with AAV DN-DLK-LZ could be attributed to the inhibition of phosphorylation of c-jun. At a population level of analysis, we found that transduction with AAV DN-DLK-LZ reduced the number of PO4-c-jun positive profiles to 38 ± 13% of those observed in AAV GFP controls at 6 d after 6OHDA (Fig. 5B,C). This percentage is not significantly different from the number of DA neurons that were not transduced by AAV DN-DLK-LZ (29 ± 6%), nor is it different from the percentage of neurons that died (25 ± 3%) (p = 0.6, NS, ANOVA). At a cellular level of analysis, we found that there were only very rare instances of c-jun phosphorylation in the presence of DN-DLK-LZ, detected by FLAG immunofluorescence staining (Fig. 5D). We therefore conclude that there are very close relationships between transduction with AAV DN-DLK-LZ, inhibition of c-jun phosphorylation and neuron survival. In other words, we found no evidence for a survival effect that exceeded the degree of inhibition of c-jun phosphorylation. Conversely, we did not detect a survival effect that was substantially less than the level of inhibition of c-jun phosphorylation, as would be anticipated if death mechanisms independent of PO4-c-jun were at work.
To further explore the role of c-jun phosphorylation in mediating cell death in this model, we examined the number of phospho-c-jun-positive profiles in SNpc after transduction with AAV DN MLK3(K144R), which did not prevent cell death. We found that DN MLK3(K144R) did not block c-jun phosphorylation after 6OHDA, in comparison with a control AAV GFP injection (Fig. 5E). We conclude that, in this model, phosphorylation of c-jun appears to be both necessary and sufficient to mediate cell death.
Discussion
Although the primary purpose of these investigations was to identify and characterize the antiapoptotic effect of MLK inhibition in vivo, we observed that the kinase-dead dominant-negative form of DLK also had trophic effects on DA neurons of the SN. These effects were observed at the level of the cell soma, and included an increase in neuron size and the activity of TH within the nigra. These observations have precedents in studies performed in tissue culture. Harris et al. (2002) observed that the MLK inhibitor CEP1347 prevented not only the cell death but also the decreases in protein synthesis and cell size that occur after withdrawal of NGF. Strikingly, CEP1347 not only prevented soma atrophy but also reversed it (Harris et al., 2002). Subsequent observations by Wang et al. (2005) demonstrated that treatment with a CEP1347 analog, CEP11004, induced increased expression of TrkA and activation of the signaling kinase Akt. These investigators postulated that inhibition of the MLKs mediated the changes in TrkA and Akt, but they recognized that a non-MLK-related mechanism could be at work. Indeed, Roux et al. (2002) also observed trophic effects of CEP1347, associated with activation of Akt, but they postulated that these effects were independent of MLK inhibition. Our results support the proposal of Wang et al. that inhibition of the MLKs alone may be sufficient.
It is uncertain why a kinase-dead dominant-negative form of DLK had trophic effects on DA neurons, whereas an LZ alone construct did not. We doubt that this difference can be attributed to quantitative differences between the two constructs in their blockade of DLK, because DN-DLK-LZ was produced at a higher titer and had a more pronounced effect in protecting DA neurons from cell death. We postulate that the different effects of the two dominant negatives are more likely to be attributable to qualitative differences in their mechanisms of action. The DN-DLK-LZ construct blocks homodimerization of DLK molecules and thereby prevents autophosphorylation and activation (Nihalani et al., 2000). DLK-LZ does not have dominant-negative effects on MLK3 kinase activity (Nihalani et al., 2000). DN-DLK(K152A), however, remains capable of homodimerization and heterodimerization, and may have a broader spectrum of dominant-negative effects.
