

Abstract
We characterized the aro arsenite oxidation system in the novel strain Ralstonia sp. 22, a β-proteobacterium isolated from soil samples of the Salsigne mine in southern France. The inducible aro system consists of a heterodimeric membrane-associated enzyme reacting with a dedicated soluble cytochrome c554. Our biochemical results suggest that the weak association of the enzyme to the membrane probably arises from a still unknown interaction partner. Analysis of the phylogeny of the aro gene cluster revealed that it results from a lateral gene transfer from a species closely related to Achromobacter sp. SY8. This constitutes the first clear cut case of such a transfer in the Aro phylogeny. The biochemical study of the enzyme demonstrates that it can accommodate in vitro various cytochromes, two of which, c552 and c554, are from the parent species. Cytochrome c552 belongs to the sox and not the aro system. Kinetic studies furthermore established that sulfite and sulfide, substrates of the sox system, are both inhibitors of Aro activity. These results reinforce the idea that sulfur and arsenic metabolism are linked.
Keywords: Bacterial Metabolism, Bioenergetics, Cytochrome c, Electron Transfer, Enzyme Kinetics, Enzyme Purification, Evolution, Arsenite Oxidase
Introduction
Arsenic is most commonly found in an insoluble, and thereby not toxic, form associated with more than 200 rock and mineral species. However, in natural environments such as geothermal springs and in sites contaminated by industries (1) or by bioleaching of arsenic minerals (see Oremland and Stolz, Ref. 2), high amounts of soluble forms can be accumulated. These forms, arsenate (AsV)5 and arsenite (AsIII) are both toxic to complex life. AsV, a phosphate analog, interferes with normal phosphorylation processes by replacing phosphate, whereas AsIII binds to sulfhydryl groups of cysteine residues in proteins, thereby inactivating them. AsIII is considered to be 100× more toxic than AsV. AsIII can be oxidized to AsV either chemically or microbially (3). Since the first report of bacterial AsIII oxidation by Green (4) in 1918, an exponential number (see Refs. 5–12) of phylogenetically diverse AsIII-oxidizing bacteria have been isolated from different environments. These bacteria can be divided into two groups: (i) chemolithoautotrophs (aerobes or anaerobes, using AsIII as the electron donor and CO2/HCO3− as the sole carbon source) or (ii) heterotrophs (growing in the presence of organic matter) (for recent reviews, see Refs. 13 and 14).
Apart from the two cases of Ectothiorhodospiraceae, Alkalilimnicola ehrlichii str. MLHE-1 (15) and PHS-1 (16), the enzyme identified as responsible for AsIII oxidation has been shown to be AsIII oxidase. Whereas Aox was the name first given to the gene cluster coding for the enzyme (17), it presents a drawback in denoting the molybdopterin subunit as AoxB in conflict with the general dimethyl sulfoxide reductase superfamily (to which the enzyme belongs; see below) nomenclature and in which the catalytic molybdopterin subunit invariably is called A. The name Aro, which was introduced later (18), is admittedly similar to a denomination already in use since the 1970s for aromatic amino acid synthesis enzymes (19) but has the advantages of i) following the dimethyl sulfoxide reductase superfamily nomenclature and ii) explicitly matching the name of AsV reductase. Aso, introduced by Silver (13), is used only scarcely. We have, therefore, chosen to use Aro in the following text.
Aro can be found either in the periplasm (20, 21) or associated to the cytoplasmic membrane (5, 13, 22–24). AroA (90–100 kDa) carries a molybdopterin cofactor together with a [3Fe-4S] center and characterizes the enzyme as a member of the dimethyl sulfoxide reductase superfamily. AroB (14 kDa), is a member of the Rieske proteins superfamily by virtue of its [2Fe-2S] center (see 25 and 66, accompanying article) and is considered to be responsible for a possible membrane attachment (23, 24). Only scant data have, so far, been reported on the enzymology of Aro. Because AsIII is a two-electron donating substrate, the catalytic turnover is assumed to start with the oxidation of AsIII by the molybdenum center (which can accept up to two electrons). Several facts indicate that the catalytic cycle of AsIII oxidation results in most cases in the reduction of a soluble cyt. First, cytochromes (cyts) have been copurified with the enzyme (21, 22). Secondly, cyt-encoding genes are often present in the aro gene clusters (10, 24, 26–28). And Finally, the AsIII oxidation process in Ochrobactrum tritici requires the cyt encoded in the aro operon (28). No detailed studies have been presented, however, addressing the electron transfer reaction between Aro and cyt. The only enzymatic data presently available on Aro have been obtained using 2,4 dichlorophenolindolphenol (DCPIP) or azurin, two nonphysiological electron acceptors of the Alcaligenes faecalis enzyme (22). Enzymatic properties of Aro deduced from these studies, therefore, do not necessarily reflect the physiological reaction. In this paper, we describe the purification and characterization of Aro from the novel strain Ralstonia sp. 22 (S22). This β-proteobacterium has been isolated from soil samples of the Salsigne mine in southern France. In addition to the enzyme, we also purified two cyts among them is the likely physiological electron acceptor of Aro, cyt c554, and we characterized the reaction of Aro with both cyts. The presented results therefore are the first detailed enzymatic data on an Aro reacting with cyts. Moreover, the use of cyts in the activity assays allowed us to screen the sensitivity of purified Aro toward sulfur compounds. The observed inhibitory effects support the idea that arsenic and sulfur metabolisms are functionally related. As stated above, a number of Aro enzymes have been studied in the past with respect to specific properties. However, in none of these cases, a complete characterization of the enzyme determining biochemical, biophysical (see Ref. 66, accompanying article), and enzymatic parameters has been obtained, hampering a comprehensive understanding of the enzyme and its comparison with other members of the dimethyl sulfoxide reductase superfamily. The present work fills in these gaps by presenting an exhaustive description of Aro in S22.
EXPERIMENTAL PROCEDURES
S22 Isolation and Growth Conditions
The strain S22 was isolated from arsenic-contaminated soil collected near the gold mine of Salsigne, Aude, France. Soil samples were inoculated at 25 ± 2 °C into a liquid chemically defined medium (CDM) (described by Muller et al. (17) for Herminiimonas arsenicoxydans), supplemented with 1.33 mm AsIII to develop enriched cultures. A pure culture was obtained by successive isolation of colonies at 25 ± 2 °C on CDM, solidified by addition of 20 g−1 liter−1 of agar-agar (Difco).
S22 was grown aerobically at 28 °C in 5-liter bottles of CDM. When included in the medium, 5 mm AsIII (NaAsO2) or 20 mm thiosulfate (Na2O3S2,5H2O) were added. The final pH was ∼ 7. Cultures were harvested during the late exponential phase.
