

Abstract
Primary microcephaly is a rare condition in which brain size is substantially diminished without other syndromic abnormalities. Seven autosomal loci have been genetically mapped, and the underlying causal genes have been identified for MCPH1, MCPH3, MCPH5, MCPH6, and MCPH7 but not for MCPH2 or MCPH4. The known genes play roles in mitosis and cell division. We ascertained three families from an Eastern Canadian subpopulation, each with one microcephalic child. Homozygosity analysis in two families using genome-wide dense SNP genotyping supported linkage to the published MCPH4 locus on chromosome 15q21.1. Sequencing of coding exons of candidate genes in the interval identified a nonconservative amino acid change in a highly conserved residue of the centrosomal protein CEP152. The affected children in these two families were both homozygous for this missense variant. The third affected child was compound heterozygous for the missense mutation plus a second, premature-termination mutation truncating a third of the protein and preventing its localization to centrosomes in transfected cells. CEP152 is the putative mammalian ortholog of Drosphila asterless, mutations in which affect mitosis in the fly. Published data from zebrafish are also consistent with a role of CEP152 in centrosome function. By RT-PCR, CEP152 is expressed in the embryonic mouse brain, similar to other MCPH genes. Like some other MCPH genes, CEP152 shows signatures of positive selection in the human lineage. CEP152 is a strong candidate for the causal gene underlying MCPH4 and may be an important gene in the evolution of human brain size.
Introduction
Primary microcephaly (PM) is defined by a head circumference more than three standard deviations below the age- and sex-adjusted mean. The reduced head size appears to be directly caused by smaller brain size, without specific structural brain abnormalities, beginning prenatally and continuing through childhood. PM may be of sporadic or familial etiology. Seven different chromosomal loci for this trait have been genetically mapped, named appropriately as MCPH1–MCPH7 (MIM 251200, 604317, 604804, 604321, 608716, 608393, and 612703, respectively). The causal genes have been identified for loci MCPH1, MCPH3, MCPH5, MCPH6, and MCPH7 (genes MCPH1 [MIM 607117], CDK5RAP2 [MIM 608201], ASPM [MIM 605481], CENPJ [MIM 609279], and STIL [MIM 181590], respectively), but not for MCPH2 or MCPH4 until now.1–6 Unlike syndromic forms of microcephaly, patients with PM do not have any other obvious organ or morphological problems. The known genes play roles in cell division, particularly in chromosome segregation and centrosome function, and some have been directly shown to be expressed in rapidly dividing cells in mouse embryonic brains. The MCPH genes have generated intense interest for their potential to explain aspects of primate and human evolution, as several studies document evidence of positive selection in the primate lineages.7–11 In this report, we document pathogenic mutations in a centrosomal protein within the published MCPH4 chromosomal region, in three patients with PM.
Subjects and Methods
Clinical Ascertainment and Consent
Patients were identified in the course of routine clinical ascertainment and treatment of developmental and behavioral disorders in the child neurology traveling clinics of New Brunswick, Canada. Approval for the research study was obtained from the research ethics board of the IWK Health Centre in Halifax, Nova Scotia, Canada. All sampled family members provided informed consent to participate in the study. DNA was obtained from blood samples via routine extraction methods.
Genotyping and Analysis
Whole-genome SNP scanning was performed at the McGill University and Genome Quebec Centre for Innovation, with the use of the Illumina Human610-Quadv1_B panel. Data were scanned with the Bead Array Reader, plate Crane Ex, and Illumina BeadLab software, on the Infinium II FastScan setting. Allele calls were generated with Beadstudio version 3.1, with the Genotyping Module used. Regions of homozygosity shared identically by state (IBS) in the two genotyped affected patients were determined by direct inspection with the use of customized scripts.
Mutation Detection and Bioinformatic Analysis
Annotated coding exons were amplified from patient genomic DNA by PCR via standard methods and were sequenced at Dalhousie University, via Sanger fluorescent sequencing and capillary electrophoresis. Control samples were sequenced at the McGill University and Genome Quebec Centre for Innovation Sequence. Traces were analyzed with MutationSurveyor (Soft Genetics).
Homologous protein sequences of the human CEP152 gene were retrieved from NCBI genome database with BLASTP. Only the genes annotated as centrosomal protein 152kDa (CEP152) or predicted protein similar to CEP152 were selected as the orthologs of human CEP152. Multiple sequence alignments (MSAs) were generated with MUSCLE.12 The effect of amino acid substitution on protein function was predicted with SIFT,13–15 PolyPhen,16–18 PANTHER,19,20 and Align-GVGD.21,22 Protein sequence of the human CEP152 gene was used as the input for SIFT, PolyPhen, and PANTHER. Default query options were used for SIFT and PolyPhen prediction. MSA of the CEP152 protein orthologs was used as the input MSA for Align-GVGD.
To study conserved domains in CEP152, reference sequence (NP_055800.2) was used as a query in the NCBI CD-Search tool with parameters Database = CDD, Maximal_hits = 250, Expect_value_threshold = 0.0001, and Apply_low_complexity_filter = yes.23 The CEP152 reference protein sequence and mutant CEP152 sequence were analyzed via several coiled-coil prediction methods: COILS, Paircoil2, and Marcoil.24–27 The following parameters were used in the analyses: COILS window_width = 28, matrix = MTIDK, weighting = yes; Paircoil2 p-score_cutoff = 0.025, minimum_search_window_length = 28; Marcoil cc_emission_prob_matrix = { 9FAM, MTIDK, MTK }, HMM_prob_matrix = MARCOIL-H. SOSUIcoil was also used to predict putative coiled-coil fragile points in both the reference and mutant protein sequences.28
Mouse Work
RT-PCR was performed with the use of RNA extracted from embryonic day 12.5 or 14.5 mouse brain via standard methods. The orthologous mouse Cep152 gene was amplified from cDNA with the use of the following PCR primers: forward, 5′-GCTGTCACTCGCACTCTCTG-3′ (in mouse exon 21, per NM_001081091); reverse, 5′-CACCCTGCTGTTCTCCTCTC-3′ (in mouse exon 24). In situ hybridization to embryonic mouse brain sections was performed as described.29 The procedures followed were in accordance with the ethical standards of the responsible committee on animal experimentation.
Functional Studies in Cells
Full-length human CEP152 cDNA corresponding to reference sequence NM_014985 was cloned between the KpnI and XhoI sites of pEGFP-c1 (Clontech). Mutations corresponding to those observed in the human microcephaly patients were introduced with the QuikChange Lightning Site-Directed Mutagenesis Kit (Stratagene) according to the manufacturer's protocol. All constructs were verified by sequencing.
Human osteosarcomas-derived U2OS cells were transfected with the wild-type or the two different mutant fusions to green fluorescent protein (GFP). Cells were fixed with ice-cold methanol for 10 min at −20°C. They were washed with PBS and blocked with 2% BSA in PBS for 30 min. Cells were incubated with mouse-anti-γ(gamma)-tubulin for 1 hr and with secondary antibody for 30 min, respectively. DNA was stained with Hoechst. Between each step, cells were washed three times with 2% BSA in PBS. All incubations took place at room temperature. Images were taken with a PerkinElmer Ultra-View spinning-disc confocal system on a Nikon Ti inverted microscope. Mouse-anti-γ-tublin (GTU-88) was purchased from Sigma-Aldrich.