The DN-DLK(K152A) and DN-DLK-LZ dominant-negative forms had equal efficacy in their ability to block apoptosis in DA neurons in the 6OHDA model. For both dominant negatives, the antiapoptotic effect provided a lasting enhanced survival. Although DN-DLK-LZ provided a greater degree of survival than DN-DLK(K152A), the parameters of these in vivo experiments do not permit a conclusion that DN-DLK-LZ is inherently more effective. These results support the interpretation of previous studies of CEP1347 and CEP11004 that their antiapoptotic effects are likely to be primarily attributable to inhibition of the MLKs rather than their apparently independent ability to stimulate the antiapoptotic kinase Akt (Roux et al., 2002). Although previous studies with these compounds in rodent models of parkinsonism (for review, see Silva et al., 2005a) do not permit analysis of molecular specificity, because of their broad inhibitory effects within the MLK family, the present study suggests that DLK may play a specific role. Whereas observations of the antiapoptotic effect of DN-DLK(K152A) alone would not permit such a conclusion, because of its known interactions with other MLKs, including MLK3 (Xu et al., 2001), similar observations with DN-DLK-LZ do support a specific role, given its molecular specificity (Nihalani et al., 2000). The role of DLK in mediating apoptosis in this context is unlikely to be attributable to a unique ability, among the MLKs, to mediate cell death. Observations in vitro in rat cells have demonstrated the ability of other members of the MLK family to mediate apoptosis (Xu et al., 2001). It is more likely that the much greater relative abundance of DLK in rodent SN (Ganguly et al., 2004) is responsible for its preponderant role. Furthermore, although we were unable to demonstrate a protective effect of DN-MLK3(K144R) in the presence of confirmed protein expression (Fig. 1), we did not quantify the level of expression in comparison with that of the DLK forms. Therefore, we cannot definitively exclude a possible role for MLK3 in cell death in this model.
In addition to its antiapoptotic effect, DN-DLK-LZ prevented atrophy of dopaminergic neurons after 6OHDA lesion. This trophic effect appears to be distinct from that of DN-DLK(K152A) on normal, unlesioned neurons, because DN-DLK-LZ did not demonstrate such effects. This ability of DN-DLK-LZ to either prevent or reverse atrophy of these neurons after lesion is similar to the ability of CEP1347 to both prevent and reverse atrophy of sympathetic neurons after NGF withdrawal (Harris et al., 2002).
The neuroprotective effects of DN-DLK(K152A) and DN-DLK-LZ did not, however, include preservation of the nigrostriatal axonal projections. This result was disappointing and unexpected, given our previous demonstration of a striking protective effect of CEP11004 on striatal dopaminergic fibers in postnatal 6OHDA model (Ganguly et al., 2004). Other investigators had also previously demonstrated protection of dopaminergic axons by blockade of downstream targets in the MAPK cascade, including c-jun in an axotomy model (Crocker et al., 2001) and c-jun N-terminal kinase (JNK) in the MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) model of parkinsonism (Xia et al., 2001). This result is not, however, unlike those of others who failed to achieve axonal protection despite effective preservation of nigral dopaminergic cell bodies because of blockade of PCD pathways (Eberhardt et al., 2000; Hayley et al., 2004; Silva et al., 2005b). This result supports the concept that the molecular pathways that mediate axonal degeneration are likely to be distinct from those of PCD, which mediate destruction of the cell soma (Raff et al., 2002; Coleman, 2005).
Transduction of SN DA neurons with dominant-negative forms of DLK was not entirely effective in blocking cell death induced by 6OHDA. In DN-DLK(K152A) and DN-DLK-LZ injected mice, there was persistent apoptosis resulting in a loss of 45 and 25%, respectively, of DA neurons. In the case of DN-DLK-LZ, the more effective of the two, this loss seemed to be simply attributable to the level of transduction efficiency. Among DN-DLK-LZ-injected mice, a mean of 71% of DA neurons were successfully transduced (range, 60–84%). This level of transduction is not significantly different from the percent survival achieved (75 ± 3%).
