

Abstract
Atherosclerosis represents the most significant risk factor for coronary artery disease (CAD), the leading cause of death in developed countries. To better understand the pathogenesis of atherosclerosis, we applied a likelihood-based model selection method to infer gene-disease causality relationships for the aortic lesion trait in a segregating mouse population demonstrating a spectrum of susceptibility to developing atherosclerotic lesions. We identified 292 genes that tested causal for aortic lesions from liver and adipose tissues of these mice, and we experimentally validated one of these candidate causal genes, complement component 3a receptor 1 (C3ar1), using a knockout mouse model. We also found that genes identified by this method overlapped with genes progressively regulated in the aortic arches of 2 mouse models of atherosclerosis during atherosclerotic lesion development. By comparing our gene set with findings from public human genome-wide association studies (GWAS) of CAD and related traits, we found that 5 genes identified by our study overlapped with published studies in humans in which they were identified as risk factors for multiple atherosclerosis-related pathologies, including myocardial infarction, serum uric acid levels, mean platelet volume, aortic root size, and heart failure. Candidate causal genes were also found to be enriched with CAD risk polymorphisms identified by the Wellcome Trust Case Control Consortium (WTCCC). Our findings therefore validate the ability of causality testing procedures to provide insights into the mechanisms underlying atherosclerosis development.
Introduction
Atherosclerosis is a progressive inflammatory disease that is characterized by hardening of the artery walls, mostly owing to depositions of lipid and fibrous elements that lead to atherosclerotic plaque formation (1, 2). Atherosclerosis represents the most significant risk factor for coronary artery disease (CAD), the leading cause of death in high-income countries. In order to better elucidate the molecular mechanisms, considerable efforts have been made to identify genes associated with the progression of atherosclerosis development using gene expression profiling of mouse models and human atherosclerotic samples (3–7). While the gene-trait correlation information obtained from these studies is important for overall understanding of the disease, it lacks the ability to distinguish genes that are causal for atherosclerosis development from those that are merely reactive to the onset and progression of the disease.
To address this limitation, we previously developed and validated a likelihood-based causality model selection (LCMS) procedure (8, 9). Briefly, for an mRNA transcript and a phenotypic trait linked to the same locus, 3 basic relationships — causal, reactive, and independent — exist. The joint probability distributions corresponding to these relationships can be calculated, and Bayesian Information Criteria (BIC) are then compared to infer the most likely of the 3 models.
In the current study, we applied LCMS to an atherosclerosis-related trait, namely aortic arch lesion size, measured in a mouse F2 population between C57BL/6J (B6) and C3H/HeJ (C3H) on the Apoe–/– background (BxH Apoe–/– cross; refs. 10, 11). We identified 292 genes that were supported as causal for aortic lesions from the adipose and liver of this cross. To validate these genes, we selected a candidate that we believe to be novel, complement component 3a receptor 1 (C3ar1), and assessed whether perturbation of this gene causes changes in lesion size in C3ar1 KO mice on an Apoe–/– background. We also performed additional validation studies in which we profiled the aortic arches of 2 mouse models, Apoe–/– and Ldlr–/– with human CETP transgene (referred to herein as Ldlr–/– huCETP tg) to identify genes that were altered during atherosclerotic lesion progression. We reason that lesion progression signature involves genes that respond to the initial perturbations (e.g., lack of Apoe or Ldlr) and then regulate the processes that result in lesion development, and therefore will be enriched for lesion candidate causal genes. Moreover, linking genes tested causal in peripheral tissues to genes tracking plaque progression in the aorta, a more physiologically relevant tissue, will provide additional support to the relevance of the genes tested causal to atherogenesis. As a third validation, we intersected the candidate causal genes with publically available human genome-wide association studies (GWAS) of CAD to test whether known CAD risk genes are among our candidate lesion causal genes and whether these candidates are enriched for genes whose functional DNA variations show evidence of association with CAD risk in humans.
Results
Identification of genes testing causal for aortic lesions in the BxH Apoe–/– F2 cross.
The BxH Apoe–/– cross was constructed using 2 strains of mice that differ dramatically in lesion susceptibility: C3H (resistant) and B6 (susceptible). The mean lesion size in F2 females was 244,524 ± 8,694 μm2, whereas the mean for F2 males was 176,424 ± 7,220 μm2 (P < 1 × 10–4, Student’s t test) (10). By integrating genotype, gene expression, and aortic lesion size measured in 334 F2 mice and testing causal, reactive, and independent models using the LCMS procedure, we identified 292 genes supported as causal for aortic lesions at a false discovery rate (FDR) of less than 5%. Among these, 89 genes were specific to the liver, 188 were specific to the adipose, and 15 were found in both tissues. We then rank-ordered the 292 genes according to the percentage of genetic variance in the aortic lesion trait that was causally explained by the variation in their transcript abundances (referred to herein as trait r2) within each tissue-gender combination (Supplemental Table 1; supplemental material available online with this article; doi: 10.1172/JCI42742DS1). Notably, all but 1 of the candidate causal genes were identified from females, supporting gender specificity of the aortic lesion trait in mice (12). The lack of identifiable causal genes from males could be due to weaker clinical quantitative trait loci (cQTLs) for aortic lesions resulting from the smaller lesion size and range in males. The 20 most highly ranked genes from adipose and 10 from liver are listed in Table 1 and represent the strongest causal candidates for the aortic lesion trait in this mouse population.
Table 1 .
Top candidate causal genes for aortic lesions identified from the liver and adipose tissues of the BxH Apoe–/– cross

As shown in Figure 1 and detailed in Supplemental Table 2, genes from adipose and liver that were supported as causal for aortic lesions were located on 9 different chromosomes — 1, 3, 5, 7, 9, 12, 13, 15, and 17 — the same chromosomes that contain the cQTLs (i.e., QTLs for aortic lesions). This is because one of the criteria we used to select candidate causal genes was the overlap between cQTLs and eQTLs; that is, only genes that have detectable eQTLs which overlap with the 10 lesion cQTLs were allowed to enter the LCMS causality test. As many of the strong detectable eQTLs are cis-eQTLs, it is less surprising that the candidate causal genes were clustered on the same chromosomes as the cQTLs. The majority of the candidate causal genes from adipose tissue mapped to chromosome 1, and the majority of the liver genes mapped to chromosome 7, suggestive of tissue specificity of lesion candidate causal genes. The observation of these hotspots can be explained by the pleiotropic effect of genetic loci on multiple local genes or strong cis-coregulation of adjacent genes around the cQTLs.
