

Abstract
Aggregation and misfolding of the prion protein (PrP) are thought to be the cause of a family of lethal neurodegenerative diseases affecting humans and other animals. Although the structures of PrP from several species have been solved, still little is known about the mechanisms that lead to the misfolded species. Here, we show that the region of PrP comprising the hairpin formed by the helices H2 and H3 is a stable independently folded unit able to retain its secondary and tertiary structure also in the absence of the rest of the sequence. We also prove that the isolated H2H3 is highly fibrillogenic and forms amyloid fibers morphologically similar to those obtained for the full-length protein. Fibrillization of H2H3 but not of full-length PrP is concomitant with formation of aggregates. These observations suggest a “banana-peeling” mechanism for misfolding of PrP in which H2H3 is the aggregation seed that needs to be first exposed to promote conversion from a helical to a β-rich structure.
Keywords: Biophysics, Neurological Diseases, Prions, Protein Conformation, Protein Stability, Protein Structure
Introduction
Transmissible spongiform encephalopathies are fatal neurodegenerative pathologies that affect humans as well as several other mammalian species. They are thought to be caused by the aggregation and misfolding of the prion protein (PrP).7 According to the “protein-only” hypothesis (1–3), PrP undergoes an α-to-β transition from its native state (PrPc) to a misfolded species (PrPsc), which is believed to act as a template to “infect” and misfold other PrP copies. As in other misfolding pathologies such as Alzheimer and Parkinson diseases, the neurotoxicity of PrPSc is thought to be associated to an oligomeric form of the protein rather than to the mature aggregates (4).
One of the crucial questions that remains unanswered concerns which region(s) of PrP promotes the polymerization process; this information would be both the key for understanding cross-species infectivity and help in decoding the bases of the aggregation process. Different regions have been proposed to be the fibrillogenic seed. PrPc consists of an unstructured N-terminal tail and a folded C-terminal domain formed by three helices (H1, H2, and H3) and a short-stranded β-sheet (formed by S1 and S2). H2 and H3 are connected through a disulfide bridge (5). A common view suggests the S1H1S2 region is crucial for β-sheet seeding and PrPSc formation (6, 7). H1 has been implicated as a primary interaction site between PrPSc and PrPc (8, 9), whereas the loop between S2 and H2, a rigid loop stabilized by its long range interactions with H3 (10), and the C terminus of H3 has been suggested to be recognized by a “Protein-X” that would affect the conversion of PrPc into PrPSc (11). A study based on intrachain distance estimation performed on tagged PrP amyloid fibrils obtained under chaotropic treatment suggests the involvement of the H2H3 domain of PrP in amyloid formation (12). H/D exchange studies of the amyloid fibrils from human PrP reveal that the β-sheet core of PrP amyloids is formed by H2, the major part of H3, and the loop between them (13, 14).
We have followed a different approach which is based on the identification of PrP fragments that reproduce the aggregation pattern and the oligomer populations observed for full-length PrP. We previously showed that the recombinant full-length ovine PrP (ovPrP) forms discrete soluble oligomeric species upon thermal unfolding through independent kinetic pathways (15) and that the fragment containing H2H3 is the only one able to reproduce this pattern of oligomerization (16). This evidence led us to suggest that H2H3 is a mini-PrP with properties that reproduce those of the full-length protein, whereas the rest of PrP does not contribute significantly to oligomer formation, stabilization, and assembly. During this study we had indications from molecular dynamics simulations that it is possible to misfold H2H3 in silico without the need of unfolding the domain first. Based on this evidence, the aggregation of H2H3 could proceed through a direct α-to-β transition because the peptide seemed to retain some secondary structure (16). If this were the case, this would imply that H2H3 hairpin is the seeding element for a native-like misfolding mechanism.
In the present work we have considered this hypothesis further and characterized the isolated H2H3 fragment to better understand the structural mechanism by which PrP misfolds. We show that the isolated H2H3 is able to form amyloid fibers with morphology similar to that of the full-length protein. Fibers can form regardless of using destabilizing conditions. By introducing a point mutation, we could obtain a species that is more resistant to aggregation and, hence, is better suited for studying its structure in solution. We prove that H2H3 retains the structure present in PrP and that the aggregation properties of the full-length protein are caused by the competition of the folding and aggregation pathways of this stable native-like element of the protein. The same behavior is observed for H2H3 from different organisms, suggesting that the properties of H2H3 are conserved despite minor species-specific mutations. Our work indicates conclusively that H2H3 is directly involved in the fibrillogenesis of PrP and sets a new paradigm of an aggregation mechanism in which self-assembly proceeds through a banana-peeling model. Our results propose a new perspective for studying the aggregation properties of PrP.
EXPERIMENTAL PROCEDURES
Protein Production
The full-length PrP from sheep (Ala-136—Arg-154—Gln-171 variant) (ovPrP) and mouse (moPrP) were expressed in Escherichia coli and purified as described previously (17). The H2H3 domain from different species was expressed and purified in the same conditions as the full-length except that refolding was performed at pH 8.5. The refolding efficiency into a monomeric and helical peptide is higher for mouse H2H3 (moH2H3) than for ovine H2H3 (ovH2H3), which hampers the production of significant amounts of ovH2H3 needed for biophysical studies. The ovH2_C182S (173–199) and ovH3_C217S (200–236) peptides were chemically synthesized by Peptide2.0. The numbering used throughout this manuscript refers to ovPrP (Swiss-Prot entry P23907).
Size Exclusion Chromatography
The oligomerization propensities of ovH2H3, ovH2H3_I208A, and ovPrP_I208A were studied by size exclusion chromatography using a TSK 4000SW column, applying the protocol for ovPrP described in a previous work (15).