The canonical model for mediation of PCD by MAPK cascade signaling has been that phosphorylation and activation of JNK by upstream kinases leads to activation of c-jun, which, in turn, results in the transcriptional activation of cell death mediators (for review, see Silva et al., 2005a). However, there is now much evidence indicating that other JNK targets, in addition to c-jun, play a role in PCD. JNK can phosphorylate and diminish the antiapoptotic effects of Bcl-2 and Bcl-XL (Maundrell et al., 1997; Kharbanda et al., 2000), and it can activate the proapoptotic effects of Bad, Bim, and Bmf (Donovan et al., 2002; Lei and Davis, 2003). In addition, a non-c-jun-dependent mechanism of cell death induction by JNK3 has been observed in an axotomy model in vivo (Keramaris et al., 2005). Conversely, phosphorylation of c-jun is not universally associated with cell death; c-jun also appears to play a vital role in axonal regeneration (Raivich et al., 2004; Waetzig et al., 2006). However, our analysis of the relationships between transduction efficiency, phosphorylation of c-jun at the cellular and population levels, and cell loss suggest that DLK and its downstream target JNK act in this model to induce cell death primarily by the phosphorylation and activation of c-jun. At the population level, percent transduction efficiencies for SN DA neurons (71 ± 6%) were not significantly different from the percent reduction in phospho-c-jun profiles induced by 6OHDA (62 ± 13%). At a cellular level, the presence of phosphorylated c-jun in AAV DN-DLK-LZ transduced neurons was exceedingly rare. Given that JNK is the principal kinase for c-jun in tissue (Kyriakis and Avruch, 2001), these results suggest that dominant-negative blockade of DLK in this context is highly likely to have also blocked JNK activation. In experiments with AAV DN-DLK-LZ, the percentage of non-transduced neurons, the residual percentage of cells expressing phosphorylated c-jun, and the percentage of neurons that died were not significantly different. These similar percentages suggest that it is unlikely that JNK acts substantially by any c-jun-independent mechanism to induce cell death.
The principal conclusion from these studies is that specific inhibition of DLK in this model blocks the phosphorylation of c-jun and thereby prevents neuronal death. In fact, our results suggest that the chief limiting factor for prevention of neuron death is transduction efficiency. These results add to the growing evidence that MLK inhibition provides protection from neuronal death (for review, see Wang et al., 2004). Although enthusiasm for this approach has been diminished by the negative results of a clinical trial in PD patients that failed to demonstrate an ability of CEP1347 to slow clinical progression (Waldmeier et al., 2006), these results cannot be taken as proof of failure of the MLK inhibition approach. One reason is that it is unknown whether kinase inhibition was achieved (Burke, 2007). A viral vector-based approach offers the advantages that intracellular blockade of MLKs is likely to be achieved, and molecular and regional specificity are possible. We therefore conclude that viral vector-based inhibition of cell death kinases merits consideration as a neuroprotective approach to PD.
Footnotes
This work was supported by National Institutes of Health, National Institute of Neurological Disorders and Stroke Grants NS26836 and NS38370, and Department of Defense Grant DAMD17-03-1-0492, and the Parkinson's Disease Foundation. We are grateful to Amy Baohan for morphometric analyses and to Dr. Lloyd Greene for his helpful comments on this manuscript.
References
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Burke RE. Inhibition of MAPK and stimulation of Akt kinase signaling pathways: two approaches with therapeutic potential in the treatment of neurodegenerative disease. Pharmacol Ther. 2007;114:261–277. doi: 10.1016/j.pharmthera.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman M. Axon degeneration mechanisms: commonality amid diversity. Nat Rev Neurosci. 2005;6:889–898. doi: 10.1038/nrn1788. [DOI] [PubMed] [Google Scholar]