Figure 1. Linkage map of lesion candidate causal genes in adipose (A) and liver (B) tissue and aortic lesion trait in the BxH Apoe–/– F2 cross.

Chromosomes are plotted on the x axis. The candidate causal genes are arranged on the y axis based on their similarities in the eQTL mapping patterns using agglomerative hierarchical clustering, whereas the cQTL for aortic lesion trait in females was plotted separately at the bottom and was not part of the hierarchical clustering. The color bar to the right shows the spectrum of lod scores.
Pathway analyses of candidate causal genes for atherosclerosis.
As shown in Table 2, the 292 genes supported as causal for aortic lesions were enriched for immune response–related pathways such as lymphocyte activation, B cell receptor signaling, and natural killer cell signaling (Bonferroni-corrected enrichment P = 5.67 × 10–5, 6.12 × 10–4, and 2.53 × 10–2, respectively; Supplemental Table 3). In concordance with these enriched functional pathways, the candidate causal genes were also enriched for genes predominantly expressed in mouse lymph nodes and spleen (Bonferroni-corrected P = 6.12 × 10–15 and P = 1.12 × 10–12, respectively) based on mouse expression body atlas (13). These lines of evidence support the causal involvement of inflammatory process in atherogenesis.
Table 2 .
Comparison of top enriched pathways and/or functionalities among the atherosclerosis-related gene sets

We have previously identified a particular coexpression subnetwork that was enriched for macrophage genes as well as genes tested causal for many metabolic traits (13), hence the name macrophage-enriched metabolic network (MEMN) module. Among the 292 aortic lesion candidate causal genes, 141 were found to be within the MEMN module, and the overlap was highly significant (P = 2.00 × 10–94, Fisher exact test), implicating macrophages as major players in lesion formation and suggesting that the aortic lesion trait shares common inflammatory causal factors with other metabolic traits.
Single-gene validation using C3ar1 Apoe double KO mice.
As a means of validating the strength of our lesion candidate causal genes identified in the above mouse F2 cross, we selected C3ar1 for characterization. C3ar1 was chosen because (a) it is within the MEMN module, a subnetwork linking to multiple metabolic traits, as described above; (b) it has been validated as a causal gene for obesity, as demonstrated by significant changes in adiposity in C3ar1–/– mice compared with littermate controls in the absence of significant changes in lipid profiles or weight (8, 9), and has also been shown to be a key mediator of insulin resistance, as evidenced by increased insulin sensitivity in C3ar1–/– mice (14), making it attractive to test its role in atherosclerosis; and (c) a C3ar1–/– mouse model was readily available. In the current study, C3ar1 was identified as a causal gene for aortic lesion size in females, but not in males. As C3ar1 is a lowly ranked candidate causal gene that most probably does not represent a strong enough perturbation by itself, we expected much slower lesion development, if any, in C3ar1–/– mice without a dyslipidemia background, limiting the power to detect differences between KO and wild-type mice. To accelerate lesion development, we constructed C3ar1 Apoe double KO mice and examined aortic root lesion susceptibility. As shown in Figure 2, whereas male mice did not exhibit any significant alterations in lesion size among the 3 genotypes, possibly due to the minimal lesions induced by the mild diet condition, female heterozygous (20,331 μm2/section, P = 0.015) and homozygous (21,877 μm2/section, P = 0.045) KO mice showed significant decreases in lesion size compared with littermate control animals (32,912 μm2/section). These results support the predictions made by our causality test, but do not exclude any potential effect of C3ar1 on lesion development in males. We did not observe any significant changes in the lipid profiles in either sex, which suggests that the alteration in lesion susceptibility is not a result of dyslipidemia. Although C3ar1 is ranked as 188 of the 292 candidate causal genes based on trait r2, the validation of this gene supports the strength of our causality test.
Figure 2. Characterization of aortic lesion size in C3ar1 Apoe double KO mice.

Data are presented as mean ± SD. C3ar1+/+Apoe–/–, C3ar1+/–Apoe–/–, and C3ar1–/–Apoe–/– mice are labeled WT, Het, and Homo, respectively. The sample sizes for male WT, male Het, male Homo, female WT, female Het, and female Homo are 10, 13, 9, 13, 12, and 12, respectively. P values for significant differences (2-sided Student’s t test) are indicated.
Identification of plaque progression signatures from 2 mouse models.
Because it is labor intensive and time consuming to validate candidate causal genes using individual KO mouse models, we used an alternative validation method, comparing our aortic lesion candidate causal genes with the genes that tracked with progressive aortic lesion development in the atherosclerotic models Apoe–/– and Ldlr–/– huCETP tg.
From the Apoe–/– mouse model, a total of 2,995 of 3,690 transcriptionally active genes (2,066 upregulated and 929 downregulated) were identified as the plaque progression signature (ANOVA P < 0.01; FDR < 3.63 × 10–5, Q-value approach; ref. 15 and Supplemental Table 4). More than 300 functional categories/pathways, with T cell activation, angiogenesis, and regulation of immune response being the top 3 pathways (Bonferroni-corrected P = 1.54 × 10–27, 2.15 × 10–25, and 2.27 × 10–25, respectively; Table 2 and Supplemental Table 5), were found to be overrepresented in the upregulated gene signatures. In the downregulated signatures, 33 functionalities/pathways, with calmodulin binding, muscle development, and muscle contraction being the top 3 pathways (Bonferroni-corrected P = 4.10 × 10–8, 7.23 × 10–8, and 2.28 × 10–6, respectively), were overrepresented.
From the Ldlr–/– huCETP tg mouse model, we identified 1,966 transcripts (1,359 upregulated and 607 downregulated) as the plaque progression signature (Supplemental Table 6). Functional categories/pathways similar to those identified from the Apoe–/– mouse model were found to be overrepresented in the upregulated gene signatures (Table 2 and Supplemental Table 7). In the downregulated signatures, 35 functionalities/pathways, including digestion, muscle development, and circulation (Bonferroni-corrected P = 3.66 × 10–11, 3.34 × 10–10, and 1.90 × 10–5, respectively), were overrepresented.
Comparison of the plaque progression signatures between the 2 mouse models showed that 1,993 transcripts were unique to the Apoe–/– mouse model, 964 transcripts were unique to Ldlr–/– huCETP tg mice, and 981 demonstrated concordant directionality between the 2 mouse models (overlap P < 1 × 10–50, Fisher exact test; Supplemental Table 8).