Molecular Evolution Analysis
Sequences were aligned via the method incorporated in the software RevTrans.30 This program generates multiple alignment of coding DNA sequences from aligned amino acid sequences, obtained in this study from NCBI: Homo sapiens centrosomal protein 152 (GI: 110347567:35-4999), Pan troglodytes centrosomal protein 152 (GI: 114656898:194-5326), Mus musculus centrosomal protein 152 (GI: 124487355:246-5456), Rattus norvegicus centrosomal protein 152 (GI: 109468754|ref|XM_230555.4), Monodelphis domestica similar to CEP152 protein (GI: 126278234|ref|XM_001380412.1), and Gallus gallus similar to rai-like protein (GI: 118095786:1-5310). The tree was computed with PhyML.31 Genomic sequences of other primates were obtained by manual alignment and inspection of the human gene with the primate genomic consensus sequences, and extraction and predicted translation of all coding exons. This was possible for chimpanzee, orangutan, rhesus, and marmoset, but not for other primate genomes, including gorilla and bonobo, which contain gaps preventing the identification of all orthologous coding-exon sequences (see Figure S7, available online, for all predicted protein sequences).
For estimation of nonsynonymous/synonymous substitution ratios, ω (= Ka/Ks = dN/dS), the method implemented in PAML, was used.32 First, ω values were calculated with the branch models implemented in PAML, in which ω is allowed to vary among all branches in the tree. With branch-site models, ω values are allowed to vary among branches and codons, for the detection of sites targeted by positive selection in specific lineages or all lineages.33
Results
Clinical Description
In the course of clinical work, we ascertained three families from a rural Maritime Canadian subpopulation, each with one child presenting with PM. In all families, both parents were of Acadian descent. None of the parents were known to be consanguineous, and none had any other children after the birth of the child with microcephaly (see Figures 1A–1C for pedigrees, Figure 1D for photograph of one patient, and Figures 1E and 1F for MRI scans of one patient).
Figure 1.
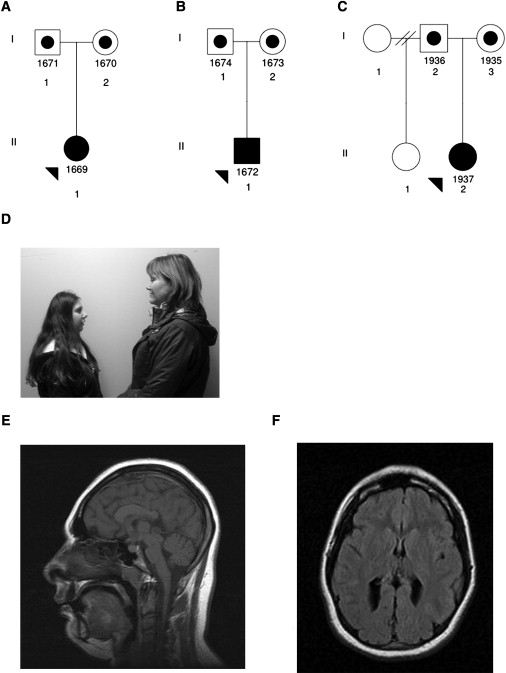
Maritime Microcephaly Cohort
(A–C) Maritime families 1–3 of patients carrying mutations in the CEP152 gene. Affected probands are represented by fully shaded symbols, noted by arrowheads.
(D) Photograph of microcephalic patient 1937 (with mother).
(E and F) MRI scans of the head of patient 1937 at the age of 18 yrs.
The patients had microcephaly, with head circumference between five and seven standard deviations below the mean (see Figures 2A and 2B for head growth charts). There were no other dysmorphic signs, and height and weight were normal. They were noted to be very visually attentive and to have fast, rather jerky movements in infancy. They all had prominent mirror movements that persisted into childhood. From an early age, they were very happy and friendly and seemed very aware socially. The patients walked alone between 14 and 15 mo of age, two of them toe walking at first. They were talking to express themselves by 3 yrs of age. All three children learned to read and attended regular classes with some modification. One patient (1937) had psychological testing, including the WISC-IV, which showed moderate cognitive impairment, with both verbal and performance scores below the 1st percentile but with visual motor skills up to the 4th percentile. One girl (1669) excelled at reading and kept up to her peers academically until the age of 11 yrs. They all responded strikingly quickly to social or verbal cues, but at times they exhibited challenging behavior, being impulsive and aggressive and having tantrums. One (1672) had tics. Another (1937) had obsessive and/or compulsive traits. She was self-conscious about her small head size and invented other reasons for her appointments: “I've got a broken leg” or “I'm very sick with asthma.” She acted these parts remarkably well, with pained expression and limping gait or rapid breathing. The three children were otherwise healthy at their last follow-up visit, at the ages of 10–18 yrs. None had epilepsy.
Figure 2.
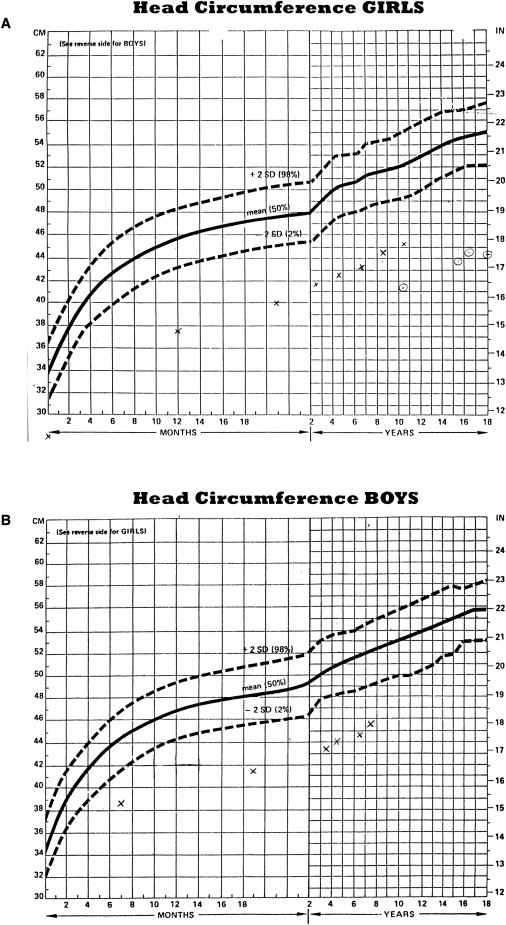
Growth Charts Tracking the Head Circumference of Our Microcephalic Patients from an Early Age Compared to Population Means for Girls and Boys
Solid lines show the mean head size at various ages. Dashed lines show the limits of two standard deviations above or below the mean size.
(A) Females: x represents patient 1669, dotted circle represents patient 1937.
(B) Males: x represents patient 1672.
An MRI scan was performed for one patient (1937), showing markedly reduced brain size with mild enlargement of the posterior horns of the lateral ventricles. The gyral pattern was simplified, in keeping with the small brain size, but the cortex appeared of normal thickness. The pituitary gland appeared large in comparison to the brain but was of normal size.