- Crocker SJ, Lamba WR, Smith PD, Callaghan SM, Slack RS, Anisman H, Park DS. c-Jun mediates axotomy-induced dopamine neuron death in vivo. Proc Natl Acad Sci USA. 2001;98:13385–13390. doi: 10.1073/pnas.231177098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan N, Becker EB, Konishi Y, Bonni A. JNK phosphorylation and activation of BAD couples the stress-activated signaling pathway to the cell death machinery. J Biol Chem. 2002;277:40944–40949. doi: 10.1074/jbc.M206113200. [DOI] [PubMed] [Google Scholar]
- Eberhardt O, Coelln RV, Kugler S, Lindenau J, Rathke-Hartlieb S, Gerhardt E, Haid S, Isenmann S, Gravel C, Srinivasan A, Bahr M, Weller M, Dichgans J, Schulz JB. Protection by synergistic effects of adenovirus-mediated X-chromosome-linked inhibitor of apoptosis and glial cell line-derived neurotrophic factor gene transfer in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson's disease. J Neurosci. 2000;20:9126–9134. doi: 10.1523/JNEUROSCI.20-24-09126.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo KA, Johnson GL. Mixed-lineage kinase control of JNK and p38 MAPK pathways. Nat Rev Mol Cell Biol. 2002;3:663–672. doi: 10.1038/nrm906. [DOI] [PubMed] [Google Scholar]
- Ganguly A, Oo TF, Rzhetskaya M, Pratt R, Yarygina O, Momoi T, Kholodilov N, Burke RE. CEP11004, a novel inhibitor of the mixed lineage kinases, suppresses apoptotic death in dopamine neurons of the substantia nigra induced by 6-hydroxydopamine. J Neurochem. 2004;88:469–480. doi: 10.1046/j.1471-4159.2003.02176.x. [DOI] [PubMed] [Google Scholar]
- Gloeckner CJ, Kinkl N, Schumacher A, Braun RJ, O'Neill E, Meitinger T, Kolch W, Prokisch H, Ueffing M. The Parkinson disease causing LRRK2 mutation I2020T is associated with increased kinase activity. Hum Mol Genet. 2006;15:223–232. doi: 10.1093/hmg/ddi439. [DOI] [PubMed] [Google Scholar]
- Greggio E, Jain S, Kingsbury A, Bandopadhyay R, Lewis P, Kaganovich A, van der Brug MP, Beilina A, Blackinton J, Thomas KJ, Ahmad R, Miller DW, Kesavapany S, Singleton A, Lees A, Harvey RJ, Harvey K, Cookson MR. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis. 2006;23:329–341. doi: 10.1016/j.nbd.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Harris CA, Deshmukh M, Tsui-Pierchala B, Maroney AC, Johnson EMJ. Inhibition of the c-Jun N-terminal kinase signaling pathway by the mixed lineage kinase inhibitor CEP-1347 (KT7515) preserves metabolism and growth of trophic factor-deprived neurons. J Neurosci. 2002;22:103–113. doi: 10.1523/JNEUROSCI.22-01-00103.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayley S, Crocker SJ, Smith PD, Shree T, Jackson-Lewis V, Przedborski S, Mount M, Slack R, Anisman H, Park DS. Regulation of dopaminergic loss by Fas in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson's disease. J Neurosci. 2004;24:2045–2053. doi: 10.1523/JNEUROSCI.4564-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keramaris E, Vanderluit JL, Bahadori M, Mousavi K, Davis RJ, Flavell R, Slack RS, Park DS. c-Jun N-terminal kinase 3 deficiency protects neurons from axotomy-induced death in vivo through mechanisms independent of c-Jun phosphorylation. J Biol Chem. 2005;280:1132–1141. doi: 10.1074/jbc.M410127200. [DOI] [PubMed] [Google Scholar]
- Kharbanda S, Saxena S, Yoshida K, Pandey P, Kaneki M, Wang Q, Cheng K, Chen YN, Campbell A, Sudha T, Yuan ZM, Narula J, Weichselbaum R, Nalin C, Kufe D. Translocation of SAPK/JNK to mitochondria and interaction with Bcl-xL in response to DNA damage. J Biol Chem. 2000;275:322–327. doi: 10.1074/jbc.275.1.322. [DOI] [PubMed] [Google Scholar]