The concordant plaque progression signatures are enriched for aortic lesion candidate causal genes.
We aimed to use the plaque progression signatures from these 2 mouse models to cross-validate the genes supported as causal for aortic lesions. We found that even though the candidate causal genes were identified from the adipose and liver tissues of the F2 mice, they significantly overlapped with the concordant plaque progression genes from the aortic arch of both Apoe–/– and Ldlr–/– huCETP tg mouse models (Table 3). Specifically, of the 292 genes supported as causal from the adipose and liver tissues, 73 (71 upregulated and 2 downregulated; Supplemental Table 9) were present in the concordant plaque progression signatures, yielding an overlap P value of 2.20 × 10–38 (Fisher exact test) and fold enrichment of 6.11. These genes included antigens Cd22, Cd48, Cd53, and Cd83 and cathepsins Ctsk and Ctsz as well as protein tyrosine phosphatases and receptors Ptpn1, Ptpn6, Ptprc, and Ptpre. Taken together, these results indicate that genes supported as causal for aortic lesions are intimately linked to plaque progression and thus support the relevance of these candidate causal genes inferred from peripheral tissues directly to atherosclerosis development in the aorta.
Table 3 .
Overlapping P values and fold enrichment among aortic lesion candidate causal genes, plaque progression signature genes, and WTCCC CAD GWAS findings at various SNP association P value cutoffs

Interestingly, among the top 30 aortic lesion candidate causal genes shown in Table 1, only those from the adipose tissue, not those from liver, overlapped with the plaque progression signatures. Similarly, when all 292 candidate causal genes were considered, the adipose genes overlapped with the plaque signatures to a much higher degree than did the liver candidate causal genes (Supplemental Figure 1). Moreover, the genes supported as causal and tracked with plaque progression were enriched for adipose candidate causal genes with higher ranks (though not necessarily among the top 30) based on trait r2. In contrast, the liver candidate causal genes showed no such trend. These results suggest greater similarity between adipose tissue and aortic plaque.
The aortic lesion candidate causal genes are genetically enriched for human CAD risk.
Because the candidate causal genes were identified in mice, it is important to address their relevance to atherosclerosis in human populations. We therefore compared our candidate lesion causal genes to risk genes of CAD and related traits as catalogued by the National Human Genome Research Institute (NHGRI) based on previous GWAS studies (16, 17). We found that 5 genes — ATP10D, SLC2A9, PIK3CG, LOXL1, and KIAA1598, previously reported to be associated with myocardial infarction (18), serum uric acid (19–21), mean platelet volume (22), aortic root size (23), and heart failure (24), respectively — were among the candidate causal genes identified in this study.
In GWAS studies, it is common that only strong hits with large genetic effects are reported and catalogued. We suspect that many more genes with smaller effects are missed in this comparison. Therefore, we used an alternative approach to assessing the overlap of the mouse lesion candidate causal genes with human GWAS findings based on the hypothesis that functional SNPs of the lesion candidate causal genes should be enriched for low P value associations to CAD. We first identified SNPs that were significantly associated with gene expression (expression SNPs; eSNPs) of the causal gene set in the liver and adipose tissues using 2 genetics of gene expression (GGE) studies (25, 26). Next, we obtained CAD association results for the eSNPs from the Wellcome Trust Case Control Consortium (WTCCC; ref. 27). We counted the proportion of eSNPs with CAD association of P < 0.001, 0.01, 0.05, or 0.1 in the lesion causal gene list and in the background gene list of the 24k mouse array and then assessed the enrichment using Fisher exact test. We found that the candidate causal genes were significantly enriched for eSNPs with low P value associations, with enrichment P values ranging from 7.15 × 10–3 to 3.39 × 10–4 and fold enrichment ranging from 1.83 to 5.04 at the 4 GWAS association P value cutoffs (Table 3). The mouse candidate causal genes whose human orthologs contain eSNPs that show evidence of CAD association at P < 0.05 in WTCCC CAD GWAS study include 3110048E14Rik (human gene FAM118A), Abcb4, BC031353 (human gene KIAA1370), BC052328 (human gene FAM105A), Cd22, Mtfmt, Plekhb1, Tbc1d1, Tmc5, Tmem71, and Usp43. The eSNPs associated with these genes in human liver and adipose tissues of the 2 GGE studies and their association P values in WTCCC CAD GWAS are shown in Supplemental Table 10.
Discussion
Integration of gene expression in the design and analysis of traditional F2 intercross studies has yielded high confidence prediction of causal genes for complex traits such as adiposity (8, 9). In this study, we applied a similar causality test to an atherosclerosis-related trait, aortic lesion size, and identified 292 candidate causal genes from 2 peripheral tissues, liver and adipose. The high number of genes that tested causal is likely due to the complexity of atherogenesis as well as the statistical inference nature of the causality test. Therefore, it is important to validate the causal calls experimentally. As independent validation approaches, we (a) tested one of these candidate causal genes — C3ar1 — using a KO mouse model, (b) compared the candidate causal genes with plaque progression signatures in the aorta of 2 mouse models with progressive development of aortic plaques, and (c) tested the overlap between the candidate causal genes and human CAD GWAS studies. We found (a) significant decreases in aortic lesion size resulting from the perturbation of C3ar1, (b) significant overlaps between the candidate causal genes and plaque progression signatures, and (c) significant enrichment of genetic associations to human CAD risk among the lesion candidate causal genes. These results support the relevance of the genes testing causal for aortic lesions to atherosclerosis development.
Enpp1 and Pik3cg, 2 of the top 30 candidate causal genes, have previously been linked to atherosclerosis (28, 29). We also experimentally validated our predictions on a relatively low-ranked causal gene for aortic lesions, C3ar1, which is involved in the complement system. Although the complement pathway has been implicated in atherosclerosis (30), our study provided direct evidence in support of a causal role for C3ar1 in altering atherosclerotic lesion susceptibility. The other candidate causal genes warrant further testing.