Molecular Mapping and Mutation Detection
Initially we were able to sample two of the affected patients (1669 and 1672) and their parents. We performed high-density genome-wide SNP genotyping, using the Illumina panel of 610,000 markers, and looked for regions of extended homozygosity shared IBS in the two affected individuals but not in their parents. As shown in Table 1, among the ten longest series of consistent SNPs, a 14 Mbp region on chromosome 15q21.1 appeared as a clear outlier, both by physical length and by number of consecutive homozygous IBS-shared SNPs. Parents of the affected children were heterozygous for many SNPs in the region, indicating that the markers were genetically informative (data not shown). The region was broken into three subsections, each separated by a single SNP, which we infer to represent false heterozygous genotype calls. The combined interval, from approximately 39–53 Mbp (February 2009 Human Genome Assembly hg19), lies within the larger published MCPH4 locus, which is defined by an anonymous marker near D15S1042 and by D15S98, equivalent to 36–60 Mbp in hg19.34 Two additional families have also been reported as linked to MCPH4, but without any further localization information.35 In our two genotyped patients, there was no evidence of homozygosity at any of the five known microcephaly genes—MCPH1, CDK5RAP2, ASPM, CENPJ, and STIL—or at the remaining MCPH2 locus. We also sequenced these five genes (total 112 coding exons) in our affected patients, without finding any potentially pathogenic variants.
Table 1.
Twelve Longest Intervals of Consecutive SNPs Homozygous and Identical by State in the Two Genotyped Affected Microcephaly Patients
| nSNPs | Chr. | BeginSNP | EndSNP | Begin | End | Size |
|---|---|---|---|---|---|---|
| 1158 | 15 | rs768269 | rs16953764 | 37 022 649 | 44 400 816 | 7 378 168 |
| 621 | 15 | rs16953764 | rs11634375 | 44 400 816 | 47 536 840 | 3 136 025 |
| 602 | 15 | rs11634375 | rs2017176 | 47 536 840 | 50 793 439 | 3 256 600 |
| 173 | 5 | rs11743309 | rs27466 | 45 158 145 | 50 125 676 | 4 967 532 |
| 129 | 6 | rs4713119 | rs12661831 | 27 820 804 | 28 445 748 | 624 945 |
| 120 | 4 | rs6531772 | rs6849320 | 33 432 805 | 34 441 990 | 1 009 186 |
| 105 | 12 | rs1265566 | rs6489848 | 110 200 759 | 111 457 037 | 1 256 279 |
| 85 | 3 | rs12631750 | rs11925493 | 131 536 341 | 131 994 964 | 458 624 |
| 83 | 4 | rs9714696 | rs4697693 | 9 094 398 | 9 504 958 | 410 561 |
| 80 | 9 | rs2564362 | rs10760302 | 125 325 467 | 125 762 468 | 437 002 |
| 74 | 17 | rs4793119 | rs9908256 | 39 979 930 | 40 406 871 | 426 942 |
| 73 | 18 | rs12457620 | rs9948246 | 64 152 547 | 64 252 757 | 100 211 |
Intervals are in descending order of number of consecutive SNPs, with the presumptive linked interval on chromosome 15 italicized and bolded. Columns give the number of consecutive homozygous SNPs identical by state, the chromosome, the first heterozygous SNP before the homozygous interval, the first heterozygous SNP after the homozygous interval, the nucleotide position of the first heterozygous SNP before, the nucleotide position of the first heterozygous SNP after, and the size of the interval spanned from the first heterozygous SNPs (none of these regions spans a centromere). Nucleotide positions are based on human genome assembly hg18.
The region defined by our patients includes more than 150 RefSeq annotated genes. On the basis of the known or hypothesized functions of the characterized gene products of MCPH1, MCPH3, MCPH5, MCPH6, and MCPH7, we prioritized genes likely to have functions related to cell division or chromosomal segregation. In all, we sequenced 261 coding exons of 14 genes (TMOD2 [MIM 602928], TP53BP1 [MIM 605230], MAP1A [MIM 600178], HAUS2, TUBGCP4 [MIM 609610], NUSAP1 [MIM 612818], BUB1B [MIM 602860], MFAP1 [MIM 600215], USP8 [MIM 603158], MYO5A [MIM 160777], STARD9, GABPB1 [MIM 600610], RAD51 [MIM 179617], and CEP152), until we observed an interesting missense variant in the gene CEP152. As shown in Figure 3A, this is a glutamine-to-proline change at residue 265 of the protein (p.Q265P), found to be homozygous in samples from the two affected patients, 1669 and 1672, and heterozygous as expected in the four parents (data not shown). This amino acid variant is not present in dbSNP build 130, nor was it detected in any of 310 local Maritime control chromosomes or 186 European control chromosomes from the Centre d'Etude du Polymorphisme Humain (CEPH). No other homozygous coding variants were detected by sequencing this set of candidate genes in the two affected patients.
Figure 3.
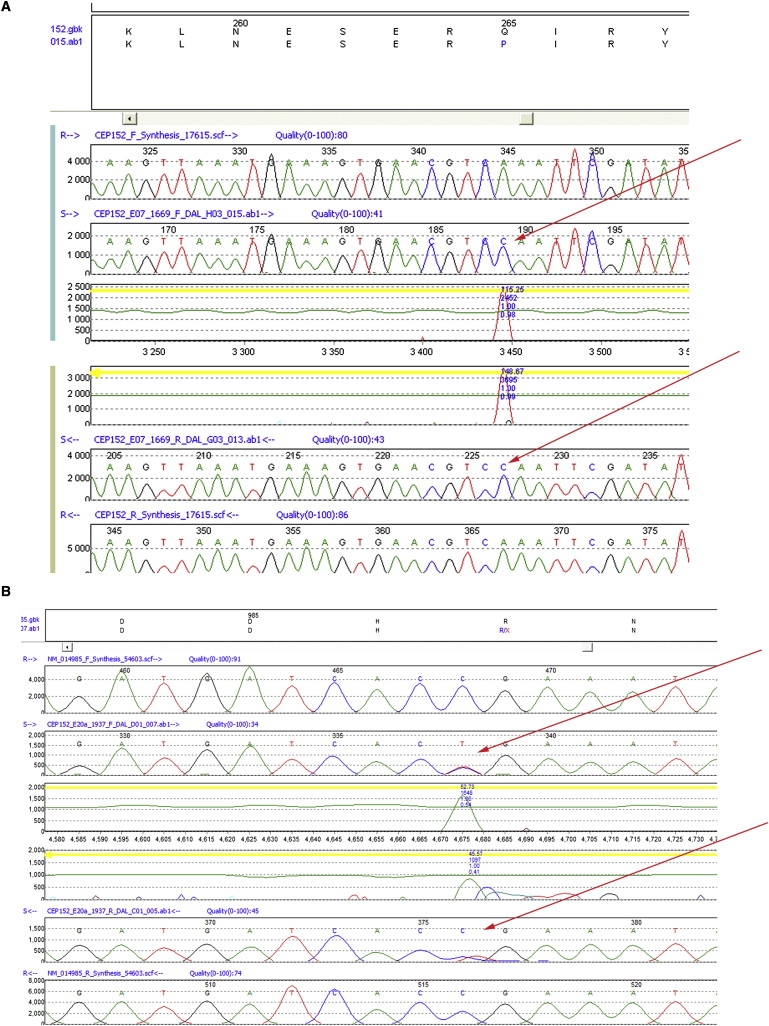
Sequences of Affected Patients Carrying Mutations in CEP152
The upper part of each panel shows wild-type amino acid sequence above, mutant amino acid sequence below. The lower part of each panel shows four chromatogram traces; uppermost and lowermost are virtual forward and reverse traces, respectively, created by the software from the consensus wild-type sequence, and inner traces are forward and reverse chromatograms, respectively, from the patient sample. The two center tracks are mutation-detection calls by MutationSurveyor for forward and reverse sequences; peaks above the green curve reach significance according to the software calling algorithm. The red arrows points to the mutations in the affected patient's forward and reverse sequences.
