

Abstract
Ste2p, a G protein-coupled receptor (GPCR), binds α-factor, WHWLQLKPGQPMY, a tridecapeptide pheromone secreted by yeast cells. Upon α-factor binding, Ste2p undergoes conformational changes activating a signal transduction system through its associated heterotrimeric G protein leading to the arrest of cell growth in the G1 phase to prepare cells for mating Previous studies have indicated that Tyr at position-13 of α-factor interacts with Arg58 on transmembrane one (TM1) of Ste2p. This observation prompted this investigation to determine whether a cation-π type interaction occurred between these residues. Tyrosine at position-13 of α-factor was systematically substituted with analogous amino acids with varying cation-π binding energies using solid phase peptide synthesis, and these analogs were modified by derivitization of their Lys7 residue with the fluorescent group 7-nitrobenz-2-oxa-1,3-diazole (NBD) to serve as a useful probe for binding determination. Saturation binding of these peptides to Ste2p was assayed using whole yeast cells and a flow cytometer. In parallel the biological activities of the peptides were determined using a growth arrest assay. The data provide evidence for the presence of a cation-π interaction between Arg58 of Ste2p and Tyr13 of α-factor.
Keywords: GPCR, Ste2p, α-factor, Cation-π interaction, 7-nitrobenz-2-oxa-1, 3-diazole (NBD), Flow cytometry
Understanding the molecular basis for the interaction of G protein-coupled receptors (GPCRs) with their cognate ligands has long been a thrust of scientific endeavor as these classes of 7-transmembrane (7-TM) receptors are pharmaceutical targets of over 40% of all approved drugs and are centrally involved in a broad spectrum of biological and pathological conditions (1). In the absence of X-ray crystal structures for all but the four GPCRs rhodopsin, β1-adrenergic, β2-adrenergic and adenosine receptors, drug discovery has relied upon ligand diversification and biological assays, receptor mutagenesis, and in silico tools such as computational and homology modeling to judge receptor-ligand interaction (2–4).
The α-factor receptor, Ste2p, is one of three GPCR’s present in the yeast Saccharomyces cervisiae (5). Despite the absence of sequence similarity between mammalian and yeast GPCRs, their mechanism of activation, signal transduction pathways and 7-TM helical structures appear to be conserved. Evidence for this comes from the fact that Ste2p can activate the mammalian Gαolf subunit (6), and that heterologously expressed mammalian receptors such as the β2-adrenergic receptor can activate the pheromone response pathway in yeast (7, 8). Two charged cationic residues Arg58 on TM1 and His94 on TM2 are predicted to reside at sites that are buried inside the hydrophobic membrane environment of Ste2p. A polar pair is highly conserved at these positions in the Ste2ps of various fungi, and molecular modeling experiments suggest that these residues project inward in the transmembrane region of these GPCRs (9).
An NMR study of the Ste2p fragment G31-T110 (TM1-TM2) in 1-palmitoyl-2-hydroxy-sn-glycero-3-[phospho-rac-(1-glycerol) micelles reveals the presence of a highly flexible kink in the G56-VRS-G60 region of TM1 which divides TM1 into two helical sections and exposes the upper end of TM1 to the extracellular domain (10). Tyr13 of α-factor is believed to penetrate into the hydrophobic region of Ste2p and bind to the receptor near the extracellular face of TM1. The role of Tyr13 on the carboxy terminal of α-factor in binding to the receptor was previously investigated by our group through alanine scanning and photoaffinity labeling experiments (11, 12). These studies showed that the presence of an aromatic residue at position-13 of α-factor is necessary for strong binding and optimal activity and that the contact point of position-13 of α-factor on the receptor involves residues Phe55-Arg58 on TM1. Recent studies using DOPA (3,4-dihydroxyphenylalanine) oxidative cross-linking with a DOPA13-labeled α-factor analog indicated that the contact point for this tyrosine surrogate was Cys59 on TM1 (13).
The cation-π interaction is a non-covalent interaction between a cation and the π-face of an aromatic ring. In biological contexts, it is usually an interaction between the cationic group of lysine or arginine and the π-face of the aromatic rings of phenylalanine, tyrosine and tryptophan. The average strength of a cation-π interaction is about 3 kcal/mol and it is now known to be an important contributor to the stabilization of protein secondary structures (reviewed in (14–16)). Cation-π interactions play prominent roles in the binding of neurotransmitters such as Ach, GABA, Dopamine, NMDA and adrenaline to their receptors (17). In the case of the well-studied nicotinic acetylcholine (nAchR) receptor, in which the binding pocket is made up of several tryptophan residues, progressive fluorination of Trp-149 of the α-subunit resulted in a linear correlation between binding and the predicted cation-π binding energies of various analogs (18, 19). Implementing similar strategies, the evaluation of the agonist binding site of the GABAc receptor, which has tyrosine at the aligning position, also revealed the presence of a cation-π type of interaction (20). Mutational studies of Tyr381 in the binding domain of the M1 muscarinic Ach receptor, along with evaluation of the affinity and signaling efficacy of three series of ligands, led to the conclusion that the Tyr381 benzene ring may form a cation-π interaction with acetylcholine ligand in the activated state but not in the ground state of this GPCR (21).
