

Abstract
With muscle wasting, caspase-3 activation and the ubiquitin-proteasome system act synergistically to increase the degradation of muscle proteins. Whether proteasome activity is also elevated in response to catabolic conditions is unknown. We find that caspase-3 increases proteasome activity in myotubes but not in myoblasts. This difference is related to the cleavage of specific 19 S proteasome subunits. In mouse muscle or myotubes, caspase-3 cleaves Rpt2 and Rpt6 increasing proteasome activity. In myoblasts, caspase-3 cleaves Rpt5 to decrease proteasome activity. To confirm the caspase-3 dependence, caspase-3 cleavage sites in Rpt2, Rpt6, or Rpt5 were mutated. This prevented the cleavage of these subunits by caspase-3 as well as the changes in proteasome activity. During differentiation of myoblasts to myotubes, there is an obligatory, transient increase in caspase-3 activity, accompanied by a corresponding increase in proteasome activity and cleavage of Rpt2 and Rpt6. Therefore, differentiation changes the proteasome type from sensitivity of Rpt5 to caspase-3 in myoblasts to sensitivity of Rpt2 and Rpt6 in myotubes. This novel mechanism identifies a feed-forward amplification that augments muscle proteolysis in catabolic conditions. Indeed, we found that in mice with a muscle wasting condition, chronic kidney disease, there was cleavage of subunits Rpt2 and Rpt6 and stimulation of proteasome activity.
Keywords: Cell/Differentiation, Diseases/Muscular, Enzymes/Proteolytic, Protease/ATP-dependent, Proteases/Caspase, Protein/Degradation, Tissue/Organ Systems/Muscle/Skeletal, Proteasome
Introduction
The ubiquitin-proteasome system (UPS)4 is responsible for the degradation of most proteins in the cytoplasm and nuclei of cells (1, 2). It is also involved in regulating many cell functions including control of the cell cycle, antigen presentation, ion transport, and muscle mass (3–6). One event that regulates proteolysis in the UPS is the rate of ubiquitin conjugation to protein substrates. For example, βTCF, the activated E3 enzyme, triggers degradation of IκB and β-catenin, whereas activation of Atrogin-1/MAFbx or MuRF1 in muscle is closely linked to increased protein degradation (7, 8).
Rates of protein degradation in the UPS could also be regulated through changes in the proteolytic activity of the proteasome. For example, treatment of lymphoid cells with γ-interferon increases the expression of proteasome subunits, LMP2 and 7, and these subunits are incorporated into proteasomes stimulating the breakdown of proteins into peptides that are more suitable for class 1 antigen presentation (9–11). Other evidence suggests that the activity of the UPS can be determined by variations in proteasome activity. In several conditions that are associated with the loss of muscle mass, proteasome subunits are expressed at a higher level but it is not proven that changes in subunit expression accelerate the rate of muscle proteolysis (12–18). Alternatively, proteasome activity can be suppressed in association with changes in subunits. Sun et al. (19) reported that caspase-3 activation in Jurkat T cells or cancer cells causes cleavage of specific subunits of the 19 S regulatory complex of the proteasome: Rpt5, Rpn10, and Rpn2. Associated with cleavage of these subunits, they found decreased proteasome activity and proposed that this forward-feed type of coordinated change in proteasome activity leads to apoptosis of Jurkat T cells. Evidence from yeast indicates that proteasome activity can be regulated by changes in the conformation of the proteasome. Kohler et al. (20) showed that Rpt2, an ATPase in the regulatory 19 S proteasome complex of yeast, functions to “gate” proteasome activity. They reported that mutation of the ATPase activity of this subunit could control both the entry of substrate into the proteasome and its release of proteolytic products. Smith et al. (21) reported that an ATPase complex (PAN) isolated from Archaea can stimulate proteasome activity by gate opening and translocation of unfolded substrates into the proteasome. These reports suggest that proteasome activity can be regulated under specific physiological conditions.
Accelerated muscle protein degradation by the UPS occurs in many catabolic disorders, leading to muscle atrophy (3). When large amounts of protein are being degraded, several adaptations occur. First, specific E3 ubiquitin-conjugating enzymes are robustly expressed in muscle; the level of Atrogin-1/MAFbx is directly related to the rate of protein degradation in muscle cells (18, 22–26). Second, there is increased expression of ubiquitin and proteasome subunits in muscle when protein degradation is accelerated in muscle (3). Third, we find that activation of caspase-3 is required to convert actomyosin and myofibril proteins into substrates of the UPS (24, 27–29). Taken together, the evidence indicates that the accelerated breakdown of the bulk of muscle proteins requires coordination of multiple events (18).
Is it possible that proteasome activity increases when large amounts of muscle protein are being degraded? We find that a caspase-3-dependent increase in proteasome activity occurs in conjunction with accelerated muscle protein degradation by a mechanism involving cleavage of two regulatory subunits of the 19 S proteasome complex. Because Fernando et al. (30) documented that activation of caspase-3 is required for differentiation from myoblasts to myotubes, we investigated how differentiation influences caspase-3-induced changes in proteasome activity and subunit cleavage during differentiation. Our results provide evidence for a novel, cell-specific mechanism that regulates proteasome activity in skeletal muscle.
EXPERIMENTAL PROCEDURES
Cell Culture
C2C12 cells (ATCC, Manassas, VA) were studied between passages 3 and 9 and differentiated by incubating in 2% horse serum (27). Jurkat T lymphocytes (ATCC) were cultured in RPMI medium with 10% fetal bovine serum, 25 mm glucose, 1 mm sodium pyruvate, 100 units/ml penicillin, 100 μg/ml streptomycin, and 2 mm l-glutamine. Cells were passaged every 2 days.
Protein Degradation
Protein degradation was measured as release of l-[U-14C]phenylalanine (Amersham Biosciences, Piscataway, NJ) from prelabeled cells (31). The rate of protein degradation was calculated as the slope of the logarithm of l- [U-14C]phenylalanine remaining in cell proteins versus time.
Immunoblotting Analysis
Detection of proteasome subunits in C2C12 muscle cells, muscle lysates or in Jurkat T lymphocytes was assessed by Western blotting (32). Primary antibodies included: anti-caspase-3 (1:1000 dilution, Cell Signaling Technology, Beverly, MA) and antibodies against different proteasome subunits. They were diluted as recommended by the manufacturer (Biomol-Affiniti, Plymouth Meeting, PA).
Proteasome Activity
Proteasomes were partially purified by differential centrifugation, and activity was measured in 2 mm ATP as described (33). Proteasome activity was 25-fold enriched in the pellet versus cell lysates, and 96% of the activity was blocked by the specific proteasome inhibitor, lactacystin (Calbiochem, La Jolla, CA). Alternatively, we separated 26 S proteasomes using glycerol density gradient (10–40%) centrifugation (34). Equal amounts of protein from the proteasome preparations were used to measure proteasome activity as the release of 7-amino-4-methylcoumarin (AMC) from the fluorogenic peptide substrate LLVY-AMC (N-Suc-Leu-Leu-Val-Tyr-AMC) (33). AMC fluorescence was measured using 380 nm excitation and 460 nm emission wavelengths. The difference between the fluorescence measured in the presence and absence of 100 μm lactacystin was used to calculate proteasome activity.