(A) Homozygous mutation p.Q265P in CEP152 in patient 1669 with PM.
(B) Heterozygous mutation p.R987X in CEP152 in patient 1937 with PM.
Subsequently, we were able to sample the remaining family. The third patient (1937) was likewise negative for mutations in the five known MCPH genes. Unexpectedly, the third affected patient was heterozygous only for the p.Q265P missense variant, as was only one parent. Resequencing of all the remaining CEP152 coding exons in this patient identified a second mutation, a stop codon at residue arginine 987, p.R987X, caused by a CGA-to-TGA change (Figure 3B). This mutation was heterozygous in the other parent, consistent with independent segregation of the two mutations in this presumably compound-heterozygous patient, and was not detected in either the local controls or the CEPH controls. This mutation would cause an obligate protein truncation eliminating 668 C-terminal amino acids of the protein. Given the sequencing results, neither the third patient nor the parents were genotyped across the entire genome.
Bioinformatic Mutation Analysis
Residue 265 is highly conserved among vertebrates (Figure 4A, Figure S1). The p.Q265P mutation is predicted to be pathogenic by SIFT and Polyphen (Table S1). PANTHER failed to give a prediction. The mutation was predicted as benign by Align-GVGD with the use of the MSA including the D. rerio sequence (which is significantly divergent from the other vertebrate orthologs), but deleterious if the Danio sequence was omitted. Residue 265 in the human gene lies in a conserved SMC_prok_B domain found in proteins that play a role in organizing chromosomes for cell division (hence, Structural Maintenance of Chromosomes) (Figure 4B, Figure S2, Table S2). More specifically, residue 265 is predicted to fall in a coiled-coiled region according to COILS, Paircoil2, Marcoil, and SOSUIcoil (Figure 4C, Figures S3 and S4). COILS, Paircoil2, and Marcoil (with the use of the MTK emission matrix) all predict that the mutant proline residue would weaken the coiled-coil structure (Figures S3 and S4, Tables S3–S5). SOSUIcoil predicts that residue 265 itself is a weak coiled-coil residue in both the wild-type and mutant sequences (Figure S5). The identification of two different pathogenic mutations in CEP152 makes it a strong candidate for the causal gene in our patients and the presumptive gene underlying MCPH4 generally.
Figure 4.
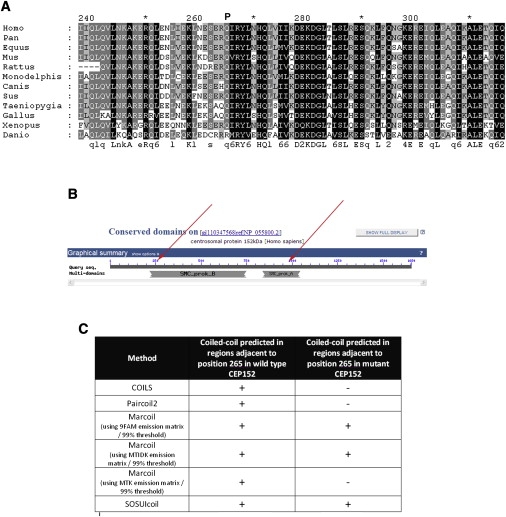
Sequence Analysis of CEP152
(A) Multiple sequence alignment of CEP152 in human and other vertebrates in the region surrounding mutated residue Q265. The mutation to proline is indicated by a P above the human sequence.
(B) Domain structures of CEP152 predicted by NCBI. Red arrows indicate the approximate location of mutations at residues 265 (Q-to-P in coiled-coil domain SMC_prok_B) and 987 (truncating domain SMC_prok_A).
(C) Coiled-coil domain predictions for wild-type and p.Q265P mutant CEP152 for four bioinformatic tools.
Functional Studies on CEP152
Expression of some microcephaly genes can be detected in dividing cells of mouse embryonic brain, particularly in the ventricular zone.7,36–38 We were able to generate a cDNA clone product of the correct size spanning exons 21–24 of the mouse Cep152 ortholog by using RNA extracted from mouse brain tissue; cDNA could be readily detected in stage 12.5 and 14.5 embryonic brains (Figure 5). By in situ hybridization with two different probes, no signal could be observed above background, suggesting a low level of Cep152 expression in those tissues.
Figure 5.
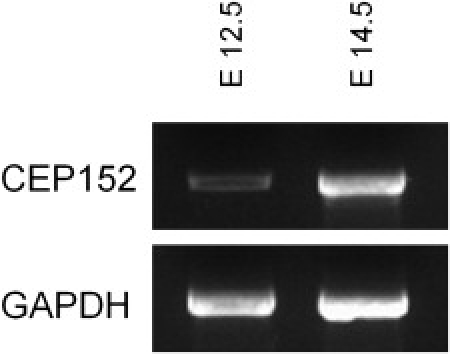
Expression of CEP152 in Embryonic Mouse Brains
RT-PCR was performed on stage 12.5 or 14.5 embryonic brain RNA with the use of primers designed to amplify across multiple splice junctions of the mouse orthologous gene.
We tested for functional effects of the two mutations by transfecting human U2OS osteosarcoma-derived cells with wild-type or mutant human CEP152 fused to GFP. Transfected GFP alone was not detectable in centrosomes, marked by costaining with antibody to γ-tubulin (Figure 6, top row), whereas wild-type CEP152-GFP was detected in centrosomes (Figure 6, middle row). The truncated CEP152 p.R987X-GFP protein was not detected in centrosomes (Figure 6, lower row). The point mutant CEP152 p.Q265P-GFP could be detected in centrosomes (data not shown).
Figure 6.
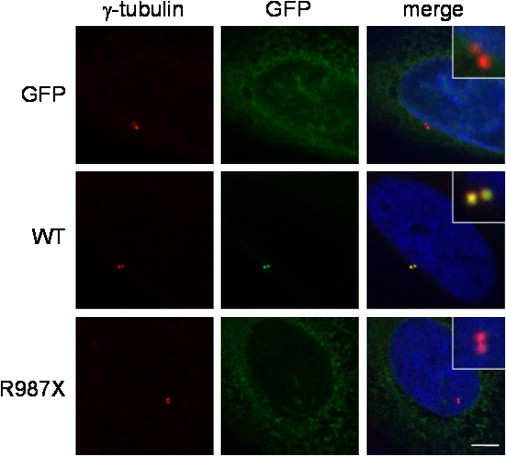
Functional Analysis of CEP152 Mutants
Human cells were transfected with vectors expressing either wild-type or mutant human CEP152 fused to GFP. In each row, the left panel shows antibody staining to γ-tubulin to mark centrosomes; the center panel shows GFP fluorescence; the right panel shows merged γ-tubulin plus GFP plus Hoechst, with insert at a higher magnification of centrosomes. Top row: cells transfected with vector alone (just GFP); middle row, cells transfected with wild-type CEP152-GFP fusion; bottom row, cells transfected with p.R987X mutant CEP152-GFP fusion. Scale bar represents 5 μm.
