

Abstract
The 2’-deoxynucleoside 5’-triphosphate (dNTP) analogs with modifications in the triphosphate chain have been used as nucleotide probes in various biochemical and structural studies. Here we report synthesis and characterization of a complete set of α,β-methylene-2’-dNTPs (α,β-m-dNTP; N = A, C, T, G, 12-15), in which the α,β-oxygen linkage of natural dNTP was replaced isosterically by a methylene group. These nucleotides were designed to be non-cleavable DNA polymerase substrates. Our synthesis process entails preparation of 2’-deoxynucleoside 5’-diphosphate precursors by nucleophilic coupling of 5’-tosyl nucleosides and methylene-diphosphate, and a subsequent enzymatic γ-phosphorylation. All four synthesized α,β-m-dNTPs were found to be potent inhibitors of polymerase β with Ki values ranging from 1-5 μM. During preparation of the dG and dT derivatives of α,β-methylene diphosphate, we isolated significant amounts of 3,5’-anhydro-2’-deoxyguanosine (cyclo-dG, 16) and 2,5’-anhydro-2’-deoxythymidine (cyclo-dT, 17), respectively. These novel 2’-deoxycyclonucleosides were formed via a base-catalyzed deprotonation of the imino proton (N1-H and N3-H), followed by an intramolecular cyclization (N3 → C5’ and O2 → C5’, respectively). In acidic solution, the cyclonucleosides underwent glycolysis at N9, followed by complete depurination at N3. In the case of cyclo-dG, there existed an equilibrium between glycolysis and deglycolysis at the glycosidic linkage prior to complete depurination. When exposed to alkaline conditions, cyclo-dG underwent an oxidative deamination at C2 to produce 3,5’-anhydro-2’-deoxyxanthosine (cyclo-dX, 19), whereas cyclo-dT was hydrolyzed exclusively to dT via cleavage at the 2,5’-ether linkage.
INTRODUCTION
Deoxy- and ribo-nucleoside 5’-triphosphates (dNTPs and rNTPs) are essential components in maintaining the integrity of living cells. Their roles as substrates for DNA or RNA polymerases as well as for many NTP binding proteins implicate them in a wide range of potential mechanistic and therapeutic applications.1,2 The NTP analogs play important roles in various cellular processes and act as the primary energy sources for the cell.
NTP derivatives with modifications in the 5’-triphosphate fragment have been used in studies examining the structures and mechanisms of NTP binding proteins and enzymes including polymerases.1,3 In particular, isosteric replacement of the α,β- or β,γ-oxygen linkage in the triphosphate chain with a methylene or an imido group produces an interesting set of NTPs with increased stability in the presence of phosphatases.3 Such modifications also yield non-hydrolyzable and stable dNTP analogs that are valuable tools for biochemical and structural studies of reactions in which hydrolysis of the P-O-P linkage and/or transfer of the phosphate group occur. For example, a series of β,γ-methylene and halogenated analogs of dGTP have recently been used to examine leaving group effects on polymerase β catalysis and fidelity.4,5 Batra et al.6,7 compared the polymerase β ternary structures with natural dNTP and non-hydrolyzable α,β-methylene-dNTP as incoming substrates and found negligible difference in active site geometry, permitting facile catalytic metal binding.
As part of our ongoing research program on sequence effects on arylamine-induced conformational heterogeneity,8 we sought the isolation of ternary polymerase intermediates involving a lesion-containing template-primer and dNTP. Strategies to trap such catalytic complex entities include utilizing a 3’-dideoxy-terminated primer (i.e., lacking the attaching O3’)9 or a non-hydrolyzable dNTP analog,3 in which the α,β-bridging oxygen has been replaced with non-cleavable isosteres (-CH2- or -NH-), in order to render the nucleotides inert toward nucleotidyl transfer. We chose to employ the latter, α,β-methylene-dNTP analogs, as they are more synthetically viable.
Here we describe the preparation of a complete set of α,β-methylene-dNTP analogs (N = A, C, T, G)(12-15, Figure 1a) and a characterization of their polymerase β binding properties. Synthetic preparation entails nucleophilic coupling of methylene-diphosphate with 5’-tosyl nucleosides and a subsequent enzymatic γ-phosphorylation. All four α,β-methylene-dNTPs (12-15) have been found to bind polymerase β as efficiently as the natural dNTPs with Ki’s in the ~1-5 μM range. We isolated significant amounts of 3,5’-anhydro-2’-deoxyguanosine (16, cyclo-dG) and 2,5’-anhydro-2’-deoxythymidine (17, cyclo-dT)(Figure 1b) during preparation of their respective diphosphate precursors. Here we present a thorough structure characterization of these novel 2’-deoxycylconucleosides and their aqueous stabilities.
Figure 1.

(a) Synthesis of α,β-methylene-2’-deoxynucleoside 5’-triphosphates (α,β-m-dNTP, N= A, C, T, G)(12 – 15). (b) Formation of novel 2’-deoxycyclonucleosides (3,5’-cyclo-dG, 16 and 2,5’-cyclo-dT, 17)
RESULTS AND DISCUSSION
Preparation of α,β-methylene-2’-deoxynucleoside 5’-triphosphates (α,β-m-dNTPs; 12-15)
As summarized in Figure 1a, our synthesis protocol entails two major steps: a nucleophilic coupling of 5’-O-tosyl-2’-deoxynucleoside with methylene-diphosphate to generate the 2’-deoxynucleoside diphosphate (dNDP) precursors and a subsequent enzymatic γ-phosphorylation. The α,β-methylene-diphosphate derivatives (α,β-m-dNDP; 8-11) were prepared as literature procedures.10-12 Briefly, individual 5’-O-tosylnucleoside was treated with 2 equivalents of tris(tetra-n-butylammonium) hydrogen methylene pyrophosphate in dry acetonitrile. The products were exchanged for tetra n-butyl ammonium cations to ammonium cations and subjected to fast performance liquid chromatography (FPLC) with a cellulose column; elution from the column was monitored by UV absorption using a diode-array detector. The 1H NMR spectra are characteristic of coupling diphosphate products, such as α,β-methylene protons at around 2.0 ppm with a coupling constant of JP-H = 19.8 Hz. 31P NMR revealed two distinct signals at around ~16 and ~20 ppm, which correspond to Pα and Pβ, respectively. As expected, an isosteric change from P-O-P to P-CH2-P resulted in large phosphorous deshieldings compared to natural dADP (-10 ppm to -15 ppm).1,12
Enzymatic γ–phosphorylation has been shown to be generally more efficient than the chemical approach for preparation of α,β-methylene-dNTPs (12-15). Although pyruvate kinase can catalyze the phosphorylation of both purine and pyrimidine α,β-imido-diphosphate, 3c as well as of azole carboxamide ribonucleoside diphosphates13 the α,β-methylene dNDP analogs examined here are poor substrates for pyruvate kinase (8-11). Therefore, we employed the substrate non-specific nucleoside diphosphate kinase (NDPK).13,14 As shown in Figure 1a, the reaction entails the incorporation of an ATP regeneration system to complete an efficient transformation and produce 2 equivalents of phosphoenol pyruvate (PEP) to completely transform ADP to ATP. Figure 2 shows such a conversion using α,β-m-dCDP (9) as a starting material. A complete conversion to α,β-m-dCTP (13) was achieved within 4 h as determined by a gradient ion pair HPLC/UV using program A. Similar conversions were achieved for the remaining cases. The α,β-methylene-dNTP products generally exhibited a longer retention time than the starting 2’-dNDP derivatives. Isolation of the desired α,β-methylene-dNTP products was achieved by a three-step process, which involved (i) addition of 1 equivalent of ADP to convert extra PEP into pyruvate, (ii) boronate affinity gel chromatography to remove the remaining cis-diol containing ATP from the mixture, and (iii) final purification by anion exchange with a Q Sepharose FF anion exchange column.
Figure 2.

HPLC chromatograms of a mixture containing α,β-m-dCDP (9) and ATP (a) before and (b) after enzymatic γ-phosphorylation (PK, PEP, ATP)(see Figure 1a). A gradient HPLC solvent program A was used.
31P NMR spectra of the final α,β-methylene dNTP products clearly showed three well separated signals at around 23, 12 and -5 ppm for Pα, Pβ and Pγ, respectively (Fig. S5-S8). In all cases, the isosteric methylene substitution at the α,β-oxygen linkage resulted in the expected large deshielding (~31-32 ppm) of Pα and Pβ and small deshielding for Pγ (~1-3 ppm).1,11 These data indicate that the structures of the final products were consistent with that predicted by the spectroscopic data (1H, 31P, HRMS)(Fig. S5-S12).
Polymerase Assays
Mammalian DNA polymerase β fills short gaps in DNA during genome maintenance. It has served as a model enzyme for the study of nucleotidyl transfer and substrate specificity.15 Strategies to capture catalytic intermediates for structure determination have utilized a 3’-dideoxy-terminated primer (without the attaching O3’)9 or a non-hydrolyzable dNTP analog,3 in which the oxygen bridge between the α– and β–phosphates has been replaced with an imido7 or methylene group6, rendering the nucleotides inert to nucleotidyl transfer. A comparison of the resulting ternary substrates complex structures indicated that structures with the non-hydrolyzable incoming nucleotides resulted in less active site distortion and permit catalytic metal binding.7 As expected, these dNTP analogs are excellent inhibitors of polymerase β-dependent DNA synthesis.6,7 Likewise, our α,β-methylene-dNTPs (12-15, Figure 1) inhibited DNA synthesis and bound tightly to polymerase β (Ki: α,β-m-dATP, 12, 5.0 μM; α,β-m-dCTP, 13, 0.9 μM; α,β-m-dGTP, 15, 1.0 μM; α,β-m-dTTP, 14, 0.6 μM). Figure 3 shows inhibition of dCTP incorporation by α,β-m-dCTP (13) as an example of the gap-filling assay. These binding constants are similar to natural dNTPs (2-10 μM) and those reported for commercially available α,β-imido-dUTP7 and α,β-m-dATP.6
Figure 3.