We also assessed proteasome activity using an ubiquitin-conjugated substrate protein, Ub5DHFR (kindly provided by Millenium Pharmaceuticals, Cambridge, MA). Proteasomes isolated by differential centrifugation were incubated with or without recombinant caspase-3 for 1h before adding the Ub5DHFR substrate for 30 min. Addition of 20 μm MG132 (Calbiochem) plus 100 μm lactacystin (Chemicon Int., Temecula, CA) was used to inhibit proteasome activity. 2.5 μm ubiquitin-aldehyde (Ubal, Boston Biochem, Cambridge, MA) was used to inhibit de-ubiquitination). Western blotting with an anti-His antibody (Morphosys Inc., Raleigh, NC) was used to detect changes in His-tagged ubiquitin conjugated to DHFR (35).
Cloning, Mutation, and in Vitro Translation of Rpt2, Rpt5, and Rpt6
RNA from C2C12 cells was used to generate cDNA templates by in vitro reverse transcription. PCR-generated fragments for full-length, mouse Rpt2, Rpt5, and Rpt6 were cloned into HindIII/XhoI (Rpt2), EcoRI/XbaI (Rpt5), and BamHI/XhoI (Rpt6) sites of pcDNA3-Myc-His-A vector (C-terminal Myc tag, Invitrogen). The pseudo-caspase-3 cleavage sites “DEID” in Rpt2 and Rpt6 were changed to WQFH by PCR. The Rpt5 caspase-3 cleavage site (DELD) at Asp-27 was mutated using the site-directed, mutagenesis kit (Agilent Technologies, La Jolla, CA). Following sequence verification, wild-type Rpt2, Rpt5, or Rpt6 clones and the mutated clones were translated in the presence of l-[35S]methionine (Amersham Biosciences) using a TnT T7 transcription/translation kit (Promega, Madison, WI) according to the manufacturer's instructions. The translated protein was incubated for 1 h at 37 °C with recombinant caspase-3; the cleaved protein was detected using 4–15% gradient SDS-PAGE followed by autoradiography.
Generation of Muscle Cells with Wild-type or Mutant Rpt2, Rpt5, or Rpt6
C2C12 cells were transfected with the expression constructs using Lipofectamine 2000 (Invitrogen). Stable clones were selected in medium containing 600 μg/ml of G418.
Animal Model of Accelerated Muscle Protein Degradation
Chronic kidney disease (CKD) was produced in mice by subtotal nephrectomy and feeding a high protein diet (36). These experiments were approved by the Animal Care Committee of Emory University School of Medicine. CKD mice were pair-fed with sham-operated, control mice (36). After 4 weeks, proteasomes were isolated from muscles of anesthetized mice to measure proteasome activity and cleaved proteasome subunits.
To inhibit caspase-3 in mouse muscle, XIAP (X-linked inhibitor of apoptosis protein) was introduced into muscle using a lentivirus expressing XIAP (37). In these mice, the same procedures were used to measure proteasome activity and subunit cleavage following creation of CKD and sham-operated controls.
Statistical Analysis
Data are expressed as the mean ± S.E. To identify significant differences between two groups, comparisons were made by using the Student's t test. One-way ANOVA followed by pair-wise comparisons with the Student, Newman Keuls test was used to determine if differences between sample groups were significant (p < 0.05).
RESULTS
Caspase-3 Increases Proteasome Activity in Differentiated C2C12 Muscle Cells
To examine if proteasome activity is linked to activated caspase-3 (24, 27–29, 38), we isolated 26 S proteasomes from differentiated C2C12 myotubes and incubated them with 100 nm recombinant caspase-3. Caspase-3 increased the proteasome activity measured as hydrolysis of LLVY-AMC. This response was blocked by 0.1 μm DEVD-CHO (39), a potent inhibitor of caspase-3 (Fig. 1A). Under these conditions, we found that DEVD-CHO alone did not change proteasome activity nor did recombinant caspase-3 cleave the LLVY-AMC substrate (data not shown). The activation of proteasomes by caspase-3 was dose- and time-dependent (supplemental Fig. S1).
FIGURE 1.

Caspase-3 stimulates proteasome activity in C2C12 myotubes. A, proteasomes isolated from myotubes were incubated with 100 nm recombinant caspase-3 with or without its inhibitor (DEVD-CHO). Proteasome activity in equal amounts of proteasome protein was measured using the fluorogenic peptide, LLVY-AMC. The results are the mean ± S.E. of replicate assays from four proteasome preparations (*, p < 0.05 versus proteasomes untreated by caspase-3). B, activity of proteasomes from A was also evaluated by measuring degradation of the ubiquitin-conjugated substrate, Ub5DHFR. Proteasomes isolated from myotubes were incubated with or without caspase-3 for 1 h; in the indicated mixtures, MG-132 plus lactacystin were used to block proteasome activity. In the left panel, ubiquitin aldehyde (Ubal) was added to all reactions to inhibit deubiquitination. In the right panel, Ubal was not added. An anti-His antibody was used in the Western blots and an antibody against Rpt1 was used to assess the loading of proteasomes. The bar graph underneath is the density analysis of Ub5DHFR based on three different Western blots. (*, p < 0.05 versus no proteasome added). C, myotubes were incubated in serum-free medium (SFM) or with 50 nm staurosporine (Stau). Pro-caspase and activated caspase-3 were assessed by Western blots. D, proteasome activity in myotubes incubated in SFM or treated with staurosporine was measured as described in A (n = 4; *, p < 0.05 versus untreated myotubes (Ctrl)).
We also examined the influence of caspase-3 on proteasome activity using an ubiquitin-conjugated protein substrate, Ub5DHFR. In the presence of Ubal added to inhibit deubiquitination, proteasomes isolated from C2C12 myotubes reduced the level of Ub5DHFR by 32% (Fig. 1B, left panel). After proteasomes were treated with caspase-3, the level of Ub5DHFR was 65% lower than in the absence of proteasomes (p < 0.05). Ub5DHFR degradation was blocked by adding the proteasome inhibitors, MG132 plus lactacystin. Even in the presence of Ubal, there was a small amount of UbDHFR, apparently due to deubiquitination of the substrate by Rpn11, which is a metalloenzyme insensitive to Ubal (40). In reactions lacking Ubal, the presence of proteasomes led to some accumulation of UbDHFR. When proteasomes that had been treated with caspase-3 were added, both UbDHFR and Ub5DHFR were lower versus proteasomes alone (Fig. 1B, right panel). Together, the results indicate that caspase-3 stimulated proteasome proteolytic activity as measured by the degradation of Ub5DHFR.