Given the existing knowledge of the importance of Tyr13 of α-factor in ligand binding and efficacy, the photoaffinity labelling investigations showing that Arg58 in Ste2p contacts Tyr13 of α-factor, the placement of Arg58 in the hydrophobic core of TM1, and the prominence of cation-π interactions in receptor-ligand recognition, we decided to explore the possibility that a cation-π interaction exists between α-factor and Ste2p. Tyr13 was substituted with various phenylalanine analogs and their agonist activities and equilibrium binding constants (Kd) were determined. Upon progressive fluorination of phenylalanine there was a linear increase in the Kd of the analogs. The fluorinated derivatives all retained some agonist activity, and wild-type α-factor efficiently competed with their binding to the receptor indicating that the analogs interacted with the pheromone binding site of the Ste2p. We conclude that there is evidence for a cation-π interaction between Tyr13 of α-factor and Arg58 of Ste2p.
EXPERIMENTAL PROCEDURES
Peptide synthesis
All peptides were synthesized on an Applied Biosystems Model 433A automated peptide synthesizer using Fmoc/OtBu protection and HBTU as the coupling agent. Fastmoc 0.1mmol chemistry designed by the manufacturer was employed for the chain assembly. The carboxyl terminal Fmoc amino acids were coupled to a Wang resin (loading 0.4mmol/g) on a 0.1mmol scale using triple coupling and the un-reacted resin was capped with acetic anhydride. Subsequent chain assembly was carried out using a single coupling followed by acetic anhydride capping. After chain assembly, peptides were cleaved using trifluoroacetic acid (9.5mL), water (0.5mL) with ethanedithiol (0.25mL) as a scavenger. The reaction mixture was stirred for 2hr at room temperature and filtered. Evaporation of the reaction mixture at reduced pressure resulted in a gummy residue which was precipitated by addition of diethyl ether and the crude peptide was isolated by centrifugation. Yield of the crude peptides ranged from 85–92% and these were usually >80% homogeneous. After isolation, the crude Fmoc-α factor analogs were purified using reverse phase HPLC and purification resulted in peptides with >90% homogeneity. The fluorescent group, 7-nitrobenz-2-oxa-1,3-diazole (NBD) was incorporated into the side chain of Lys7 of α-alpha factor using NBD-fluoride and the Fmoc-α-factor analogs following a previously described procedure (22), the Fmoc protection was removed in situ, and the crude reaction mixture was purified by reverse phase preparative HPLC. The isolated yield of the Lys7(NBD),Nle12,Tyr13(substituted) α-factors was about 50% after purification and all peptides used in binding analyses were >99% pure as judged by analytical HPLC and had the calculated molecular weights as determined by electrospray ionization mass spectrometry.
Saturation binding assays
Ligand stock solution was freshly prepared by dissolving a small amount of the [Lys7(NBD),Nle12]α-factor analog in 200 μL of methanol. Water was added to give a final volume of 400 μL of the methanol/water (1:1) stock. The UV absorbance at 470 nm of the ligand stock was then measured and the concentration of the stock solution was calculated using a molar extinction coefficient (ε) of the peptide of 23000 M−1cm−1 at 470nm (23). Subsequent ligand dilutions were made using the concentration of this quantified stock solution as the starting peptide-ligand concentration.
Receptor binding assays were carried out using Saccharomyces cerevisiae A3365 which contains a multicopy vector coding for expression of truncated Ste2p (Met1-K304) as reported previously (23) and S. cerevisiae A454 (ste2-Δ) which does not contain Ste2p was used as a negative control. Strain A454 contains a multicopy URA3 vector (pMD228) with no insert in host strain A232 (ste2-Δ ura3− leu2− bar1). Both pMD228 and A232 are described earlier (24). Strain A3365 contains pMD1422, which encodes a truncated STE2 allele in A232. pMD1422 was created by ligating a SacI to SphI fragment from pMD803 (25) containing a truncated STE2 into plasmid pMD1383 also cut with SacI and SphI. pMD1383 was created by using site directed mutagenesis to eliminate XbaI and HpaI sites in pMD228 (see above) using oligonucleotides ON622 (AGATGCTTCGTTGACAAAGATAT) and ON623(TCCTATTCTCTGGAAAGTATAGGA).