- Kholodilov N, Yarygina O, Oo TF, Zhang H, Sulzer D, Dauer WT, Burke RE. Regulation of the development of mesencephalic dopaminergic systems by the selective expression of glial cell line-derived neurotrophic factor in their targets. J Neurosci. 2004;24:3136–3146. doi: 10.1523/JNEUROSCI.4506-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RL, Dayton RD, Leidenheimer NJ, Jansen K, Golde TE, Zweig RM. Efficient neuronal gene transfer with AAV8 leads to neurotoxic levels of tau or green fluorescent proteins. Mol Ther. 2006;13:517–527. doi: 10.1016/j.ymthe.2005.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81:807–869. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- Lei K, Davis RJ. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc Natl Acad Sci USA. 2003;100:2432–2437. doi: 10.1073/pnas.0438011100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroney AC, Finn JP, Connors TJ, Durkin JT, Angeles T, Gessner G, Xu Z, Meyer SL, Savage MJ, Greene LA, Scott RW, Vaught JL. Cep-1347 (KT7515), a semisynthetic inhibitor of the mixed lineage kinase family. J Biol Chem. 2001;276:25302–25308. doi: 10.1074/jbc.M011601200. [DOI] [PubMed] [Google Scholar]
- Mata IF, Wedemeyer WJ, Farrer MJ, Taylor JP, Gallo KA. LRRK2 in Parkinson's disease: protein domains and functional insights. Trends Neurosci. 2006;29:286–293. doi: 10.1016/j.tins.2006.03.006. [DOI] [PubMed] [Google Scholar]
- Maundrell K, Antonsson B, Magnenat E, Camps M, Muda M, Chabert C, Gillieron C, Boschert U, Vial-Knecht E, Martinou JC, Arkinstall S. Bcl-2 undergoes phosphorylation by c-Jun N-terminal kinase/stress-activated protein kinases in the presence of the constitutively active GTP-binding protein Rac1. J Biol Chem. 1997;272:25238–25242. doi: 10.1074/jbc.272.40.25238. [DOI] [PubMed] [Google Scholar]
- Murakata C, Kaneko M, Gessner G, Angeles TS, Ator MA, O'Kane TM, McKenna BA, Thomas BA, Mathiasen JR, Saporito MS, Bozyczko-Coyne D, Hudkins RL. Mixed lineage kinase activity of indolocarbazole analogues. Bioorg Med Chem Lett. 2002;12:147–150. doi: 10.1016/s0960-894x(01)00690-4. [DOI] [PubMed] [Google Scholar]
- Nihalani D, Merritt S, Holzman LB. Identification of structural and functional domains in mixed lineage kinase dual leucine zipper-bearing kinase required for complex formation and stress-activated protein kinase activation. J Biol Chem. 2000;275:7273–7279. doi: 10.1074/jbc.275.10.7273. [DOI] [PubMed] [Google Scholar]
- Olson VG, Heusner CL, Bland RJ, During MJ, Weinshenker D, Palmiter RD. Role of noradrenergic signaling by the nucleus tractus solitarius in mediating opiate reward. Science. 2006;311:1017–1020. doi: 10.1126/science.1119311. [DOI] [PubMed] [Google Scholar]
- Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, Lopez de Munain A, Aparicio S, Gil AM, Khan N, Johnson J, Martinez JR, Nicholl D, Carrera IM, Pena AS, de Silva R, Lees A, Marti-Masso JF, Perez-Tur J, Wood NW, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- Raff MC, Whitmore AV, Finn JT. Axonal self-destruction and neurodegeneration. Science. 2002;296:868–871. doi: 10.1126/science.1068613. [DOI] [PubMed] [Google Scholar]
- Raivich G, Bohatschek M, Da Costa C, Iwata O, Galiano M, Hristova M, Nateri AS, Makwana M, Riera-Sans L, Wolfer DP, Lipp HP, Aguzzi A, Wagner EF, Behrens A. The AP-1 transcription factor c-Jun is required for efficient axonal regeneration. Neuron. 2004;43:57–67. doi: 10.1016/j.neuron.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Ries V, Henchcliffe C, Kareva T, Rzhetskaya M, Bland RJ, During MJ, Kholodilov N, Burke RE. Oncoprotein Akt/PKB: trophic effects in murine models of Parkinson's disease. Proc Natl Acad Sci USA. 2006;103:18757–18762. doi: 10.1073/pnas.0606401103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux PP, Dorval G, Boudreau M, Angers-Loustau A, Morris SJ, Makkerh J, Barker PA. K252a and CEP1347 are neuroprotective compounds that inhibit mixed-lineage kinase-3 and induce activation of Akt and ERK. J Biol Chem. 2002;277:49473–49480. doi: 10.1074/jbc.M203428200. [DOI] [PubMed] [Google Scholar]