The candidate causal genes for aortic lesions were enriched for immune response–related genes and pathways. Furthermore, these candidate causal genes highly overlapped with the previously identified MEMN module, which was linked to multiple metabolic disorders. These lines of evidence support the inflammatory component of plaque formation as well as the connection between inflammation and other metabolic diseases, in particular obesity and diabetes (31). The fact that C3ar1 has been confirmed experimentally as a causal gene for obesity (8, 9), diabetes (14), and aortic lesions here further supports the shared etiology among these metabolic disorders. Although weight and adiposity data were not collected from the C3ar–/– mice on an Apoe–/– background, we suspect that these traits do not play a major role in the development of atherosclerosis due to the lack of dyslipidemia in these animals. It is possible that the effect of C3ar1 on macrophage infiltration and activation in adipose tissue (14) mediates lesion development in the double KO mice.
Validating candidate causal genes by perturbing individual candidate genes can be labor intensive and time consuming. Therefore, we profiled the aortic arches of 2 mouse models that develop progressive atherosclerotic lesions upon diet challenge to test the relevance of the lesion candidate causal genes to atherosclerosis in a pathologically more relevant tissue. The purpose of using 2 models was to avoid potential bias from a single model, as pathophysiological differences exist in different atherosclerosis models (32). Although only 1 pool per study group was used for the microarray analysis of the Ldlr–/– huCETP tg model, each pool was composed of 10 mice, and a total of 40 mice were studied, which substantially reduces within-group variance and can still provide meaningful and reliable signatures (33). Moreover, the reliability of the Ldlr–/– huCETP tg signature genes was supported by highly significant overlaps between the signatures from this mouse model and the signatures from (a) the Apoe–/– mouse model (P < 1 × 10–50; fold enrichment 4.08), (b) the Ldlr–/–ApoB100/100Mttpfl/flMx1-Cre mouse model reported in the literature (P < 1 × 10–50, Fisher exact test; fold enrichment 8.68; ref. 7), and (c) the candidate lesion causal genes (P = 8.75 × 10–34; fold enrichment 4.01).
We found significant overlaps between the aortic lesion candidate causal genes from peripheral tissues and the concordant upregulated plaque progression signatures from aorta, supporting the relevance of our candidate causal genes to atherogenesis. We also found greater overlap between the candidate causal genes from adipose tissue and the plaque progression signatures from the aortic arch, which suggests that different tissues are linked in a disease setting and adipose could be a surrogate tissue for plaque. Although this link may not be too surprising, given that the vessel wall and adipose share similar inflammatory changes in response to hyperlipidemia (34), it is nonetheless important, as having a surrogate tissue to track a hard-to-access tissue such as plaque will be critical for biomarker studies.
Among the 73 candidate causal genes that were confirmed in the plaque progression signature, 14 were found to contain druggable domains, and therefore may serve as drug targets. These include genes that have been previously linked to atherosclerosis, such as Abcb4, Ctsk, Ptpn1, Ptpn6, Ptprc, and Msr1 (35–40), and genes that we believe to be novel, like Atgr1a, Gpr65, Inpp5d, Mcoln2, Ctsz, Ptpre, Havcr2, and Galns. C3ar1, although not in the common plaque progression signature, is a G protein–coupled receptor and may also serve as a candidate drug target. Some of the candidate causal genes that track with plaque progression may also be potential imaging biomarkers. By requiring (a) high expression level at advanced lesion stage, (b) greater than 5-fold change in expression levels between the most advanced lesion stage tested and the baseline, and (c) extracellular localization, we propose that 7 genes, namely Cd53, Ptprc, Plek, Cd48, Ctsk, Pld3, and Il10ra, could be biomarkers through imaging. Because good markers do not need to be causal for an endpoint, we applied similar criteria to the concordant plaque progression signatures without considering lesion causality and found that Spp1 (>100-fold change in expression level during plaque progression), Vcam1, Cd53, and Abcg1 were top ranked. Among these, Vcam1 has been used as an imaging biomarker for atherosclerosis in practice (41).
Although it is encouraging to identify and validate the candidate causal genes in mice via independent experiments, it is ultimately critical to link the findings to human disease pathophysiology. The past few years have seen promises in human GWAS studies in identifying genetic variants with strong effects on common diseases, including CAD. A comparison between our candidate causal genes and GWAS genes associated with CAD and other related disease traits revealed 5 overlapping genes — ATP10D, SLC2A9, PIK3CG, LOXL1, and KIAA1598 — that have been previously linked to conditions or traits associated with atherosclerosis and/or CAD, namely myocardial infarction (18), serum uric acid (19–21), mean platelet volume (22), aortic root size (23), and heart failure (24), respectively. Moreover, 11 additional candidate causal genes were found to harbor eSNPs that show evidence of CAD association (P < 0.05) in the WTCCC CAD GWAS. These SNPs and genes warrant further testing in future studies. The link between the mouse genes tested causal for aortic lesions and genetic variants associated with human CAD risk not only validates the relevance of our mouse candidate causal genes to atherogenesis in humans, but supports the notion that numerous genes with genetic effects ranging from subtle to strong play a role in disease onset. Although the candidate causal genes were mainly identified from female mice, whereas sexual dimorphism was not reported as a significant factor in human GWAS studies, we consider the comparison conducted here appropriate based on the following reasons. First, compared with mouse crosses, human populations are less well controlled and are poorly powered to examine sex differences; it is therefore not uncommon to use different strategies and approaches to identify susceptibility genes in the 2 species. Second, a large proportion of findings on atherogenesis in mice is derived from females and not only has been replicated in humans, but also helped to advance our understanding of atherosclerosis.
It is important to note that because the current study uses gene expression levels to reflect activities of genes in the LCMS causality procedure, our findings are limited to genes whose expression levels, but not necessarily protein or biological activity levels, are affected by genetic perturbations. Incorporation of additional intermediate traits derived from proteomic and metabolic data into the LCMS procedure will provide a more comprehensive view of causal factors for complex diseases. Similarly, only eSNPs that affect gene expression were used to represent functional SNPs when conducting overlap analysis between the candidate causal genes and GWAS findings. We chose eSNPs because experimental data are available to support their putative functional role in regulating gene function. However, this approach has its limitations in the coverage of functional SNPs, because other types of functional SNPs, such as those that alter posttranscriptional mechanisms, will be missed and thus cause loss of power. A more comprehensive functional annotation of SNPs that incorporates alternative splicing, noncoding RNA, proteomics, metabolomics, and possibly other biological processes, such as epigenetics, in all key physiological tissues is needed to improve this coverage.
