

Cecropin B derived from the hemolymph of Bombyx mori has been crystallized by the hanging-drop vapour-diffusion method. The crystal diffracted to 1.43 Å resolution using X-ray radiation.
Keywords: cecropin B, Bombyx mori, antimicrobial peptides
Abstract
Cecropin B is a 37-residue cationic antimicrobial peptide derived from the haemolymph of Bombyx mori. The precise mechanism by which cecropins exert their antimicrobial and cytolytic activities is not well understood. Crystals of cecropin B were obtained by the hanging-drop vapour-diffusion method using polyethylene glycol as a precipitant at 289 K. The crystal diffracted to 1.43 Å resolution using X-ray radiation and belonged to the orthorhombic space group P1, with unit-cell parameters a = 15.08, b = 22.75, c = 30.20 Å, α = 96.9, β = 103.1, γ = 96.5°. The asymmetric unit contained only one molecule of cecropin B, with a calculated Matthews coefficient of 2.48 Å3 Da−1 and a solvent content of 50.4%.
1. Introduction
In the past decade, a large number of cationic antimicrobial peptides (AMPs) have been isolated from various animal, plant and bacterial species (Bals & Wilson, 2003 ▶; Ganz, 2003 ▶; Jenssen et al., 2006 ▶; van Dijk et al., 2007 ▶). Most of these AMPs have cationic and amphipathic properties that do not target specific molecular receptors of pathogens but rather interact with and permeabilize microbial membranes (Lehrer & Ganz, 1999 ▶; Zasloff, 2002 ▶). The development of AMPs as novel therapeutic agents could potentially overcome the serious problem of the increasing resistance of pathogens to chemical antibiotics (Hancock & Patrzykat, 2002 ▶).
In insects, AMPs such as cecropins, defensins, proline-rich antimicrobial peptides and antifungal proteins are mainly synthesized in the fat body and secreted into the haemolymph to combat invading microorganisms (Lehrer & Ganz, 1999 ▶). In particular, cecropins have been well documented to play important roles in innate immunity in species from insects to vertebrates. Initially discovered in Hyalophora cecropia (Steiner et al., 1981 ▶), cecropins now refer to a group of peptides, designated A, B, C, D, E and F, with highly homologous sequences of 35–39 amino acids in length (Hultmark et al., 1980 ▶, 1982 ▶).
These peptides are of low molecular weight and are soluble and heat-stable; importantly, they exhibit a broad spectrum of antimicrobial activities against both Gram-positive and Gram-negative bacteria but are unable to lyse normal eukaryotic cells (Sato & Feix, 2006 ▶). Moreover, cecropins have been shown to specifically target certain cancer cells and are therefore potentially useful in selective therapy (Boman, 1995 ▶; Chen et al., 1997 ▶; Steiner et al., 1981 ▶).
Previous studies have demonstrated that AMPs primarily act as self-defence systems against microorganisms or pathogens by forming channels or pores in microbial cell membranes, causing cell permeation and disruption of cellular physiology. Several mechanisms have been proposed for AMP-mediated membrane disruption. For example, melittin and pandinin can insert into the membrane hydrophobic core with their average helical axis perpendicular to the membrane surface and rotate around the membrane (Naito et al., 2000 ▶; Nomura et al., 2004 ▶; Toraya et al., 2004 ▶). Additionally, magainin 2 and mastoparan X form ‘toroidal pores’ in the lipid bilayer, in which the inner and outer monolayers are associated via phospholipid lining and the pores are formed by the lipid polar head groups and the helix polar face (Matsuzaki et al., 1998 ▶). In contrast, cecropin A and pandinin lie on the surface of the lipid bilayer and self-associate in a ‘carpet-like’ manner (Marassi et al., 1999 ▶; Nomura et al., 2005 ▶).
The sequences of natural cecropins have basic residues in their N-terminal segments and hydrophobic residues in their C-terminal segments; the N-terminal sequence of cecropin B was found to be more important than the C-terminal sequence from a functional point of view (Wu et al., 2009 ▶).
The solution structures of natural cecropin A and cecropin P1 and the custom peptides cecropin B1, cecropin B1a and cecropin B3 have been determined by nuclear magnetic resonance (NMR) spectroscopy (Holak et al., 1988 ▶; Sipos et al., 1992 ▶; Srisailam et al., 2000 ▶, 2001 ▶; Wu et al., 2009 ▶). There is about 50% sequence identity between cecropin B and cecropin B1a, which is the homologue with the highest sequence similarity to cecropin B.
Although many reports have shown that AMPs may interact with the lipid bilayers of the cell membrane and consequently lead to cell death by membranous pore perforation or in a carpet-like manner (Oren & Shai, 1998 ▶; Zasloff, 2002 ▶), the mechanism by which cationic antimicrobial peptides cause cell death are not yet fully understood. Furthermore, the structure–function relationship of cecropin B remains unclear and its three-dimensional structure is unknown. Structure determination of cecropin B will help to clarify the mechanisms of action of these peptides. In this communication, we report the crystallization and preliminary X-ray diffraction analysis of cecropin B from Bombyx mori.