NMR Measurements
The 15N- and 13C,15N-labeled ovH2H3_I208A samples used for NMR studies were typically ∼0.4 mm in 5 mm sodium citrate buffer at pH 3.5 containing 10% (v/v) D2O. All the NMR experiments were performed at 25 °C on a Bruker Avance spectrometer (equipped with cryoprobe) or on a Varian Inova spectrometer, both operating at 14.1 T (600 MHz). 15N-HSQC, HNCA, HN(CO)CA, 15N-NOESY-HSQC, and CC(CO)NH experiments were recorded and analyzed to obtain sequence specific 1HN, 15N, and 13Cαassignments. Side chain assignments were obtained by a combination of CC(CO)NH, HCCH-TOCSY, CBARO, 15N-NOESY-HSQC, and 1H,1H-TOCSY experiments. Interproton distance restraints were derived from 15N-NOESY-HSQC (100 ms) and 13C-NOESY-HSQC (100 ms) spectra acquired at 25 °C. All spectra were processed using NMRPipe/NMRDraw (18) and analyzed by Xeasy/Cara software (19). A set of 54 backbone ϕ and ψ dihedral angles was obtained using the backbone torsion angle prediction package Talos (20). T1, T2, and heteronuclear NOE measurements were performed using standard pulse sequences.
Structure Calculations
Structure calculations were performed using the Aria program (Version 2.2) (21). A typical run consisted of nine iterations. Twenty structures were calculated at each iteration by simulated annealing using the Parallhdg force field and the standard CNS protocol (22). Floating assignment and correction for spin diffusion during iterative NOE assignment were applied (23). At the end of each iteration the best seven structures in terms of lowest global energy were taken and used for assignment of additional NOEs during the following iterations. In the final iteration the number of structures generated was increased to 100. After refinement by molecular dynamics simulation in water of the 50 lowest-energy structures (24), the 12 lowest energy structures were selected as representative of the ovH2H3_I208A structure and used for statistical analysis. In total, 850 NOEs were assigned, 485 of which were unambiguous, and 365 were ambiguous. Of them, 338 were intraresidual, 202 were sequential, 141 were short range, 64 were medium range, and 105 were long range. Structure quality was evaluated using the programs Whatif (25) and Procheck (26). The program Molmol (27) was used to analyze the results of the structure calculations, and the program Pymol (28) was used to prepare figures. The coordinates are deposited in the Protein Data Bank (code 2ktm).
Circular Dichroism Studies
Circular dichroism (CD) measurements were carried out on a Jasco-715 spectropolarimeter equipped with a thermostatted cell holder controlled by a Jasco Peltier element. Far-UV CD spectra of ovH2H3_I208A, ovPrP, ovH2_C182S, and ovH3_C217S were recorded from 260 to 195 nm at 25 °C in a 1-mm path length quartz cuvette at a protein concentration of 20 μm in 5 mm citrate buffer at pH 3.5. Each CD spectrum was obtained by averaging 15 scans collected at a scan rate of 200 nm/min. Base-line spectra obtained with buffer were subtracted for all spectra. The thermal denaturation of ovH2H3_I208A was tested by heating a 20 μm sample from 5 to 97 °C at 2 °C/min and by monitoring the ellipticity at 220 nm with a 4-s averaging time and a 0.2 °C data pitch. The thermogram was analyzed assuming a two-state model. To study the conformational change accompanying aggregation for moH2H3 and moPrP, we incubated samples (350 μm in 20 mm MOPS and 150 mm NaF at pH 7.0) at 37 °C under shaking at 600 rpm. Far UV-CD spectra were collected at different incubation times upon a 7-fold dilution. Reduction experiments on ovH2H3_I208A were performed at pH 3.5 by the addition of 5 mm Tris(2-carboxyethyl)phosphine to the sample and recording CD spectra between 195 and 260 nm at 50 nm/min with a 4-s averaging time, each 5 min.
Light-scattering/Thioflavin T Fluorescence Correlation Experiments
The static light-scattering fluorescence kinetics experiments were performed on a homemade device using a 407-nm laser beam for the excitation of ThT and three other lasers (473, 533, and 633 nm) for light-scattering measurements. moPrP and moH2H3 samples (80 μm in 50 mm MOPS at pH 7.0) were incubated at 37 °C in a 2-mm cuvette in the presence of 150 μm ThT. The fluorescence at 485 nm and light-scattered signals were recorded at a 112° angle. Signal processing was achieved by a homemade MatLab program.
Transmission Electron Microscopy
Samples for electron microscopy analysis were prepared by incubating solutions of goat H2H3, moH2H3, ovPrP, and moPrP prepared at 80–300 μm under classical amyloid forming conditions (4 m urea, 200 mm NaCl in 5 mm acetate buffer at pH 5.0) with continuous shaking at 1000 rpm at 37 °C. Samples of ovH2H3_I208A, moH2H3, ovPrP, and moPrP were also prepared under native-like conditions (20 mm MOPS at pH 7.0 in presence of 150 mm NaCl) and stored at 37 °C under shaking conditions (1000 rpm). A droplet of 4 μl of the sample (diluted in H2O to ∼1 μm) was placed on freshly prepared carbon-coated and glow-discharged copper grids and left to adsorb for 30 s. After adsorption to the grid surface, the sample was washed briefly in milli-Q water subsequently stained with 1% (w/v) uranyl acetate for 30 s. Samples aggregated under native-like conditions for a longer time were diluted 1:2 with 8 m urea and sonicated for 1 min in a water bath sonicator before spotting on the grids. Micrographs of negatively stained areas were taken with a JEOL 1200 transmission electron microscope operating at 100 KV and at a magnification of 27,800.
RESULTS
H2H3 Is Sufficient for Fibrillization
We have shown elsewhere that H2H3 has kinetics of aggregation similar to those of PrP (16). To check whether it is also sufficient for fibrillization, we incubated different H2H3 variants in parallel to full-length PrP under conditions that are known to promote aggregation (200 mm NaCl and 4 m urea in 5 mm sodium acetate at pH 5.0 at 37 °C) (29, 30).