Molecular Evolution Studies
Molecular evolution studies of other microcephaly genes have suggested that some of these are under positive selection in primates. We asked whether CEP152 has been subject to adaptive changes in humans. We used models allowing different levels of heterogeneity in the nonsynonymous/synonymous substitution ratio ω = Ka/Ks. Ratio estimates of ω greater than 1 indicate an excess of amino acid changes relative to the silent or neutral expectation and suggest adaptive evolution. Estimates of ω less than 1 are suggestive of selective constraints. Our analysis is similar to that employed in an evolutionary study of the microcephaly gene ASPM.39
By comparison of the human gene with other primate and vertebrate orthologs, we found an elevated value (greater than 1) of ω = 1.43 only in the human branch (Figure 7A), significantly different from the other ratios throughout the tree, consistent with positive (adaptive) selection in this gene specific to that lineage (p = 1.23 × 10−2, by comparing models B and D in Figure 7B). By detailed comparison across the coding region of CEP152 among vertebrate sequences, some segments are demonstrably more conserved than others (Figure S6). To incorporate this into selection models, we allowed ω ratios to vary both across lineages and among sites in the protein. This provides greater resolution, because sites of potential positive or negative selection may be interspersed with domains undergoing only neutral variation. The increased resolution is at the expense of power, because the number of available sites is decreased by testing subdomains rather than the entire protein. Nonetheless, we detected the presence of sites subject to adaptive evolution in the human branch (H) (p = 1.23 × 10−2). Specifically eight sites in the protein are potentially under positive selection (Figure 7C).
Figure 7.
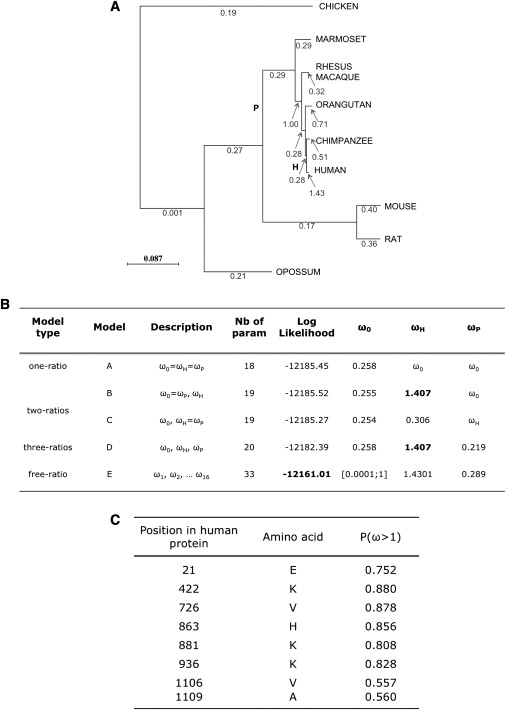
Selection Analysis of CEP152 in Vertebrates
(A) Phylogenetic tree and ω ratios for CEP152 coding sequences. For all branches, the ω ratio is set free to vary. The maximum-likelihood estimate of ω is shown along each branch.
(B) Log likelihood values and parameter estimates under different branch models. The parameters in each model are the 16 branch lengths of the tree (number of nucleotide substitutions per codon), the transition/ transversion rate ratio κ, and the ω ratio(s). Estimates of branch lengths and κ are not shown. To test whether the human ratio is significantly different from the others, we focused specifically on comparisons among the human (H), primates (P), and other (O) lineages. ωH, ωP, and ω0 are thus the dN/dS ratios for branches H, P, and all other branches, respectively. Significant model comparison: B (H0) and D (H1): p = 1.23 × 10−2, fixed-ratio models (H0) and free-ratio (H1): p < 10−10.
(C) Sites of CEP152 under potential positive selection in humans. The log likelihood ratio test supports the branch-site model of positive selection with p = 2.62 × 10−2 for the general test. P(ω > 1) are the posterior probabilities of specific potential sites under positive selection in the human lineage.
Discussion
By homozygosity mapping in patients from an Eastern Canadian subpopulation affected with PM, we identified two independent rare mutations in the gene encoding CEP152. Our three affected patients were either homozygous or compound heterozygous for these mutations. One mutation is a nonconservative change of glutamine to proline at a highly conserved residue and is predicted to be pathogenic, disturbing a potential coiled-coil domain of the protein. The second mutation is an obligate protein truncation removing a large C-terminal proportion of the predicted coding region. In comparing the phenotypes of the two females, the compound heterozygous individual 1937 showed greater reduction in expected head size than the missense homozygous individual 1669. The truncating allele may thus be more severe than the missense, although this must be considered tentative, given that there were only two affected individuals to compare. The truncation could conceivably cause nonsense-mediated decay of the mutated transcript; however, fresh tissue from the compound-heterozygous patient was not available for testing of this directly. In a functional assay for subcellular localization, the wild-type CEP152-GFP fusion protein could be detected in structures costaining for γ-tubulin. These are either centrioles or centrosomes (or both), given that the tubulin antibody does not allow a formal distinction between these two related structures. Using a new antibody specific for CEP152 N-terminal sequence, we have also detected the protein in bona fide centrioles in untransformed serum-starved hTERT-RPE cells (M.A., unpublished data). The mutant CEP152 protein fusing the equivalent truncated human protein to GFP failed to colocalize with γ-tubulin, verifying pathogenicity of the mutation. Localization is only a partial assay for the functional gene product. It was not possible to assess full functionality of the mutant CEP152 proteins in the absence of the endogenously expressed gene; thus, the p.Q265P missense mutation may have a more subtle effect on cell division despite being physically present in the centrosome in the qualitative immunohistochemical assay. Expression of the CEP152 gene could be detected by RT-PCR in embryonic mouse brains.
Our molecular evolution studies suggest that CEP152 is under positive selection in the human lineage. This is consistent with reports of positive selection for other characterized human microcephaly genes, and it suggests that these genes may play an important role in the rapid and disproportionate increase in total brain size in primates and especially in humans. By dividing the protein into segments, we could detect selection signatures for specific residues in the human gene. The individual sites of CEP152 potentially under evolutionary selection do not include either of the residues mutated in our human microcephaly patients. This is not particularly surprising, because residues of the protein mutable to a loss-of-function phenotype could potentially include any functionally important positions, whereas sites under positive selection are expected to be only a subset of functionally important sites for which adaptive variation arose by chance and survived to achieve fixation. There were too few variable sites in the gene among human sequences to measure selection across human subpopulations. By inspection of Neandertal genomic sequence reads submitted to GenBank, the modern human amino acid residues are predicted at each of the specific sites under potential selection.