In summary, we have identified 292 candidate causal genes for aortic lesions in an F2 mouse cross and confirmed their relevance to atherosclerosis development via independent validation in mice and humans. That so many genes were supported as causal for this complex trait, and that these candidate causal genes formed components of gene networks linking to metabolic traits, continued to support the concept that complex traits like atherosclerosis are emergent properties of networks rather than individual genes (42). The identification of atherosclerosis candidate causal genes that tracked with plaque progression and showed evidence of association to human CAD risk provides insights into the mechanisms underlying the development of atherosclerosis and may help uncover potential therapeutic targets and diagnostic biomarkers.
Methods
Identification of candidate causal genes for aortic lesion size from BxH Apoe–/– cross.
The construction of the BxH Apoe–/– mouse cross was described previously (10, 11). Briefly, 334 (169 female, 165 male) F2 mice were fed Purina Chow containing 4% fat until 8 weeks of age and then transferred to a Western diet containing 42% fat and 0.15% cholesterol for 16 weeks. Upon euthanization, liver and white adipose tissues were collected and flash frozen in liquid N2. The tissues were profiled using microarrays containing 23,574 probes (Agilent Technologies) as described previously (10, 11), and transcript intensities were normalized and reported as the mean log10 ratio (mlratio) of an individual experiment relative to a pool composed of equal aliquots of RNA from 150 F2 and parental samples (43, 44). The microarray data from liver and adipose have been deposited to GEO under accession numbers GSE2814 and GSE3086.
To score aortic lesion size, the aortae of all mice were sectioned, and every fifth 10-μm section starting from the aortic valve throughout the aortic sinus was stained with hematoxylin and oil red O to specifically stain lipids. An ocular with a micrometer-squared grid was used to quantify lesions by measuring the average areas of fatty streaks and the adjacent fibrous cap, necrotic core, and extracellular matrix throughout the aortic sinus; the average lesion area was normalized to 40 sections. All procedures were done in accordance with the current National Research Council Guide for the Care and Use of Laboratory Animals and were approved by the UCLA Animal Research Committee.
The LCMS causality test and its variation have been previously described (8) and are further detailed here. For 2 quantitative traits (an mRNA transcript and a phenotypic trait), T1 and T2, which are linked to the same locus L in the BxH Apoe–/– F2 cross, there are 3 basic relationships between the 2 traits relative to the DNA locus. Either DNA variations at locus L lead to changes in trait T1 that in turn lead to changes in trait T2, DNA variations at locus L lead to changes in trait T2 that in turn lead to changes in trait T1, or DNA variations at locus L independently lead to changes in traits T1 and T2. Assuming standard Markov properties for these basic relationships, the joint probability distributions corresponding to these 3 models are (a) P(L,T1,T2) = P(L)P(T1|L)P(T2|T1), (b) P(L,T1,T2) = P(L)P(T2|L)P(T1|T2), and (c) P(L,T1,T2) = P(L)P(T2|L)P(T1|T2,L), respectively, where T1|T2,L in (c) reflects that the correlation between T1 and T2 may be explained by other shared loci or common environmental influences, in addition to locus L. We assume Markov equivalence between T1 and T2 for model (c), so that P(T2|L) P(T1|T2,L) = P(T1|L) P(T2|T1,L), where P(L) is the genotype probability distribution for locus L, based on a previously described recombination model (45). The random variables T1 and T2 are taken to be normally distributed about each genotypic mean at the common locus L, so that the likelihoods corresponding to each of the joint probability distributions are then based on the normal probability density function, with respective mean and variance for each component given as follows: (a) for P(T1|L), E(T1|L) = μT1L and Equation 1; (b) for P(T2|L), E(T2|L) = μT2L and Equation 2; and (c) for P(T1,T2), Equations 3 and 4, where ρ represents the correlation between T1 and T2 and μT1L and μT2L are the respective genotypic-specific means for T1 and T2. The mean and variance for P(T2|T1) follow similarly from that given for P(T1|T2).
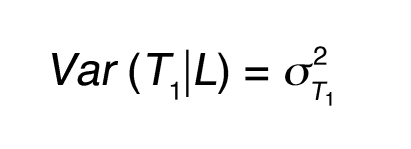
(Equation 1)
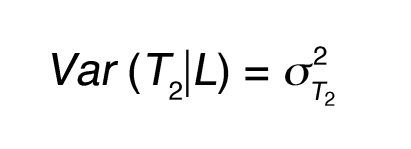
(Equation 2)
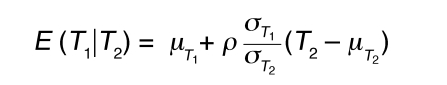
(Equation 3)
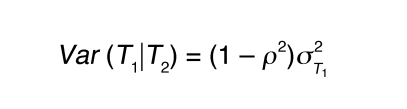
(Equation 4)
The likelihoods for each model are formed by multiplying the densities for each of the component pieces across all individuals in the population (8). The likelihoods are then compared among the different models in order to infer the most likely of the 3. Because the number of model parameters among the models differs, a penalized function of the likelihood was used to avoid bias against parsimony. The model with the smallest value of the penalized statistic –2log Li (θi|L,R,C) + k × pi was chosen, where Li (θi|L,R,C) is the maximum likelihood for the ith model, pi is the number of parameters in the ith model, and k is a constant. We took the penalized statistic to be the BIC where k is set to logn, where n denotes the number of observations.
To ease the computational burden in computing all pairwise tests, we carried out a regression-based version of the LCMS procedure described above to identify the model best supported by the data, in which the conditional likelihood arguments were replaced with equivalent conditional regression arguments. For example, if model (a) holds, then the correlation between T2 and L conditional on T1 will not be significantly different from 0. On the other hand, if model (b) holds, then the correlation between T1 and L conditional on T2 will not be significantly different from 0. If the conditional correlations in both cases are significantly greater than 0, then we can conclude that model (c) is best supported by the data. If the conditional correlations are statistically indistinguishable from 0, then the results are inconclusive.