The morphology of the resulting aggregates was studied by transmission electron microscopy. The micrographs obtained after 48–72 h of incubation showed the appearance of linear unbranched fibrillar assemblies in all H2H3 and PrP variants (Fig. 1, A and B). Various lengths could be detected, with a uniform diameter and an average width of 7 nm. Upon prolonged incubation (1–3 weeks), we observed that the fibrils formed by H2H3 and PrP have a tendency for lateral association-forming fibers with a remarkable higher order assembly and a detectable twisting repeat (Fig. 1, C and D) with a diameter of ∼120 nm. The amyloid character of the fibrils formed from H2H3 and PrP was confirmed by ThT fluorescence (data not shown). These results demonstrate that the H2H3 fragment is sufficient to form amyloid fibers similar to those formed by PrP, suggesting that PrP fibrillization may be guided by H2H3.
FIGURE 1.
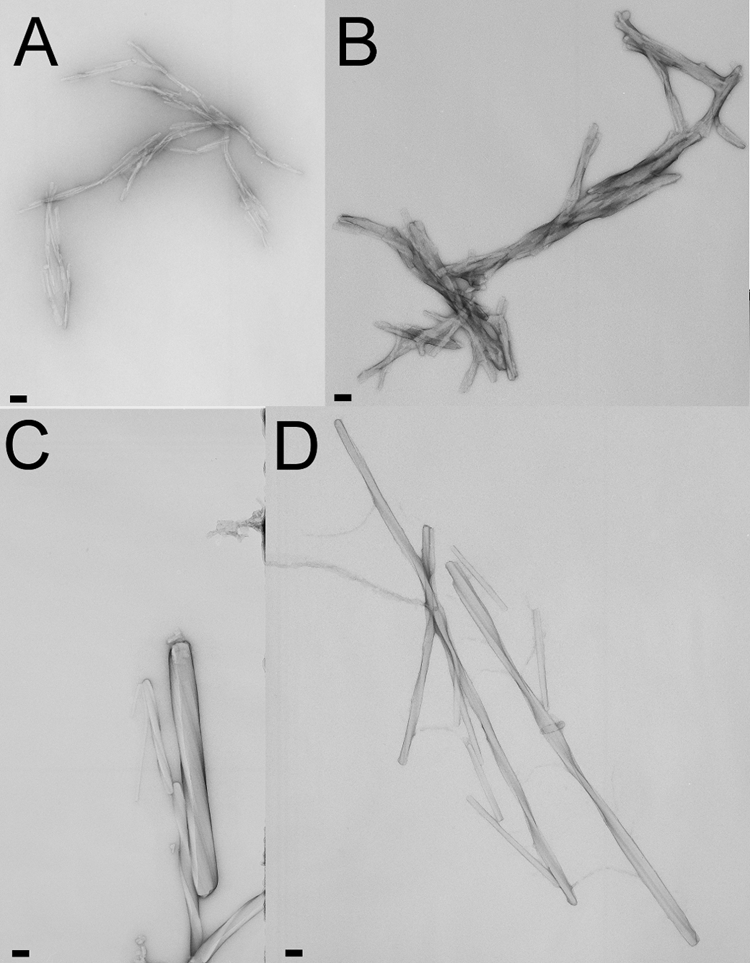
Amyloid character of H2H3 and full-length PrP fibers. Electron micrographs of negatively stained amyloid fibrils from goat H2H3 (A), moPrP (B), ovPrP (C) and moH2H3 (D) incubated in 5 mm sodium acetate (pH 5.0) with 200 mm NaCl and 4 m urea at 37 °C under constant shaking at 1000 rpm. The pictures where taken after 48–72h (A and B) and after 100 h (C and D). Bars correspond to 100 nm.
Identification of an H2H3 Variant Amenable to Structural Studies
We wanted to understand the structure of H2H3 in isolation of the rest of the domain to appreciate its involvement in fibrillization. However, because the aggregation kinetics are too fast for structural studies, we screened for mutations that would slow down the kinetics. We systematically mutated the hydrophobic residues of ovH2H3 and studied the effects on the polymerization kinetics using size exclusion chromatography. We found that introduction of a single I208A point mutation in either full-length ovPrP (ovPrP_I208A) or ovH2H3 (ovH2H3_I208A) is sufficient to drastically reduce oligomerization at pH 3.5 (Fig. 2A), although it does not abolish it altogether. We used this mutant to characterize the structure of isolated H2H3.
FIGURE 2.

Characterization of H2H3. A, size exclusion chromatogram of ovH2H3 (black), ovH2H3_I208A (red), and ovPrP_I208A (blue) after heating the samples for 60 min at 50 °C. The samples were prepared at 80 μm protein concentration in 20 mm citrate buffer at pH 3.5. B, secondary structure monitored by CD of ovH2H3_I208A (red) and ovPrP (black) at 20 μm protein concentration in 5 mm citrate buffer at pH 3.5. C, thermal denaturation scan of ovH2H3_I208A at 20 μm concentration in 5 mm MOPS at pH 7.0 monitored at 220 nm from 5 to 97 °C. The first scan is shown in red, whereas the second and the third scans recorded after refolding are shown in blue and black, respectively.
H2H3 Is Rich in α-Helical Structure and Unfolds Cooperatively
The far-UV CD spectra of freshly prepared ovH2H3_I208A is typical of an α-helical structure displaying two minima at 208 and 222 nm and compares well with that of ovPrP (Fig. 2B). The estimated α-helical content for ovH2H3_I208A is 39.9%, whereas ovPrP has an α-helix content of 22.7% as derived by using CDPro software. Thermal denaturation of ovH2H3_I208A at pH 7.0 shows a cooperative transition at 47.3 ± 0.8 °C (Fig. 2C). The same behavior was observed at pH 3.5, 5.0, and 7.0 and is independent from the protein concentration (data not shown). These results suggest that the isolated monomeric H2H3 retains its secondary structure.
H2H3 Retains the Structure Observed in the Full-length Protein
The structure of ovH2H3_I208A was solved by classical NMR methods and has excellent Whatif (25) and Procheck (26) scores (Table 1). This protein fragment gives well resolved 15N-HSQC spectra with an excellent dispersion in the two dimensions (supplemental Fig. S1).
TABLE 1.