To examine whether activation of endogenous caspase-3 in C2C12 muscle cells would increase proteasome activity, we deprived cells of serum or incubated them with staurosporine. In both cases, caspase-3 was activated (Fig. 1C) and proteasome activity increased (Fig. 1D).
Caspase-3 Cleaves Specific 19 S Proteasome Subunits in C2C12 Muscle Cells
To explore whether the caspase-3-induced increase in proteasome activity in differentiated muscle cells involves cleavage of proteasome subunits, we isolated proteasomes from myotubes and incubated them with recombinant caspase-3 before Western blotting with antibodies against 26 S subunits. Three “base” subunits of the 19 S component of 26 S proteasome (Rpt2 (49 kDa), Rpt6 (46 kDa), and Rpn2 (110 kDa) were cleaved by caspase-3 (Fig. 2). The caspase-3 inhibitor, DEVD-CHO, blocked these responses.
FIGURE 2.

Caspase-3 cleaves certain 19 S proteasome regulatory subunits. Proteasomes were isolated from C2C12 myotubes and treated without or with 100 nm recombinant caspase-3 or caspase-3 plus its inhibitor (DEVD-CHO) for 2 h. Eight antibodies against proteasome 19 S regulatory subunits (base subunits) were examined by Western blotting and a representative blot from three separate experiments is shown.
Mutation of Caspase-3 Cleavage Sites in Rpt2 and Rpt6 Decreases Staurosporine-induced Proteasome Activity in Differentiated C2C12 Cells
We cloned Rpt2 and Rpt6 in pcDNA3-myc-His-A and translated the proteins in vitro in the presence of l-[35S]methionine. Recombinant caspase-3 cleaved Rpt2 (49 kDa) into fragments of 31 and 18 kDa (Fig. 3A) and Rpt6 (46 kDa) into 28-kDa and 17-kDa fragments (Fig. 3B). Based on the size of the fragments and the amino acid sequences of Rpt2 and Rpt6, we identified potential cleavage sites, DEID, at 288 amino acids in Rpt2 and 248 amino acids in Rpt6. We mutated these sites into WQFH and found that the mutations blocked the ability of caspase-3 to cleave the Rpt2 and Rpt6 proteasome subunits (Fig. 3, A and B). The cleavage of Rpt6 by caspase-3 was dose- and time-dependent (supplemental Fig. S2).
FIGURE 3.

In Rpt2 and Rpt6, mutation of caspase-3 cleavage sites suppresses staurosporine-induced proteasome activity in C2C12 myotubes. A and B, recombinant caspase-3 cleaved l-[35S]methionine- labeled, in vitro translated Rpt2 or Rpt6. Mutation of the caspase-3 cleavage site in Rpt2 and Rpt6 abolished caspase-3-induced cleavage. C, C2C12 cell lysates were subjected to glycerol density gradient centrifugation and proteasome fractions were assayed for proteasome activity using LLVY-AMC. D, proteasomes from wild-type cells and cells expressing mutant Rpt6 (mRpt6) were separated by glycerol density centrifugation and fractions 13–15 were subjected to native PAGE (left) or SDS-PAGE (right). Using anti-Myc or anti-Rpt6 antibodies, the mutant Rpt6 was found to be present in 26 S proteasomes by detection of Myc. E, wild-type cells and cells stably transfected to express mutated Rpt2 (mRpt2) or Rpt6 (mRpt6) were treated with or without 50 nm staurosporine for 1, 5, or 8 h. Proteasomes isolated from these cells were used to measure proteasome activity using LLVY-AMC as a substrate (n = 3; *, p < 0.05 versus proteasomes subunits not exposed to staurosporine). F, Proteasomes from myotubes treated with staurosporine for 5 or 8 h (see E) were incubated with Ub5DHFR in the presence of Ubal. The anti-His antibody (see Fig. 1) was used to detect His-tagged ubiquitins conjugated to DHFR. The staurosporine-induced increase in proteasome activity was blocked in cells expressing the mutated subunits (E and F).
The two mutated subunit genes were stably transfected in C2C12 muscle cells. To demonstrate incorporation of the mutated subunits into the proteasome, we isolated proteasomes using 10–40% glycerol gradient centrifugation (34). From the 22 fractions of the gradient, we found that fractions 13–15 exhibited the highest proteasome activity (Fig. 3C). These proteasome fractions from wild-type and mutant cells were separated by both native PAGE (Fig. 3D, left panel) and SDS-PAGE (Fig. 3D, right panel) gels. Myc expression in 26 S proteasomes from cells expressing mutant Rpt6 (mRpt6) was validated by Western blotting in native PAGE using anti-Myc and anti-Rpt6 antibodies (Fig. 3D).
Myotubes expressing wild-type or mutated Rpt2 or Rpt6 subunits were treated with or without 50 nm staurosporine for 1, 5, or 8 h. Proteasomes were isolated from these cells and their activity was measured using LLVY-AMC (Fig. 3E) or Ub5DHFR (Fig. 3F) substrates. After 1 h of staurosporine treatment, proteasome activity was unchanged but after 5 or 8 h, proteasome activity in wild-type cells increased. The staurosporine-induced increase in proteasome activity was blocked in cells expressing the mutated subunits (Fig, 3, E and F).
Because the caspase-3 cleavage site is present within the ATPase domain of Rpt subunits, we assessed whether the ATP-dependence of peptide hydrolysis would be impaired in proteasomes from cells expressing mutant Rpt6. Proteasomes from both wild-type cells and cells expressing mRpt6 exhibited an ATP-dependence of proteasome activity even though proteasome activity was reduced in proteasomes expressing mutated Rpt6. Staurosporine increased the activity of proteasomes from wild-type cells but this response was eliminated in proteasomes from mRpt6-expressing cells (supplemental Fig. S3). We also found that caspase-3 activity did not change in cells expressing the mutant subunits (data not shown).
Regulation of Proteasome Activity by Caspase-3 Is Cell-specific
The increase in proteasome activity in myotubes differs from the caspase-3-induced decrease in proteasome activity occurring in Jurkat T or cancer cells (19). An explanation for this difference could be that caspase-3 cleaves regulatory subunits of the proteasome in a cell-specific manner. Consistent with the report of Sun et al. (19), etoposide (25 μm) caused a 54% reduction in proteasome activity in Jurkat T cells and this response was blocked by Z-VAD-FMK (Fig. 4A, left panel). Their finding that etoposide stimulated cleavage of Rpt5 and Rpn2 subunits in these cells was also confirmed (Fig. 4A, right panel). In undifferentiated C2C12 myoblasts, 50 nm staurosporine treatment decreased proteasome activity by 12% (Fig. 4B) and caused a subunit cleavage pattern similar to that induced in Jurkat T cells by etoposide (data not shown). To verify that proteasomes from myoblasts differ from those of myotubes, we isolated proteasomes from myoblasts and incubated them with recombinant caspase-3 with or without its inhibitor DEVD-CHO. Caspase-3 decreased proteasome activity, a response that was inhibited by DEVD-CHO (supplemental Fig. S4).