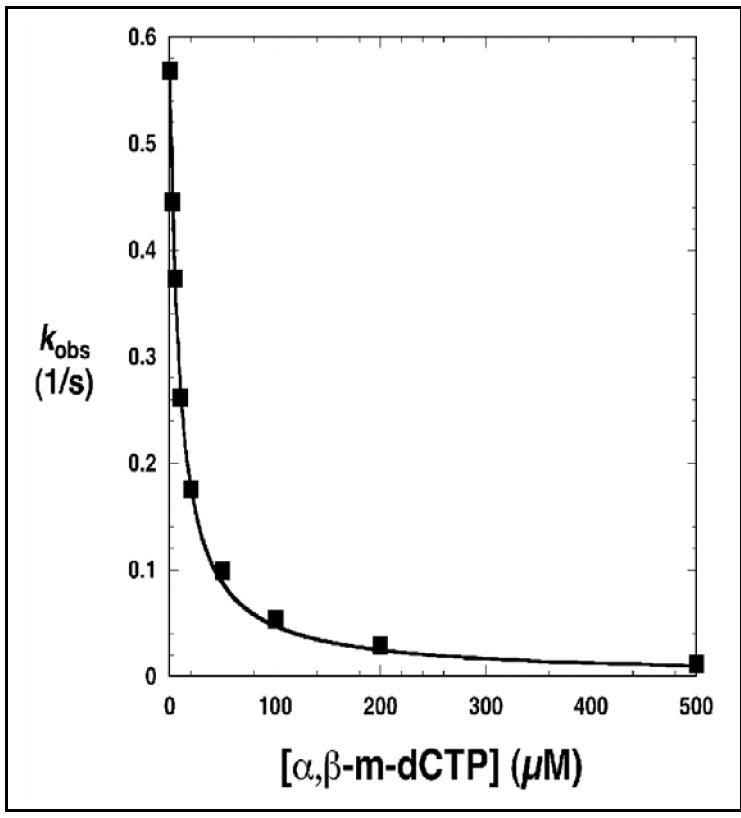
Inhibition of dCTP incorporation by α,β-m-dCTP (13). DNA synthesis was assayed on a single-nucleotide gapped DNA substrate where the templating nucleotide in the gap was dG. In this assay, the concentration of dCTP was 20 μM. Dixon analysis for competitive inhibition indicates that the Ki for 13 is 0.9 μM (solid line).
Isolation of novel 2’-deoxycyclonucleosides
The nucleophilic coupling reactions involving the 5’-O-tosyl derivatives of dA (1) and dC (2) yielded α,β-m-dADP (8) and α,β-m-dCDP (9), respectively, as exclusive products. By contrast, similar reactions with tosyl-dG (4) and -dT (3) produced two major fractions in FPLC chromatograms (not shown), with a late eluting peak corresponding to the desired diphosphates 11 and 10, respectively. The early eluting fractions yielded white fluffy solids, which showed no signals in the 31P NMR and no α,β-methylene signals in the 1H NMR spectra. As elaborated below, two non-polar compounds were identified as novel 2’-deoxycyclonucleosides: 3,5’-anhydro-dG (cyclo-dG, 16) and 2, 5’-anhydro-dT (cyclo-dT, 17). The proposed structures were confirmed by comparing the spectroscopic (UV, NMR, MS) data of these compounds to those of cyclic nucleoside standards prepared by another method (see below). As shown in Figure 1b, these results clearly implicated an initial base-catalyzed deprotonation of the imino proton (N1 and N3 respectively for 11 and 10), followed by an intramolecular cyclization.
NMR
Figure 4 shows 1H NMR spectra of cyclo-dG and dG in DMSO-d6. Assignment of the 1H signals was aided by analyses of COSY, long-range-COSY, and TOCSY spectra (S13,15,16). Cyclo-dG showed all of the characteristics of dG except for the imino proton at N1 (10.63 ppm) and 5’-OH (4.95 ppm). Another notable difference was significant deshielding of H5’,5”with a large difference in chemical shifts (4.45 and 3.89 ppm) relative to dG (3.5 ppm).17 By contrast, the H2’,2” resonances were merged at 2.16 ppm. All of the remaining sugar protons were shifted downfield (~0.4-0.6 ppm). Interestingly, however, H8 was shielded by about 0.15 ppm. A similar comparison of cyclo-dT and dT in D2O revealed that all protons, except H1’, were shifted downfield (Figure 5, also S14,17). As with cyclo-dG, a significant deshielding with signal split was observed for H5’,5” (4.65 and 4.26 ppm) relative to dT (3.75 ppm). In both cases, H4’ showed the greatest extent of deshielding (~0.8 ppm), implicating dramatic change in the electronic environment of H4’ for the cyclonucleosides.
Figure 4.
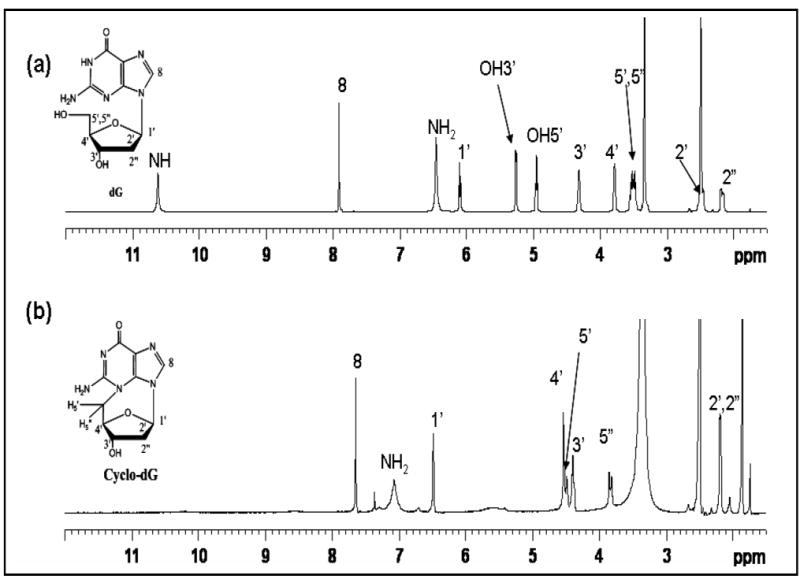
Comparison of 1H NMR spectra of (a) dG and (b) cyclo-dG (16) recorded in DMSO-d6.
Figure 5.
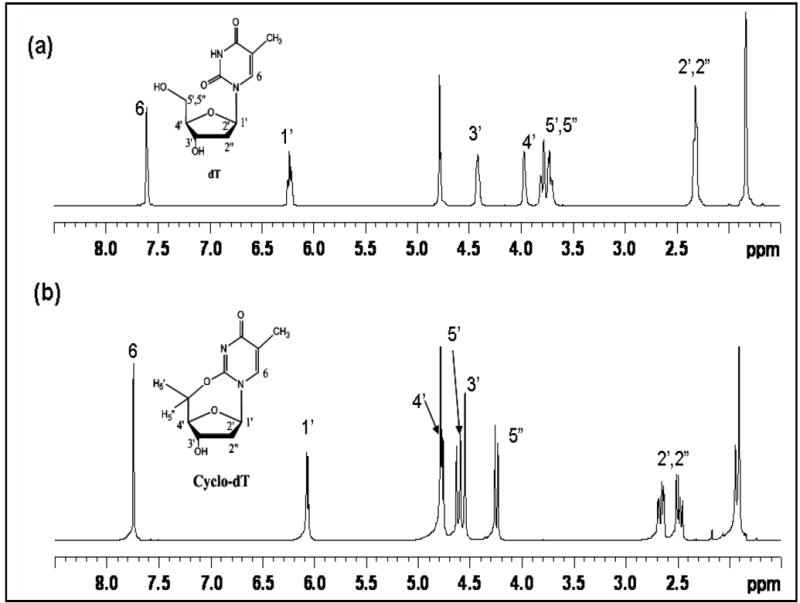
Comparison of 1H NMR spectra of (a) dT and (b) cyclo-dT (17) recorded in D2O.
Both of the cyclonucleosides isolated are rigid cage-like molecules. Energy minimized model structures (Figure 6) clearly indicated that they exist in the well-defined syn glycosidyl conformation, 40.8° (χ ∠O4’- C1’- N9-C8) and 61.6° (χ ∠O4’- C1’- N1-C6), respectively for cyclo-dG and cyclo-dT. This is contrasted with their noncyclic counterparts dG and dT, which are known to exist in flexible, but mostly anti conformation (χ =170-300°).18 The rigidity of cyclo-dG and cyclo-dT allows the H1’ and H8 protons to be in a close spatial proximity, 2.5 and 2.2 Å, respectively (Figure 6). This is evidenced by observations of strong NOEs in the NOESY spectrum of cyclo-dG (Figure 7). Such NOEs are absent in the anti-glycosidic dG and dT (not shown).
Figure 6.
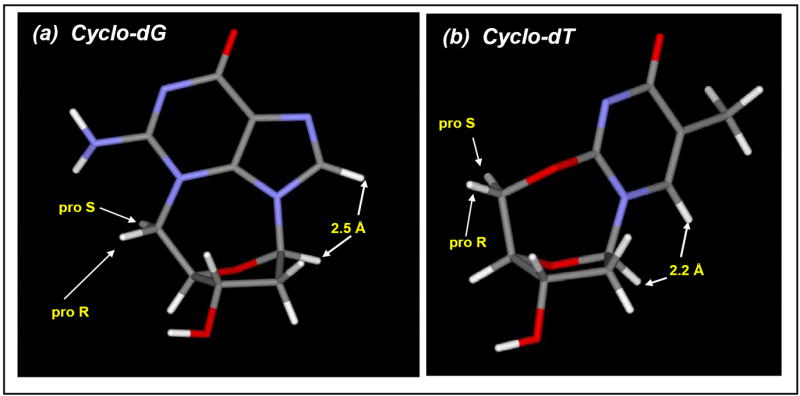
Energy-minimized models (Spartan) of (a) cyclo-dG (16) and (b) cyclo-dT (17).
Figure 7.