Strain A3365 was cultured overnight at 30oC in dropout SD-ura medium (26) until an OD600 of 0.8–1.2A was reached. A volume of culture corresponding to 1.5 × 106 cells (an OD600 of 1.0 corresponds to approximately 1.0 × 107 cells/ml) was transferred to microfuge tubes which were maintained on ice. The cultures were then diluted with ice-cold 20mM sodium acetate/acetic acid buffer (pH 4.6) containing the desired concentration of ligand and the final volume was adjusted to 400 μL. The methanol concentration in the final binding experiment never exceeded 3 percent by volume. The samples were incubated for 60 min (in the dark at 4°C) and analyzed in triplicate over 30 min on an Accuri-c6 flow cytometer (Accuri cytometers, Inc., Ann Arbor, MI). The samples were excited at 488 nm (Blue laser) and the fluorescence emitted at 530±15 nm on the FL1-A channel was recorded. The fluorescence data were corrected for autofluorescence and were analyzed using Sigma Plot 8.0. The equilibrium binding constants Kd were obtained by fitting the data to a single site specific binding equation, which included a non-specific component N. The Kd values reported herein are the average of at least three independent experiments run on separate days using independently grown yeast cells.
Competition binding assay
Competition binding assays were performed on yeast strain A3365 expressing truncated Ste2p receptor (Δ304-431). The cells were streaked out from yeast master plates into 4mL of SD-ura media and were grown at 30°C overnight until they attained an OD600 of 1.0. NBD-labeled and unlabeled α-factor ligand solutions were prepared by dissolving a small amount of peptide in 500μL of 1:1 MeOH:H2O and were then quantified at 470nm and 280nm, respectively. After quantification, appropriate volumes of Lys7(NBD),Nle12,Phe13(analog) and unlabeled Lys7,Nle12,Tyr13(WT) α-factor were placed in 20mM sodium acetate buffer (pH 4.6) in a microfuge tube. The solution was maintained at 4°C and the competition binding assay was started by the addition of yeast culture corresponding to1.5 × 106 cells/mL. The total volume of reaction mixture was 400μL. The concentration of the NBD-labeled analog used in the binding assays were 1.5 times their respective Kd and was 40nM for Lys7(NBD),Nle12,Phe13 (4-F), 60nM for Lys7[NBD],Nle12,Phe13 (3,4-F2) and 300nM for Lys7[NBD],Nle12,Phe13 (2,3,4,5,6-F5). The concentration of unlabeled α-factor, Lys7,Nle12,Tyr13(WT) used was 0nM, 0.3nM, 0.5nM, 1.0nM, 3.0nM, 5.0nM, 12nM, 50nM, 100nM, 250nM, 600nM, 1000nM and 2000nM. After incubation of samples for 60min on an ice-bath they were run on an Accuri-c6 flow cytometer in triplicate and the mean fluorescence on the FL1A channel was measured. The mean fluorescence obtained was converted into % Bound and was plotted against the concentration of unlabeled Lys7,Nle12,Tyr13(WT) on a log scale.
Growth arrest halo assay
Yeast strains A3365 (expressing truncated Ste2p receptor Δ304-431) and A454 (ste2-Δ) were grown to OD600 of 1.0 in 4mL of SD-ura medium at 30oC. The construction of these strains has been described earlier (23). A volume (1mL) of culture corresponding to 6.0 × 106 cells was gently mixed with 1.0mL of melted SD-ura medium containing 2% difco agar and quickly spread over plates containing a bed of 2% SD-ura/agar. The plates were incubated for 2h at 30oC, then 6μL portions of α-factor or α-factor analogs at various concentrations in 1:1 methanol:H2O were spotted on top of the evenly spread yeast lawn. The plates were then incubated at 30oC for 24–26h until clearly demarcated halos were observed. Six μL of 1:1 methanol:H2O spotted as a control did not give rise to any halo formation. The assay was carried out in triplicate and the reported values are the mean value of the halo diameters in millimeters. The halo sizes for the triplicate assays were within 1.0 mm for a given concentration. The concentrations of α-factor spotted for all the analogs were 0μM, 1.56μM, 3.12μM, 6.25μM, 12.5μM, 25.0μM and 50μM. [Phe13(2,3,4,5,6-F5)]α-factor which did not give halos using the lower amount of the above concentrations was repeated using concentrations of 6.25μM, 12.5μM, 25.0μM, 50.0μM, 100.0μM, 200.0 μM and 400μM concentrations. The halo diameters were plotted against the log of the concentration of α-factor. The data were fit to a linear regression curve using Sigma Plot 8.0. Relative activities were defined as the concentration of α-factor analogs giving rise to a halo diameter of 18 mm.