CEP152 was originally identified as a component of mammalian centrosomes through direct proteomic analysis.40,41 Other centrosomal or chromosomal organizing proteins mutate to cause PM types 1, 3, 5, 6, and 7. CEP152 is the likely mammalian ortholog of the Drosophila gene asterless, which has been studied intensively for its role in cell division and development.42,43 Mutations in asterless cause arrest of embryogenesis in the fly, or else male infertility, and the protein product of asterless was directly localized to centrioles,42 consistent with our results for CEP152 in human cells. Antisense knockdown of the putative CEP152 asterless ortholog in zebrafish led to a curly-tail phenotype typical of centriolar or ciliary defects.43 These model-organism studies support a role for CEP152 in mitosis consistent with other known PM genes. Together, our findings strongly suggest that we have identified the causal gene for microcephaly type MCPH4 in our three affected patients. It remains to be seen whether CEP152 is also mutated in other published families linked to this chromosomal region.
Although we could not document parental consanguinity in our families, strong founder effects have been observed in Acadian francophonic populations. In other studies, we used dense SNP genotyping together with homozygosity analysis to map and identify causal mutations in patients with congenital sideroblastic anemia and cutis laxa type II in families from this region of eastern Canada.44,45 The occurrence of a compound heterozygote among our three patients was unanticipated but not unusual. Such individuals presumably arise from a more prevalent deleterious mutation, rising to moderately increased frequency through an early founder effect, combining with a second mutation arising more recently and thus found only in the heterozygous state. We similarly identified two different mutations in the sacsin gene (SACS [MIM 604490]) in two Maritime Canadian ataxia families, in which one family was homozygous and the other compound heterozygous for the same mutation plus a different second mutation.46
Our three patients studied showed overall phenotypic similarity, including severe microcephaly from birth and absence of other dysmorphic features. They shared a similar developmental pattern, with mild delay in early motor development, moderate cognitive impairment, difficult behavior patterns, but no other neurological abnormalities. This clinical picture, as well as the findings on MRI scanning of one patient, is consistent with the findings reported in other children with autosomal-recessive PM.
According to the published mapping, the as-yet-unidentified MCPH2 gene lies in a 9.7 Mbp region of chromosome 19q13. This region contains several interesting candidates, including genes potentially involved in cell-growth regulation (PDCD2L, WTIP, MAP4K1 [MIM 601983], MAP3K10 [MIM 600137], LTBP4 [MIM 604710], CIC [MIM 612082]) or brain ciliary function (B9D2 [MIM 611951], putative ortholog of the mouse gene stumpy, which has brain and renal phenotypes) and the gene HAUS5, encoding a component of the multiprotein Augmin or HAUS complex, which is essential for centrosome function in dividing cells.47–49
With respect to PM, a major outstanding question is why mutations of the MCPH genes cause this phenotype and not a more general developmental or syndromic condition. Some centriolar and centrosomal proteins are implicated in a variety of different human-disease syndromes.50 For example, mutations in the SMC1A gene (MIM 300040), whose product contains an SMC domain similar to CEP152, can cause Cornelia de Lange syndrome (CdLS [MIM 300590]).51 The phenotype of CdLS includes microcephaly together with dysmorphologies. Mutations in the gene encoding pericentrin2 (MIM 605925), another mitotic gene, cause microcephaly together with dwarfism and other abnormalities.52 One possibility is that the MCPH genes are expressed specifically in developing brain cells. However, this appears unlikely on the basis of centrosomal proteomic studies carried out with cells of lymphoblastoid origin,41 as well as reported expression patterns in BioGPS. Conceivably, there might be alternatively spliced protein isoforms specific to embryonic brain cells, or cell type specific functional protein domains. For the MCPH5 gene (ASPM), however, there are 69 reported point or small indel mutations scattered throughout the gene from residue 117 to 3353 out of 3477 (according to the HGMD Professional database), suggesting that there is not a specific region of the gene required uniquely in brain cells; moreover, the gene is reportedly expressed in many tissues.37 There are only small numbers of mutations reported for other MCPH genes (MCPH1, CDKRAP, STIL, CENPJ), so this hypothesis is perhaps more viable for those genes. In the case of CEP152, we found a missense mutation early in the gene and a termination codon removing approximately the last third of the coding sequence; thus, these fall in quite different regions of the protein's primary sequence.
It appears most likely that the developing brain has a special requirement for the functions of the MCPH genes. The reasons for this are unknown, although cell division in embryonic brains may be different than other cell divisions during early development.53 There are probably additional PM loci to be found (besides the gene underlying MCPH2), but probably not in large numbers. This is in contrast to the case of more general intellectual disability, in which the number of genes with potentially causal alleles is rising rapidly as larger segments of the genome are queried with the use of high-throughput sequencing and other structural genomics technologies.54,55 Apparently isolated PM is a more specific phenotype than intellectual disability, with fewer potentially causal genes or mutations.52,56–58
Acknowledgments
Foremost, we thank the families who generously contributed their time and materials to this research study. Thanks to Monica Bettencourt-Dias for helpful discussions regarding centrosome function and evolution. D.L.G. and M.E.S. were supported by Genome Canada, Genome Atlantic, Nova Scotia Health Research Foundation, Nova Scotia Research and Innovation Trust, IWK Health Centre Foundation, Dalhousie Medical Research Fund, and Capital Health Research Fund. M.E.S. was supported by the Centre de Recherche du CHU Ste-Justine. J.L.M. is a senior scientist of the Fonds pour la Recherche en Santé du Québec and was supported by an operating grant from the Canadian Institutes of Health Research. P.A. was supported by a Ministry of Economic Development, Innovation and Export Trade (PSR-SIIRI-195) grant and a Genome Quebec Chair of Population and Medical Genomics. I.H. was supported by the Deutsche José Carreras-Leukämie Stiftung (DJCLC R07/21v).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
BioGPS gene expression portal, http://biogps.gnf.org/#goto=welcome
COILS server, http://www.ch.embnet.org/software/COILS_form.html