FIGURE 4.
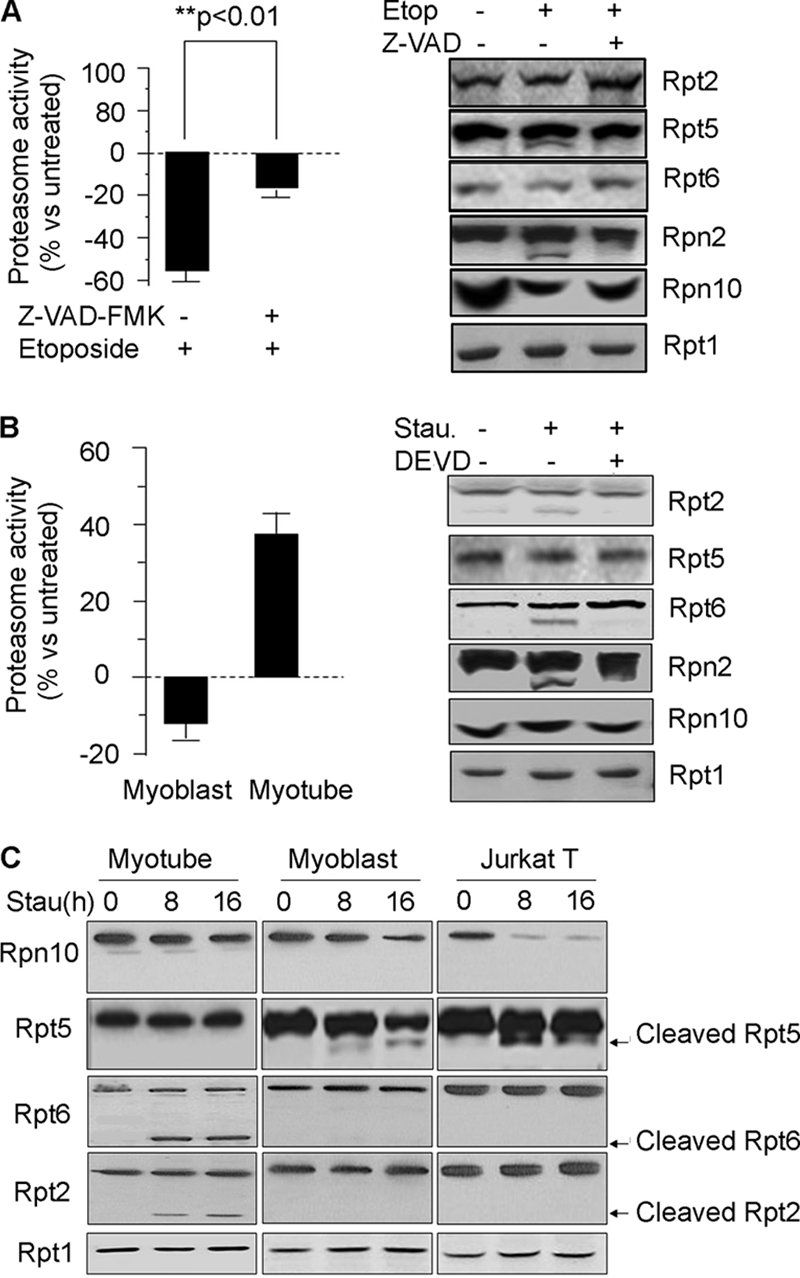
Caspase-3 activation cleaves proteasome subunits and changes proteasome activities in a cell-specific manner. A, proteasomes were isolated from Jurkat T cells following treatment with 25 μm etoposide (Etop) for 8h without or with 25 μm of the pan-caspase inhibitor, Z-VAD-FMK. Their activity (left panel) and subunit cleavage (right panel) are presented. The percentage differences in proteasome activity measured with LLVY-AMC was calculated based on values in cells not treated with etoposide (n = 4; p < 0.01). B, myoblasts and myotubes were treated with 50 nm staurosporine (Stau) for 8 h without or with the caspase-3 inhibitor, DEVD-CHO. Activity of isolated proteasomes was measured as in Fig. 1A and the percentage change induced by staurosporine was calculated based on values in cells not treated with staurosporine (left panel). Cleaved proteasome subunits from myotubes were detected by Western blot (right panel). C, myotubes or myoblasts were treated with 50 nm staurosporine (Stau) for 8 or 16 h and cleavage of proteasome subunits was assessed by Western blot. Results from Jurkat T cells were used as a control.
In myotubes, staurosporine increased proteasome activity 38% (Fig. 4B, left panel) and stimulated cleavage of proteasome subunits, Rpt2, Rpt6, and Rpn2; Rpt5 and Rpn10 subunits were not cleaved in staurosporine-treated myotubes. The cleavage responses were effectively blocked by DEVD-CHO (Fig. 4B, right panel).
The time dependence of changes in subunit cleavage because of staurosporine was examined in Jurkat T cells, C2C12 myoblasts, and myotubes (Fig. 4C). Staurosporine led to cleavage of Rpt5 in myoblasts or Jurkat T cells but in myotubes, activation of caspase-3 led to cleavage of Rpt 6 and Rpt 2 rather than Rpt5. We conclude that the stage of muscle cell differentiation determines not only the caspase-3-induced subunit cleavage patterns but also the changes in proteasome activity.
The Differentiation Process Accounts for Changes in Proteasome Activity and the Subunit Cleavage
To explore why responses in myoblasts and myotubes differ, 26 S proteasomes isolated from the two types of cells were treated with 0.01% SDS to disassociate the 19 S regulatory proteasome complex into subunits so that caspase-3 could cleave individual subunits (41). Caspase-3 did not cleave Rpt2 and Rpt6 in proteasomes isolated from undifferentiated myoblasts unless 0.01% SDS was present. Likewise, subunit Rpt5 in myotubes was not cleaved by caspase-3 unless SDS was present (Fig. 5A). Thus, all 3 subunits are present in proteasomes of both myoblasts and myotubes but their susceptibility to caspase-3 cleavage differs sharply, suggesting that the form of proteasomes in differentiated and undifferentiated muscle cells determines the subunit sensitivity or resistance to caspase-3 cleavage.
FIGURE 5.

Differentiation from myoblasts to myotubes changes the sensitivity of proteasomes to caspase-3. A, proteasomes isolated from myotubes or myoblasts were incubated with recombinant caspase-3 in the presence of 0.01% SDS and cleavage of Rpn10, Rpt5, Rpt6, and Rpt2 was assessed by Western blot. B, proteasomes were isolated from C2C12 cells at different stages of differentiation and incubated for 1 h with recombinant caspase-3. Cleavage of Rpt5 and Rpt6 were detected by Western blot. C, proteasomes from experiments of panel B were examined for cleavage of Rpt6. D, proteasome activity at each differentiation time in panel C was measured (*, p < 0.05 versus values in undifferentiated C2C12 cells).