Preparation of Spheroplast and Periplasmic Fractions
Spheroplasts were prepared as published previously (29) with some modifications. Bacteria were incubated for 1 h at 30 °C (instead of 30 min) with lysozyme 1 mg−1 ml−1 (instead of 0.5 mg−1 ml−1). After incubation, cells were centrifuged at 4000 × g for 30 min, and spheroplasts were retrieved in the pellet, whereas the supernatant constitutes the periplasmic fraction. Spheroplasts were resuspended in 100 mm phosphate buffer at pH 7.4 for subsequent characterization.
Purification of the Aro
Cells were suspended in 50 mm Tricine at pH 8 (buffer A) and broken by passing twice through a French press. Unbroken cells were eliminated by centrifugation at 10,000 × g, and a subsequent ultracentrifugation (280,000 × g) separated the “total soluble fraction” (in the supernatant) from the “membrane fraction” (in the pellet). Enzyme purification was performed from the total soluble fraction at 4 °C. The sample, once oxidized with ferricyanide, was loaded on a DEAE Sephacel column equilibrated with buffer A. Aro eluted at 50 mm NaCl from this column. The sample was then dialyzed to eliminate NaCl and subsequently loaded on a monoQ DEAE column (fast protein liquid chromatography (FPLC) system) equilibrated with buffer A. This second DEAE was eluted at 1 ml−1 min−1 with a 0–100 mm NaCl gradient and Aro eluted at ∼30 mm NaCl. The Aro fraction was then concentrated by centrifugation in Amicon Ultra-5 concentrators. The sample was then loaded onto a Superdex 200 gel filtration column (FPLC system), which was equilibrated with buffer A/NaCl 100 mm and eluted at 0.4 ml−1 min−1. Only freshly purified enzyme was used for enzymatic analyses.
Purification of Cytochromes
The fraction containing almost all the soluble cyts eluted during washing of the DEAE Sephacel used for the Aro purification. This “DEAE-cyt fraction,” was then loaded on a CM-52 column equilibrated with buffer A. Because binding of the cyt c552 on the CM column depends on its oxidation state, we systematically oxidized the fraction before loading. Cyts c551 and c554 eluted together during washing of the CM. The c552 eluted from the CM column at 25 mm NaCl. Both cyt fractions were then separately concentrated by centrifugation in an Amicon Ultra-5 concentrator and loaded separately onto a Superdex 75 gel filtration column (FPLC system), which was equilibrated with buffer A/NaCl 100 mm and eluted at 0.4 ml−1 min−1. Pure cyts c554 and c552 were obtained after this step. The enriched cyt c551 obtained from this step was not further purified.
Aro Activity Assays
Aro activity was routinely measured optically in 50 mm MES, pH 6, at 37 °C, using 200 μm sodium AsIII as an electron donor, 150 μm DCPIP as an electron acceptor, and 20 μm phenazine methosulfate as an electron mediator between Aro and DCPIP. The activity was followed as the reduction of DCPIP, i.e. decreasing absorption monitored at 600 nm (ϵ600 pH 6 experimentally determined at 12 mm−1 cm−1). The reaction was initiated by addition of AsIII. In the specific enzymatic studies, sulfite (sodium sulfite) and thiosulfate (sodium thiosulfate) were tested as electron donors, whereas azurin from Pseudomonas aeruginosa and cyt c from bovine heart (commercially available) and cyt c555 purified from Aquifex aeolicus as described in Ref. 30, or cyts c552 and c554 purified from S22 (see above) were tested as electron acceptors. The pH optimum was assayed by using mixed buffers MES/MOPS/Tricine/AMPSO/CAPS at 15 mm each. Finally NaN3, sulfite, sulfide, thiosulfate, and AsV were tested as potential inhibitors. In these cases, the kinetics were followed as reduction of cyt, i.e. increasing absorption monitored at the α band maximum. Aro activity was also detected on native polyacrylamide gels. Total soluble fraction and membrane fraction from French press treatment on one side or periplasm and spheroplasts from lysozyme treatment on the other side were analyzed by native gel electrophoresis. Equivalent samples (∼30 μg of total proteins) from each cell-breaking treatment were loaded on the gel. The electrophoresis was done on a native 10% polyacrylamide Laemmli gel system (31) containing 0.1% Triton X-100. The gel was then equilibrated in 50 mm MES, pH 6, for 15 min and subsequently incubated for 30 min in the dark in the same buffer supplemented with 300 μm DCPIP and 100 μm phenazine methosulfate. Addition of 200 μm sodium AsIII allowed the detection of the Aro band by its destaining activity.
Biochemical Protein Analyses
Protein concentrations were determined by the BCA method using bovine serum albumin as standard. The subunit composition was determined by SDS-PAGE following the procedure of Laemmli (31) on a 5–15% gradient polyacrylamide gel. The cyt composition of the “total soluble fraction” (see above) was analyzed by electrophoresis following the procedure of Judd (32) on a 18% polyacrylamide gel. The molecular weight of native Aro and cyts were estimated by gel filtration in buffer A/100 mm NaCl on Superdex 200 or 75, respectively, using apoferrin, amylase, alcohol dehydrogenase, bovine serum albumin, carbonic anhydrase, and bovine heart cyt as molecular weight standards.
DNA Works and Sequencing
DNA manipulations were carried out according to standard protocols as described by Sambrook et al. (33). Total DNA of strain S22 was isolated using the Wizard Genomic DNA purification kit (Promega). 16S rDNA fragments were amplified by PCR on DNA extract using the eubacterial universal primers specific for 16S rDNA (P8, 5′-AGAGATTTGATCCTGGCTCAG-3′ and Pc1544, 5′- AAGGAGGTGATCCAGCCGCA-3′). The amplified 16S rDNA fragment was purified via phenol extraction and 2-propanol precipitation and sequenced. Based on the aro operon of Achromobacter sp. SY8 (SY8) (GenBankTM accession no. EF523515), oligonucleotides were designed that amplify a DNA stretch of 3508 bp using PCR, covering the aroA, aroB, and aroC (cyt c554) genes of S22, in three overlapping fragments. For fragment 1 (1190 bp), forward primer 5′-CGTCCGAAAGCTACTTGG-3′ and reverse primer 5′-GCAGTGAACATCAGCTTCC-3′ were used; for fragment 2 (1246 bp), forward primer 5′-TTCTCCTGTTTCGACCACG-3′ was used; for reverse primer, 5′- CCTTTAGCGTGTTGGCG-3′ was used; and for fragment 3 (1241 bp), forward primer 5′-ACGCATCCGCCTATCTC-3′ and reverse primer 5′-CATTAAGCGCGAACCCG-3′ were used. PCR amplification was done using Pfu DNA polymerase (Promega) on DNA extract, and the amplified DNA fragments were cloned into vector pSTBlue-1 using the Perfectly Blunt cloning kit (Novagen). Then, DNA sequencing of the vector inserts was performed (GATC Biotech) using the T7 and SP6 primers, and the resulting sequences were assembled into the complete 3508-bp sequence.