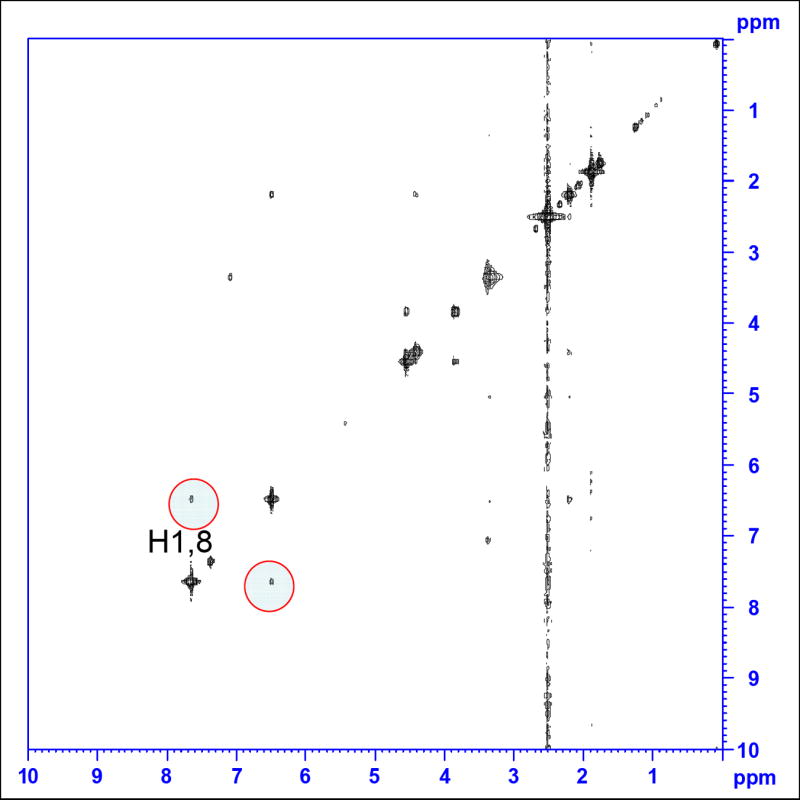
NOESY spectrum of cyclo-dG (16). The cross peaks observed for the H1’ and H8 protons (highlighted in red circles) indicate their spatial proximity (i.e., syn-glycosidyl conformation).
It is noteworthy that cyclo-dG and cyclo-dT adopted rare C1’-endo sugar puckering (Figure 6).19,20 This structural feature distinguishes these cyclonucleosides from dG and dT, which are known to exist primarily with C2’-endo puckering. This difference in sugar conformation is clearly indicated in dramatic changes in chemical shifts (H4’) and the unique coupling patterns of sugar protons, particularly with respect to H1’ and H2’,2” (Figures 4 and 5). Finally, the rigidity of the cage-like cyclonucleoside structures places the H5’ (pro-S) and H5” (pro-R) protons in a well-defined electronic environment (Figure 6), presumably explaining the large differences in chemical shifts and coupling patterns mentioned above. The pro-S H5’ signal was assigned tentatively downfield to the pro-R H5” signal because it is closer to O4’ than the pro-R H5”. The large deshielding (~15 ppm) observed for the C5’ carbon relative to dT21 in the 13C NMR spectrum (Fig. S18) can be attributed to the presence of a 2,5’-anhydo ether linkage in cyclo-dT.
UV
The UV maxima for cyclo-dG and cyclo-dT were shifted significantly toward lower wavelengths relative to the diphosphate products 15 and 14 (Fig. S19). A distinct cyclo-dG band at 215 nm most likely reflects the major change in the electronic configuration of the guanine base. The UV spectra of cyclo-dG were not affected by exposure to neutral and basic pHs, but were altered by exposure to highly acidic pHs below 3.4 (Fig. S23). However, the UV spectra of dG remained steady across acidic and neutral pH range, but were altered by exposure to alkaline pHs above 11.9 (Fig. S22). These results are consistent with the supposition that the N-1 imino proton appears to be absent in cyclo-dG. The UV spectra of cyclo-dT changed dramatically with shifting pH through the range of 1.7 – 11.9 range with a well-defined isosbestic point at 263 nm, possibly implicating multiple hydrolytic species (see below)(Fig. S26).
CD
The circular dichroism (CD) characteristics of cyclo-dG and cyclo-dT differed qualitatively from dG and dT, respectively (Fig. S20,21,24). The syn-conformeric cyclo-dG exhibited well-defined large negative ellipticities at 214 nm and 248 nm (Fig. S24). This behavior precisely complemented that of anti-conformeric dG, which showed large positive and small negative ellipticities at 212 and 248 nm, respectively (Fig. S20).22 The CD spectra of cyclo-dG remained unchanged from pH 5.1 to 11.9, but underwent a dramatic change when exposed to pHs below 3.4 (Fig. S24). This sensitivity to acidity suggests that the protonated base may change conformation. The CD spectra observed for cyclo-dT were similar to that of dT (Fig. S21), but exhibited a strong negative ellipticity at 228 nm.
Stability of cyclo-dG (16) and cyclo-dT (17)
Cyclo-dG in H2O exhibited virtually no change in CD after 6 h at 90 °C, whereas cyclo-dT under the same conditions showed dramatic reduction in absorption (Fig. S20,21). These results demonstrate the lesser stability of cyclo-dT relative to cyclo-dG in aqueous solution, which motivated us to conduct a systematic hydrolysis study. To this end, samples of each cyclonucleoside were placed in an acidic (1.0 M HCl) or basic (1.0 M NaOH) solution. Figures 8 and 9 illustrate the hydrolysis schemes of cyclo-dG and cyclo-dT, respectively, at specified conditions and their HPLC profiles as a function of time.
Figure 8.
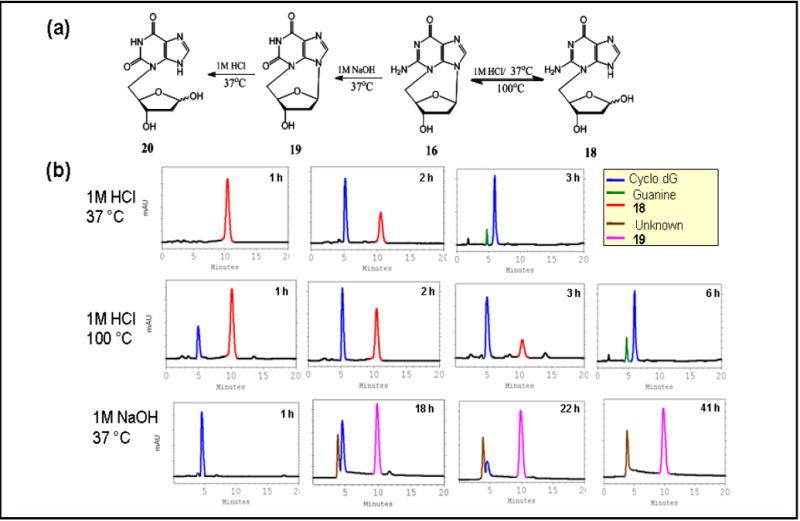
(a) Hydrolysis schemes of cyclo-dG (16) and (b) the corresponding HPLC profiles in 1 M HCl, 37 °C (b) 1 M HCl, 100 °C, and 1 M NaOH, 37 °C using gradient program B.
Figure 9.
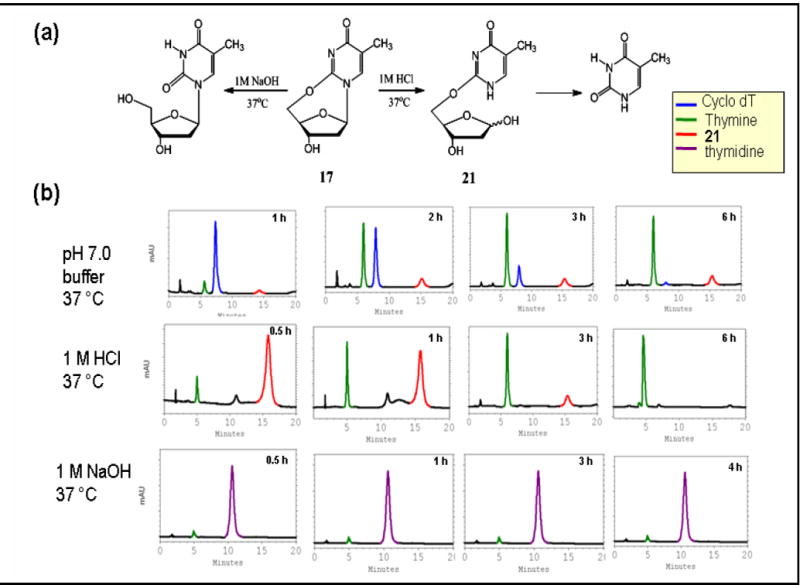
(a) Hydrolysis schemes of cyclo-dT (17) and (b) the corresponding HPLC profiles in pH 7.0 buffer, 37 °C (b) 1 M HCl, 37 °C, and 1 M NaOH, 37 °C using gradient program B.
Acid hydrolysis of cyclo-dG
After 1 h, a refluxing acid solution of cyclo-dG produced the hydrolyzed product 18 (RT = 10.3 min, Figure 8b), which arose from the cleavage of the glycosidic linkage at N9. Additional heating resulted in reduction of 18 with a concomitant increase of the starting cyclo-dG. It was apparent that the glycolytic 18 was formed first and subsequently converted back to cyclo-dG. The latter deglycolysis process was complete by 6 h and was accompanied by a small amount of guanine at 4.9 min. Continued heating (48 h) resulted in complete depurination (not shown), involving hydrolysis at both positions 9 and 3. Interestingly, acid hydrolysis at 37 °C yielded an exclusive formation of 18 in less than 1h (Figure 8a). Additional incubation at 37 °C for 3 h resulted in a complete deglycosylation (Figure 8b). This is in contrast to experiments with the ribose analog of cyclo-dG, which produced a glycolytic product with a half life of >122 min under similar conditions. Note the similarity of the HPLC profiles between the 3-h reaction at 37 °C and 6-h reaction at 100 °C (Figure 8b). The equilibrium between glycolysis and deglycolysis was clearly better controlled at 37 °C.