We also studied how differentiation affects the caspase-3 sensitivity of subunit cleavage and proteasome activity. In myoblasts, caspase-3 cleaved Rpt5 (Fig. 5B) but during differentiation Rpt5 cleavage decreased and Rpt6 is cleaved; this increases proteasome activity. Because caspase-3 activation is required for muscle cell differentiation (30), we evaluated how differentiation affects Rpt6 cleavage. Over 48 h, Rpt6 was progressively cleaved but at 72 h, there was no more Rpt6 cleavage (Fig. 5C). These events corresponded to increases in proteasome activity (Fig. 5D). Therefore, during muscle cell differentiation, the activation of caspase-3 cleaves susceptible proteasome subunits to stimulate proteasome activity. The increase in proteasome activity could function as a means of removing extra or abnormal proteins produced during the process of differentiation, providing potential biological benefit.
In myoblasts, Rpt5 is sensitive to caspase-3-induced cleavage but in myotubes, it is not sensitive. To assess whether Rpt5 sensitivity influences the form of proteasomes in myoblasts and myotubes, we mutated Rpt5 to remove the caspase-3 cleavage site (Asp-27 to Ala); this blocked caspase-3 cleavage of Rpt5 (Fig. 6A). Myoblasts were then transfected with the mutated Rpt5-myc. It was detected in cell lysates and in proteasomes (Fig. 6B), confirming that the Myc-tagged mutant Rpt5 was incorporated into proteasomes. In myotubes expressing the mutant Rpt5, there was reduced cleavage of Rpt6 in proteasomes by recombinant caspase-3 (Fig. 6C). Therefore, caspase-3 cleavage of Rpt5 is responsible for the difference in the patterns of proteasome subunit cleavage in myoblasts compared with myotubes. Notably, myotubes expressing the mutated Rpt5 did not respond with an increase in proteasome activity when intracellular caspase-3 was activated by staurosporine (Fig. 6D). Finally, stable expression of mutant Rpt5 in myoblasts prevented the expected decrease in proteasome activity occurring with activated caspase-3 (Fig. 6E).
FIGURE 6.

Mutation of the caspase-3 cleavage site in Rpt5 reduces Rpt6 cleavage, proteasome activity and protein degradation in myotubes. A, mutation of the caspase-3 cleavage site in Rpt5 abolished the ability of caspase-3 to cleave the in vitro translated Rpt5. B, expression of mutated Rpt5 by stably transfected cells was detected in cell lysates by Western blots (top panel). Proteasomes isolated from myotubes expressing mRpt5 were used to detect Myc-tagged Rpt5 by Western blotting with anti-Myc (middle panel) or anti-Rpt5 (lower panel). C, proteasomes isolated from myotubes of wild-type or those expressing mRpt5 were incubated with caspase-3 for 1 h and Rpt6 cleavage was detected by Western blot. D and E, expression of mutant Rpt5 prevents caspase-3-mediated changes in proteasome activity in myotubes (panel D) and myoblasts (panel E). Myoblasts or myotubes expressing wild-type or mRpt5 were treated with staurosporine and proteasome activity was measured as described in Fig. 1A. (n = 4, *, p < 0.05 versus wild-type cells untreated with staurosporine). F, expression of mRpt5 prevents the increase in protein degradation in myotubes stimulated by serum starvation (n = 4, *, p < 0.05 versus WT cells treated with 0.05% horse serum (HS)).
To determine if the increase in proteasome activity will stimulate proteolysis in muscle cells, we compared protein degradation in wild-type myotubes with that in myotubes expressing mutant Rpt5. In wild-type cells, serum starvation stimulated protein degradation (Fig. 6F); this did not occur in expressing mRpt5. We note that in myotubes expressing mRpt5, proteasome activity and the rate of protein degradation differed somewhat. We presume this reflects the fact that protein degradation includes proteolysis by proteasomes, lysosomes, and other proteases versus proteolysis in isolated proteasomes.
CKD Increases Muscle Proteasome Activity in a Caspase-3-dependent Fashion
As expected (38), proteasome activity in muscle of CKD mice was increased versus values in muscles of sham-operated, control mice (Fig. 7A). Moreover, cleavage of proteasome subunits Rpt2 and Rpt6 in muscles of CKD mice was similar to that found in C2C12 myotubes treated with staurosporine (Figs. 7B and 4C). These results are consistent with our finding that CKD activates caspase-3 in skeletal muscle (46). However, in mice that overexpress the caspase-3 inhibitor, XIAP, in muscle, CKD did not increase proteasome activity or subunit cleavage in muscle. We conclude that caspase-3 in muscle regulates the activity of the proteasome in a catabolic condition, CKD.
FIGURE 7.
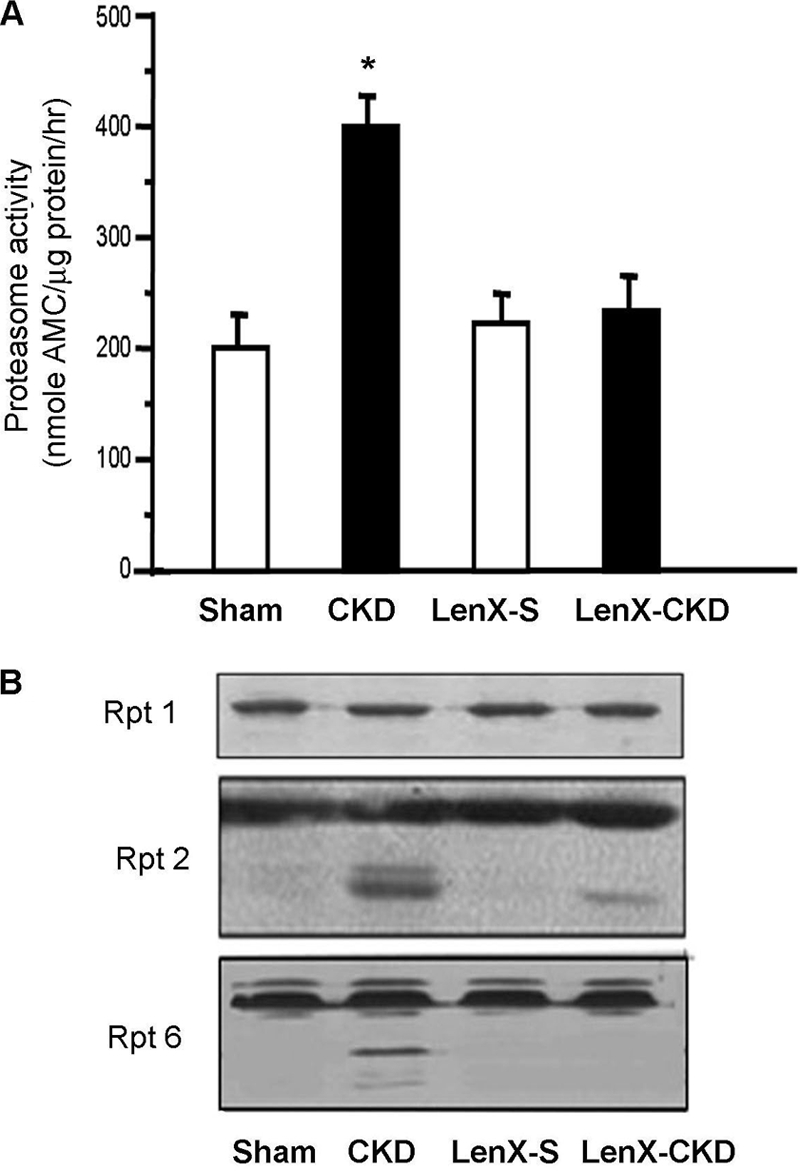
Inhibition of caspase-3 by XIAP in mouse muscle suppresses CKD-induced proteasome activation. Proteasomes were isolated from muscles of CKD and sham-operated, pair-fed control (Sham) mice or from mice that expressed XIAP and had undergone subtotal nephrectomy (LenX-CKD) or sham operation (LenX-S). A, proteasome activity was measured as described in Fig. 1A (n = 4 pairs; *, p < 0.01 versus sham-operated control). B, cleavage of Rpt2 and Rpt6 was examined by Western blot. Rpt1 was used as a loading control.