Structural statistics for the calculations of the ovH2H3_I208A structure
Statistics were calculated for the 12 lowest energy structures after water refinement.
| Final NMR restraints | |
|---|---|
| Total distance restraints | 850 |
| Unambiguous/ambiguous | 485/365 |
| Intraresidue | 338 |
| Sequential | 202 |
| Short (i to i+2 to 3) | 141 |
| Medium (i to i +4 to 5) | 64 |
| Long range (i to i + >5) | 105 |
| Dihedral angle restraintsa | |
| Φ | 54 |
| Ψ | 54 |
| Deviation from idealized geometry | |
| Bond lengths (Å) | 0.005 ± 0.000 |
| Bond angles (°) | 0.703 ± 0.034 |
| Improper dihedrals (°) | 1.930 ± 0.128 |
| Restraint violations | |
| Distance restraint violation >0.5 Å | 0.0 |
| Dihedral restraint violation >5 Å | 0.3 |
| Coordinate precision (Å) with respect to the mean structureb | |
| Backbone of structure regions | 0.49 ± 0.12 |
| Heavy atoms of structured regions | 1.07 ± 0.19 |
| Whatif quality checkc | |
| First generation packing quality | 0.415 |
| Second generation packing quality | 0.230 |
| Ramachandran plot appearance | −2.743 |
| χ1−χ2 rotamers normality | −3.510 |
| Backbone conformation | −0.767 |
| Procheck Ramachandran statisticsc | |
| Most favored region (%) | 92.7 |
| Additional allowed regions (%) | 5.6 |
| Generously allowed regions (%) | 0.9 |
| Disallowed regions (%) | 0.9 |
The final representative family of the 12 lowest-energy structures after water refinement is superimposable on the average structure with overall root mean square deviation values of 0.49 ± 0.12 and 1.07 ± 0.19 Å for backbone and heavy atoms, respectively, in the region 168–229 (Fig. 3A). It consists of a short α-helix in the N-terminal region comprising the residues 170–173 followed by two longer α-helices that encompass the residues 175–196 (H2) and 203–228 (H3) (Fig. 3B). H2 is connected with H3 via a disulfide bridge formed between Cys-182 and Cys-217. The H2-H3 packing induces an inter-helix angle of ∼42°, which is observed also in most of the PrP structures (10, 31, 32).
FIGURE 3.

Solution structure of the ovH2H3_I208A. A, NMR bundle of the 12 lowest energy structures. The backbone is shown in blue, and the disulfide bridge formed between Cys-182 and Cys-217 is shown in red. B, average structure obtained by the Wheatsheaf algorithm (33). Two different views are shown with a 90° rotation. C, superposition of the corresponding NMR ovH2H3_I208A structure (in green) and crystallographic ovPrP_ARQ structure obtained in complex with the VRQ14 antibody (in red, Protein Data Bank code 1tpx).
When we submitted the average structure of H2H3 to structural similarity servers, we picked up the crystal structure of the ARQ polymorphism of ovPrP (1tpx.pdb) as the structure with the highest Z score (∼3.5). The two structures superpose with a root mean square deviation of 1.88 Å in the region 167–288 (Fig. 3C). The largest deviations are observed in the region T191-K197, where ovPrP is bound to an antibody (34). These results indicate that H2H3 is as an independently folded unit of PrP.
H2H3 Is Stabilized by Tertiary Interhelical Interactions and by the Disulfide Bridge
We then wondered that factors determine the fold stability of H2H3. We used the AGADIR software (35) to assess the helical propensity of H2H3 in silico on the basis of local stabilizing interactions in the sequence. The results showed that the sequences of H2 and H3 do not have any detectable tendency to be helical (data not shown). This was experimentally verified by individually probing the secondary structure of two synthetic peptides, ovH2_C182S (173–199) and ovH3_C217S (200–236), by CD (Fig. 4A). The spectra display the typical random coil profile, in agreement with the AGADIR output. This indicates that the α-helical structure of H2H3 must be stabilized by tertiary rather than secondary interactions, a phenomenon also observed in other all α-helical proteins (36, 37).
FIGURE 4.

Factors that stabilize the H2H3 fold. A, far UV-CD spectra of the ovH2H3_C182S (black) and ovH2H3_C217S (red) at 25 °C and 20 μm protein concentration in 5 mm citrate buffer pH 3.5. B, a schematic diagram of H2H3 with the observed long range interactions. C, time-dependent far UV-CD spectra of ovH2H3_I208A at 20 μm in 5 mm citrate buffer, pH 3.5, at 25 °C in the presence of 5 mm Tris(2-carboxyethyl)phosphine. Decreasing blue tonalities are plotted as a function of time from 0 to 4 h at 1-h intervals.
A detailed analysis of the H2H3 structure identified several specific hydrophobic and salt-bridge long range interactions in the helical interface that are directly supported by strong unambiguous NOEs (Fig. 4B). Many of them are highly conserved, as expected from their role in fold stabilization. Their importance is also supported by the observation that some of them are absent in disease cases. It has been shown for instance that mutation of V183I, E203K, R211H, V213I, and E214Q is associated with the development of Creutzfeldt-Jakob disease (38–42). Mutations of F201S (a residue important for inter-helical packing) and D205N are responsible for the development of Gerstmann-Straussler-Scheinker disease (43, 44).
Finally we verified the contribution of the disulfide bridge between Cys-182 and Cys-217 to the hairpin conformation. Upon reduction of the disulfide bond of ovH2H3_I208A with Tris(2-carboxyethyl)phosphine, we observed a decrease of the signal intensity with the simultaneous conversion of the α-helical CD profile of H2H3 to a typically β-strand spectrum (Fig. 4C). In the course of this experiment, the protein solution became turbid with protein aggregates visibly detected in the cuvette. This instantaneous α-to-β conversion upon reducing agent addition reflects the importance of the covalent link between H2 and H3 to maintain the structural integrity of the α-helical hairpin. Taken together these results indicate that both the long range interactions and the disulfide bond are crucial for H2H3 to maintain its autonomous monomeric α-helical hairpin structure.