HGMD public database, http://www.hgmd.cf.ac.uk/ac/index.php
HGNC gene nomenclature, http://www.genenames.org/index.html
Marcoil server, http://www.isrec.isb-sib.ch/webmarcoil/webmarcoilC1.html
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/omim
Paircoil2 server, http://groups.csail.mit.edu/cb/paircoil2/
SOSUI Coil server, http://bp.nuap.nagoya-u.ac.jp/sosui/coil/submit.html
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Mochida G.H., Walsh C.A. Molecular genetics of human microcephaly. Curr. Opin. Neurol. 2001;14:151–156. doi: 10.1097/00019052-200104000-00003. [DOI] [PubMed] [Google Scholar]
- 2.Roberts E., Hampshire D.J., Pattison L., Springell K., Jafri H., Corry P., Mannon J., Rashid Y., Crow Y., Bond J., Woods C.G. Autosomal recessive primary microcephaly: an analysis of locus heterogeneity and phenotypic variation. J. Med. Genet. 2002;39:718–721. doi: 10.1136/jmg.39.10.718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Woods C.G. Human microcephaly. Curr. Opin. Neurobiol. 2004;14:112–117. doi: 10.1016/j.conb.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 4.Woods C.G., Bond J., Enard W. Autosomal recessive primary microcephaly (MCPH): a review of clinical, molecular, and evolutionary findings. Am. J. Hum. Genet. 2005;76:717–728. doi: 10.1086/429930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abuelo D. Microcephaly syndromes. Semin. Pediatr. Neurol. 2007;14:118–127. doi: 10.1016/j.spen.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 6.Kaindl A.M., Passemard S., Kumar P., Kraemer N., Issa L., Zwirner A., Gerard B., Verloes A., Mani S., Gressens P. Many roads lead to primary autosomal recessive microcephaly. Prog Neurobiol. 2010;90:363–383. doi: 10.1016/j.pneurobio.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 7.Jackson A.P., Eastwood H., Bell S.M., Adu J., Toomes C., Carr I.M., Roberts E., Hampshire D.J., Crow Y.J., Mighell A.J. Identification of microcephalin, a protein implicated in determining the size of the human brain. Am. J. Hum. Genet. 2002;71:136–142. doi: 10.1086/341283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ponting C., Jackson A.P. Evolution of primary microcephaly genes and the enlargement of primate brains. Curr. Opin. Genet. Dev. 2005;15:241–248. doi: 10.1016/j.gde.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 9.Cox J., Jackson A.P., Bond J., Woods C.G. What primary microcephaly can tell us about brain growth. Trends Mol. Med. 2006;12:358–366. doi: 10.1016/j.molmed.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 10.Evans P.D., Vallender E.J., Lahn B.T. Molecular evolution of the brain size regulator genes CDK5RAP2 and CENPJ. Gene. 2006;375:75–79. doi: 10.1016/j.gene.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 11.Vallender E.J., Mekel-Bobrov N., Lahn B.T. Genetic basis of human brain evolution. Trends Neurosci. 2008;31:637–644. doi: 10.1016/j.tins.2008.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Edgar R.C. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ng P.C., Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11:863–874. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ng P.C., Henikoff S. Accounting for human polymorphisms predicted to affect protein function. Genome Res. 2002;12:436–446. doi: 10.1101/gr.212802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ng P.C., Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ramensky V., Bork P., Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sunyaev S., Ramensky V., Bork P. Towards a structural basis of human non-synonymous single nucleotide polymorphisms. Trends Genet. 2000;16:198–200. doi: 10.1016/s0168-9525(00)01988-0. [DOI] [PubMed] [Google Scholar]
- 18.Sunyaev S., Ramensky V., Koch I., Lathe W., 3rd, Kondrashov A.S., Bork P. Prediction of deleterious human alleles. Hum. Mol. Genet. 2001;10:591–597. doi: 10.1093/hmg/10.6.591. [DOI] [PubMed] [Google Scholar]
- 19.Thomas P.D., Campbell M.J., Kejariwal A., Mi H., Karlak B., Daverman R., Diemer K., Muruganujan A., Narechania A. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 2003;13:2129–2141. doi: 10.1101/gr.772403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thomas P.D., Kejariwal A., Campbell M.J., Mi H., Diemer K., Guo N., Ladunga I., Ulitsky-Lazareva B., Muruganujan A., Rabkin S. PANTHER: a browsable database of gene products organized by biological function, using curated protein family and subfamily classification. Nucleic Acids Res. 2003;31:334–341. doi: 10.1093/nar/gkg115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mathe E., Olivier M., Kato S., Ishioka C., Hainaut P., Tavtigian S.V. Computational approaches for predicting the biological effect of p53 missense mutations: a comparison of three sequence analysis based methods. Nucleic Acids Res. 2006;34:1317–1325. doi: 10.1093/nar/gkj518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tavtigian S.V., Byrnes G.B., Goldgar D.E., Thomas A. Classification of rare missense substitutions, using risk surfaces, with genetic- and molecular-epidemiology applications. Hum. Mutat. 2008;29:1342–1354. doi: 10.1002/humu.20896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marchler-Bauer A., Bryant S.H. CD-Search: Protein domain annotations on the fly. Nucleic Acids Res. 2004;32:W327–W331. doi: 10.1093/nar/gkh454. (Web Server issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Delorenzi M., Speed T. An HMM model for coiled-coil domains and a comparison with PSSM-based predictions. Bioinformatics. 2002;18:617–625. doi: 10.1093/bioinformatics/18.4.617. [DOI] [PubMed] [Google Scholar]
- 25.Lupas A. Prediction and analysis of coiled-coil structures. Methods Enzymol. 1996;266:513–525. doi: 10.1016/s0076-6879(96)66032-7. [DOI] [PubMed] [Google Scholar]
- 26.Lupas A. Predicting coiled-coil regions in proteins. Curr. Opin. Struct. Biol. 1997;7:388–393. doi: 10.1016/s0959-440x(97)80056-5. [DOI] [PubMed] [Google Scholar]
- 27.McDonnell A.V., Jiang T., Keating A.E., Berger B. Paircoil2: improved prediction of coiled coils from sequence. Bioinformatics. 2006;22:356–358. doi: 10.1093/bioinformatics/bti797. [DOI] [PubMed] [Google Scholar]
- 28.Tanizawa H., Taniguchi M., Ghimire G.D., Mitaku S. Prediction of fragile points of coiled coils. Chem-Biol. Info. J. 2009;9:12–29. [Google Scholar]
- 29.Khan S.H., Aguirre A., Bobek L.A. In-situ hybridization localized MUC7 mucin gene expression to the mucous acinar cells of human and MUC7-transgenic mouse salivary glands. Glycoconj. J. 1998;15:1125–1132. doi: 10.1023/a:1006955604501. [DOI] [PubMed] [Google Scholar]
- 30.Wernersson R., Pedersen A.G. RevTrans: Multiple alignment of coding DNA from aligned amino acid sequences. Nucleic Acids Res. 2003;31:3537–3539. doi: 10.1093/nar/gkg609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guindon S., Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 32.Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 33.Zhang J., Nielsen R., Yang Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol. Biol. Evol. 2005;22:2472–2479. doi: 10.1093/molbev/msi237. [DOI] [PubMed] [Google Scholar]