The UV spectra of 18 (Fig. S28) do not differ substantially from those of cyclo-dG (Fig. S28). Like cyclo-dG, the UV and CD spectra were particularly sensitive to extreme pHs (Fig. S27). These results collectively indicate that the overall electronic structures of cyclo-dG remain intact despite cleavage of the glycosidic bond. ESI-MS of 18 showed (M+H)+, at 268.1, which is 18 units (H2O) greater than that of cyclo-dG. The product ion spectrum of m/z 268.1 ion exhibited fragment ions at 250 and 232.1, corresponding to successive loss of one and two H2O molecules from the sugar. As expected, the m/z 152.2 ion corresponds to the protonated guanine formed by a cleavage of the 3,5’-cyclic linkage. The characteristic patterns of the H1’ and H5’,5” protons that existed in the 1H NMR of cyclo-dG disappeared. Also, the appearance of the N9-H and H8 protons as a doublet at 7.27 and 7.85 ppm, respectively, indicate the prototropic tautomerism exhibited by N(7)H and N(9)H in 18 (Fig. S29,30). The C1’ signal was shifted downfield (-9.4 ppm) relative to that of cyclo-dG (89.1 ppm) (Fig. S31). These results are indicative of a broken glycosidic linkage and a hydroxyl at C1’.
Base hydrolysis of cyclo dG
Cyclo-dG was slow to react in 1.0 M NaOH, but it transformed completely to a 4:1 mixture of products after 41 h at 37 °C (Figure 8b). The major peak at 10.0 min was identified as 3,5’-anhydro-2’-deoxyxanthosine (cyclo-dX, 19), which arose via an oxidative deamination at C2 position (see below). Holmes and Robins23 reported a similar deamination reaction on 2’,3’-O-isoprolylidene-3,5’-anhydroguanosine under similar conditions.
ESI-MS of the major product showed an (M+H)+ at 251.0, which is one more unit than the starting cyclo-dG. The product ion spectra show a fragment ion at 233.0 due to loss of a water molecule involving the 3’-OH and a hydrogen on the adjacent C2’ (Fig. S38). The UV and CD spectra of 19 are quite different from those of cyclo-dG, suggesting that there is a change in the chromophore purine base (Fig. S34,35). The 1H NMR profile of 19 (Fig. S34,35) was essentially similar to that of the starting cyclo-dG 16, except for appearance of a new imide proton at 11.1 ppm. All 1H NMR signals were assigned and confirmed by COSY/TOCSY spectra. A strong cross peak between H8 and H1’ in the NOESY spectrum (Fig. S37) confirmed the inheritance of the well-defined syn-glycosidic conformation from cyclo-dG. The 13C NMR of 19 (Fig. S36) exhibited two distinctive carbonyl signals at 157.9 and 151.7 ppm, which confirms an additional new keto functionality at C2. The dramatic UV changes that were observed in both acidic and basic pHs are consistent with the presence of an imino proton at N1 (Fig. S33).
Treatment of 19 with 1.0 M HCl (HPLC data not shown) resulted in a single fraction whose ESI-MS and 1H NMR data are consistent with the glycolytic product 20. ESI-MS showed an (M+H)+ at m/z 269.0, which represents the addition of a H2O molecule. The base fragment at m/z 153.2 corresponds to the protonated xanthine base formed by cleavage of the 3,5’-cyclic linkage of a protonated 19. Both the mass and NMR spectra are similar to those observed for the glycolytic product 18 isolated from cyclo-dG 16.
Acid hydrolysis of cyclo-dT
Acid treatment of cyclo-dT for 30 min at 37 °C produced a single product 21 at 15.5 min (Figure 9) with a small amount of thymine at 4.8 min. The depurination reaction was complete in 6 h. A similar hydrolysis profile was obtained in a neutral buffer solution. Interestingly, however, 21 never exceeded that of cyclo-dT (assuming they have similar molar absorptivity coefficients) throughout the reaction. It appears that an initial glycolysis was followed immediately by the cleavage of the 2,5’-anhydro-linkage. Unlike in the case of cyclo-dG, no recyclization (deglycolysis) of cyclo-dT was observed. These results indicate a greater fragility of the 2,5’-O-linkage in acidic conditions. Attempts to isolate the intermediate 21 in its pure form in order to purse a detailed characterization were not successful. Nonetheless, the ESI-MS spectra show an (M+H)+ at 243.1, which is 18 units greater than starting (pure) cyclo-dT, indicating that there is an addition of one H2O to the parent compound. The product ion spectrum of the m/z 243.1 ion exhibited fragment ions at 225.0, and 207.0 for losses of one and two H2O molecules from the sugar. The base fragment at 127.1 corresponds to the protonated thymine base. The mass spectrum of 21 is similar to that obtained for 18, thus 21 was assigned as a glycolysis product, as suggested by their exact masses. The possibility of an initial cleavage of the 2,5’-O-anhydrous linkage, followed by a glycolysis, was ruled out because no dT was detected in the reaction mixture.
Base Hydrolysis of Cyclo-dT
The base hydrolysis of cyclo-dT at 37 °C occurred within 30 min to give rise to dT exclusively (Figure 9). This reaction represents an exclusive cleavage of the 2,5′-anhydrous linkage and no additional decomposition occurred in 4 h. This was in clear contrast to cyclo-dG, which underwent an oxidative deamination to 19. The greater susceptibility to base hydrolysis of cyclo-dT versus cyclo-dG can be explained by the nature of the 5’-bridge linkage. That is, the 5′,2-O linkage is more vulnerable to basic hydrolysis than is the 5’,3-N-linkage.
Previous studies on the corresponding ribonucleoside hydrolysis showed that the rate of glycosidic hydrolysis in 0.1 N HCl at 25 °C followed the order 3,5’- ≪ 5’,8-cyclonucleoside < non-cyclic nucleoside.23-26 The present findings indicate that the loss of ring strain during hydrolysis of cyclic nucleosides does not increase the rate of hydrolysis contrary to what might have been expected.24,25 Reese et al.26 reported that exclusive glycosidic hydrolysis of 3,5’-cyclo-guanosine occurred after 27 h at 37 °C in 0.9 N HCl. That result was based on a loss of starting material and no product characterization was reported. In the present study, we found that hydrolysis at the glycosidic bond was complete in 1h at 37 °C in 1.0 M HCl. Thus it appears that the rate of hydrolysis is much greater for 2’-deoxycyclonucleosides.
In summary, we described the preparation of a complete set of α,β-methylene dNTPs (12-15, N = A, C, T, G, Figure 1) and their substrate binding affinities for polymerase β. The synthesis entails a nucleophilic coupling reaction between a 5’-O-tosyl-2’-deoxynucleoside and methylene diphosphate, followed by an enzymatic γ-phosphorylation. In the cases of dG and dT, significant amounts of cyclo-dG (16) and cyclo-dT (17) were also isolated. Under acidic conditions, these novel 2’-deoxycyclonucleosides undergo a glycolysis reaction, followed by complete depurination. In the case of cyclo-dG, there existed an equilibrium between glycolysis and deglycolysis prior to a complete depurination. Under basic conditions, cyclo-dG underwent an oxidative deamination at C2 to give cyclo-dX (19), while cyclo-dT was transformed to dT via an exclusive cleavage of the ether linkage between positions 2 and 5’. Gap-filling DNA synthesis assays showed that all four α,β-m-dNTPs (12-15) behave as non-cleavable substrates of polymerase β with binding affinities (Ki 1-5 μM) similar to natural dNTPs.16
EXPERIMENTAL SECTION
All solvents, reagents, and standards were purchased from Aldrich (Milwaukee, WI) and Fisher Scientific (Pittsburgh, PA) and used without further purification. Purification of cyclonucleosides and their hydrolysis products were conducted on silica gel (200-400 mesh) column chromatography (Scientific Adsorbents Inc., Atlanta, GA). Flash cellulose chromatography was performed on Whatman CF-11 fibrous cellulose. DOWEX AG-50W column (H+ form) was purchased from Fisher Scientific. Affi-Gel® Boronate affinity gel, Q sepharose FF anion exchange resin and associated columns were purchased from Bio-RAD laboratories, Inc. Nucleoside diphosphate kinase (NDPK) and pyruvate kinase were purchased from Sigma-Aldrich.
All NMR (1H, 13C, and 31P) spectra were recorded using an inverse 5-mm inverse probe on a Bruker DPX400 Avance spectrometer operating at 400.1, 100.6, and 161.9 MHz, respectively. 1H and 13C spectra are referenced to TMS. 31P spectra are referenced using an external 85% aqueous phosphoric acid solution. The energy-minimized structures of cyclo-dG and cyclo-dT were obtained using Spartan modeling software (Essential Edition. Version 3.0.2).
HPLC analyses were carried out on Hitachi EZChrom Elite HPLC unit with a L2450 diode array detector and a Phenomenex Luna C18 column (150 cm × 4.5 mm, 5 μm) with flow rate of 1 mL/min. The following two solvent programs were used: (A) a step gradient involving buffer I (10 mM tetrabutylammonium hydroxide, 10 mM KH2PO4, 0.25% methanol, pH 7.00) and buffer II (2.8 mM tetrabutyl ammonium hydroxide, 100 mM KH2PO4, 30% methanol, pH 5.50); 0-15 min 100% I, 15-20 min 0 to 10% II, 20-25 min 10 to 30% II; 25-40 min 30 to 37% II, 40-55 min 37 to 45% II, 55-70 min 45 to 55% II, 70-80 min 55 to 75% II, 80-90 min 70 to 100% II; (B) a 30-min isocratic program involving 2% of solvent I (an equal mixture of acetonitrile and 0.10 M ammonium acetate) and 98% of solvent II (0.10 M ammonium acetate).
CD experiments were performed on a Jasco J-810 spectropolarimeter equipped with a variable temperature controller. In a typical run, samples (~2 ODS) were dissolved in 400 μL of water or a buffer containing 0.2 M NaCl, 10 mM sodium phosphate, pH = 6.9. Spectra were recorded at 15 °C in a 1 mm path length cell. The spectropolarimeter was scanned from 200 to 400 nm at a rate of 50 nm/min. Data points were acquired every 0.2 nm with a 2-s response time. Spectra were the averages of 10 accumulations and smoothed using 17 point adaptive smoothing algorithms provided by Jasco. UV absorption spectra were obtained on a Beckman DU 800 UV/VIS spectrophotometer using a 1 cm path length cell. Samples (~0.25 ODS) were prepared in 350 μL of a pH 7.0 buffer solution containing 0.2 M NaCl, and 10 mM sodium phosphate.