RESULTS
The Kd values for the binding of the various fluorophenylalanine analogs of α-factor to Ste2p were determined using a flow cytometer and the NBD labelled pheromones. All analogs were synthesized by solid state methods and were purified to high homogeneity using reversed phase HPLC. All peptides were greater than 99% homogeneous as judged by HPLC and mass spectroscopy (data not shown).
Fluorescent Binding Analysis of Position13 analogs of α-Factor
In previous investigations we reported the use of the flow cytometric method for determining the binding affinity of [Lys7(NBD),Nle12]α-factor to various mutant forms of Ste2p (23). In these investigations 5- to 8-fold higher fluorescence values were obtained with truncated Ste2p as compared to full length Ste2p because the truncated receptor is deficient in endocytosis resulting in higher receptor numbers on the cell membrane. However, both the truncated and full length receptor exhibit nearly identical binding affinities for α-factor. We therefore decided to use the truncated receptor in the present investigation. Using this binding assay we found that the α-factor analogs with fluorinated phenylalanines at position 13 exhibited saturable binding to truncated Ste2p (A3365 strain) while the negative control experiments using a ste2-Δ deficient strain (A454) gave low background fluorescence which did not exhibit saturation (Fig. 1). The results were highly reproducible and the data could be fit to a single site binding equation which allowed us to account for a nonspecific binding component. Using this method we determined the binding affinities for fluorescently labeled wild-type sequence α-factor, [Lys7(NBD),Phe13]α-factor, [Lys7(NBD),Phe13(3-F)]α-factor, [Lys7(NBD),Phe13(4-F)]α-factor, [Lys7(NBD),Phe13(3,4-F2)]α-factor and [Lys7(NBD),Phe13(2,3,4,5,6-F5)]α-factor (Table 1).
Figure 1.

Representative saturation binding curve for [Lys7(NBD),Nle12,Phe13(3,4-F2)]α-factor with yeast strains A3365 (with receptor) and A454 (without receptor).
Table 1.
Effect of substitution at position-13 of α-factor on binding affinity
| Entry No. | Analogs of α-factor position-13 residue [Lys7(NBD),X13]α-factor | Cation-π binding energy kcal/mol (27, 28) | Average Kd [nM] |
|---|---|---|---|
| 1 | Tyrosine (wild type) | 26.9 | 15.9 ± 3.5 [n=3] |
| 2 | Phenylalanine | 27.1 | 20.4 ± 4.4 [n=4] |
| 3 | 3-fluoro Phenylalanine | 22.0 | 31.4 ± 7.3 [n=4] |
| 4 | 4-fluoro Phenylalanine | 22.0 | 26.2 ± 5.9 [n=3] |
| 5 | 3,4-Difluoro Phenylalanine | 17.4 | 38.1 ± 4.7 [n=3] |
| 6 | Pentafluoro Phenylalanine | 3.9 | 177.3 ± 59.9 [n=3] |
| 7 | Alaninea | 0 | 534.0 ± 137.3 [n=3] |
| 8 | 4-Methyl Phenylalanine | 28.6 | 53.2 ± 11.3 [n=3] |
| 9 | 4-Methoxy Phenylalanine | 28.6 | 37.8 ± 20.9 [n=5] |
π-ring is absent, hence cation-π binding energy is assumed to be zero
Following approaches developed by Dougherty, Lester and co-workers (18) we plotted the log of the Kd versus the previously-published, calculated cation-π energies (27, 28) for the fluorinated phenylalanine derivatives (Fig. 2A). The data showed a linear correlation between Log Kd and the cation-π binding energies. To further explore the possibility of a cation-π interaction we synthesized three additional analogs; [Lys7(NBD),Ala13]α-factor, [Lys7(NBD),Phe13(4-CH3)]α-factor, and [Lys7(NBD),Phe13(4-OCH3)]α-factor, compounds 7, 8, and 9, respectively, in Table 1, and determined their binding constants (Table 1). These analogs either completely lacked cation-π interaction capabilities at position 13 (analog 7) or had slightly stronger predicted cation-π binding energies (analogs 8 and 9) than α-factor. The [Lys7(NBD),Ala13]α-factor analog showed extremely weak binding exhibiting at least a 30-fold decrease in affinity compared to [Lys7(NBD)]α-factor, with significant variability in the results. The [Lys7(NBD),Phe13(4-CH3)]α-factor showed a decrease in affinity of approximately 3-fold compared to normal alpha factor whereas the [Lys7(NBD),Phe13(4-OCH3)]α-factor had an affinity quite similar to the difluorophenylalanine analogs accompanied by a large deviation between individual assays. Inclusion of the latter two compounds in the log Kd versus cation-π binding energy resulted in considerable scattering of the data and a weaker correlation between Kd and binding energy (Fig. 2B).