H2H3 Is a Relatively Rigid Helical Hairpin
The dynamical properties of ovH2H3_I208A were evaluated from T1, T2, and {1H}-15N heteronuclear NOE relaxation parameters (Fig. 5). The T1 values are largely invariant, having on average relaxation values of 650 ms except for the last three C-terminal residues which exhibit larger values. The T2 and {1H}-15N heteronuclear NOE values are also very similar, with values around 100 ms and 0.8, respectively. Deviations from these values are observed for the first and the last four residues in the N and C termini, respectively, and for loop residues (197–202). The T2 and the heteronuclear NOE values of residues in the regions 193–197 (TTTTK) and 203–208 (ETDIK) are slightly different from those obtained for other α-helical residues. This suggests that the C terminus of H2 and the N terminus of H3 have a relatively higher mobility and/or are prone to conformational exchange, in agreement with H/D exchange experiments (48) and molecular dynamics simulations on PrP (49).
FIGURE 5.
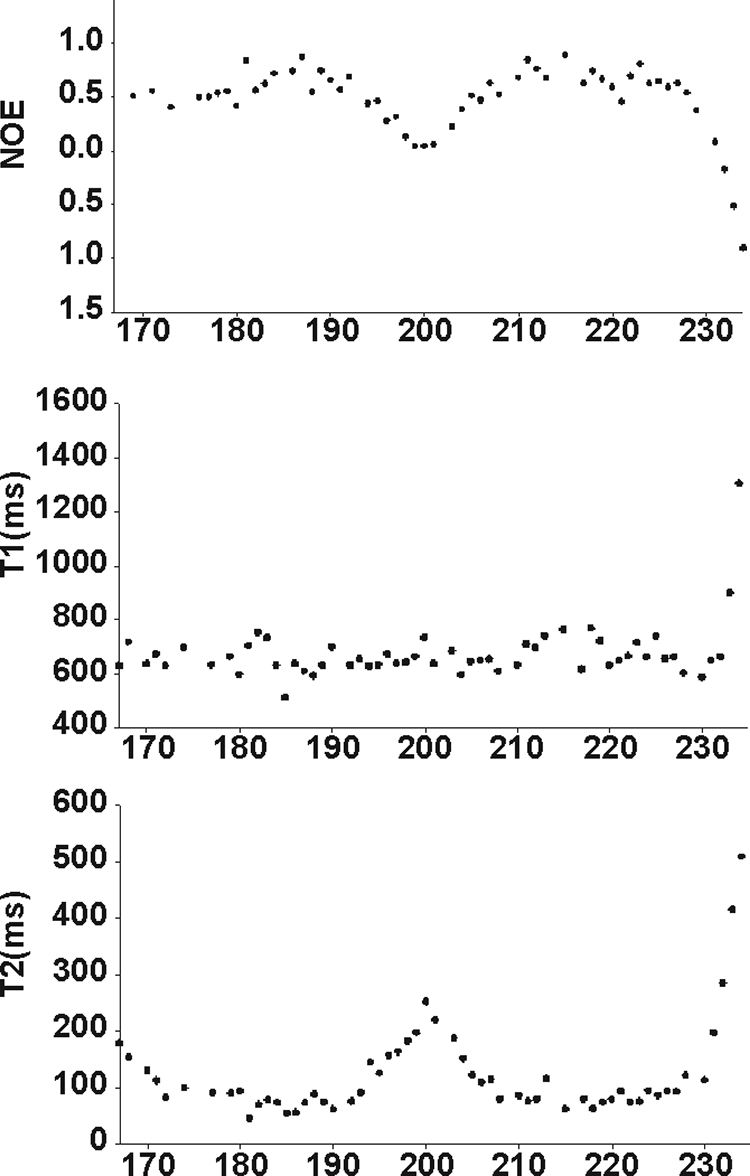
NMR relaxation data along the sequence of the ovH2H3_I208A. The data were recorded at 25 °C and 600 MHz.
Overall, these results indicate a relatively more rigid structure of the hairpin, leaving the interhelix loop and the termini more flexible. A similar dynamical behavior is observed in most mammalian PrPs structures (45–47).
H2H3 Forms Fibers Also under Native-like Conditions
Most studies of PrP aggregation make use of chemical denaturants and mildly acidic pH conditions (13, 50, 51). No specific rationale is usually given for these protocols, although it is reasonable that these conditions destabilize the fold and expose aggregation-prone regions. We reasoned that destabilizing conditions could in fact be needed to expose H2H3 from the remaining of the structure. In such a case, given the stability of this structural element, fibrillogenesis could occur also under native-like conditions in the absence of denaturing agents.
To test this hypothesis we incubated moH2H3 and moPrP in 20 mm MOPS and 150 mm NaF at pH 7.0 at 37 °C. Far UV-CD spectra revealed that both proteins undergo a conformational change with an α-to-β transition under native-like conditions (Fig. 6, A and B). After an overnight incubation, aggregates were observed in the cuvette, indicating that the α-to-β transition is linked to the aggregation process.
FIGURE 6.

H2H3 and PrP aggregates under native-like conditions. A and B, far UV-CD spectra of moH2H3 and moPrP, respectively, after 0 h (black) and 14 h (red) of incubation at 37 °C in 20 mm MOPS, pH 7.0, and 150 mm NaF under constant shaking. C and D, electron micrographs of negatively stained amyloid fibrils of moPrP and ovH2H3_I208A, respectively, incubated at 37 °C in 20 mm MOPS at pH 7.0 and 150 mm NaCl. The pictures where taken after 100 h of incubation. Scale bars represent 100 nm.