We applied the above procedure to the aortic lesion and gene expression data from the liver and adipose tissue derived from the BxH Apoe–/– cross to identify candidate causal genes of aortic lesion trait for each tissue (liver and adipose tissue) and gender group (females and males). The first step involved the identification of cQTLs for aortic lesions using a previously established stepwise regression procedure (46, 47). As a result, 10 and 9 cQTLs with lod scores greater than 2 were identified from the female and male groups, respectively. In the second step, we searched for eQTLs that underlie each of the 23,574 expression traits, and a total of 56,588 eQTLs with lod score greater than 2 were identified. If an expression trait tests causal for the aortic lesion trait, then at least 1 QTL underlying the aortic lesion trait must also underlie the expression trait. Thus, as a third step, we identified those genes whose eQTLs coincided with the aortic lesion cQTLs as determined by a 15-cM distance range. For each overlapping eQTL/cQTL, we fitted the corresponding QTL genotypes, gene expression data, and aortic lesion data to the independent, causal, and reactive models using conditional correlation and identified the model with the highest probability. Based on the rationale that expression traits that causally explain a significant proportion of the correlation between variations in DNA and the aortic lesion trait should also correlate with the trait itself, we subsequently filtered the 1,543 causal calls by requiring significant correlations between genes tested causal and the aortic lesion trait at an FDR less than 5% based on permutation tests per tissue and gender group.
Characterization of C3ar1 Apoe double KO mouse model.
A C3ar1 KO mouse model was originally obtained from Deltagen as described previously (8) and was backcrossed to B6 for 5 generations. To produce C3ar1 Apoe double KO animals for lesion studies, C3ar1+/–Apoe+/+ animals were backcrossed to B6 Apoe–/– mice for 10 generations to generate C3ar1+/–Apoe+/– animals, which were then intercrossed to generate C3ar1+/+Apoe–/–, C3ar1+/–Apoe–/–, and C3ar1–/–Apoe–/– mice. Mice were fed a 4% fat, 0% cholesterol chow diet (diet 7017; Harlan Teklad) ad libitum for 12 weeks and then were fasted overnight and sacrificed using CO2 asphyxiation. Aortic lesions were scored using the same procedures described above for the BxH Apoe–/– F2 cross.
Identification of plaque progression signatures from Apoe–/– mouse model.
B6 Apoe–/– mice were purchased from the Jackson Laboratory and fed a high-fat diet (HFD; 21% fat, 0.15% cholesterol; diet TD.88137; Harlan Teklad) starting at 8 weeks of age for 8, 16, and 24 weeks. B6 mice fed HFD for 16 weeks starting at 8 weeks of age were used as controls. 9 mice per group were fasted overnight and euthanized, and whole aortae (from aortic root to renal bifurcation) were collected and flash-frozen in liquid N2.
Due to the small tissue size of individual arches and the lack of robust methods to profile very low amounts of RNA at the time the experiments were carried out, the aortic arches of 3 mice in the same study group were pooled, resulting in 3 pools per study group. Tissues were profiled using microarrays containing 23,574 probes (Agilent Technologies) as described previously (10, 11). The control RNA pool was composed of equal aliquots of RNA derived from the 9 pools (27 individual mice) from the HFD 8-, 16-, and 24-week time points. Transcript intensities were reported as mlratio of each pool relative to the control pool of 27 samples.
In order to identify genes that were significantly differentially expressed among the B6 control mice and Apoe–/– mice on HFD for 8, 16, and 24 weeks, we first selected genes that showed significant differential expression with fold change greater than 1.2 and error model–derived P < 0.01 (43, 48) compared with the control pool in at least 3 profiled sample pools (9 mice) to determine the set of most transcriptionally active genes. A 1-way ANOVA test was then applied to those selected genes to identify signature genes (see below).
Identification of plaque progression signatures from Ldlr–/– huCETP tg mouse model.
The Ldlr–/– huCETP tg mouse strain was initially constructed at Merck Research Laboratories by cross-breeding B6 SJL-Tg(APOA-CETP) mice from Xenogen Biosciences and B6 129S7-Ldlrtm1Her/J mice from The Jackson Laboratory, then rederived at Taconic Farms. B6 SJL-Tg(CETP) mice express the human CETP transgene under the control of the human ApoA1 promoter and are on greater than 99% B6 background.
Homozygous Ldlr–/– huCETP tg mice were fed a low-fat, cholesterol-containing diet (LFCD; 9% fat, 0.15% cholesterol) starting at 8 weeks of age for 4, 8, 12, and 16 weeks. 10 mice per group were studied. Due to the same issues as described above for the Apoe–/– mouse model, the aortic arches of 10 mice in each of the study groups were pooled for profiling, resulting in 1 pool per group. Tissues were profiled using microarrays containing 23,574 probes as described above. The control RNA pool was composed of equal aliquots of RNA derived from the 4 pools (40 samples) from the LFCD 4-, 8-, 12-, and 16-week time points. Transcript intensities were reported as mlratio of each pool relative to the control pool of 40 mice. As only 1 pool per study group was available, no formal statistical test could be applied. We treated the mlratios of the earliest time point, in this case the LFCD 4-week group, as baseline, and took the ratio of the expression values from the remaining groups against the baseline and then used K-means clustering based on cosine correlation to look for gene clusters that were progressively up- or downregulated across the 4 time points. The microarray data for both Apoe–/– and Ldlr–/– huCETP tg have been deposited to GEO under SuperSeries accession number GSE18479.
Pathway annotation of candidate causal genes and signature gene sets.
Each set of candidate causal genes or signature genes identified above was classified using Gene Ontology (GO) database (49), Panther pathway database (50), Ingenuity pathway (Ingenuity Systems), KEGG database (http://www.genome.ad.jp/kegg/pathway.html), and mouse body gene expression atlas (13) assignments. Rosetta TGI gene set annotator was used to identify overrepresented gene categories in each gene list. Only categories consisting of 10–500 genes were used in the test to reduce multiple testing.
Test for overlap between candidate lesion candidate causal genes and human CAD risk associations.
We used 2 approaches to compare the candidate lesion candidate causal genes with findings from human CAD GWAS studies. In the first approach, we downloaded the GWAS catalog from NHGRI (17) and compiled genes reported to be associated with disease and/or trait, including aortic aneurysm, aortic root size, blood lipid traits, blood pressure traits, cardiac structure and function, CAD, coronary disease, endothelial function, heart failure, major cardiovascular disease, mean platelet volume, myocardial infarction, serum uric acid, and stroke. These candidate risk genes were compared with the human orthologs of the mouse genes tested causal for lesion size.