2. Materials and methods
2.1. Synthesis of cecropin B
Cecropin B (NP-001037460) was synthesized chemically. The synthesis of cecropin B (4.1 kDa) was carried out by solid-phase peptide synthesis using Fmoc chemistry and purification by reversed-phase high-performance liquid chromatography (RP-HPLC). The purity of the peptide synthesized was verified by analytical RP-HPLC and was further characterized by mass spectrometry and amino-acid analysis.
2.2. Crystallization
Prior to crystallization, the protein was dissolved to 40 mg ml−1 in 10 mM Tris–HCl pH 8.0, 150 mM NaCl, 5 mM DTT and centrifuged for 30 min at 13 000g and 277 K to clarify the solution. Screening of crystallization conditions was carried out by the hanging-drop vapour-diffusion method using Crystal Screen and Crystal Screen 2 (Hampton Research) at 289 K. The drop size was 2 µl, with a 1:1 protein:reservoir ratio. The volume of the reservoir solution was 20 µl. Crystals were obtained using Crystal Screen condition No. 24 [0.2 M calcium chloride dihydrate, 0.1 M sodium acetate trihydrate pH 4.6, 20%(v/v) 2-propanol] after five months. However, these twinned crystals were unsuitable for data collection. Alternative crystals appeared using Crystal Screen 2 condition No. 7 [10%(w/v) polyethylene glycol 1000, 10%(w/v) polyethylene glycol 8000] after six months (Fig. 1 ▶).
Figure 1.

Crystals of cecropin B from B. mori grown in 10%(w/v) polyethylene glycol 1000 and 10%(w/v) polyethylene glycol 8000. The scale bar represents 0.1 mm.
2.3. Data collection
A complete data set was collected at 100 K on a Rigaku R-AXIS IV++ (Rigaku, Japan) image-plate detector equipped with Osmic MaxFlux confocal optics using X-rays with a wavelength of 1.54 Å (Cu Kα) from a Rigaku rotating-anode generator operated at 40 kV and 100 mA. Prior to data collection, the crystal was dehydrated for a few seconds in cryoprotectant solution [20%(w/v) polyethylene glycol 1000, 20%(w/v) polyethylene glycol 8000]. The crystal was then mounted in a 0.5 mm cryoloop (Hampton Research) and flash-frozen in a nitrogen stream at 100 K. Data collection was performed with a total oscillation range of 360°, a step size of 1.0° and an exposure time of 30 s. All diffraction data were processed using the HKL-2000 program package (Otwinowski & Minor, 1997 ▶).
3. Results and discussion
Diffraction data were obtained from the crystal in the resolution range 50–1.43 Å (Fig. 2 ▶) and were processed using the HKL-2000 program package. Data-collection and processing statistics are shown in Table 1 ▶. The crystals belonged to space group P1, with unit-cell parameters a = 15.08, b = 22.75, c = 30.20 Å, α = 96.9, β = 103.1, γ = 96.5°. Based on the molecular weight of 4.1 kDa, packing considerations indicated the presence of one molecule in the asymmetric unit, which corresponds to a Matthews coefficient (Matthews, 1968 ▶) of 2.48 Å3 Da−1 and a solvent content of 50.4%. Space group P1 is inherently less likely to accumulate multiple measurements of an individual reflection than other space groups because of the lack of symmetry. Furthermore, during the scaling procedure many spots were rejected by HKL-2000 because very strong diffraction saturated the detector, so that the overall completeness and that in the last resolution shell are low, while the mean I/σ(I) is large.
Figure 2.

X-ray diffraction image from a cecropin B crystal.
Table 1. Crystal data and data-collection statistics.
Values in parentheses are for the highest resolution shell.
| Space group | P1 |
| Unit-cell parameters (Å, °) | a = 15.08, b = 22.75, c = 30.20, α = 96.9, β = 103.1, γ = 96.5 |
| Matthews coefficient (Å3 Da−1) | 2.48 |
| Solvent content (%) | 50.4 |
| No. of protomers per asymmetric unit | 1 |
| Temperature (K) | 100 |
| X-ray wavelength (Å) | 1.54 |
| Oscillation range (°) | 1.0 |
| Crystal-to-detector distance (mm) | 70 |
| Resolution range (Å) | 50–1.43 (1.45–1.43) |
| Completeness (%) | 88.5 (69.4) |
| Rmerge† (%) | 2.3 (4.3) |
| Mean I/σ(I) | 44.6 (38.1) |
| Redundancy | 3.8 (3.0) |
R
merge = 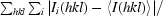
 , where 〈I(hkl)〉 is the mean intensity of symmetry-equivalent reflections.
, where 〈I(hkl)〉 is the mean intensity of symmetry-equivalent reflections.
Since there is about 50% sequence identity between cecropin B and cecropin B1a (PDB code 2igr; Wu et al., 2009 ▶), one might expect the cecropin B1a structure to be useful in structure solution by the molecular-replacement method. Regrettably, molecular-replacement calculations using the cecropin B1a structure as the model did not yield a solution.