- Saporito MS, Hudkins RL, Maroney AC. Discovery of CEP-1347/KT-7515, an inhibitor of the JNK/SAPK pathway for the treatment of neurodegenerative diseases. Prog Med Chem. 2002;40:23–62. doi: 10.1016/s0079-6468(08)70081-x. [DOI] [PubMed] [Google Scholar]
- Sauer H, Oertel WH. Progressive degeneration of nigrostriatal dopamine neurons following intrastriatal terminal lesions with 6 hydroxydopamine a combined retrograde tracing and immunocytochemical study in the rat. Neuroscience. 1994;59:401–415. doi: 10.1016/0306-4522(94)90605-x. [DOI] [PubMed] [Google Scholar]
- Silva RM, Kuan CY, Rakic P, Burke RE. Mixed lineage kinase-c-jun N-terminal kinase signaling pathway: a new therapeutic target in Parkinson's disease. Mov Disord. 2005a;20:653–664. doi: 10.1002/mds.20390. [DOI] [PubMed] [Google Scholar]
- Silva RM, Ries V, Oo TF, Yarygina O, Jackson-Lewis V, Ryu EJ, Lu PD, Marciniak SM, Ron D, Przedborski S, Kholodilov N, Greene LA, Burke RE. CHOP/GADD153 is a mediator of apoptotic death in substantia nigra dopamine neurons in an in vivo neurotoxin model of parkinsonism. J Neurochem. 2005b;95:974–986. doi: 10.1111/j.1471-4159.2005.03428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith WW, Pei Z, Jiang H, Dawson VL, Dawson TM, Ross CA. Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci. 2006;9:1231–1233. doi: 10.1038/nn1776. [DOI] [PubMed] [Google Scholar]
- Tank AW, Ham L, Curella P. Induction of tyrosine hydroxylase by cyclic AMP and glucocorticoids in a rat pheochromocytoma cell line: effect of the inducing agents alone or in combination on the enzyme levels and rate of synthesis of tyrosine hydroxylase. Mol Pharmacol. 1986;30:486–496. [PubMed] [Google Scholar]
- Waetzig V, Zhao Y, Herdegen T. The bright side of JNKs—multitalented mediators in neuronal sprouting, brain development and nerve fiber regeneration. Prog Neurobiol. 2006;80:84–97. doi: 10.1016/j.pneurobio.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Waldmeier P, Bozyczko-Coyne D, Williams M, Vaught JL. Recent clinical failures in Parkinson's disease with apoptosis inhibitors underline the need for a paradigm shift in drug discovery for neurodegenerative diseases. Biochem Pharmacol. 2006;72:1197–1206. doi: 10.1016/j.bcp.2006.06.031. [DOI] [PubMed] [Google Scholar]
- Wang LH, Besirli CG, Johnson EM., Jr Mixed-lineage kinases: a target for the prevention of neurodegeneration. Annu Rev Pharmacol Toxicol. 2004;44:451–474. doi: 10.1146/annurev.pharmtox.44.101802.121840. [DOI] [PubMed] [Google Scholar]
- Wang LH, Paden AJ, Johnson EM., Jr Mixed-lineage kinase inhibitors require the activation of Trk receptors to maintain long-term neuronal trophism and survival. J Pharmacol Exp Ther. 2005;312:1007–1019. doi: 10.1124/jpet.104.077800. [DOI] [PubMed] [Google Scholar]
- Waymire JC, Bjur R, Weiner N. Assay of tyrosine hydroxylase by coupled decarboxylation of DOPA formed from 1–14 C-l-tyrosine. Anal Biochem. 1971;43:588–600. doi: 10.1016/0003-2697(71)90291-0. [DOI] [PubMed] [Google Scholar]
- West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM. Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci USA. 2005;102:16842–16847. doi: 10.1073/pnas.0507360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia XG, Harding T, Weller M, Bieneman A, Uney JB, Schulz JB. Gene transfer of the JNK interacting protein-1 protects dopaminergic neurons in the MPTP model of Parkinson's disease. Proc Natl Acad Sci USA. 2001;98:10433–10438. doi: 10.1073/pnas.181182298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Maroney AC, Dobrzanski P, Kukekov NV, Greene LA. The MLK family mediates c-Jun N-terminal kinase activation in neuronal apoptosis. Mol Cell Biol. 2001;21:4713–4724. doi: 10.1128/MCB.21.14.4713-4724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Muller-Myhsok B, Dickson DW, Meitinger T, Strom TM, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]