ESI-MS and MS/MS spectra were acquired with a ThermoFinnigan Advantage (San Jose, CA) linear ion trap (LTQ) interfaced to a Fourier transform mass spectrometer (FTMS) at the Washington University Mass Spectrometry Research Resource in St. Louis, MO. Molecular weight and product ion spectra of all the protonated molecule ions were acquired by direct infusion of low picomolar amounts of sample into the LTQ. The relative collision energy for MS/MS experiments was set to 35% of its maximum value for fragmentation of all the molecule ions in this study. A 70:30 mixture of methanol and water that was 0.1% in formic acid was used as the mobile phase. Exact masses of all protonated molecule ions and product ions formed in the LTQ were acquired in the FTMS. The capillary temperature was 230° C and the spray voltage was 4.5 kV. The mass resolution (m/Δm) was 100,000 at m/z 400.
Synthesis of 5’-O-tosylnucleosides
5’-O-Tosylation of dA, dC, and dT was carried out in a similar fashion to the literature procedures10,12,24,27 and is summarized as follows. Briefly, 2’-deoxynucleoside (2.0 mmol) was treated with a 1.2 equivalent of p-TsCl in 15 ml dry pyridine at -5 °C. After 16 h at 0 °C, the reaction was quenched with ice-water and extracted with chloroform. Combined chloroform extracts were washed consecutively with saturated NaHCO3 and water and dried over anhydrous MgSO4. Flash chromatography on silica (methanol/chloroform) yielded pure 5’-O-tosyl derivatives (1-3) in 30~40% yields.
A direct 5’-O-tosylation of dG was problematic due to its low solubility in pyridine. Instead, 5’-tosyl-2’-deoxyguanosine (4) was obtained by the three-step indirect procedure shown in Figure 1.
5’,3’-O-(bis-tert-butyldimethylsilyl)-2’-dG(5)
A mixture of dG (500 mg, 1.86 mmol), TBMSCl (670 mg, 4.46 mmol) and imidazole (316 mg, 4.65 mmol) was stirred in 3 mL of dry DMF at room temperature under nitrogen for 24 h. The solvent was removed under reduced pressure, and water was added to the reaction mixture. The precipitated solid was filtered and dried under vacuum. Recrystallization from 95% ethanol gave 5 as shiny white crystals (800 mg, 87% yield). 1H NMR (DMSO-d6): δ 10.62 (1H, s, CONH), 7.90 (1H, s, H8), 6.49 (2H, s, NH2), 6.11 (1H, t, J = 7.0 Hz, H1’), 4.49 (1H, m, H3’), 3.81 (1H, m, H4’), 3.68 (2H, m, H5’,H5”), 2.25 (2H, m, H2’,H2”), 0.90 (18H, s, 2 t-C4H9), 0.13 (12H, s, 4 CH3)(Fig. S1); LR-MS (ESI) (M+H)+ 496.34.
3’-O-(tert-butyldimethylsilyl)-2’-dG (6)
5 (800 mg, 1.6 mmol) was added to a solution of acetic acid, water and THF (13:7:3, 160 mL). The mixture was stirred at room temperature under nitrogen for 18 h. The solvent was reduced to one-fifth of the original volume under vacuum and lyophilized to dryness. Elution with methylene chloride: methanol (95:5) on silica yielded 380 mg of 6 (63% yield). 1H NMR (DMSO-d6): δ 10.66 (1H, s, CONH), 8.00 (1H, s, H8), 7.27 (1H, t, OH), 6.50 (2H, s, NH2), 6.11 (1H, t, J = 7.0 Hz, H1’), 4.51 (1H, m, H3’), 3.80 (2H, m, H5’,H5”), 2.21 (2H, m, H2’,H2”), 0.92 (s, 9H, t-C4H9), 0.13 (6H, s, 2 CH3)(Fig. S2); LR-MS (ESI) (M+H)+ 382.23.
5’-O-tosyl-3’-O-(tert-butyldimethylsilyl)-2’-dG (7)
A solution of 6 (380 mg, 1.0 mmol) in 18 mL of dry pyridine was cooled to 0 °C in an ice-salt mixture. Then 1.0 mmol of p-TsCl in 10 mL of dry pyridine was added dropwise to the reaction mixture. The mixture was stirred at 0 °C for 24 h. The reaction was quenched with ice water, and the mixture was extracted with chloroform. The organic layer was washed consecutively with saturated NaHCO3 and ice-water, and dried over anhydrous MgSO4. Silica column chromatography (methylenechloride: methanol 96:4) gave 340 mg of 7 (64 %). 1H NMR (DMSO-d6): δ 10.68 (1H, s, CONH), 7.83 (1H, s, H8), 7.75 (2H, d, J = 8.0, tosyl), 7.41 (2H, d, J = 8.0 Hz, tosyl), 6.48 (2H, s, NH2), 6.09 (1H, t, J = 7.0 Hz, H1’), 4.44 (1H, m, H3’), 4.17 (1H, m, H4’), 3.8 (2H, m, H5’,H5”), 2.29 (2H, m, H2’, H2”), 1.28 (3H, s, tosyl-CH3) 0.84 (s, 9H, t-C4H9), 0.05 (6H, s, 2 CH3)(Fig. S3); LR-MS (ESI) (M+H)+ 536.29.
5’-O-Tosyl-2’-dG (4)
A 15 mL of 2% aqueous hydrofluoric acid was added to 15 mL of acetonitrile containing 7 (340 mg, 0.64 mmol). After stirring for 24 h at room temperature, a homogeneous solution was neutralized with excess solid ammonium bicarbonate. The acetonitrile was removed under vacuum and the water was removed by lyophilization, leaving a white solid. The solid was extracted with 9:1 chloroform/methanol and the extract was eluted on silica gel with with dichloromethane:methanol (90:10) and yielded 160 mg of 4 (70%). 1HNMR (DMSO-d6): δ 10.62 (1H, s, CONH), 8.64 (1H, s, OH), 8.03 (1H, s, H8), 7.77 (2H, d, tosyl), 7.32 (2H, d, tosyl), 6.41 (2H, s, NH2), 6.01 (1H, s, H1’), 4.25 (1H, m, H3’), 4.14-3.84 (3H, m, H4’, H5’,H5”), 2.25 (2H, m, H2’,H2”), 1.16 (3H, s, Ar-CH3)(Fig. S4); LR-MS (ESI) (M+H)+ 421.99.
Synthesis of α,β-methylene 2’-deoxynucleoside 5’-diphosphate (α,β-m-dNDP)
α,β-methylene 2’-deoxyadenosine 5’-diphosphate (8)
Freshly prepared tris(tetra-n-butylammonium) hydrogen methylene pyrophosphate (472 mg, 0.53 mmol) was added to Compound 1 (140 mg, 0.35 mmol) in 0.2 mL of acetonitrile. This viscous solution was allowed to stir under N2 at room temperature for 48 h. The solution was diluted with water (10 ml). The tetra-n-butylammonium cation was exchanged for an ammonium cation by passing the solution through a DOWEX AG-50W column (NH4 +) form with 2 column volumes of deionized water. The eluent was lyophilized and the solid was extracted with acetonitrile/100 mM NH4HCO3/NH4OH (7:3:2). The extract was loaded onto CF-11 cellulose column (5 × 23 cm) connected to a FPLC and eluted with the same solution. The elution was monitored using UV detector set at 230 nm. The major UV absorbing fractions were pooled, and the acetonitrile was removed by rotary evaporation. The resulting aqueous solution was lyophilized to give 90 mg of 8 (63%) as a white fluffy solid. HPLC RT 79.4 min (program A); 1H NMR (D2O) δ 8.39 (1H, s, H8), 8.13 (1H, dd, J = 6.6 Hz, H2), 6.37 (1H, t, J= 6.6 Hz, H1’), 4.65 (1H, m, H3’), 4.16 (1H, m, H4’) 3.99 (2H, m, H5’,H5”), 2.72-2.48 (2H, m, H2’,H2”) 2.04 (2H, t, J = 20.3 Hz, Pα–CH2–Pβ); 31P NMR (D2O) δ 19.70 (m, Pα), 16.13 (m, Pβ). LR-MS (ESI) (M+H)+ 410.29.
α,β-methylene 2’-deoxycytidine 5’-diphosphate (9) was prepared in a similar manner from 2 (134 mg, 0.35 mmol) and tris(tetra-n-butylammonium) hydrogen methylene pyrophosphate (472 mg, 0.53 mmol). Compound 9 (74 mg (55%) was obtained as a white fluffy solid. HPLC RT 59.7 min (program A); 1H NMR (D2O) δ 7.85 (1H, d, J = 7.8 Hz, H6), 6.19 (1H, t, J = 6.5 Hz, H1’), 5.98 (1H, d, J = 7.6 Hz, H5), 4.47 (1H, m, H3’), 4.03 (1H, m, H4’), 3.99 (2H, m, H5’,H5”), 2.25-2.35 (2H, m, H2’,H2”), 1.99 (2H, t, J = 19.6 Hz, Pα–CH2–Pβ ); 31P NMR (D2O) δ 19.92 (m, Pα), 16.54 (m, Pβ). LR-MS (ESI) (M+H)+ 386.29.
α,β-methylene 2’-deoxyguanosine 5’-diphosphate (11) and 3, 5’-anhydro-2’-deoxyguanosine (16)
The coupling reaction between 4 (147 mg, 0.35 mmol) and tris(tetra-n-butylammonium) hydrogen methlene pyrophosphate (472 mg, 0.53 mmol) gave two main fractions with an approximately equal intensity after an FPLC. An early eluting major fraction was collected and purified by HPLC to afford 3, 5’-anhydro-2’-deoxyguanosine (cyclo-dG, 16) (14 mg, 16%) as a white fluffy solid. 16: 1H NMR (DMSO-d6) δ 7.64 (1H, s, H8), 7.04 (2H, bs, NH2), 6.50 (1H, dd, J = 5.2 Hz, H1’), 5.53, (1H, bs, 3’-OH), 4.54 (1H, m, H4’), 4.50 (1H, d, J = 13.6 Hz, H5’), 4.40 (1H, m, H3’), 3.84 (1H, d, J = 13.6 Hz, H5”), 2.16 (2H, m, H2’,H2”); 13C NMR (D2O): 89.15 (C1’), 84.39 (C4’), 70.50 (C3’), 53.80 (C5’), 42.97 (C2’). LR-MS (ESI) [M+H]+ 250.13. HRMS (ESI): calcd. 250.0935 (M + H)+. found. 250.0925 (M + H)+. The late eluting fractions were identified as the desired diphosphate product 11 (44 mg, 30%) as a white fluffy solid. 11: HPLC RT 61.8 min (program A); 1H NMR (D2O) δ 8.28 (1H, s, H8), 6.32 (1H, t, J = 6.9 Hz, H1’), 4.72 (1H, m, H3’), 4.25 (1H, m, H4’), 4.10 (2H, m, H5’), 2.80-2.52 (2H, m, H2’,H2’); 2.13 (2H, t, J = 19.8 Hz, Pα–CH2–Pβ); 31P NMR (D2O) δ 19.47 (m, Pα), 16.08 (m, Pβ). LR-MS (ESI) (M+H)+ 425.83.