Our efforts are now to aimed at growing crystals of heavy-atom derivatives and thus determining the structure of cecropin B using the multiple isomorphous replacement (MIR) method. In addition, attempts are being made to solve the crystal structure by combining molecular replacement with direct methods.
Acknowledgments
We would like to thank the Institute of Biophysics, Chinese Academy of Sciences for assistance with data collection. We are grateful to Dr Zhiyong Lou and Kai Zhang for their technical support. This work was supported by grants from the Xinjiang Science and Technology Aiding Project of China (Nos. 200821120 and 200991130), the National Drug Discovery Program (No. 2008ZX09401-05) and the National Natural Science Foundation of China (No. 30770439).
References
- Bals, R. & Wilson, J. M. (2003). Cell. Mol. Life Sci.60, 711–720. [DOI] [PMC free article] [PubMed]
- Boman, H. G. (1995). Annu. Rev. Immunol.13, 61–92. [DOI] [PubMed]
- Chen, H. M., Wang, W., Smith, D. & Chan, S. C. (1997). Biochim. Biophys. Acta, 1336, 171–179. [DOI] [PubMed]
- Dijk, A. van, Veldhuizen, E. J. A., Kalkhove, S. I. C., Johanna, L. M., Bokhoven, T. V., Romijn, R. A. & Haagsman, H. P. (2007). Antimicrob. Agents Chemother.51, 912–922. [DOI] [PMC free article] [PubMed]
- Ganz, T. (2003). Nature Rev. Immunol.3, 710–720. [DOI] [PubMed]
- Hancock, R. E. W. & Patrzykat, A. (2002). Curr. Drug Targets Infect. Dis.2, 79–83. [DOI] [PubMed]
- Holak, T. A., Engstrom, A., Kraulis, P. J., Lindeberg, G., Bennich, H. & Jones, T. A. (1988). Biochemistry, 27, 7620–7629. [DOI] [PubMed]
- Hultmark, D., Engstrom, A., Bennich, H., Kapur, R. & Boman, H. G. (1982). Eur. J. Biochem.127, 207–217. [DOI] [PubMed]
- Hultmark, D., Steiner, H., Rasmuson, T. & Boman, H. G. (1980). Eur. J. Biochem.106, 7–16. [DOI] [PubMed]
- Jenssen, H., Hamill, P. & Hancock, R. E. W. (2006). Clin. Microbiol. Rev.19, 491–511. [DOI] [PMC free article] [PubMed]
- Lehrer, R. I. & Ganz, T. (1999). Curr. Opin. Immunol.11, 23–27. [DOI] [PubMed]
- Marassi, F. M., Opella, S. J., Juvvadi, P. & Merrifield, R. B. (1999). Biophys. J.77, 3152–3155. [DOI] [PMC free article] [PubMed]
- Matsuzaki, K., Sugishita, K., Ishibe, N., Ueha, M., Nakata, S., Miyajima, K. & Epand, R. M. (1998). Biochemistry, 37, 11856–11863. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Naito, A., Nagao, T., Norisada, K., Mizuno, T., Tuzi, S. & Saito, H. (2000). Biophys. J.78, 2405–2417. [DOI] [PMC free article] [PubMed]
- Nomura, K., Corzo, G., Nakajima, T. & Iwashita, T. (2004). Biophys. J.87, 2497–2507. [DOI] [PMC free article] [PubMed]
- Nomura, K., Ferrat, G., Nakajima, T., Darbon, H., Iwashita, T. & Corzo, G. (2005). Biophys. J.89, 4067–4080. [DOI] [PMC free article] [PubMed]
- Oren, Z. & Shai, Y. (1998). Biopolymers, 7, 451–463. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Sato, H. & Feix, J. B. (2006). Biochim. Biophys. Acta, 1758, 1245–1256. [DOI] [PubMed]
- Sipos, D., Andersson, M. & Ehrenberg, A. (1992). Eur. J. Biochem.209, 163–169. [DOI] [PubMed]
- Srisailam, S., Arunkumar, A. I., Wang, W., Yu, C. & Chen, H. M. (2000). Biochim. Biophys. Acta, 1479, 275–285. [DOI] [PubMed]
- Srisailam, S., Kumar, T. K., Arunkumar, A. I., Leung, K. W., Yu, C. & Chen, H. M. (2001). Eur. J. Biochem.268, 4278–4284. [DOI] [PubMed]
- Steiner, H., Hultmark, D., Engstrom, A., Bennich, H. & Boman, H. G. (1981). Nature (London), 292, 246–248. [PubMed]
- Toraya, S., Nishimura, K. & Naito, A. (2004). Biophys. J.87, 3323–3335. [DOI] [PMC free article] [PubMed]
- Wu, J.-M., Jan, P.-S., Yu, H.-C., Haung, H.-Y., Fang, H.-J., Chang, Y.-I., Cheng, J.-W. & Chen, H. M. (2009). Peptides, 30, 839–848. [DOI] [PubMed]
- Zasloff, M. (2002). Nature (London), 415, 389–395. [DOI] [PubMed]