Sequence Analyses
ClustalX (34) was used to obtain multiple sequence alignments of proteins or 16S rDNA. Phylogenetic trees were reconstructed from these alignments using the NJ algorithm implemented in ClustalX or using the parsimony method (PHYLYP package). The nucleotide sequences described in this study have been deposited in GenBankTM with the following accession numbers: EU304284 (S22 16S rRNA), EU304273 (partial aoxB gene), and GQ904715 (total 3508-bp aro cluster).
Determination of Arsenic Speciation in the Growth Medium and Minimal Inhibitory Concentration
Qualitative AsIII oxidation activity from bacteria was followed by visualizing the AsIII concentration in the growth medium by a colorimetric method as described by Simeonova et al. (35). When needed, arsenic species were more precisely quantified by HPLC-ICP AES as described by Weeger et al. (36). Minimal inhibitory concentration of AsIII and AsV were determined by following the procedure described by Lim and Cooksey (37).
Protein Identification Techniques
Intact protein mass analyses were performed on a MALDI-TOF mass spectrometer UltraflexII from Bruker Daltonik. External calibration was made on the singly charged ion [M + H]+ at 16,951.56 of apomyoglobin, at 12,361.96 of cyt, and the doubly charged ion [M + 2H]2+/2 at 8476.28 of apomyoglobin. N-terminal sequence determination was performed by Edman degradation using an automatic sequencer model Procise 494 from Applied Biosystems on bands from blotted protein onto 0.2 μm polyvinylidene fluoride membrane stained with Ponceau Red.
Optical spectra were recorded on a Carry 5E spectrophotometer. Redox titrations were performed on purified cytochromes at 15 °C as described by Dutton (38) in the presence of the following redox mediators at 10 μm: 1,4-p-benzoquinone, 2,5-dimethyl-p-benzoquinone, and 2-hydroxy-1,2-naphthoquinone. Reductive titrations were carried out using sodium dithionite, and oxidative titrations were carried out using ferricyanide.
RESULTS
Isolation and Phylogenetic Characterization of the Novel Strain Ralstonia sp. 22
A soil sample contaminated by gold mine wastes rich in arsenic (Salsigne, Aude, France) was cultured on CDM supplemented with AsIII (1.33 mm). A pure culture was obtained by successive isolation of colonies at 25 ± 2 °C on AsIII-supplemented CDM. Isolate S22 showed AsIII oxidase activity when grown on CDM supplemented with 1.33 mm AsIII. HPLC-ICP AES experiments demonstrated the progressive disappearance of AsIII in parallel to the appearance of AsV in the supernatant of the strain S22 culture (data not shown). These findings suggested the oxidation of AsIII by strain S22. Phylogenetic analyses based on the 16S rDNA (1,455 bp) sequence indicated that the strain belongs to the class of the β-proteobacteria and that the nearest phylogenetic relatives are members of the Burkholderiaceae family in the order Burkholderiales. Analysis of binary similarity data showed that the 16S rRNA sequence of strain S22 displays 97% identity to sequences of representatives of Ralstonia genera (solanacearum species), suggesting that the strain S22 is a new species member of the genus Ralstonia, a group of bacteria frequently found in soils.
The minimal inhibitory concentration value, defined as the ion concentration that inhibited confluent growth on plates after 3 days at 30 °C, was determined for AsIII and AsV. Interestingly, S22 showed resistance up to 30 mm AsIII, which is a 5× higher resistance level than that exhibited by the arsenic oxidizing bacterium H. arsenicoxydans (17) (Table 1) and comparable to the recently characterized SY8, Pseudomonas sp. TS44 and O. tritici bacteria (13, 23, and 50 mm, respectively) (10, 28). The Aro enzyme from S22 subsequently was further characterized.
TABLE 1.
Comparison of S22 to H. arsenicoxydans for AsIII and AsV Minimal Inhibitory Concentrations
| H. arsenicoxydans | Ralstonia sp. 22 | |
|---|---|---|
| MIC AsIII | 6 mm | 30 mm |
| MIC AsV | 200 mm | 200 mm |
Inducibility and Cellular Localization of the Aro
In contrast to A. faecalis (22), H. arsenicoxydans (36), and Rhizobium NT-26 (NT-26) (27), S22 shows a basic, although weak, activity when grown in the absence of AsIII, as already observed for Thiomonas 3As (3As) (24), but features a 20× enhanced activity when grown with 5 mm AsIII (data not shown).
Because conflicting results have been published previously concerning the localization of the Aro enzyme, we addressed this question for the S22 enzyme after treatment of the bacteria with French press or lysozyme. As found previously for A. faecalis (22), H. arsenicoxydans (17), Chloroflexus aurantiacus (23), and 3As (24), the Aro of S22 is membrane-associated as evidenced by the detection of the major part of the activity in the fraction of spheroplasts on native gel (Fig. 1). However, as already observed for A. faecalis (22), the enzyme could be released to the soluble fraction by changing the cell rupture method (Fig. 1). As previously published (20), the Aro from NT-26 is dominantly retrieved from the soluble fraction, even after lysozyme treatment (data not shown), whereas the Aro from H. arsenicoxydans, even after French press treatment, remained up to 65% membrane-associated.6 The “localization” of the enzyme, therefore, seems to depend not only on the breaking conditions but also on the specific organism.
FIGURE 1.
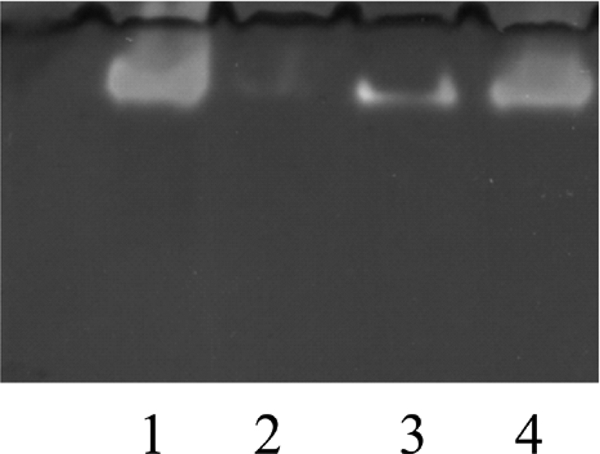
Polyacrylamide gel electrophoresis and in-gel Aro activity staining of subcellular fractions of S22. Lane 1, spheroplasts; lane 2, periplasm; lane 3, membrane fraction after French press treatment; lane 4, soluble fraction after French press treatment.
Purification and Preliminary Characterization of Aro
As almost all the Aro from S22 is found in the soluble fraction after French press treatment, this fraction was used for purification. As first noticed for A. faecalis (22), Aro is particularly thermostable in all organisms tested so far (39). The enzyme from S22 retained 95% activity after heating to 60 °C for 5 min (data not shown). This property allowed us to routinely measure Aro activity at 37 °C with a better signal to noise ratio (see “Experimental Procedures”).