α,β-methylene 2’-deoxythymidine 5’-diphosphate (10) and 2,5’-anhydro-2’-deoxythymidine (17)
The usual coupling reaction of 3 (100 mg, 0.25 mmol) with tris(tetra-n-butylammonium) hydrogen methlene pyrophosphate (340 mg, 0.38 mmol) gave two main fractions of equal abundance by FPLC. An early eluting major fraction was collected and purified by HPLC to give 2,5’-anhydro-dT 17 (cyclo-dT; 167 mg, 31%). 1H NMR (D2O) δ 7.75 (1H, s, H6), 6.1 (1H, dd, J = 8.0, 1.8 Hz, H1’), 4.82 (1H, m, H4’), 4.62 (1H, d, J = 12.9 Hz, H5’), 4.55 (1H, m, H3’), 4.26 (1H, d, J = 12.9 Hz, H5”), 2.67 (1H, m, H2’), 2.49 (1H, m, H2’’), 1.92 (3H, s, CH3); 13C NMR (DMSO-d6) δ 171.90 (C4), 158.09 (C2), 141.42 (C6), 118.69 (C5), 94.64 (C1’), 86.34 (C4’), 75.36 (C5’), 71.96 (C3’), 41.95 (C2’), 12.36 (CH3); LR-MS (ESI) [M+H]+ 225.07. HRMS (ESI): calcd. 225.0870 [M + H]+. found. 225.0859 [M + H]+. The late eluting fractions were identified as the desired diphosphate product 10 (30 mg, 30%) as a white fluffy solid. HPLC RT 45.4 min (program); 1H NMR (D2O) δ 7.60 (1H, s, H6), 6.19 (1H, dd, J = 6.7 Hz, H1’), 4.48 (1H, m, H3’), 4.03 (1H, m, H4’), 3.98 (2H, m, H5’,H5”); 2.15-2.45 (2H, m, H2’,H2’’), 2.03 (2H, t, J = 19.8 Hz, Pα–CH2–Pβ); 1.78 (3H, s, CH3); 31P NMR (D2O) δ 19.90 (m, Pα), 16.45 (m, Pβ): LR-MS (ESI) (M+H)+ 400.99.
Synthesis of α,β-methylene 2’-deoxynucleoside 5’-triphosphate (α,β-m-dNTP; 12-15)
α,β-methylene-dATP (12)
A mixture of 8 (40 mg, 0.1 mmol), ATP (55 mg, 0.1 mmol), and phosphoenol pyruvate (PEP, 38 mg, 0.2 mmol) were placed in a 10 mL solution of triethanolamine (83 mM), MgCl2 (17 mM), KCl (67 mM). The pH of the mixture was adjusted to 7.6 with 6.0 M NaOH. Nucleoside di-phosphate kinase (NDPK) (200 unit) and pyruvate kinase (400 unit) were added to this solution and the mixture was incubated at 37 °C for 4h. The reaction progress was monitored by HPLC (program A) until γ-phosphorylation was complete. Then ADP (45 mg, 0.1 mmol) was added to remove the excess PEP in the reaction mixture. After 1 h, the mixture was filtered through a 0.2-μm syringe disk and lyophilized. The residual solid on the disk was dissolved in 10 mL of 1 M NH4HCO3 and adjusted to pH 8.5 with concentrated NH4OH. The mixture was then applied to a Bio-Rad boronate affinity gel column (2.5 × 5.5 cm, in 1 M NH4HCO3, pH 8.5). The column was eluted with the same buffer at a flow of 1 mL/min and the fractions containing the first two-column volumes were pooled and lyophilized. Excess NH4HCO3 was removed by repeated lyophilization from deionized water after adjustment to pH 7.2 with CO2.
Final purification of the triphosphate product was performed on FPLC with a Q Sepharose FF anion exchange column (2.5 × 20 cm, HCO3 - form) by a linear gradient elution with NH4HCO3 (from 0.05 ~ 0.5 M, pH 7.8) at a flow rate of 2 mL/min monitored at 230 nm. The desired fractions were pooled and lyophilized to give a white fluffy solid 12 (25 mg, NH4 + salt, 52%). HPLC RT 89.3 min (program A); 1H NMR (D2O) δ 8.37 (1H, s, H8), 8.10 (1H, dd, J = 6.6 Hz, H2), 6.35 (1H, t, J = 6.6 Hz, H1’), 4.70 (1H, m, H3’), 4.13 (1H, s, H4’), 3.97 (2H, m, H5’), 2.71-2.44 (1H, m, H2’, H2’’), 2.20 (2H, t, J = 20.3 Hz, , Pα–CH2–Pβ); 31P NMR (D2O) δ 23.33 (m, Pα), 12.42 (m, Pβ) -4.82 (d, JP-P = 24.7 Hz, Pγ); HRMS (ESI): calcd 488.0137 (M – H)-, found 488.0143 (M – H)-.
α,β-methylene-dCTP (13) was prepared in a similar manner to 12 from 9 (30 mg, 0.08 mmol) to yield 18 mg (NH4 + salt, 49%) of 13 as a white fluffy solid. HPLC RT 69.1 min; 1H NMR (D2O) δ 7.83 (1H, d, J = 7.8 Hz, H6), 6.18 (1H, m, J = 6.1 Hz, H1’), 5.96 (1H, d, J = 7.8, H5) 4.48 (1H, m, H3’), 4.04 (1H, m, H4’), 3.96 (2H, m, H5’,H5”), 2.15-2.32 (2H, m, H2’,H2’’), 2.24 (2H, t, J = 20.4 Hz, Pα–CH2–Pβ); 31P NMR (D2O) δ 23.29 (m, Pα), 12.27 (m, Pβ), -4.87 (d, JP-P = 25.8 Hz, Pγ). HRMS (ESI): calcd 464.0025 (M – H)-, found 464.0032 (M – H)-.
α,β-methylene-dTTP (14) was prepared from 10 (25 mg 0.06 mmol) as described above to yieled 13 mg (NH4 + salt, 45%) of 14 as a white fluffy solid. HPLC RT 82.5 min (program A); 1H NMR (D2O) δ 7.72 (1H, s, H6), 6.31 (1H, dd, J = 6.8 Hz, H1’), 4.61 (1H, m, H3’), 4.2 – 4.1 (3H, m, H4’, H5’,H5”), 2.37 (2H, m, H2’, H2’’), 2.34 (2H, t, J = 20.4Hz, Pα–CH2–Pβ); 1.90 (3H, s, CH3); 31P NMR (D2O) δ 21.82 (m, Pα), 11.42 (m, Pβ), -6.04 (d, JP-P = 16.7 Hz, Pγ). HRMS (ESI): calcd 479.0022 (M – H)-, found 479.0028 (M – H)-.
α,β-methylene-dGTP (15) was prepared from 11 (30 mg 0.08 mmol) to give 21 mg (NH4 + salt, 52%) of 15 as a white fluffy solid. 15: HPLC RT 69.4 min; 1H NMR (D2O) δ 8.47 (1H, s, H8), 6.31 (1H, t, J = 6.5 Hz, H1’), 4.84 (1H, m, H3’), 4.23 (1H, m, H4’), 4.15 (2H, m, H5’,H5”), 2.85-2.50 (2H, m, H2’, H2’’), 2.32 (2H, t, J = 19.8 Hz, , Pα-CH2-Pβ); 31P NMR (D2O) δ 22.19 (m, Pα), 11.37 (m, Pβ), -6.07 (t, JP-P = 23.8 Hz, Pγ). HRMS (ESI): calcd 504.0087 (M – H)-, found 504.0092 (M – H)-.
Alternative synthesis of cyclo-dG (16) and cyclo-dT (17)
Cyclo-dG (16) was prepared according to a modified literature procedure.28,29 A solution of DIAD (690 mg, 3 mmol) in dioxane (1.0 mL) was added dropwise to a suspension of dG (268 mg, 1.0 mmol) and triphenyl phosphine (786 mg, 3.0 mmol) in dioxane (2.0 mL) at 70 °C. dG was solubilized as DIAD was added. The resulting homogeneous solution was stirred at 70 °C for 3 h and then allowed to cool to ambient temperature. The resulting precipitate was filtered and washed with dioxane. Purification by silica gel column chromatography (ethyl acetate: methanol 60:40) gave 16 (80 mg, 30%). The chromatographic (TLC, HPLC) and spectroscopic (1H NMR, UV, CD, ESI-HRMS) properties of this sample match to those obtained from the coupling reaction between 5’-O-tosyl-dG 4 and methylene diphosphate described above.
Cyclo-dT (17) was prepared according to a general cyclization procedure.24 To a solution of 3 (1.13 g, 2.54 mmol) in acetonitrile (70 mL) was added DBU (0.7 g, 4.6 mmol). The reaction mixture was refluxed for 1 h, and was monitored for starting material by TLC (CH2Cl2-CH3OH (80:20)). After 90 minutes, TLC indicated the reaction was complete. The hot solution was immediately filtered through a Celite pad, and the filtrate was evaporated to dryness. The residue was purified by silica gel column chromatography (CH2Cl2-CH3OH (80:20)) to give 285 mg (50%) of pure 17. The chromatographic (TLC, HPLC) and spectroscopic (1H NMR, UV, CD, ESI-HRMS) properties of this sample match to those obtained from the coupling reaction between 3 and methylene diphosphate described above.