DISCUSSION
We find that caspase-3 regulates the proteolytic activity of muscle proteasomes in a cell-specific fashion (Fig. 8). In myoblasts, Rpt5 is sensitive to caspase-3-induced cleavage, leading to decreased proteasome activity (Fig. 4). In sharp contrast, Rpt5 in myotubes is resistant to caspase-3 but Rpt2 and Rpt6 become caspase-3-sensitive. Their cleavage leads to increased proteasome activity (Fig. 4). The cell-specific difference occurs because during differentiation the transient increase in caspase-3 activity converts the myoblast type of proteasome to the myotube type. When caspase-3 in mouse muscle was activated by, CKD, there was cleavage of Rpt2 and Rpt6 which would increase muscle protein degradation by stimulating proteasome activity (Figs. 7 and 1). Thus, caspase-3 is at the center of muscle cell metabolism and exerts a feed-forward amplification of protein degradation in muscle. We recognize that proteasome activity could be altered further by other modifications of the subunits besides caspase-3. Alternatively, proteasome-associated proteins might influence proteasome forms in undifferentiated and differentiated muscle cells. These possibilities remain to be determined.
FIGURE 8.

A suggested model depicting the influence of caspase-3 on changes in proteasome activity. In myoblasts, Rpt5 is sensitive to caspase-3-induced cleavage, decreasing proteasome activity. In myotubes, Rpt5 is resistant to caspase-3 but Rpt2 and Rpt6 are sensitive to it. Cleavage of Rpt2 and Rpt6 increases proteasome activity. During differentiation, there is an obligatory, transient increase in caspase-3 activity, which can convert the myoblast type of proteasome to the myotube type. In catabolic conditions such as CKD, caspase-3 is activated and cleaves Rpt2 and Rpt6, increasing proteasome activity.
Catabolic conditions stimulating muscle wasting involve events including activation of caspase-3, which converts myofibrillar proteins to substrates that are rapidly degraded by the UPS (24, 27, 28, 38). The present results identify another important role for caspase-3 activation in degrading muscle protein: it stimulates proteasome activity in differentiated muscle cells. A direct link between activated caspase-3 and the increase in proteasome activity was demonstrated when we treated proteasomes isolated from differentiated myotubes with recombinant caspase-3; there was increased proteasome activity (Fig. 1, A and B). This response differs sharply from the report that activation of caspase-3 in immune or cancer cells decreases proteasome activity (19). Notably, we found that caspase-3 activation in myoblasts also reduces proteasome activity as it does in Jurkat T cells (Fig. 4, A and B and supplemental Fig. S3). Thus, there is a cell-specific change in proteasome activity induced by activation of caspase-3.
How does caspase-3 activate proteasomes differently in myoblasts, myotubes and Jurkat T cells? Our results indicate that caspase-3 cleaves Rpt5 in myoblasts/Jurkat T cells to suppress proteasome activity. In myotubes, however, caspase-3 cleaves Rpt2 and Rpt6 but not Rpt5, and stimulates proteasome activity (Fig. 4, B and C). Caspase-3 cleavage of different subunits in myoblasts and myotubes was not due to loss of the subunits during differentiation because gentle SDS treatment of proteasomes isolated from both types of muscle cells led to cleavage of all three subunits by caspase-3 (Fig. 5A). We conclude that the cleavage of Rpt2 and Rpt6 in myotubes is responsible for the increase in proteasome activity because mutation of their caspase-3 recognition sites decreased proteasome activity (Fig. 3, E and F). Our results uncover a novel mechanism that can augment muscle protein degradation via activation of caspase-3 (12, 27). The pathophysiologic relevance of these results is that cleavage of proteasome subunits and increased proteasome activity in muscle of CKD mice exhibited the same pattern as we uncovered in differentiated myotubes (Fig. 7). This is consistent with our earlier finding that CKD accelerates muscle protein degradation (29, 38).
What occurs during differentiation to induce different types of proteasomes? A transient activation of caspase-3 is required for the conversion of myoblasts to myotubes (30). The transient activation is accompanied by a post-translational change in proteasomes from myoblast type to a myotube type. Indeed, when we added caspase-3 to proteasomes from myoblasts, there was a decrease in proteasome activity accompanied by cleavage of Rpt5 (Figs. 4B and 5B). After 24 h of differentiation, Rpt6 was cleaved, and this was increased further at 48 h. As we predicted, the cleavage of Rpt6 at 48 h was accompanied by increased proteasome activity. After 72 h of differentiation, both Rpt6 cleavage and proteasome activity decreased although proteasome activity still remained significantly greater than that measured in proteasomes from myoblasts (Fig. 5, C and D). Thus, the increase in proteasome activity during differentiation of myoblasts to myotubes is transient. These results must reflect a structural change in proteasomes occurring during differentiation of myoblasts to myotubes. Defining structural changes underlying the difference in proteasome activity in different cells will be the focus of future studies.