MoH2H3, moPrP, ovH2H3_I208A, and ovPrP samples were individually incubated while shaking at 37 °C to analyze the nature of the final aggregated state by transmission electron microscopy. The micrographs reveal the presence of fibrils after 100 h (Fig. 6, C and D), the amyloid nature of which was confirmed by ThT fluorescence. The amyloid fibrils seem to be generally similar to those grown under classical aggregation conditions (i.e. in the presence of NaCl and urea). Some morphological differences can, however, be detected. As compared with the fibers grown in the presence of chaotropic agents, fibrillogenesis seems to be much more massive. Upon prolonged incubation (1–3 weeks), the fibrils tend to cluster together into deposits too large to be characterized satisfactorily (data not shown). The fibers formed under native-like conditions are also overall more heterogeneous than those formed in the presence of urea. Our results clearly demonstrate that both full-length PrP and the α-helical H2H3 domain are capable of converting into insoluble, linear, and unbranched β-sheet-rich amyloid fibrils under native-like conditions without the need of unfolding the H2H3 domain.
Contribution of Regions outside H2H3 to PrP Aggregation
To determine the contribution of residues 23–167 in polymerization, we monitored the variation of size through multiwavelength light scattering and ThT fluorescence. Incubation of full-length moPrP (we used this variant because it has the kinetics most favorable for these studies) at 37 °C led to a rapid increase of the light scattering, whereas the ThT signal increased with a significant delay (Fig. 7A). This suggests that aggregation and fibrillization are uncoupled events. The behavior of moH2H3 is completely different; increase of the light-scattering signal is concomitant with ThT appearance, indicating that fibrillization and aggregation occur simultaneously (Fig. 7B). These results confirm the role of H2H3 as a central seeding element, which determines the kinetics of aggregation and fiber formation of PrP.
FIGURE 7.

Aggregation kinetics of the moPrP (A) and moH2H3 (B) monitored by static light scattering (SLS) (in blue) and ThT fluorescence (in red).
DISCUSSION
One of the critical challenges in the transmissible spongiform encephalopathy field is the determination of the aggregation pathway of PrP, the protein that is central to the pathology. A popular model, based on EM observations, proposes a β-helical structure that involves the PrP N- terminus and the S1H1S2 region whereby the H2 and H3 helices remain largely preserved (7, 52). An alternative model based on molecular dynamics postulates a structure with a β-core consisting of parallel and antiparallel β-strands formed by the regions 127–141 and 161–167 with H1, H2, and H3 retaining the native conformation (53). Both models agree with the idea that breaking the S1S2 interactions (54) and detachment of the S1H1S2 region from the rigid H2H3 (55) are the barrier of PrPc/PrPSc conversion. In apparent contrast to these models, site-directed spin labeling studies (12), mass spectrometry (14), and NMR H/D exchange experiments (13) on the fibers of recombinant PrP indicate that the core of the amyloid fibrils is formed mainly by H2H3 (residues 167–227).
Our results reconcile both hypotheses by suggesting that H2H3 is the central element for the fold of the PrP globular domain and explain which role other regions of PrP could play in aggregation. We have studied the structure of H2H3 and shown that despite a strong competing tendency toward aggregation, it retains the same α-hairpin conformation as observed in the full-length protein. Accordingly, we concluded that H2H3 is an autonomous structural element that is able to fold independently in the absence of the rest of the sequence. Our conclusions are fully supported by the observation that, in studies of full-length PrP, H2H3 is the only region that retains its fold even in the presence of 5 m urea (56). These findings are not trivial; it is rather unusual for a helical hairpin that is part of a globular domain to retain so completely its structural features when studied in isolation, especially considering that the two individual helices are not strongly amphipathic and have no intrinsic helical propensity. Although certainly stabilized by the disulfide bridge, the fold is also held together by several interhelix interactions whose mutation correlates with disease.
An obvious but important consequence of our results is the hypothesis that H2H3 constitutes one of the main if not the main seeding element for the conformational transition and the fibrillogenesis of the multiple aggregation site PrP. We have demonstrated in a previous paper that isolated H2H3 is able to reproduce the oligomerization pathway observed in full-length PrP, thus providing direct evidence in favor of H2H3 as the key seeding element of aggregation (16). Here we prove conclusively that although other regions might contribute to aggregation, H2H3 is per se highly fibrillogenic also in the absence of the remaining sequence.
We can go further in hypothesizing, on the basis of the results reported here, that aggregation is caused by competition between the aggregation propensity and the fold stability of H2H3 and occurs through a direct α-to-β transition without the need of complete unfolding of this region. This is in agreement with the observations that H2H3 forms fibers also in the absence of destabilizing conditions and that fibrillization of H2H3 but not of PrP is concomitant with aggregation. Accordingly, Cobb et al. (57) have recently described PrP amyloidogenesis in the absence of denaturants although still using acidic conditions (i.e. pH 4).
We can, thus, suggest a simplified and yet general model (a banana-peeling model) for the aggregation and fibrillogenesis of PrP (Fig. 8). The process would start with the “pealing-off” (like a banana) of the interactions between H2H3 and the rest of the globular domain, exposing the highly fibrillogenic seeding element H2H3. The competition between fold and aggregation would then be shifted toward promotion of self-assembly, triggering the conformational change to a β-rich structure that further matures into amyloid fibers. This model explains the advantages of using destabilizing agents to facilitate fiber formation. It is also in excellent agreement with the H/D exchange experiments, which indicate faster exchange of regions outside H2H3 in the fiber and with the idea of the central role played by breaking the S1S2 interactions and detaching this region from H2H3 proposed in previous work (54, 55). In vivo destabilization of the fold may be caused by mutations and/or environmental determinants, in agreement with our observation that even a point mutation may strongly affect the PrP tendency to aggregate. An important point to be addressed in the future is whether H2H3 is infective. This point is not necessarily consequent to our results because, although amyloid fibrils are a hallmark of neurodegenerative diseases, the relationship between PrP infectivity and the structure of PrPSc remains largely unclear (4, 58). Prusiner and co-workers (59, 60) have shown for instance that PrP amyloid fibrils are not required for propagation of scrapie infectivity by discovering highly infectious PrP fractions that contained only amorphous structures. Non-fibrillar particles with masses equivalent to 14–28 PrP molecules have been identified as the most infectious species (61). Further studies will be needed to validate our model and clarify its consequences for understanding the PrP related pathologies. Future work in PrP-infected cells is also specifically necessary to determine the relation between the current results and prion infectivity.