Two anion-exchange chromatographic steps and one gel filtration chromatography step were applied to achieve the purification of the enzyme. This protocol led to a 145-fold final enrichment of the enzyme with a yield of 38% (Table 2). As shown in Fig. 2, the obtained pure enzyme consists of two subunits with apparent molecular masses of 97 kDa and 16 kDa. They correspond to AroA and AroB, respectively, as confirmed by N-terminal sequencing (Fig. 2). The native molecular mass of the isolated enzyme, based on gel filtration chromatography, was found to be 110 kDa (data not shown) suggesting a α1β1 configuration, similar to what was reported for the A. faecalis enzyme (22).
TABLE 2.
Purification of Aro from S22
All kinetics were performed using DCPIP as electron acceptor.
| Purification step | Total proteins | Total activity | Specific activity | Purification | Yield |
|---|---|---|---|---|---|
| mg | μm DCPIP/min | μmol/min/mg | -fold | % | |
| Cell extract | 266.6 | 10.5 | 0.0392 | 1 | 100 |
| DEAE-Sephacel | 11.9 | 9.6 | 0.802 | 20.5 | 91 |
| MonoQ | 1.368 | 5.7 | 4.189 | 106.9 | 55 |
| Gel filtration | 0.684 | 3.9 | 5.7 | 145.4 | 38 |
FIGURE 2.

Coomassie Blue-stained SDS-PAGE on purified Aro from S22. Lane 1, molecular weight standards; lane 2, purified enzyme. Identification of subunits was confirmed by N-terminal sequencing.
The complete DNA sequence of the aroA and aroB genes has been obtained. The deduced amino acid sequence of AroA displays 99, 81, 72, and 71% identity with AroA from SY8, A. faecalis, H. arsenicoxydans, and Burkholderia multivorans, respectively (supplemental Fig. S1). The deduced amino acid sequence of AroB displays 99, 69, 57, and 54% identity with SY8, A. faecalis, B. multivorans, and H. arsenicoxydans AroB, respectively (supplemental Fig. S2).
Purification and Characterization of Two Cyts Able to React with Aro
During the first DEAE chromatographic step, we detected the presence of c-type cyts in the unretained fraction. Because in vitro enzymatic experiments revealed that purified Aro was able to reduce this fraction in the presence of AsIII, we decided to purify its constituent heme proteins. We isolated three cyts using a CM-52 cation exchange column and a gel filtration column successively and named them cyts c551, c552, and c554 according to their respective α-bands in the reduced state. In contrast to cyt c551, cytochromes c552 and c554 were both able to react with isolated Aro (Fig. 3) and were therefore further characterized.
FIGURE 3.

Spectra of c552 and c554 cytochromes enzymatically reduced by purified Aro. Oxidized cyts (black lines) were incubated for 3 min with the enzyme (dotted lines); c552 (A) and c554 (B). Insets show the α-bands of cytochrome c552 and c554 absorbing at 552 and 554 nm, respectively.
The N-terminal sequence (54 residues) of cyt c552 (Fig. 4A) revealed a high identity (75%) with cyts of Cupriavidus necator (formerly Ralstonia eutropha) H16, Cupriavidus taiwanensis and Cupriavidus metallidurans (formerly Ralstonia metallidurans) CH34 (for which genome sequence are available) encoded by genes located in the sox cluster. The N-terminal sequence (56 residues) of cyt c554 (Fig. 4B) revealed this protein to be the product of the aroC gene located in the S22 aro cluster. Its sequence is astonishingly similar to that translated from the cyt gene present in the arsenite oxidase aox operon (10) of SY8 (99%) and shares only 45 and 43% identity with cyts c551/552 of Burkholderia multivorans and Burkholderia cenocepacia, respectively. Between each other, the two cyts c554 and c552 from S22 are <40% identical.
FIGURE 4.

Sequence alignment of cytochromes purified from S22. The N-terminal sequence of purified cyt c552 (c552_S22; A) is compared with the sequences of cyts from Cupriavidus necator H16 (cyt_Cnec; GenBankTM accession no. YP_727996), C. taiwanensis (cyt_Ctai; GenBankTM accession no. YP_002007005) and C. metallidurans CH34 (cyt_Cmet; GenBankTM accession no. YP_585565), whereas the N-terminal sequence of c554 (c554_S22; B) is compared with the sequences of cyts from Achromobacter sp. SY8 (cyt_SY8; GenBankTM accession no. ABP63661), Burkholderia multivorans (cyt_Bmul; GenBankTM accession no. YP_001578392) and B. cenocepacia (cyt_Bcen; GenBankTM accession no. YP_002229329).
The molecular masses, determined by MALDI-TOF mass spectrometry, of cyts c552 and c554 are 9615 ± 2 and 9648 ± 2 Da, respectively. The calculated molecular masses of the cyts retrieved from the Cupriavidus genomes and from the S22 aro operon, however, are both higher, around 11,600 Da. Indeed, in both the c552 and c554 precursor sequences, a 20-amino acid stretch was predicted, using the SignalP program (40), to be a signal peptide characteristic of the Sec secretion pathway (see (41) for review). The prediction of the cleavage site was in perfect agreement with the determined N-terminal sequences VDA and APD (Fig. 4) for c552 and c554, respectively.
Both cyts have been purified in the monomeric state as judged from size exclusion chromatography (data not shown). The redox potentials Em of cyts c552 and c554 at pH 8 were determined at +230 ± 5 mV and +250 ± 5 mV, respectively (data not shown), close to the value determined for cyt c552 in NT-26 (27).
The fact that the aroC gene in the aro operon of S22 codes for cyt c554 strongly argued in favor of c554, rather than c552, being involved in AsIII oxidation. However, both cyts were found in approximately equal amounts when purified from cells grown on 5 mm AsIII. We therefore analyzed which of the cyts was induced by the presence of AsIII. Because the cyt c552 homologs in the sequenced Cupriavidus genomes are encoded by genes localized in the sulfur oxidation sox cluster, we also analyzed the cyt contents of cells grown with and without thiosulfate (known to be the regulator of the sox operon (42–45)). Cyt contents under these conditions were then compared with those obtained from cells grown with 5 mm AsIII. SDS-PAGE (Fig. 5A) detected an increased total amount of 10-kDa cyts in the cells grown either on thiosulfate or AsIII but could not discriminate between cyt c552 and c554 (Fig. 5A, lanes 5 and 6), due to their very similar molecular masses, as mentioned above. We therefore spectroscopically quantified each of the cyts contained in the DEAE-cyt fraction, under the tested growth conditions (Fig. 5B). In the absence of AsIII or thiosulfate, cyt c552 represents 75% of the DEAE-cyt fraction content (again spectroscopically quantified), whereas cyt c554 amounts to ∼5% and c554 accounts for <2%. In the presence of 5 mm AsIII, the c552 fraction fell to 39% of the DEAE-cyt fraction content, whereas the c554 complement increased to at least 43%. The relative increase of cyt c554 quantity in response to AsIII therefore approximately parallels that observed for Aro activity (see above). In the presence of 20 mm thiosulfate, the contribution of c552 fell to 67% in relative cyt content but increased in absolute quantity as compared with the control conditions (Fig. 5A, lane 4 versus lane 2, and Fig. 5B, Thio versus N.A.). The proportion of c552 decreased due to the increase of c551 from 2 to 12%. Altogether, these results established not only that the accumulation of c551 is linked to the AsIII oxidation process but also suggested that production of c552 and c551 is linked to the thiosulfate oxidation pathway.