How could caspase-3 influence changes in proteasome forms? We find that Rpt5 cleavage is responsible for the cell-specific change in proteasome activity in muscle. Evidence for this conclusion is that: 1) expression of mutated Rpt5 in myoblasts suppresses the caspase-3-dependent decrease in proteasome activity (Fig. 6E); 2) in myotubes, the increase in proteasome activity following caspase-3 activation is significantly suppressed when mutant Rpt5 is expressed (Fig. 6D); and 3) in myotubes, cleavage of Rpt6 depends on cleavage of Rpt5 (Fig. 6C). These results suggest that during differentiation (which activates caspase-3), the myoblast form of proteasomes undergoes a dynamic change: Rpt2 and Rpt6 become exposed and Rpt5 is no longer sensitive to caspase-3.
Regulation of proteasome activity by subunit-mediated gate opening of the 20 S particle has been reported by others (42). Specifically, individual subunits of the AAA family (i.e. Rpt1-Rpt6) play different roles in regulating proteasome activity. For example, it was shown that C-terminal peptides of Rpt2 or Rpt5 can bind to the 20 S proteasome, stimulating proteasome activity. Rpt 5 functions to open the gate so substrate proteins can be degraded in the 20 S particle of the 26 S proteasome. Rpt2 enhances this mechanism suggesting that proteasome activation is a multistate process. Because binding of Rpt2 and Rpt5 increased proteasome activity in an additive manner, it was suggested that each binds to distinct sites in the 20 S proteasome. These results suggest that Rpt subunits bind to dedicated sites on the proteasome and play specific role in activation of 26 S proteasome (43). Our results are consistent with these reports: proteasome subunits of the AAA family exert different roles in the regulation of proteasome activity and the function of these subunits operates in a cell-specific fashion. Specifically, in myoblasts, cleavage of Rpt5 by activated caspase-3, decreases activity of the 26 S proteasome, indicating an essential role of Rpt5. In myotubes, caspase-3 activation cleaves subunits Rpt2 and Rpt6, increasing proteasome activity, confirming a cell-specific role of AAA subunits in the regulation of proteasome activity.
How could limited cleavage of Rpt subunits affect proteasome activity? The answer is not clear but since the cleavage is incomplete, many of the proteasomes must not be contributing to the changes in activity, and those in which the ATPases are cleaved must have quite large increases in activity (greater than these overall changes would suggest). In addition, it is noteworthy, that the in vivo cleavage of subunits in muscle was greater than the in vitro cleavage measured by incubating recombinant caspase-3 with proteasomes (there was a higher amount of cleaved Rpt6 in muscle of uremic mice compared with incubation of proteasomes with caspase-3). This indicates that changes induced in vitro underestimate the larger changes that were occurring in vivo (Figs. 4, 5, and 7). We speculate that cleavage of the 19 S proteasome subunits induces changes in the structure of the proteasome leading to stimulation of its activity. A similar explanation was proposed by Sun et al. (19); they concluded that limited cleavage of proteasome subunits in Jurkat T cells induced by activation of caspase-3 affected the integrity and function of proteasomes. The ATPases are known to contribute in multiple ways to protein degradation by: unfolding protein substrates; translocating the substrates; and opening the gate in the 20S proteasomes for substrate entry. The latter effect on gate opening stimulates degradation of LLVY-AMC and other peptides, as well as proteins (44). An enhancement of this process after caspase treatment has appeal for future research especially since it is now clear that gate-opening is not at a maximal level and can be activated physiologically by binding of ubiquitinated proteins, and in pathological conditions by caspase cleavages (45).
There was variability in the degree of caspase-3-induced cleavage of subunits. In part, the variability depended on the duration of incubating proteasomes with recombinant caspase-3 or the length of time cells were treated with staurosporine. For example, when we extended the duration of the incubation of recombinant caspase-3 with in vitro translated Rpt2 and Rpt6 to 12 h there was accumulation of subunit fragments (supplemental Fig. S2). In contrast, incubation of isolated proteasomes with recombinant caspase-3 or prolonged treatment of cells with staurosporine resulted in variability in the amounts of cleaved fragments of subunits Rpt2 and Rpt6. We suppose that the cleaved fragments of Rpt2 and Rpt6 could have been degraded in cells or cell lysates contributing to variability in the amounts of cleaved subunit proteins. Indeed, we have found that activated caspase-3 will cleave actin and actomyosin leaving a specific 14-kDa fragment that is degraded by the UPS (27).
Physiologically, these processes can play a prominent role in catabolic conditions causing loss of muscle proteins. In muscle, the positive feed-forward amplification of protein degradation by caspase-3 has two roles, activation of the proteasome plus conversion of actomyosin and myofibrils into substrates for the UPS (27).
Supplementary Material
This work was supported, in whole or in part, by the National Institutes of Health Grants NIH R37 DK37175 and P50 DK64233 (to W. E. M.) and R01HL70762 (to J. D.). This work was also supported by the University Research Committee of Emory University and a Norman S. Coplon extramural research grant from Satellite Health (to X. H. W. and L. Z.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S4.
- UPS
- ubiquitin-proteasome system
- Rpt
- regulatory particle ATPase subunit
- Rpn
- regulatory particle non-ATPase subunit
- WT
- wild type
- Stau
- staurosporine
- Ub5DHFR
- polyubiquitinated dihydrofolate reductase
- Ubal
- ubiquitin-aldehyde
- CKD
- chronic kidney disease
- XIAP
- X-linked inhibitor of apoptosis protein
- LLVY-AMC
- N-Suc-Leu-Leu-Val-Tyr 7-amino-4-methylcoumarin.