FIGURE 8.

A simplified model of H2H3-mediated PrP aggregation. In normal circumstances the stably folded H2H3 (indicated as a white ribbon) is sheltered by interactions with other regions of PrP (generically indicated as yellow shapes). When destabilizing factors intervene, these interactions are “peeled off,” and the fibrillogenic H2H3 is exposed. This seed triggers aggregation, which can be reinforced by other regions of the molecule with consequent growth of fibers.
Supplementary Material
Acknowledgments
We thank the MRC Biomedical NMR center for technical support and Lesley Calder for generous assistance with EM analysis.
This work was supported by MRC Grant U117584256.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
The atomic coordinates and structure factors (code 2ktm) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
The assigned chemical shifts of ovH2H3_I208A are deposited in the BioMagResBank (http://www.bmrb.wisc.edu) under accession number 16706.
- PrP
- prion protein
- ov-
- ovine
- mo-
- mouse
- go-
- goat
- HSQC
- heteronuclear single quantum correlation
- NOESY
- two-dimensional nuclear Overhauser effect (NOE) spectroscopy
- CD
- circular dichroism
- TOCSY
- two-dimensional total correlation spectroscopy
- MOPS
- 4-morpholinepropanesulfonic acid
- ThT
- thioflavin T.
REFERENCES
- 1.Prusiner S. B. (1982) Science 216, 136–144 [DOI] [PubMed] [Google Scholar]
- 2.Prusiner S. B., McKinley M. P., Bowman K. A., Bolton D. C., Bendheim P. E., Groth D. F., Glenner G. G. (1983) Cell 35, 349–358 [DOI] [PubMed] [Google Scholar]
- 3.Pan K. M., Baldwin M., Nguyen J., Gasset M., Serban A., Groth D., Mehlhorn I., Huang Z., Fletterick R. J., Cohen F. E. (1993) Proc. Natl. Acad. Sci. U.S.A. 90, 10962–10966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Simoneau S., Rezaei H., Salès N., Kaiser-Schulz G., Lefebvre-Roque M., Vidal C., Fournier J. G., Comte J., Wopfner F., Grosclaude J., Schätzl H., Lasmézas C. I. (2007) PLoS Pathog. 3, e125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pastore A., Zagari A. (2007) Prion 1, 185–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wille H., Michelitsch M. D., Guenebaut V., Supattapone S., Serban A., Cohen F. E., Agard D. A., Prusiner S. B. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 3563–3568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Govaerts C., Wille H., Prusiner S. B., Cohen F. E. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 8342–8347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prusiner S. B. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 13363–13383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heller J., Kolbert A. C., Larsen R., Ernst M., Bekker T., Baldwin M., Prusiner S. B., Pines A., Wemmer D. E. (1996) Protein Sci. 5, 1655–1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Christen B., Hornemann S., Damberger F. F., Wüthrich K. (2009) J. Mol. Biol. 389, 833–845 [DOI] [PubMed] [Google Scholar]
- 11.Kaneko K., Zulianello L., Scott M., Cooper C. M., Wallace A. C., James T. L., Cohen F. E., Prusiner S. B. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 10069–10074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cobb N. J., Sönnichsen F. D., McHaourab H., Surewicz W. K. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 18946–18951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu X., Wintrode P. L., Surewicz W. K. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 1510–1515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nazabal A., Hornemann S., Aguzzi A., Zenobi R. (2009) J. Mass. Spectrom. 44, 965–977 [DOI] [PubMed] [Google Scholar]
- 15.Eghiaian F., Daubenfeld T., Quenet Y., van Audenhaege M., Bouin A. P., van der Rest G., Grosclaude J., Rezaei H. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 7414–7419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chakroun N., Prigent S., Dreiss C. A., Noinville S., Chapuis C., Skrelic D., Fraternali F., Rezaei H. (2010) FASEB J. 10.1096/fj.09-153924 [DOI] [PubMed] [Google Scholar]
- 17.Rezaei H., Marc D., Choiset Y., Takahashi M., Hui, Bon, Hoa G., Haertlé T., Grosclaude J., Debey P. (2000) Eur. J. Biochem. 267, 2833–2839 [DOI] [PubMed] [Google Scholar]
- 18.Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) J. Biomol. NMR. 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 19.Bartels C., Xia T. H., Billeter M., Güntert P., Wüthrich K. (1995) J. Biomol. NMR 6, 1–10 [DOI] [PubMed] [Google Scholar]
- 20.Cornilescu G., Delaglio F., Bax A. (1999) J. Biomol. NMR 13, 289–302 [DOI] [PubMed] [Google Scholar]
- 21.Rieping W., Habeck M., Bardiaux B., Bernard A., Malliavin T. E., Nilges M. (2007) Bioinformatics 23, 381–382 [DOI] [PubMed] [Google Scholar]
- 22.Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 23.Linge J. P., Habeck M., Rieping W., Nilges M. (2004) J. Magn. Reson. 167, 334–342 [DOI] [PubMed] [Google Scholar]
- 24.Linge J. P., Williams M. A., Spronk C. A., Bonvin A. M., Nilges M. (2003) Proteins 50, 496–506 [DOI] [PubMed] [Google Scholar]
- 25.Vriend G. (1990) J. Mol. Graph. 8, 52–56 [DOI] [PubMed] [Google Scholar]
- 26.Laskowski R. A., Rullmannn J. A., MacArthur M. W., Kaptein R., Thornton J. M. (1996) J. Biomol. NMR 8, 477–486 [DOI] [PubMed] [Google Scholar]
- 27.Koradi R., Billeter M., Wüthrich K. (1996) J. Mol. Graph. 14, 51–55 [DOI] [PubMed] [Google Scholar]