FIGURE 5.

A, 3,3′,5,5′-tetramethyl-benzidine- stained SDS-PAGE of cytochromes present in the S22 periplasm under different growing conditions. Lane 1, molecular weight standards; lane 2, cyts expressed without any addition; lane 3, cyts expressed in the presence of 5 mm AsIII; lane 4, cyts expressed in the presence of 20 mm thiosulfate; lane 5, purified cyt c554; lane 6, purified cyt c552. B, schematic representation of cyt content of the DEAE-cyt fraction under different growing conditions. Lane N.A., cyt content without any addition; lane As, cyt content with 5 mm AsIII; lane Thio, cyt content with 20 mm thiosulfate. Note that the DEAE-cyt fraction represent only part of the total cyt fraction (see “Experimental Procedures”) and that ∼20% of cyt content in this DEAE-cyt fraction remains unidentified under each of the growth conditions.
Enzymatic Study of the AsIII Oxidation System
As detailed above, the enzyme from S22 was able to reduce cyts c552 and c554 from S22 (Fig. 3). Several further cyts were tested for their reactivity with Aro. Bovine heart cyt c or Rhodobacter sphaeroides cyt c2 were not reduced by the S22 enzyme (data not shown), whereas cyt c555 from A. aeolicus and azurin from P. aeruginosa were (data not shown). Due to the limited quantities of purified c552 and c554, in-depth kinetic studies were performed using cyt c555 from A. aeolicus and yielded a Km of 6 μm for AsIII (Table 3) as well as substrate inhibition at concentrations higher than 100 μm. We therefore analyzed the affinity for the reacting cyts at a concentration of 100 μm AsIII. Kinetic data, analyzed by reciprocal plots, yielded a Km value of 50 μm for cyt c555 (Table 3). With the aim to compare the efficiency of cyts and DCPIP as electron acceptors, we first measured kinetics at pH 6, i.e. the pH value determined by Anderson et al. (22) and ourselves (data not shown) to be optimal with this electron acceptor. The Vm obtained using c555 at this pH value is an order of magnitude higher than the Vm determined using DCPIP at the same pH. However, kinetics measured at other pH values yielded the range of pH 8–9 as optimal for electron transfer from Aro to the cyts and led to a further doubling of Vm. Preliminary kinetic studies with cyts c552 and c554 purified from S22 yielded Km values of 13 μm and 7 μm, respectively, at a pH of 8.5.
TABLE 3.
Kinetic properties of the Aro from S22
*Vmax is expressed as μm Asoxidized × min−1 mg−1. It is noteworthy that 1 mole of AsIII reduces 1 mole of DCPIP but 2 moles of cyt. N.D., not determined.
| e− donor | AsIII | |||
| Km (μm) | 6 | |||
| Inhibitors | Sulfite | Sulfide | ||
| I50 (μm) | 10 | 70 | ||
| e− acceptor | c555 | c552 | c554 | DCPIP |
| Km (μm) | 50 | 13 | 7 | N.D. |
| Vmax* | 152 | 140 | 140 | 5.7 |
Because no data have been reported to identify potential inhibitors of Aro, we screened the effect of selected chemicals on the Aro activity. We first tested the product of the physiological reaction, AsV, as potential inhibitor and found no effect. We observed any inhibitory effect by NaN3, a well known inhibitor of several other molybdopterin enzymes (46, 47). Using cyts as electron acceptors allowed us to test sulfur compounds that otherwise would reduce DCPIP directly. Because H2S has been reported to strongly inhibit AsIII oxidation of Hydrogenobaculum whole cells (48), we tested this compound on Aro and indeed found an inhibitory effect on our enzyme. We did not perform a detailed enzymatic analysis in the presence of sulfide but observed an I50 (sulfide concentration yielding 50% inhibition) of ∼70 μm. A further sulfur compound, i.e. sulfite (but not thiosulfate), appeared to strongly inhibit Aro. A more detailed enzymatic analysis in the presence of sulfite showed a “mixed mode of inhibition” (data not shown), i.e. with not only an effect on affinity (Km) but also on catalysis (Km/Vm), with an I50 of 10 μm. Precise kinetics parameters were obtained with the A. aeolicus cyt but were always verified using the S22 cyts.
As cyt c552 potentially participates in thiosulfate oxidation, we assayed whether the Aro featured measurable thiosulfate or sulfite oxidase activity. This was not the case (data not shown).
DISCUSSION
As mentioned in the introduction, many different aspects of the Aro enzymes have been studied in a variety of species, although an exhaustive characterization of a single case is still lacking. We took advantage of the availability of a new species of AsIII oxidizer, the β-proteobacterium S22, to perform a comprehensive study of its Aro covering its phylogenetic positioning, expression properties, biochemical and biophysical (see Ref. 66, accompanying article), as well as enzymatic parameters and its interaction with potential redox partners.
Aro from S22, a Clear Cut Case of Lateral Gene Transfer
Previous phylogenetic studies on the molybdopterin subunit of Aro (23, 49) suggested this enzyme to have evolved with its parent species. These conclusions have later been confirmed by the study of its Rieske subunit (50). Analysis of binary similarity data showed the 16S rDNA sequence of S22 to be more closely related to H. arsenicoxydans (91% identity) and B. multivorans (92%) species than to SY8 (86%) and A. faecalis (89%) species. As described above, however, both the sequences of the molybdopterin and the Rieske subunits of Aro from S22 cluster with those of SY8 and A. faecalis enzymes instead of those from B. multivorans and H. arsenicoxydans species (see “Results”). This evolutionary relationship is further corroborated by the sequence that we determined for cytochrome c554. The deduced identity of the AroA, AroB, and AroC sequences from S22 with the AoxB, AoxA, and AoxC sequences from SY8 were so suspiciously high (99%) that we were led to verify by 16S rDNA sequencing that our S22 strain was pure even when grown in 5 mm AsIII. These results therefore suggest that the entire aro operon of S22 has been acquired from an Achromobacter-related species by horizontal gene transfer. A detailed examination of the AroA-based phylogenetic tree (Fig. 2 in Ref. 49) pinpoints several abnormal branches as compared with the phylogenetic tree of the parent species. In some of these cases, discrepancies may be due to misalignments. The high homology between the SY8 and S22 sequences, however, does not allow for such a possibility for S22. This case, therefore, constitutes the first unambiguous example of lateral gene transfer in the evolutionary pathway of Aro. It is noteworthy, though, that this lateral gene transfer occurred within the β-proteobacterial subclass and thus does not significantly blur the picture of an overall coevolution of the enzyme and its parent species.