Stability studies of 2’-cyclonucleosides
General procedure
Cyclonucleosides (0.2 mmol) were placed in 5.0 mL of either 1.0 M HCl or 1.0 M NaOH for acid and base hydrolysis, respectively. Reaction progress at either 37 or 100 °C was monitored by HPLC/UV using gradient program B. After appropriate time, the reaction mixture was neutralized, lyophilized, and purified by silica column chromatography.
Acid hydrolysis of cyclo-dG (16)
Complete disappearance of 16 (RT = 5.0 min) occurred after 1 h at 37 °C with an exclusive formation of a new peak at 10.5 min (Figure 8b). The product was identified as the glycosidic cleavage product 18. 1H NMR (DMSO-d6): δ 7.85 (1H, d, J = 8.0 Hz, H8), 7.27 (1H, d, J = 8.0 Hz, NH), 6.81 (2H, s, NH2), 6.22 (1H, bs, OH), 5.43 (1H, t, H1’), 4.41-4.33 (1H, m, H3’), 4.25-4.04 (2H, m, H5’, H4’), 4.01-3.93 (1H, m, H5’’), 2.33 (1H, m, H2’), 2.05 (1H, m, H2’’); 13C NMR (DMSO-d6): δ 140.19 (C8), 98.54 (C1’), 82.96 (C4’), 71.81 (C3’), 48.26 (C5’), 40.59 (C2’); HRMS (ESI): calcd. 268.1045 (M +(H)+, found. 268.1039 (M + H)+ Continued incubation at 37 °C triggered a reverse cyclization reaction (e.g., deglycosylation). After 3h, entire 18 was reverted back to the starting cyclo-dG along with trace of the free base guanine (Figure 8b). The formation of cyclo-dG and guanine was confirmed by cochromatography with standards on HPLC. When the above reaction was carried out at 100 °C, a similar HPLC profile was obtained after 6h (Figure 8b). Prolonged reaction time (48 h) at 100 °C resulted in a complete depurination.
Base hydrolysis of cyclo-dG
No decomposition of cyclo-dG 16 (RT 4.9 min, Figure 8b) occurred in 1.0 M NaOH after stirring for 1h at 37 °C. A longer reaction time (18 h), however, led to formation of one major product at 10.0 min. The base hydrolysis completed after 41 h. An acid work-up, followed by a silica chromatography (ethyl acetate:methanol 30:70%), yielded 3,5’-anhydro-2’-deoxyxanthosine 19 (cyclo-dX) as a main product. A polar minor product at 4.0 min decomposed on passing through a reverse phase column. 19: 1H NMR (DMSO-d6): δ 11.11 (1H, s, CONH), 7.71 (1H, s, H8), 6.47 (1H, m, H1’), 5.46 (1H, bs, 3’-OH), 4.65 (1H, J =16 Hz, H5’), 4.60 (1H, m, H3’), 4.36 (1H, m, H4’), 3.65 (1H, m, J = 16.0 Hz, H5’’), 2.21 (2H, m, H2’,H2’’); 13C NMR (DMSO-d6): δ 157.91 (C6), 151.78 (C2), 141.58 (C8), 118.56 (C5), 88.43 (C1’), 86.08 (C4’), 70.37 (C3’), 51.59 (C5’), 44.79 (C2’); HRMS (ESI): calcd. 251.0780 (M + H)+. found. 251.0775 (M + H)+.
Acid hydrolysis of cyclo-dX (19)
A solution of 19 (25 mg, 0.10 mmol) in 1.0 M HCl (5 mL) was placed in a shaker at 37 °C. After 1h, the reaction was complete with formation of an unstable intermediate whose HRMS (ESI) 269.0880 (M + H)+ is consistent with the hydrolysis product structure 20.
Acid hydrolysis of cyclo dT (17)
Complete disappearance of 17 (RT = 7.8 min) in 1.0 M HCl occurred within 30 min at 37 °C and was accompanied with formation of intermediate 21 (RT = 15.4 min) along with a trace of thymine (RT = 6.0 min, Figure 9). Increasing reaction time (6h) led to complete decomposition of 21 to thymine. The intermediate 21 was not stable enough to allow 1H NMR characterization, however the eaxt mass at the ions at 243.1 (M + H)+ ESI-MS suggests that has a structure specified by the glycosidic cleavage product 20.
Hydrolysis of cyclo-dT (17) in a pH 7.0 buffer
A solution of 17 (20 mg, 0.09 mmol) in a pH 7.0 buffer containing 0.2 M NaCl, 10 mM sodium phosphate, and 0.2 mM EDTA was placed in a shaker at 37 °C. The HPLC profile was similar to that in 1.0 M HCl above, yielding complete hydrolysis within 6 h (Fig. 9). However, the concentration of X was small throughout the reaction.
Base hydrolysis of cyclo-dT (17)
The hydrolysis of cyclo-dT to dT (RT = 10.5 min, Figure 9), by cleavage of the 2,5’-anhydro bridge was complete in 30 minutes at 37 °C. No other products were formed on increasing the reaction time up to 4h.
DNA Synthesis Assays
Human DNA polymerase β was purified as described previously.30 DNA synthesis was assayed on four single-nucleotide gapped DNA substrates where the templating base in the gap was varied. The template sequence was 3’–GACGTCGACTACGCGXCATGCCTAGGGGCCCATG–5’ where X represents A, C, G or T. Complementary 15- and 18-mer oligonucleotides where annealed to the template strand as described. 31 The upstream 15-mer primer was 5’-labeled with [γ-32P]ATP using T4 polynucleotide kinase. The radioactive ATP was removed with a MicroSpin G-25 column. The downstream 18-mer oligonucleotide was synthesized with a 5’-phosphate. Enzyme activities were determined using a reaction mixture containing 50 mM Tris-HCl, pH 7.4, 20 mM MgCl2, 200 nM single-nucleotide gapped DNA, at various dNTP concentrations. Reactions were initiated with 0.5 nM enzyme at room temperature and stopped with EDTA mixed with formamide dye. The substrates/products were separated on 15% denaturing polyacrylamide gels and quantified in the dried gels by phosphorimagery. Steady-state kinetic parameters (kcat, Km) were determined by fitting the rate data to the Michaelis equation. Inhibition of dNTP insertion by various α,β-methylene-dNTPs was determined at different concentrations of inhibitor (I) and sub-saturating dNTP concentration (S). The inhibitor equilibrium binding constant, Ki, was determined by fitting the inhibition data to Equation 1 for competitive inhibition by nonlinear regression methods.
| (Eq. 1) |
Supplementary Material
Acknowledgments
This research was supported by the NIH Grant CA098296 and the Intramural Research Program of the NIH, NIEHS in association with the NIH Grant 1U19CA105010. This research was also made possible in part by the use of the INBRE Research Core Facility, which is supported by the NCRR/NIH (P20 RR016457). Mass spectrometry was provided by Washington University Mass Spectrometry Resource with support from the NCRR/NIH (P41RR0954).
Footnotes
Supporting Information Available: 1H (1D, NOESY, TOCSY), 13C, and 31P NMR spectra; ESI-LQT-HRMS spectra; UV and CD Spectra. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Vaghefi Morteza., editor. Nucleoside Triphosphates and their Analogs: Chemistry Biotechnology and Biological Applications. CRC Taylor & Francis; 2005. [Google Scholar]
- 2.Burgess K, Cook D. Synthesis of Nucleoside Triphosphates. Chem Rev. 2000;100:2047–2059. doi: 10.1021/cr990045m. [DOI] [PubMed] [Google Scholar]
- 3.(a) Shen Y, Guo Q, Zhukovskaya NL, Drum CL, Bohm A, Tang W-J. Structure of Anthrax Edema Factor–calmodulin–adenosine 5′-(α,β-methylene)-triphosphate Complex Reveals an Alternative Mode of ATP Binding to the Catalytic Site. Biochem Biophy Res Commun. 2004;317:309–314. doi: 10.1016/j.bbrc.2004.03.046. [DOI] [PubMed] [Google Scholar]; (b) Bell MA, Gough GR, Gilham PT. The Analogue of Thymidine Triphosphate Containing a Methylene Group in Place of the 5’ Oxygen Can Serve as a Substrate for Reverse Transcriptase. Nucleoside Nucleotides & Nucleic Acids. 2003;22:405–417. doi: 10.1081/NCN-120022031. [DOI] [PubMed] [Google Scholar]; (c) Li R, Muscate A, Kenyon GL. Synthesis, Characterization, and Inhibitory Activities of Nucleoside α,β-Imido Triphosphate Analogues on Human Immunodeficiency Virus-1 Reverse Transcriptase. Bioorg Chem. 1996;24:251–261. [Google Scholar]; (d) Job C, Soulié JM, Job D. Kinetic co-operativity of Wheat-germ RNA polymerase II with adenosine 5’-[beta gamma-imido]triphosphate as Substrate. Biochem J. 1988;252:55–63. doi: 10.1042/bj2520055. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Alexandrova LA, Skoblov LY, Jasko MV, Victorova LS, Krayevsky AA. 2’-Deoxynucleoside 5’-triphosphates modified at alpha-, beta- and gamma- phosphates as Substrates for DNA Polymerases. Nucleic Acids Res. 1998;26:778–786. doi: 10.1093/nar/26.3.778. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Dixit VM, Poulter CD. Convenient Synthesis of Adenosine 5’-Diphosphate, Adenosine 5’-Methylduphosphate, and Adenosine 5-Triphosphate. Tetrahedron Lett. 1984;25:4055–4058. [Google Scholar]; (g) Ma Q-F, Bathurst IC, Barr PJ, Kenyon GL. New Thymidine Triphosphate Analog Inhibitors Of Human Immunodeficiency Virus-1 Reverse Transcriptase. J Med Chem. 1992;35:1938–1941. doi: 10.1021/jm00089a002. [DOI] [PubMed] [Google Scholar]; (h) Shipitsin AV, Victorova LS, Shirokova EA, Dyatkina NB, Goryunova LE, Beabealashvilli RS, Hamilton CJ, Roberts SM, Krayevsky A. New Modified Nucleoside 5’-Triphosphates: Synthesis, Properties Towards DNA Polymerases, Stability in Blood Serum and Antiviral Activity. J Chem Soc Perkin Trans. 1999;1:1039–1050. [Google Scholar]; (i) Ma Q-F, Babbitt PC, Kenyon GL. Adenosine 5’-[a,b-imido]triphosphate, a Substrate For T7 RNA Polymerase And Rabbit Muscle Creatine Kinase. J Am Chem Soc. 1988;110:4060–4061. Erratum: 110, 8267. [Google Scholar]; (j) Krug F, Parikh I, Illiano G, Cuatrecasa P. α,β-Methylene-adenosine 5’-phosphate. J Biol Chem. 1973;248:1203–1206. [PubMed] [Google Scholar]; (k) Breaker RR, Gough GR, Gilham PT. Synthesis and Properties of Adenosine Oligonucleotide Analogs Containing Methylene Groups in Place of Phosphodiester 5’-Oxygens. Biochemistry. 1993;32:9125–9128. doi: 10.1021/bi00086a017. [DOI] [PubMed] [Google Scholar]