In the second approach, we first mapped the mouse genes tested causal for lesions and all 23,574 mouse genes on the gene expression array to human orthologs and then derived eSNPs of the human orthologs using 2 GGE studies (25, 26). The first GGE study profiled more than 400 liver samples from people of European descent. The second multitissue GGE cohort was composed of approximately 1,000 patients of European descent who underwent Roux-en-Y gastric bypass surgery. Liver, subcutaneous adipose, and omental adipose tissues were collected from each patient. RNA samples of individual tissues from both cohorts were profiled on a custom 44K Agilent array, and each DNA sample was genotyped at 782,476 unique SNPs. These 2 GGE cohorts allowed us to identify a total of 20,563 distinct eSNPs associated with 9,964 known genes. The CAD association P values for the eSNPs associated with the human orthologs of the mouse lesion candidate causal genes were extracted from the WTCCC study (27). The number of eSNPs of the lesion candidate causal genes that reached P values less than 0.001, 0.01, 0.05, or 0.1 for WTCCC CAD association, and the number of eSNPs corresponding to all 23,574 mouse array transcripts that reached the same levels of CAD association, were counted. The enrichment of low P value associations was analyzed as described below.
Statistics.
To assess the differences in aortic lesions between C3ar1 Apoe double KO and Apoe–/– mice, 2-sided Student’s t test was used, and P values less than 0.05 were considered significant. To identify plaque progression signature genes in the Apoe–/– mouse model, 1-way ANOVA was used, P values less than 0.01 were considered significant, and FDR was estimated using the Q-value approach (15). To identify overrepresented pathways or gene categories in each signature gene set, 1-sided Fisher exact test was used, and P values less than 0.05 (after Bonferroni correction for multiple category comparisons) were considered significant. For human CAD risk enrichment analysis, 1-sided Fisher exact test was used to assess the enrichment of low P value associations within the causal gene set compared with the 23,574 transcripts on the mouse Agilent array at each of the 4 GWAS association significance levels (P < 0.001, 0.01, 0.05, or 0.1). Enrichment significance level was set to P < 0.05 from the Fisher exact test.
Supplementary Material
Acknowledgments
The study was funded by NIH grants HL30568 and HL28481 (to A.J. Lusis). The authors thank Archie Russell, Xavier Schidwachter, Solly Sieberts, Jason Eglin, and Robert Kleinhanz for technical assistance.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2010;120(7):2414–2422. doi:10.1172/JCI42742.
References
- 1.Libby P. Inflammation in atherosclerosis. Nature. 2002;420(6917):868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 2.Lusis AJ. Atherosclerosis. Nature. 2000;407(6801):233–241. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wuttge DM, Sirsjo A, Eriksson P, Stemme S. Gene expression in atherosclerotic lesion of ApoE deficient mice. Mol Med. 2001;7(6):383–392. [PMC free article] [PubMed] [Google Scholar]
- 4.Tabibiazar R, et al. Signature patterns of gene expression in mouse atherosclerosis and their correlation to human coronary disease. Physiol Genomics. 2005;22(2):213–226. doi: 10.1006/geno.1994.1365. [DOI] [PubMed] [Google Scholar]
- 5.Seo D, et al. Gene expression phenotypes of atherosclerosis. Arterioscler Thromb Vasc Biol. 2004;24(10):1922–1927. doi: 10.1161/01.ATV.0000141358.65242.1f. [DOI] [PubMed] [Google Scholar]
- 6.Karra R, et al. Molecular evidence for arterial repair in atherosclerosis. Proc Natl Acad Sci U S A. 2005;102(46):16789–16794. doi: 10.1073/pnas.0507718102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Skogsberg J, et al. Transcriptional profiling uncovers a network of cholesterol-responsive atherosclerosis target genes. PLoS Genet. 2008;4(3):e1000036. doi: 10.1371/journal.pgen.1000036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schadt EE, et al. An integrative genomics approach to infer causal associations between gene expression and disease. Nat Genet. 2005;37(7):710–717. doi: 10.1038/ng1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang X, et al. Validation of candidate causal genes for obesity that affect shared metabolic pathways and networks. Nat Genet. 2009;41(4):415–423. doi: 10.1038/ng.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang SS, et al. Identification of pathways for atherosclerosis in mice: integration of quantitative trait locus analysis and global gene expression data. Circ Res. 2007;101(3):e11–e30. doi: 10.1161/CIRCRESAHA.107.152975. [DOI] [PubMed] [Google Scholar]
- 11.Yang X, et al. Tissue-specific expression and regulation of sexually dimorphic genes in mice. Genome Res. 2006;16(8):995–1004. doi: 10.1101/gr.5217506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Daugherty A, Rateri DL. Development of experimental designs for atherosclerosis studies in mice. Methods. 2005;36(2):129–138. doi: 10.1016/j.ymeth.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 13.Chen Y, et al. Variations in DNA elucidate molecular networks that cause disease. Nature. 2008;452(7186):429–435. doi: 10.1038/nature06757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mamane Y, et al. The C3a anaphylatoxin receptor is a key mediator of insulin resistance and functions by modulating adipose tissue macrophage infiltration and activation. Diabetes. 2009;58(9):2006–2017. doi: 10.2337/db09-0323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A. 2003;100(16):9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hindorff LA, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A. 2009;106(23):9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Office of Population Genomics, National Human Genome Research Institute. A catalog of published genome-wide association studies. http://www.genome.gov/gwastudies . Accessed January 2, 2010. [Google Scholar]
- 18.Hicks AA, et al. Genetic determinants of circulating sphingolipid concentrations in European populations. PLoS Genet. 2009;5(10):e1000672. doi: 10.1371/journal.pgen.1000672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dehghan A, et al. Association of three genetic loci with uric acid concentration and risk of gout: a genome-wide association study. Lancet. 2008;372(9654):1953–1961. doi: 10.1016/S0140-6736(08)61343-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doring A, et al. SLC2A9 influences uric acid concentrations with pronounced sex-specific effects. Nat Genet. 2008;40(4):430–436. doi: 10.1038/ng.107. [DOI] [PubMed] [Google Scholar]