Is Aro a Membrane-bound Enzyme?
Muller et al. (17) observed a twin arginine translocation signal peptide in the H. arsenicoxydans AroB protein, and the presence of such a signal has now been detected in all available AroB sequences (18 and Fig. 6). This observation raises the question of whether this signal peptide is cleaved after the protein has been transported across the cytoplasmic membrane or whether it serves to anchor Aro to the membrane. Cleavage sites were predicted (via the method developed by Bendtsen et al. (51)) for all Aro-Rieske proteins but not for the Rieske subunit of Rieske-cyt b complexes (a homologous protein, see Ref. 66, accompanying article). The latter result is in line with the following. i) All Rieske/cyt b Rieskes characterized so far have been demonstrated to be membrane bound with their uncleaved twin arginine translocation signal peptide serving as an anchor (52). ii) The predicted cleavage sites in Aro-Rieskes correspond to the N terminus determined in the isolated enzymes. Nevertheless, three types of Aro can be found. The C. aurantiacus and 3As enzymes correspond to the “rather membranous type” i.e. retained in the membrane fraction even after harsh treatment (23, 24). The NT-26 and Hydrogenophaga NT-14 (NT-14) enzymes belong to the class of “rather soluble type” Aros present in the soluble fraction even after mild treatment (18, 21). Intermediate between these extreme cases are the H. arsenicoxydans, A. faecalis, Arthrobacter, and S22 cases, for which the relative abundance in the membrane and the soluble fractions varies as a function of the harshness of cell disruption (5, 17, 22 and present work).
FIGURE 6.

Prediction of twin arginine translocation signal cleavage site in the N-terminal sequences of the Rieske subunit from Aros and Rieske-cyt b complex. Prediction has been performed using the method developed by Bendtsen et al. (51). The first residue after the cleavage site is colored in gray, whereas the observed first residue in the purified enzyme is boxed.
A scenario reconciling all results obtained so far, already proposed by Santini and vanden Hoven (18), consists in the attachment of AroAB to the membrane via another protein. This attachment must be of variable strength to explain the ensemble of the data and structurally specific as the Rieske protein has been observed to be oriented in a defined geometry (23) on Chloroflexus membranes. This question is reminiscent of the problem arising from the study of the AsV reductase enzyme (49). Some of the representatives of this family have been isolated as membrane-associated and others as soluble. However, in all characterized AsV reductase enzymes, a membrane-associated component, which can be variable proteins, has been identified (53–55). We can imagine a similar scenario for the Aro enzyme, with the corresponding membrane-attached partner still to be identified. To address this question, we are currently studying the case of the H. arsenicoxydans Aro, as this enzyme can indeed be easily obtained in a membrane-associated form (see “Results”).
Aro Displays Strong Selectivity toward Its Electron Transfer Partners
Whereas the Aro enzyme may be more or less tightly membrane-associated in different species, it invariably appears to reduce soluble periplasmic electron carrier proteins. To the exception of one previous study (27), nonphysiological electron acceptors have been employed in activity tests, and the physiological electron acceptors have been deduced merely from genetic arguments. To place conclusions concerning the interaction of Aro with its redox partners on firm ground, we have studied coexpression profiles and electron transfer activities of soluble cyts interacting with Aro in S22. The obtained results strongly suggest the physiological electron carrier of the S22 Aro to be cyt c554, i.e. the cyt present in the aro cluster. Spectroscopic quantification clearly established a strong increase of the content of cyt when cells were grown on AsIII. This result suggests that c554 gene expression is induced by AsIII. It is of note that only four species were shown to have a cyt gene co-transcribed with the aroA and aroB genes and hence induced by AsIII: Agrobacterium tumefaciens, H. arsenicoxydans, 3As, and O. tritici (26, 28, 56, 57).
As we have shown, cyt c554 is not the only cyt reacting with Aro in S22. Another cyt, c552, dominant in the absence of AsIII but still as abundant as c554 in the presence of AsIII, accepts electrons from Aro and therefore mediates high turnover of the enzyme. Although no equivalent study has been performed on other bacteria, this result correlates well with circumstantial observations made in other organisms. For example, the Δc552 mutant of NT-26 is still capable of autotrophic growth on AsIII (27), suggesting that at least one other carrier may accept electrons from Aro. Finally, the recent sequencing and characterization of the aro operon in 3As identified two cyts as being co-transcribed with aroAB (57). The Aro enzyme therefore appears to often be able to interact with several different electron carriers in the same organism and the production of these carriers can either be regulated by AsIII or not. In stark contrast to this stands the clear cut and strong discrimination against a specific subgroup of type I cyt as exemplified by horse heart or R. sphaeroides cyts. This selectivity is striking and not related to the redox potential of the carrier because all of the above cited carriers have Em pH 8 values ∼+240 mV. We therefore have initiated an in-depth study of this phenomenon in a range of different Aro enzymes. The corresponding results and a structural rationalization for this selectivity, observed in all examined Aros, will be published elsewhere.6
Inhibitory Effects of Sulfur Compounds and Metabolic Significance Thereof
Arsenic and sulfur often coexist in the environment and share similar microbial transformations. The study of the effect of sulfur compounds on AsIII oxidation is therefore indispensable to understand how both metabolisms interact. Our work revealed inhibitory effects of both sulfide and sulfite, with apparent I50 of 70 μm and 10 μm, respectively. Are these compounds true enzymatic inhibitors? A concentration of 60 μm sulfide has been observed to completely stop AsIII oxidation in whole cells of Hydrogenobaculum at low pH (48), whereas sulfide has been reported to strongly enhance the AsIII oxidation in Mono Lake samples at high pH (58). However, did sulfide act on the Aro directly in these cases? Sulfide has been shown to react with AsIII, forming orpiment at low pH (59) and thioarsenic species at high pH (58, 60). In these three cited works, sulfide was added in equal if not higher quantities compared with AsIII, allowing the product of the reaction between AsIII and sulfide to significantly modify the AsIII quantity available for Aro-mediated oxidation. The observed effects are therefore potentially nonenzymatic. In our case, sulfide shows significant inhibitory effect at concentrations where, whatever the sulfide/AsIII reaction product, free AsIII is always saturating. Our work is therefore the first to clearly establish a true inhibitory effect of sulfide on the enzymatic AsIII oxidation.
Sulfite is another sulfur compound revealed by our work to be a strong inhibitor of the Aro enzyme from S22. A conflicting result was published by Phillips (61) on A. faecalis whole cells, but we observed a similar inhibition also on the NT-26 enzyme. In both cases, the inhibition appears to be of a mixed-type inhibition, i.e. both competitive and noncompetitive. The competitive character, i.e. the effect on the Km for AsIII suggests that sulfite reacts at the molybdopterin center. Because dialysis restores the activity, the binding of sulfite seems to be transient. This effect appears, therefore, to be distinct from the irreversible effect described for sulfite oxidase (a molybdopterin enzyme) in presence of AsIII (62).