- 34.Jamieson C.R., Govaerts C., Abramowicz M.J. Primary autosomal recessive microcephaly: homozygosity mapping of MCPH4 to chromosome 15. Am. J. Hum. Genet. 1999;65:1465–1469. doi: 10.1086/302640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gul A., Hassan M.J., Mahmood S., Chen W., Rahmani S., Naseer M.I., Dellefave L., Muhammad N., Rafiq M.A., Ansar M. Genetic studies of autosomal recessive primary microcephaly in 33 Pakistani families: Novel sequence variants in ASPM gene. Neurogenetics. 2006;7:105–110. doi: 10.1007/s10048-006-0042-4. [DOI] [PubMed] [Google Scholar]
- 36.Bond J., Roberts E., Mochida G.H., Hampshire D.J., Scott S., Askham J.M., Springell K., Mahadevan M., Crow Y.J., Markham A.F. ASPM is a major determinant of cerebral cortical size. Nat. Genet. 2002;32:316–320. doi: 10.1038/ng995. [DOI] [PubMed] [Google Scholar]
- 37.Kouprina N., Pavlicek A., Collins N.K., Nakano M., Noskov V.N., Ohzeki J., Mochida G.H., Risinger J.I., Goldsmith P., Gunsior M. The microcephaly ASPM gene is expressed in proliferating tissues and encodes for a mitotic spindle protein. Hum. Mol. Genet. 2005;14:2155–2165. doi: 10.1093/hmg/ddi220. [DOI] [PubMed] [Google Scholar]
- 38.Bond J., Roberts E., Springell K., Lizarraga S.B., Lizarraga S., Scott S., Higgins J., Hampshire D.J., Morrison E.E., Leal G.F. A centrosomal mechanism involving CDK5RAP2 and CENPJ controls brain size. Nat. Genet. 2005;37:353–355. doi: 10.1038/ng1539. [DOI] [PubMed] [Google Scholar]
- 39.Kouprina N., Pavlicek A., Mochida G.H., Solomon G., Gersch W., Yoon Y.H., Collura R., Ruvolo M., Barrett J.C., Woods C.G. Accelerated evolution of the ASPM gene controlling brain size begins prior to human brain expansion. PLoS Biol. 2004;2:E126. doi: 10.1371/journal.pbio.0020126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nogales-Cadenas R., Abascal F., Díez-Pérez J., Carazo J.M., Pascual-Montano A. CentrosomeDB: a human centrosomal proteins database. Nucleic Acids Res. 2009;37(Database issue, Database issue):D175–D180. doi: 10.1093/nar/gkn815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Andersen J.S., Wilkinson C.J., Mayor T., Mortensen P., Nigg E.A., Mann M. Proteomic characterization of the human centrosome by protein correlation profiling. Nature. 2003;426:570–574. doi: 10.1038/nature02166. [DOI] [PubMed] [Google Scholar]
- 42.Varmark H., Llamazares S., Rebollo E., Lange B., Reina J., Schwarz H., Gonzalez C. Asterless is a centriolar protein required for centrosome function and embryo development in Drosophila. Curr. Biol. 2007;17:1735–1745. doi: 10.1016/j.cub.2007.09.031. [DOI] [PubMed] [Google Scholar]
- 43.Blachon S., Gopalakrishnan J., Omori Y., Polyanovsky A., Church A., Nicastro D., Malicki J., Avidor-Reiss T. Drosophila asterless and vertebrate Cep152 Are orthologs essential for centriole duplication. Genetics. 2008;180:2081–2094. doi: 10.1534/genetics.108.095141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guernsey D.L., Jiang H., Campagna D.R., Evans S.C., Ferguson M., Kellogg M.D., Lachance M., Matsuoka M., Nightingale M., Rideout A. Mutations in mitochondrial carrier family gene SLC25A38 cause nonsyndromic autosomal recessive congenital sideroblastic anemia. Nat. Genet. 2009;41:651–653. doi: 10.1038/ng.359. [DOI] [PubMed] [Google Scholar]
- 45.Guernsey D.L., Jiang H., Evans S.C., Ferguson M., Matsuoka M., Nightingale M., Rideout A.L., Provost S., Bedard K., Orr A. Mutation in pyrroline-5-carboxylate reductase 1 gene in families with cutis laxa type 2. Am. J. Hum. Genet. 2009;85:120–129. doi: 10.1016/j.ajhg.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guernsey D.L., Dubé M.P., Jiang H., Asselin G., Blowers S., Evans S., Ferguson M., Macgillivray C., Matsuoka M., Nightingale M. Novel mutations in the sacsin gene in ataxia patients from Maritime Canada. J. Neurol. Sci. 2010;288:79–87. doi: 10.1016/j.jns.2009.09.034. [DOI] [PubMed] [Google Scholar]
- 47.Goshima G., Mayer M., Zhang N., Stuurman N., Vale R.D. Augmin: a protein complex required for centrosome-independent microtubule generation within the spindle. J. Cell Biol. 2008;181:421–429. doi: 10.1083/jcb.200711053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lawo S., Bashkurov M., Mullin M., Ferreria M.G., Kittler R., Habermann B., Tagliaferro A., Poser I., Hutchins J.R., Hegemann B. HAUS, the 8-subunit human Augmin complex, regulates centrosome and spindle integrity. Curr. Biol. 2009;19:816–826. doi: 10.1016/j.cub.2009.04.033. [DOI] [PubMed] [Google Scholar]
- 49.Uehara R., Nozawa R.S., Tomioka A., Petry S., Vale R.D., Obuse C., Goshima G. The augmin complex plays a critical role in spindle microtubule generation for mitotic progression and cytokinesis in human cells. Proc. Natl. Acad. Sci. USA. 2009;106:6998–7003. doi: 10.1073/pnas.0901587106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nigg E.A., Raff J.W. Centrioles, centrosomes, and cilia in health and disease. Cell. 2009;139:663–678. doi: 10.1016/j.cell.2009.10.036. [DOI] [PubMed] [Google Scholar]
- 51.Musio A., Selicorni A., Focarelli M.L., Gervasini C., Milani D., Russo S., Vezzoni P., Larizza L. X-linked Cornelia de Lange syndrome owing to SMC1L1 mutations. Nat. Genet. 2006;38:528–530. doi: 10.1038/ng1779. [DOI] [PubMed] [Google Scholar]
- 52.Rauch A., Thiel C.T., Schindler D., Wick U., Crow Y.J., Ekici A.B., van Essen A.J., Goecke T.O., Al-Gazali L., Chrzanowska K.H. Mutations in the pericentrin (PCNT) gene cause primordial dwarfism. Science. 2008;319:816–819. doi: 10.1126/science.1151174. [DOI] [PubMed] [Google Scholar]
- 53.Fish J.L., Kosodo Y., Enard W., Pääbo S., Huttner W.B. Aspm specifically maintains symmetric proliferative divisions of neuroepithelial cells. Proc. Natl. Acad. Sci. USA. 2006;103:10438–10443. doi: 10.1073/pnas.0604066103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ropers H.H. New perspectives for the elucidation of genetic disorders. Am. J. Hum. Genet. 2007;81:199–207. doi: 10.1086/520679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Raymond F.L., Whibley A., Stratton M.R., Gecz J. Lessons learnt from large-scale exon re-sequencing of the X chromosome. Hum. Mol. Genet. 2009;18(R1, R1):R60–R64. doi: 10.1093/hmg/ddp071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Graham J.M., Jr., Hennekam R., Dobyns W.B., Roeder E., Busch D. MICRO syndrome: an entity distinct from COFS syndrome. Am. J. Med. Genet. A. 2004;128A:235–245. doi: 10.1002/ajmg.a.30060. [DOI] [PubMed] [Google Scholar]
- 57.Faivre L., Le Merrer M., Lyonnet S., Plauchu H., Dagoneau N., Campos-Xavier A.B., Attia-Sobol J., Verloes A., Munnich A., Cormier-Daire V. Clinical and genetic heterogeneity of Seckel syndrome. Am. J. Med. Genet. 2002;112:379–383. doi: 10.1002/ajmg.10677. [DOI] [PubMed] [Google Scholar]
- 58.Hall J.G., Flora C., Scott C.I., Jr., Pauli R.M., Tanaka K.I. Majewski osteodysplastic primordial dwarfism type II (MOPD II): natural history and clinical findings. Am. J. Med. Genet. A. 2004;130A:55–72. doi: 10.1002/ajmg.a.30203. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.