- 28.DeLano W. L. (2002) The PyMOL Molecular Graphics System, DeLano Scientific LLC, San Carlos, CA [Google Scholar]
- 29.Morillas M., Vanik D. L., Surewicz W. K. (2001) Biochemistry 40, 6982–6987 [DOI] [PubMed] [Google Scholar]
- 30.Apetri A. C., Surewicz W. K. (2003) J. Biol. Chem. 278, 22187–22192 [DOI] [PubMed] [Google Scholar]
- 31.Calzolai L., Lysek D. A., Pérez D. R., Güntert P., Wüthrich K. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 651–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lysek D. A., Schorn C., Nivon L. G., Esteve-Moya V., Christen B., Calzolai L., von Schroetter C., Fiorito F., Herrmann T., Güntert P., Wüthrich K. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 640–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thomas D., Pastore A. (2005) Acta Crystallogr. D Biol. Crystallogr. 61, 112–116 [DOI] [PubMed] [Google Scholar]
- 34.Eghiaian F., Grosclaude J., Lesceu S., Debey P., Doublet B., Tréguer E., Rezaei H., Knossow M. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 10254–10259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muñoz V., Serrano L. (1997) Biopolymers 41, 495–509 [DOI] [PubMed] [Google Scholar]
- 36.Xian W., Connolly P. J., Oslin M., Hausrath A. C., Osterhout J. J. (2006) Protein Sci. 15, 2062–2070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kamtekar S., Hecht M. H. (1995) FASEB J. 9, 1013–1022 [DOI] [PubMed] [Google Scholar]
- 38.Kitamoto T., Ohta M., Doh-ura K., Hitoshi S., Terao Y., Tateishi J. (1993) Biochem. Biophys. Res. Commun. 191, 709–714 [DOI] [PubMed] [Google Scholar]
- 39.Chapman J., Ben-Israel J., Goldhammer Y., Korczyn A. D. (1994) Neurology 44, 1683–1686 [DOI] [PubMed] [Google Scholar]
- 40.Mastrianni J. A., Iannicola C., Myers R. M., DeArmond S., Prusiner S. B. (1996) Neurology 47, 1305–1312 [DOI] [PubMed] [Google Scholar]
- 41.Pocchiari M., Salvatore M., Cutruzzolá F., Genuardi M., Allocatelli C. T., Masullo C., Macchi G., Alemá G., Galgani S., Xi Y. G. (1993) Ann. Neurol. 34, 802–807 [DOI] [PubMed] [Google Scholar]
- 42.Peoc'h K., Manivet P., Beaudry P., Attane F., Besson G., Hannequin D., Delasnerie-Laupretre N., Laplanche J. L. (2000) Hum. Mutat. 15, 482. [DOI] [PubMed] [Google Scholar]
- 43.Hsiao K., Dlouhy S. R., Farlow M. R., Cass C., Da Costa M., Conneally P. M., Hodes M. E., Ghetti B., Prusiner S. B. (1992) Nat. Genet. 1, 68–71 [DOI] [PubMed] [Google Scholar]
- 44.Piccardo P., Dlouhy S. R., Lievens P. M., Young K., Bird T. D., Nochlin D., Dickson D. W., Vinters H. V., Zimmerman T. R., Mackenzie I. R., Kish S. J., Ang L. C., De Carli C., Pocchiari M., Brown P., Gibbs C. J., Jr., Gajdusek D. C., Bugiani O., Ironside J., Tagliavini F., Ghetti B. (1998) J. Neuropathol. Exp. Neurol. 57, 979–988 [DOI] [PubMed] [Google Scholar]
- 45.López Garcia F., Zahn R., Riek R., Wüthrich K. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 8334–3839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Viles J. H., Donne D., Kroon G., Prusiner S. B., Cohen F. E., Dyson H. J., Wright P. E. (2001) Biochemistry 40, 2743–2753 [DOI] [PubMed] [Google Scholar]
- 47.O'Sullivan D. B., Jones C. E., Abdelraheim S. R., Brazier M. W., Toms H., Brown D. R., Viles J. H. (2009) Protein Sci. 18, 410–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hosszu L. L., Baxter N. J., Jackson G. S., Power A., Clarke A. R., Waltho J. P., Craven C. J., Collinge J. (1999) Nat. Struct. Biol. 6, 740–743 [DOI] [PubMed] [Google Scholar]
- 49.Dima R. I., Thirumalai D. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 15335–15340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bocharova O. V., Breydo L., Salnikov V. V., Gill A. C., Baskakov I. V. (2005) Protein Sci. 14, 1222–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baskakov I. V., Bocharova O. V. (2005) Biochemistry 44, 2339–2348 [DOI] [PubMed] [Google Scholar]
- 52.Wille H., Bian W., McDonald M., Kendall A., Colby D. W., Bloch L., Ollesch J., Borovinskiy A. L., Cohen F. E., Prusiner S. B., Stubbs G. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 16990–16995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.DeMarco M. L., Daggett V. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 2293–2298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barducci A., Chelli R., Procacci P., Schettino V., Gervasio F. L., Parrinello M. (2006) J. Am. Chem. Soc. 128, 2705–2710 [DOI] [PubMed] [Google Scholar]
- 55.DeMarco M. L., Daggett V. (2007) Biochemistry 46, 3045–3054 [DOI] [PubMed] [Google Scholar]
- 56.Julien O., Chatterjee S., Thiessen A., Graether S. P., Sykes B. D. (2009) Protein Sci. 18, 2172–2182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cobb N. J., Apetri A. C., Surewicz W. K. (2008) J. Biol. Chem. 283, 34704–34711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shindoh R., Kim C. L., Song C. H., Hasebe R., Horiuchi M. (2009) J. Virol. 83, 3852–3860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McKinley M. P., Meyer R. K., Kenaga L., Rahbar F., Cotter R., Serban A., Prusiner S. B. (1991) J. Virol. 65, 1340–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wille H., Zhang G. F., Baldwin M. A., Cohen F. E., Prusiner S. B. (1996) J. Mol. Biol. 259, 608–621 [DOI] [PubMed] [Google Scholar]
- 61.Silveira J. R., Raymond G. J., Hughson A. G., Race R. E., Sim V. L., Hayes S. F., Caughey B. (2005) Nature 437, 257–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.