An increasing number of studies find relationships between sulfur and arsenic metabolism. Interest has been focused on the interaction between AsV and sulfate reduction (see Refs. 63, 64 for original works). More recently, the interaction between AsIII oxidation and sulfur metabolism was addressed (48, 58, 65), but it has not been studied at the molecular level prior to this work. Although ability of Aro to use cyt c552 as electron acceptor may be interpreted as a possible cross-talk between sulfur and arsenic metabolisms, this merging of electron transfer pathways is not obligatory because Aro possesses a second, dedicated, acceptor, i.e. cyt c554. The more straightforward link between AsIII and sulfur oxidation processes revealed by our work consists in the sensitivity of Aro to two substrates of the sox system, i.e. sulfite and sulfide. The sox system, well characterized in Paracoccus pantotrophus and coded by 15 genes, catalyzes thiosulfate-, sulfite-, sulfur-, and sulfide-dependent cyt c reduction (43, 44). We can therefore imagine that the sox system, will enhance the AsIII oxidation rate of S22, by promoting the consumption of sulfite and sulfide.
Supplementary Material
Acknowledgments
We are indebted to Maya Belghazi from CAPM-IFR Jean Roche for access to the Ultraflex II mass spectrometer. We furthermore thank Pascale Infonsi for the gift of A. aeolicus cyt c555.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
The nucleotide sequence(s) reported in this paper has been submitted to the DDBJ/GenBankTM/EBI Data Bank with accession number(s) EU304284, EU304273, and GQ904715.
S. Duval and B. Schoepp-Cothenet, unpublished data.
- AsV
- arsenate
- Aro
- arsenite oxidase
- 3As
- Thiomonas sp. 3As
- AMPSO
- 3-((1,1-dimethyl-2-hydroxyethyl)amino)-2-hydroxypropanesulfonic acid
- AsIII
- arsenite
- CAPS
- N-cyclohexyl-3-aminopropanesulfonic acid
- CDM
- chemically defined medium
- cyt
- cytochrome
- DCPIP
- 2,4-dichlorophenolindophenol
- FPLC
- fast protein liquid chromatography
- HPLC-ICP AES
- high performance liquid chromatography-inductively coupled plasma atomic emission spectrophotometry
- MES
- 2-(N-morpholino)ethanesulfonic acid
- MOPS
- 3-(N-morpholino)propanesulfonic acid
- NT-14
- Hydrogenophaga NT-14
- NT-26
- Rhizobium NT-26
- S22
- Ralstonia sp. 22
- SY8
- Achromobacter sp. SY8
- MALDI-TOF
- matrix-assisted laser desorption ionization time-of-flight.
REFERENCES
- 1.Nriagu J. O. (1994) Arsenic in the Environment. John Wiley and Sons, Inc, New York [Google Scholar]
- 2.Oremland R. S., Stolz J. F. (2005) Trends Microbiol. 13, 45–49 [DOI] [PubMed] [Google Scholar]
- 3.Inskeep W. P., McDermott T. R., Fendorf S. (2002) Environmental Chemistry of Arsenic (Frankenberger W. T. ed) pp. 183–215, Marcel Dekker, Inc. New York [Google Scholar]
- 4.Green H. H. (1918) S. Afr. J. Sci. 14, 465–467 [Google Scholar]
- 5.Prasad K. S., Subramanian V., Paul J. (2009) Biometals 22, 711–721 [DOI] [PubMed] [Google Scholar]
- 6.Hamamura N., Macur R. E., Korf S., Ackerman G., Taylor W. P., Kozubal M., Reysenbach A. L., Inskeep W. P. (2009) Environ. Microbiol. 11, 421–431 [DOI] [PubMed] [Google Scholar]
- 7.Chang J. S., Yoon I. H., Lee J. H., Kim K. R., An J., Kim K. W. (2010) Environ. Geochem. Health 32, 95–105 [DOI] [PubMed] [Google Scholar]
- 8.Valenzuela C., Campos V. L., Yañez J., Zaror C. A., Mondaca M. A. (2009) Bull. Environ. Contam. Toxicol. 82, 593–596 [DOI] [PubMed] [Google Scholar]
- 9.Sun W., Sierra-Alvarez R., Fernandez N., Sanz J. L., Amils R., Legatzki A., Maier R. M., Field J. A. (2009) FEMS. Microbiol. Ecol. 68, 72–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cai L., Rensing C., Li X., Wang G. (2009) Appl. Microbiol. Biotechnol. 83, 715–725 [DOI] [PubMed] [Google Scholar]
- 11.Cai L., Liu G., Rensing C., Wang G. (2009) BMC Microbiol. 9, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Handley K. M., Héry M., Lloyd J. R. (2009) Int. J. Sys. Evol. Microbiol. 59, 886–892 [DOI] [PubMed] [Google Scholar]
- 13.Silver S., Phung L. T. (2005) Appl. Environ. Microbiol. 71, 599–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stolz J. F., Basu P., Santini J. M., Oremland R. S. (2006) Annu. Rev. Microbiol. 60, 107–130 [DOI] [PubMed] [Google Scholar]
- 15.Hoeft S. E., Blum J. S., Stolz J. F., Tabita F. R., Witte B., King G. M., Santini J. M., Oremland R. S. (2007) Int. J. Syst. Evol. Microbiol. 57, 504–512 [DOI] [PubMed] [Google Scholar]
- 16.Kulp T. R., Hoeft S. E., Asao M., Madigan M. T., Hollibaugh J. T., Fisher J. C., Stolz J. F., Culbertson C. W., Miller L. G., Oremland R. S. (2008) Science 321, 967–970 [DOI] [PubMed] [Google Scholar]
- 17.Muller D., Lièvremont D., Simeonova D. D., Hubert J. C., Lett. M. C. (2003) J. Bacteriol. 185, 135–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Santini J. M., vanden Hoven R. N. (2004) J. Bacteriol. 186, 1614–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nasser D., Nester E. W. (1967) J. Bacteriol. 94, 1706–1714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Santini J. M., Sly L. I., Schnagl R. D., Macy J. M. (2000) Appl. Environ. Microbiol. 66, 92–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.vanden Hoven R. N., Santini J. M. (2004) Biochim. Biophys. Acta 1656, 148–155 [DOI] [PubMed] [Google Scholar]
- 22.Anderson G. L., Williams J., Hille R. (1992) J. Biol. Chem. 267, 23674–23682 [PubMed] [Google Scholar]
- 23.Lebrun E., Brugna M., Baymann F., Muller D., Lièvremont D., Lett M. C., Nitschke W. (2003) Mol. Biol. Evol. 20, 686–693 [DOI] [PubMed] [Google Scholar]
- 24.Duquesne K., Lieutaud A., Ratouchniak J., Muller D., Lett M. C., Bonnefoy V. (2008) Environ. Microbiol. 10, 228–237 [DOI] [PubMed] [Google Scholar]