- 21.Kolz M, et al. Meta-analysis of 28,141 individuals identifies common variants within five new loci that influence uric acid concentrations. PLoS Genet. 2009;5(6):e1000504. doi: 10.1371/journal.pgen.1000504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Soranzo N, et al. A genome-wide meta-analysis identifies 22 loci associated with eight hematological parameters in the HaemGen consortium. Nat Genet. 2009;41(11):1182–1190. doi: 10.1038/ng.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vasan RS, et al. Genetic variants associated with cardiac structure and function: a meta-analysis and replication of genome-wide association data. Jama. 2009;302(2):168–178. doi: 10.1001/jama.2009.978-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Larson MG, et al. Framingham Heart Study 100K project: genome-wide associations for cardiovascular disease outcomes. BMC Med Genet. 2007;8(suppl 1):S5. doi: 10.1186/1471-2350-8-S1-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schadt EE, et al. Mapping the genetic architecture of gene expression in human liver. PLoS Biol. 2008;6(5):e107. doi: 10.1371/journal.pbio.0060107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhong H, Yang X, Kaplan LM, Molony C, Schadt EE. Integrating pathway analysis and genetics of gene expression for genome-wide association studies. Am J Hum Genet. 2010;86(4):581–591. doi: 10.1016/j.ajhg.2010.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447(7145):661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bacci S, et al. The K121Q polymorphism of the ENPP1/PC-1 gene is associated with insulin resistance/atherogenic phenotypes, including earlier onset of type 2 diabetes and myocardial infarction. Diabetes. 2005;54(10):3021–3025. doi: 10.2337/diabetes.54.10.3021. [DOI] [PubMed] [Google Scholar]
- 29.Fougerat A, et al. Genetic and pharmacological targeting of phosphoinositide 3-kinase-gamma reduces atherosclerosis and favors plaque stability by modulating inflammatory processes. Circulation. 2008;117(10):1310–1317. doi: 10.1161/CIRCULATIONAHA.107.720466. [DOI] [PubMed] [Google Scholar]
- 30.Oksjoki R, et al. Receptors for the anaphylatoxins C3a and C5a are expressed in human atherosclerotic coronary plaques. Atherosclerosis. 2007;195(1):90–99. doi: 10.1016/j.atherosclerosis.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 31.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444(7121):860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 32.Zadelaar S, et al. Mouse models for atherosclerosis and pharmaceutical modifiers. Arterioscler Thromb Vasc Biol. 2007;27(8):1706–1721. doi: 10.1161/ATVBAHA.107.142570. [DOI] [PubMed] [Google Scholar]
- 33.Peng X, Wood CL, Blalock EM, Chen KC, Landfield PW, Stromberg AJ. Statistical implications of pooling RNA samples for microarray experiments. BMC Bioinformatics. 2003;4:26. doi: 10.1186/1471-2105-4-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lohmann C, et al. Atherosclerotic mice exhibit systemic inflammation in periadventitial and visceral adipose tissue, liver, and pancreatic islets. Atherosclerosis. 2009;207(2):360–367. doi: 10.1016/j.atherosclerosis.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 35.Boilson BA, Kiernan TJ, Harbuzariu A, Nelson RE, Lerman A, Simari RD. Circulating CD34+ cell subsets in patients with coronary endothelial dysfunction. Nat Clin Pract Cardiovasc Med. 2008;5(8):489–496. doi: 10.1038/ncpcardio1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burdon KP, et al. Association of protein tyrosine phosphatase-N1 polymorphisms with coronary calcified plaque in the Diabetes Heart Study. Diabetes. 2006;55(3):651–658. doi: 10.2337/diabetes.55.03.06.db05-0058. [DOI] [PubMed] [Google Scholar]
- 37.Manning-Tobin JJ, et al. Loss of SR-A and CD36 activity reduces atherosclerotic lesion complexity without abrogating foam cell formation in hyperlipidemic mice. Arterioscler Thromb Vasc Biol. 2009;29(1):19–26. doi: 10.1161/ATVBAHA.108.176644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pennings M, et al. Bone marrow-derived multidrug resistance protein ABCB4 protects against atherosclerotic lesion development in LDL receptor knockout mice. Cardiovasc Res. 2007;76(1):175–183. doi: 10.1016/j.cardiores.2007.05.016. [DOI] [PubMed] [Google Scholar]
- 39.Samokhin AO, Wong A, Saftig P, Bromme D. Role of cathepsin K in structural changes in brachiocephalic artery during progression of atherosclerosis in apoE-deficient mice. Atherosclerosis. 2008;200(1):58–68. doi: 10.1016/j.atherosclerosis.2007.12.047. [DOI] [PubMed] [Google Scholar]
- 40.Sugano M, Tsuchida K, Maeda T, Makino N. SiRNA targeting SHP-1 accelerates angiogenesis in a rat model of hindlimb ischemia. Atherosclerosis. 2007;191(1):33–39. doi: 10.1016/j.atherosclerosis.2006.04.021. [DOI] [PubMed] [Google Scholar]
- 41.Nahrendorf M, et al. 18F-4V for PET-CT imaging of VCAM-1 expression in atherosclerosis. JACC Cardiovasc Imaging. 2009;2(10):1213–1222. doi: 10.1016/j.jcmg.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schadt EE. Molecular networks as sensors and drivers of common human diseases. Nature. 2009;461(7261):218–223. doi: 10.1038/nature08454. [DOI] [PubMed] [Google Scholar]
- 43.He YD, et al. Microarray standard data set and figures of merit for comparing data processing methods and experiment designs. Bioinformatics. 2003;19(8):956–965. doi: 10.1093/bioinformatics/btg126. [DOI] [PubMed] [Google Scholar]
- 44.Schadt EE, et al. Genetics of gene expression surveyed in maize, mouse and man. Nature. 2003;422(6929):297–302. doi: 10.1038/nature01434. [DOI] [PubMed] [Google Scholar]
- 45.Jiang C, Zeng ZB. Multiple trait analysis of genetic mapping for quantitative trait loci. Genetics. 1995;140(3):1111–1127. doi: 10.1093/genetics/140.3.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zeng ZB, Liu J, Stam LF, Kao CH, Mercer JM, Laurie CC. Genetic architecture of a morphological shape difference between two Drosophila species. Genetics. 2000;154(1):299–310. doi: 10.1093/genetics/154.1.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Laurie CC, et al. The genetic architecture of response to long-term artificial selection for oil concentration in the maize kernel. Genetics. 2004;168(4):2141–2155. doi: 10.1534/genetics.104.029686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weng L, Dai H, Zhan Y, He Y, Stepaniants SB, Bassett DE. Rosetta error model for gene expression analysis. Bioinformatics. 2006;22(9):1111–1121. doi: 10.1093/bioinformatics/btl045. [DOI] [PubMed] [Google Scholar]
- 49.Ashburner M, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mi H, et al. The PANTHER database of protein families, subfamilies, functions and pathways. Nucleic Acids Res. 2005;33(Database issue):D284–D288. doi: 10.1093/nar/gki418. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.