REFERENCES
- 1.Rock K. L., Gramm C., Rothstein L., Clark K., Stein R., Dick L., Hwang D., Goldberg A. L. (1994) Cell 78, 761–771 [DOI] [PubMed] [Google Scholar]
- 2.Glickman M. H., Ciechanover A. (2002) Physiol. Rev. 82, 373–428 [DOI] [PubMed] [Google Scholar]
- 3.Mitch W. E., Goldberg A. L. (1996) N. Engl. J. Med. 335, 1897–1905 [DOI] [PubMed] [Google Scholar]
- 4.Malik B., Schlanger L., Al-Khalili O., Bao H. F., Yue G., Price S. R., Mitch W. E., Eaton D. C. (2001) J. Biol. Chem. 276, 12903–12910 [DOI] [PubMed] [Google Scholar]
- 5.Rock K. L., York I. A., Saric T., Goldberg A. L. (2002) Adv. Immunol. 80, 1–70 [DOI] [PubMed] [Google Scholar]
- 6.Hershko A. (2005) Cell Death Differ. 12, 1191–1197 [DOI] [PubMed] [Google Scholar]
- 7.Chen Z. J. (2005) Nat. Cell Biol. 7, 758–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Angers S., Thorpe C. J., Biechele T. L., Goldenberg S. J., Zheng N., MacCoss M. J., Moon R. T. (2006) Nat. Cell Biol. 8, 348–357 [DOI] [PubMed] [Google Scholar]
- 9.Brown M. G., Driscoll J., Monaco J. J. (1991) Nature 353, 355–357 [DOI] [PubMed] [Google Scholar]
- 10.Yang Y., Waters J. B., Früh K., Peterson P. A. (1992) Proc. Natl. Acad. Sci. U.S.A. 89, 4928–4932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goldberg A. L., Cascio P., Saric T., Rock K. L. (2002) Mol. Immunol. 39, 147–164 [DOI] [PubMed] [Google Scholar]
- 12.Bailey J. L., Wang X., England B. K., Price S. R., Ding X., Mitch W. E. (1996) J. Clin. Invest. 97, 1447–1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mansoor O., Beaufrere B., Boirie Y., Ralliere C., Taillandier D., Aurousseau E., Schoeffler P., Arnal M., Attaix D. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 2714–2718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Price S. R., Bailey J. L., Wang X., Jurkovitz C., England B. K., Ding X., Phillips L. S., Mitch W. E. (1996) J. Clin. Invest. 98, 1703–1708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tiao G., Hobler S., Wang J. J., Meyer T. A., Luchette F. A., Fischer J. E., Hasselgren P. O. (1997) J. Clin. Invest. 99, 163–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mitch W. E., Bailey J. L., Wang X., Jurkovitz C., Newby D., Price S. R. (1999) Am. J. Physiol. 276, C1132–1138 [DOI] [PubMed] [Google Scholar]
- 17.Williams A., Sun X., Fischer J. E., Hasselgren P. O. (1999) Surgery 126, 744–749 [PubMed] [Google Scholar]
- 18.Lecker S. H., Jagoe R. T., Gilbert A., Gomes M., Baracos V., Bailey J., Price S. R., Mitch W. E., Goldberg A. L. (2004) FASEB J. 18, 39–51 [DOI] [PubMed] [Google Scholar]
- 19.Sun X. M., Butterworth M., MacFarlane M., Dubiel W., Ciechanover A., Cohen G. M. (2004) Mol. Cell 14, 81–93 [DOI] [PubMed] [Google Scholar]
- 20.Köhler A., Cascio P., Leggett D. S., Woo K. M., Goldberg A. L., Finley D. (2001) Mol. Cell 7, 1143–1152 [DOI] [PubMed] [Google Scholar]
- 21.Smith D. M., Kafri G., Cheng Y., Ng D., Walz T., Goldberg A. L. (2005) Mol. Cell 20, 687–698 [DOI] [PubMed] [Google Scholar]
- 22.Sacheck J. M., Ohtsuka A., McLary S. C., Goldberg A. L. (2004) Am. J. Physiol. Endocrinol. Metab. 287, E591–E601 [DOI] [PubMed] [Google Scholar]
- 23.Bodine S. C., Latres E., Baumhueter S., Lai V. K., Nunez L., Clarke B. A., Poueymirou W. T., Panaro F. J., Na E., Dharmarajan K., Pan Z. Q., Valenzuela D. M., DeChiara T. M., Stitt T. N., Yancopoulos G. D., Glass D. J. (2001) Science 294, 1704–1708 [DOI] [PubMed] [Google Scholar]
- 24.Lee S. W., Dai G., Hu Z., Wang X., Du J., Mitch W. E. (2004) J. Am. Soc. Nephrol. 15, 1537–1545 [DOI] [PubMed] [Google Scholar]
- 25.Sandri M., Sandri C., Gilbert A., Skurk C., Calabria E., Picard A., Walsh K., Schiaffino S., Lecker S. H., Goldberg A. L. (2004) Cell 117, 399–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stitt T. N., Drujan D., Clarke B. A., Panaro F., Timofeyva Y., Kline W. O., Gonzalez M., Yancopoulos G. D., Glass D. J. (2004) Mol. Cell 14, 395–403 [DOI] [PubMed] [Google Scholar]
- 27.Du J., Wang X., Miereles C., Bailey J. L., Debigare R., Zheng B., Price S. R., Mitch W. E. (2004) J. Clin. Invest. 113, 115–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Song Y. H., Li Y., Du J., Mitch W. E., Rosenthal N., Delafontaine P. (2005) J. Clin. Invest. 115, 451–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang X., Hu Z., Hu J., Du J., Mitch W. E. (2006) Endocrinology 147, 4160–4168 [DOI] [PubMed] [Google Scholar]
- 30.Fernando P., Kelly J. F., Balazsi K., Slack R. S., Megeney L. A. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 11025–11030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Isozaki U., Mitch W. E., England B. K., Price S. R. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 1967–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou Q., Du J., Hu Z., Walsh K., Wang X. H. (2007) Endocrinology 148, 5696–5705 [DOI] [PubMed] [Google Scholar]
- 33.Kisselev A. F., Goldberg A. L. (2005) Methods Enzymol. 398, 364–378 [DOI] [PubMed] [Google Scholar]
- 34.Koulich E., Li X., DeMartino G. N. (2008) Mol. Biol. Cell 19, 1072–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fabunmi R. P., Wigley W. C., Thomas P. J., DeMartino G. N. (2000) J. Biol. Chem. 275, 409–413 [DOI] [PubMed] [Google Scholar]
- 36.Wang X. H., Du J., Klein J. D., Bailey J. L., Mitch W. E. (2009) Kidney Int. 76, 751–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang X. H., Hu J., Du J., Klein J. D. (2007) Gene. Ther. 14, 711–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bailey J. L., Zheng B., Hu Z., Price S. R., Mitch W. E. (2006) J. Am. Soc. Nephrol. 17, 1388–1394 [DOI] [PubMed] [Google Scholar]
- 39.Nicholson D. W., Ali A., Thornberry N. A., Vaillancourt J. P., Ding C. K., Gallant M., Gareau Y., Griffin P. R., Labelle M., Lazebnik Y. A. (1995) Nature 376, 37–43 [DOI] [PubMed] [Google Scholar]
- 40.Verma R., Aravind L., Oania R., McDonald W. H., Yates J. R., 3rd, Koonin E. V., Deshaies R. J. (2002) Science 298, 611–615 [DOI] [PubMed] [Google Scholar]
- 41.Minetti C. A., Blake M. S., Remeta D. P. (1998) J. Biol. Chem. 273, 25329–25338 [DOI] [PubMed] [Google Scholar]
- 42.Rabl J., Smith D. M., Yu Y., Chang S. C., Goldberg A. L., Cheng Y. (2008) Mol. Cell 30, 360–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gillette T. G., Kumar B., Thompson D., Slaughter C. A., DeMartino G. N. (2008) J. Biol. Chem. 283, 31813–31822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith D. M., Chang S. C., Park S., Finley D., Cheng Y., Goldberg A. L. (2007) Mol. Cell 27, 731–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peth A., Besche H. C., Goldberg A. L. (2009) Mol. Cell 36, 794–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hu J., Du J., Zhang L., Price S. R., Klein J. D., Wang H. X. (2010) J. Am. Soc. Nephrol., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.