- 25.Ellis P. J., Conrads T., Hille R., Kuhn P. (2001) Structure 9, 125–132 [DOI] [PubMed] [Google Scholar]
- 26.Kashyap D. R., Botero L. M., Franck W. L., Hassett D. J., McDermott T. R. (2006) J. Bacteriol. 188, 1081–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Santini J. M., Kappler U., Ward S. A., Honeychurch M. J., vanden Hoven R. N., Bernhardt P. V. (2007) Biochim. Biophys. Acta 1767, 189–196 [DOI] [PubMed] [Google Scholar]
- 28.Branco R., Francisco R., Chung A. P., Morais P. V. (2009) App. Environ. Microbiol. 75, 5141–5147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaback H. L. (1971) Methods Enzymol. 22, 99–122 [Google Scholar]
- 30.Baymann F., Tron P., Schoepp-Cothenet B., Aubert C., Bianco P., Stetter K. O., Nitschke W., Schütz M. (2001) Biochemistry 40, 13681–13689 [DOI] [PubMed] [Google Scholar]
- 31.Laemmli U. K. (1970) Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 32.Judd R. C. (2002) in The Protein Protocols Handbook, (Walker J. M. ed) 2nd Ed., pp. 73–76, Humana Press, Inc., Ottawa, Ontario, Canada [Google Scholar]
- 33.Sambrook J., Fritsch E. F., Maniatis T. (1989) in Molecular Cloning: A Laboratory Manual, 2nd Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 34.Thompson J. D., Higgins D. G., Gibson T. J. (1994) Nucleic Acids Res. 22, 4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Simeonova D. D., Lièvremont D., Lagarde F., Muller D. A., Groudeva V. I., Lett M. C. (2004) FEMS Microbiol. Lett. 237, 249–253 [DOI] [PubMed] [Google Scholar]
- 36.Weeger W., Lièvremont D., Perret M., Lagarde F., Hubert J. C., Leroy M., Lett M. C. (1999) Biometals 12, 141–149 [DOI] [PubMed] [Google Scholar]
- 37.Lim C. K., Cooksey D. A. (1993) J. Bacteriol. 175, 4492–4498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dutton P. L. (1971) Biochim. Biophys. Acta 226, 63–80 [DOI] [PubMed] [Google Scholar]
- 39.Duval S. (2008) Les enzymes du métabolisme de l'arsenic chez les procaryotes, aspects fonctionnels, phylogénétiques et structuraux. Ph.D. thesis, Université de Provence, Marseilles, France [Google Scholar]
- 40.Bendtsen J. D., Nielsen H., von Heijne G., Brunak S. (2004) J. Mol. Biol. 340, 783–795 [DOI] [PubMed] [Google Scholar]
- 41.Pugsley A. P. (1993) Microbiol. Rev. 57, 50–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kelly D. P., Shergill J. K., Lu W. P., Wood A. P. (1997) Antonie Leeuwenhoek 71, 95–107 [DOI] [PubMed] [Google Scholar]
- 43.Friedrich C. G., Quentmeier A., Bardischewsky F., Rother D., Kraft R., Kostka S., Prinz H. (2000) J. Bacteriol. 182, 4677–4687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rother D., Henrich H. J., Quentmeier A., Bardischewsky F., Friedrich C. G. (2001) J. Bacteriol. 183, 4499–4508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hensen D., Sperling D., Trüper H. G., Brune D. C., Dahl C. (2006) Mol. Microbiol. 62, 794–810 [DOI] [PubMed] [Google Scholar]
- 46.Enoch H. G., Lester R. L. (1975) J. Biol. Chem. 250, 6693–6705 [PubMed] [Google Scholar]
- 47.Forget P. (1971) Eur. J. Biochem. 18, 442–450 [DOI] [PubMed] [Google Scholar]
- 48.Donahoe-Christiansen J., D'Imperio S., Jackson C. R., Inskeep W. P., McDermott T. R. (2004) Appl. Environ. Microbiol. 70, 1865–1868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Duval S., Ducluzeau A. L., Nitschke W., Schoepp-Cothenet B. (2008) BMC Evol. Biol. 8, 206–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lebrun E., Santini J. M., Brugna M., Ducluzeau A. L., Ouchane S., Schoepp-Cothenet B., Baymann F., Nitschke W. (2006) Mol. Biol. Evol. 23, 1180–1191 [DOI] [PubMed] [Google Scholar]
- 51.Bendtsen J. D., Nielsen H., Widdick D., Palmer T., Brunak S. (2005) BMC Bioinformatics 6, 167–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kallas T. (1994) in The Molecular Biology of Cyanobacteria (Bryant D. A. ed.) pp. 259–317, Kluwer Academic Publishers, the Netherlands [Google Scholar]
- 53.Saltikov C. W., Newman D. K. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 10983–10988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Malasarn D., Keeffe J. R., Newman D. K. (2008) J. Bacteriol. 190, 135–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Murphy J. N., Saltikov C. W. (2007) J. Bacteriol. 189, 2283–2290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Muller D. (2004) Analyse génétique et moléculaire du stress arsenic de souches bactériennes isolées d'environments contaminés par l'arsenic. Ph.D. thesis, Université de Strasbourg, France [Google Scholar]
- 57.Slyemi D., Ratouchniak J., Bonnefoy V. (2007) Adv. Mat. Res. 20–21, 427–430 [Google Scholar]
- 58.Fisher J. C., Wallschläger D., Planer-Friedrich B., Hollibaugh J. T. (2008) Environ. Sci. Technol. 42, 81–85 [DOI] [PubMed] [Google Scholar]
- 59.Rochette E. A., Bostick B. C., Li G. C., Fendorf S. (2000) Environ. Sci. Technol. 34, 4714–4720 [Google Scholar]
- 60.Stauder S., Raue B., Sacher F. (2005) Environ. Sci. Technol. 39, 5933–5939 [DOI] [PubMed] [Google Scholar]
- 61.Phillips S. E., Taylor M. L. (1976) Appl. Environ. Microbiol. 32, 392–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gardlik S., Rajagopalan K. V. (1991) J. Biol. Chem. 266, 16627–16632 [PubMed] [Google Scholar]
- 63.Dowdle P. R., Laverman A. M., Oremland R. S. (1996) Appl. Environ. Microbiol. 62, 1664–1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Newman D. K., Kennedy E. K., Coates J. D., Ahmann D., Ellis D. J., Lovley D. R., Morel F. M. (1997) Arch. Microbiol. 168, 380–388 [DOI] [PubMed] [Google Scholar]
- 65.Langner H. W., Jackson C. R., McDermott T. R., Inskeep W. P. (2001) Environ. Sci. Technol. 35, 3302–3309 [DOI] [PubMed] [Google Scholar]
- 66.Duval S., Santini J. M., Nitschke W., Hille R., Schoepp-Cothenet B. (2010) J. Biol. Chem. 285, 20442–20451 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.