- 4.Sucato CA, Upton TG, Kashemirov BA, Batra VK, Martínek V, Xiang Y, Beard WA, Pedersen LC, Wilson SH, McKenna CE, Florián J, Warshel A, Goodman MF. Modifying The Beta, Gamma Leaving-Group Bridging Oxygen Alters Nucleotide Incorporation Efficiency, Fidelity, and the Catalytic Mechanism of DNA Polymerase β. Biochemistry. 2007;46:461–471. doi: 10.1021/bi061517b. [DOI] [PubMed] [Google Scholar]
- 5.Mckenna CE, Kashemirov BA, Upton TG, Batra VK, Goodman MF, Pedersen LC, Beard WA, Wilson SH. (R)-β,γ-Fluoromethylene-dGTP-DNA Ternary Complex with DNA Polymerase β. J Am Chem Soc. 2007;129:15412–15413. doi: 10.1021/ja072127v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Batra VK, Beard WA, Shock DD, Pedersen LC, Wilson SH. Structures of DNA Polymerase B With Active Site Mismatches Suggest a Transient Abasic Site Intermediate During Misincorporation. Mol Cell. 2008;30:315–24. doi: 10.1016/j.molcel.2008.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Batra VK, Beard WA, Shock DD, Krahn JM, Pedersen LC, Wilson SH. Magnesium Induced Assembly of a Complete DNA Polymerase Catalytic Complex. Struct (Camb) 2006;14:757–766. doi: 10.1016/j.str.2006.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Liang F, Cho BP. Probing the Thermodynamics of Aminofluorene-Induced Translesion DNA Synthesis by Differential Scanning Calorimetry. J Am Chem Soc. 2007;129:12108–12109. doi: 10.1021/ja075271p. [DOI] [PubMed] [Google Scholar]; (b) Meneni S, Liang F, Cho BP. Examination of the Long-Range Effects of Aminofluorene-Induced Conformational Heterogeneity and its Relevance to the Mechanisms of Translesion DNA Synthesis. J Mol Biol. 2007;366:1387–1400. doi: 10.1016/j.jmb.2006.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Meneni S, Shell SM, Gao Jurecka P, Lee W, Sponer J, Zou Y, Chiarelli MP, Cho BP. Spectroscopic and Theoretical Insights into Sequence Effects of Aminofluorene-induced Conformational Heterogeneity and Nucleotide Excision Repair. Biochemistry. 2007;40:11263–11278. doi: 10.1021/bi700858s. [DOI] [PubMed] [Google Scholar]
- 9.(a) Pelletier H, Sawaya MR, Kumar A, Wilson SH, Kraut J. Structures of ternary complexes of rat DNA polymerase ß, a DNA template-primer, and ddCTP. Science. 1994;264:1891–1903. [PubMed] [Google Scholar]; (b) Doublie S, Tabor S, Long AM, Richardson CC, Ellenberger T. Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 A resolution. Nature. 1998;391:251–258. doi: 10.1038/34593. [DOI] [PubMed] [Google Scholar]
- 10.Davisson VJ, Davis DR, Dixit VM, Poulter CD. Synthesis of Nucleotide 5’-Diphosphates from 5’-O-Tosyl Nucleosides. J Org Chem. 1987;52:1794–1801. [Google Scholar]
- 11.Stock JA. Synthesis of Phosphonate Analogues of Thymidine Di- and Triphosphate from 5’-O-Toluenesulfonylthymidine. J Org Chem. 1979;44:3997–4000. [Google Scholar]
- 12.Blackburn GM, Langston SP. Novel P1,P2-substituted phosphate analogs of 2’-deoxyadenosine and 2’-deoxythymidine 5’-phosphates. Tetrahedron Lett. 1991;32:6425–6428. [Google Scholar]
- 13.Wu W, Bergstrom D, Davisson VJ. A Combination Chemical and Enzymatic Approach for the Preparation of Azole Carboxamide Nucleoside Triphophate. J Org Chem. 2003;68:3860–3865. doi: 10.1021/jo020745i. [DOI] [PubMed] [Google Scholar]
- 14.Kamiya H, Kasai H. Preparation of 8-Hydroxy-dGTP and 2-Hydroxy-dATP by a Phosphate Transfer Reaction by Nucleoside-Diphosphate Kinase. Nucleosides Nucleotides and Nucleic Acids. 1999;18:307–310. [Google Scholar]
- 15.Beard WA, Wilson SH. Structure And Mechanism of DNA Polymerase β. Chem Rev. 2006;106:361–382. doi: 10.1021/cr0404904. [DOI] [PubMed] [Google Scholar]
- 16.Ahn J, Kraynov VS, Zhong X, Werneburg BG, Tsai M-D. DNA polymerase beta: effects of gapped DNA substrates on dNTP specificity, fidelity, processivity and conformational changes. Biochem J. 1998;331:79–87. doi: 10.1042/bj3310079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Remin M, Shugar Conformation of the Exocyclic 5’-CH2OH in Nucleosides and Nucleotides in Aqueous Solution From Specific Assignments of the H5’ and H5” Signals in the NMR Spectra. Biochem Biophy Res Commun. 1972;48:636–642. doi: 10.1016/0006-291x(72)90395-6. [DOI] [PubMed] [Google Scholar]
- 18.Cho BP, Evans FE. Correlation Between NMR Spectral Parameters of Nucleosides and its Implication of the Conformation About the Glycosyl Bond. Biochem Biophy Res Commun. 1991;180:272–278. doi: 10.1016/s0006-291x(05)81288-4. [DOI] [PubMed] [Google Scholar]
- 19.Harmony TP, Raleigh J, Sundaralingam M. Enzyme-Bound Conformations of Nucleotide Substrates. X-Ray Structure and Absolute Configuration of 8,5’-Cycloadenosine Monohydrate. Biochemistry. 1980;19:1718–1722. doi: 10.1021/bi00549a031. [DOI] [PubMed] [Google Scholar]
- 20.Frick DN, Weber DJ, Abeygunawardana C, Gittis AG, Bessman MJ, Mildvan AS. NMR Studies of the Conformations and Location of Nucleotides Bound to the Escherichia coli MutT Enzyme. Biochemistry. 1995;34:5577–5586. doi: 10.1021/bi00016a032. [DOI] [PubMed] [Google Scholar]
- 21.Czernek J, Sklenar V. Ab Initio Calculations of 1H and 13C Chemical Shift in Anhydrodeoxythymidines. J Phys Chem A. 1999;103:4089–4093. [Google Scholar]
- 22.Guschlbauer W, Courtois Y. pH Induced Changes in Optical Activity of Guanine Nucleosides. FEBS Letters. 1968;1:183–186. doi: 10.1016/0014-5793(68)80055-9. [DOI] [PubMed] [Google Scholar]
- 23.Holmes RE, Robins RK. Preparation and Reactions of Some 9β-D-Ribofuranosyl- 3,5’-purine Cyclonucleosides. J Org Chem. 1963;28:3483–3486. [Google Scholar]
- 24.Lin T-S, Shen Z-Y, August EM, Brankovan V, Yang H, Ghazzouli I, Prusoff WH. Synthesis and Antiviral Activity of Several 2,5’-Anhydro Analogues of 3’-Azido-3’deoxythymidine, 3’Azido-2’,3’-dideoxyuridine, 3’-Azido-2’,3’-dideoxy-5-halouridines, and 3’-Deoxythymidine against Human Immunodeficiency Virus and Rauscher-Murine Leukemia Virus. J Med Chem. 1989;32:1891–1895. doi: 10.1021/jm00128a034. [DOI] [PubMed] [Google Scholar]
- 25.Lin L-G, Bakthavachalam V, Cherian XM, Czarnik AW. Effects of Cyclonucleoside Formation on the Rates of Glycosidic Hydrolyses in Purine Ribonucleosides. J Org Chem. 1987;52:3113–3119. [Google Scholar]
- 26.Reese CB, Whittall N. An Acid-stable Analogue of the 3-β-D-ribofuranoside of Y-base. Nucleic Acids Res. 1976;3:3439–3445. doi: 10.1093/nar/3.12.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reist EJ, Benitez A, Goodman L. The synthesis of some 5-thiopentofuranosyl-pyridines. J Org Chem. 1964;29:554. [Google Scholar]
- 28.Kimura J, Yagi K, Suzuki H, Mitsunobu O. Study on Nucleosides and Nucleotides. VIII. Preparation and Reactions of Triphenylphosphoranediylnucleosides. Bull Chem Soc Jpn. 1980;53:3670–3677. [Google Scholar]
- 29.Li H, Miller MJ. Synthesis of 5’-deoxy-5’-N-hydroxylaminopyrimidine and Purine Nucleosides: Building Blocks for Novel Antisense Oligonucleosides With Hydroxaminate Linkage. J Org Chem. 1999;64:9289–9293. [Google Scholar]
- 30.Beard WA, Wilson SH. Purification and domain-mapping of Mammalian DNA Polymerase β. Methods Enzymol. 1995;262:98–107. doi: 10.1016/0076-6879(95)62013-3. [DOI] [PubMed] [Google Scholar]
- 31.Beard WA, Shock DD, Yang X-P, DeLauder SF, Wilson SH. Loss of DNA Polymerase β Stacking Interactions With Templating Purines, But Not Pyrimidines, Alters Catalytic Efficiency and Fidelity. J Biol Chem. 2002;277:8235–8242. doi: 10.1074/jbc.M107286200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.