Figure 2.

Plot of log[Kd analog/Kd WT] vs cation-π binding energy. A] For [Lys7(NBD),Tyr13] α-factor, [Lys7(NBD),Phe13]α-factor, [Lys7(NBD),Phe13(3-F)]α-factor, [Lys7(NBD),Phe13(4-F)]α-factor, [Lys7(NBD),Phe13(3,4-F2)]α-factor and [Lys7(NBD),Phe13(2,3,4,5,6-F5)]α-factor. The data were fitted into a straight line y = 1.186−0.0425x [B] Plot involving all position 13 analogs synthesized except [Lys7(NBD),Ala13]α-factor. The data were fitted into a straight line y= 1.021−0.0296x. The Kd values were determined using saturation binding curves for the fluorescent analogs measured with the fluorescent activated flow cytometer as described in the Methods.
Competition Binding Assay
It was important to ascertain whether the position 13 analogs bound to Ste2p at the same site as normal α-factor. We investigated this by determining whether normal unlabelled α-factor could compete with the fluorescently-labeled fluorophenylalanine α-factors for binding to the receptor. Fluorescence binding analyses for three labeled α-factor analog ([Lys7(NBD),Phe13(4-F)]α-factor, [Lys7(NBD),Phe13(3,4-F2)]α-factor, and [Lys7(NBD),Phe13(2,3,4,5,6-F5)]α-factor were conducted in the presence of increasing concentrations of unlabeled α-factor. Measurements were made using a concentration of the NBD labeled α-factor analog that was 1.5 times the Kd of the labeled ligand so that a strong fluorescence was initially measured but the receptor was not saturated by the analog. The cells were incubated for 60 min on ice and run on an Accuri-C6 flow cytometer. The presence of unlabelled α-factor caused a sharp decrease in the fluorescence of the NBD-fluorophenylalanine α-factor analogs (Fig. 3). While greater than 97% of the fluorescence from bound [Lys7(NBD)Phe13(4-F)]α-factor and [Lys7(NBD)Phe13(3,4-F2)]α-factor could be competed out with unlabeled α-factor, only 83% of the fluorescence from bound [Lys7(NBD)Phe13(2,3,4,5,6-F5)]α-factor was removed by this wild-type pheromone, (Fig. 3). The amount of unlabeled α-factor required to compete 50% of the bound labeled ligand is 30nM for [Lys7(NBD)Phe13(4-F)]α-factor, 32nM for [Lys7(NBD)Phe13(3,4-F2)]α-factor and 10.5nM in the case of [Lys7(NBD)Phe13(2,3,4,5,6-F5)]α-factor
Figure 3.

Competition between [Lys7,Nle12,Tyr13]α-factor (WT) and fluorescently labeled fluorophenylalanine analogs for binding to Ste2p. Plot of percentage of mean fluorescence bound vs log of α-factor concentration [nM].
Biological Activities of the Position13 analogs of α-Factor: Growth arrest halo assay
The biological activities of the position 13 α-factor analogs were determined using a halo assay of growth arrest in response to ligand. Increasing concentrations of α-factor or the various fluorophenylalanine analogs resulted in increased diameters for growth arrest on lawns of cultured S. cerevisiae strain A3365 expressing C-terminally truncated Ste2p (Fig. 4B–D). No halo formation was observed with the ste2-Δ strain A454 (Fig. 4A). All of the unlabeled fluorophenylalanine α-factor analogs except for [Lys7,Phe13(2,3,4,5,6-F5)]α-factor gave sharply defined halos using ligand concentrations ranging from 1.56μM to 50μM. When assays with the pentafluorophenylalanine α-factor, which did not form a halo until 12.5μM, were repeated with higher concentrations of ligand (12.5μM to 400μM) the resulting halos were comparable with those observed at lower concentrations of the other α-factor analogs (Fig. 4B–D).
Figure 4.

Growth arrest halo assay studied at different pheromone concentrations. (Panel A) [Lys7,Phe13(3,4-F2)]α-factor with yeast strain A454 and ligand concentration ranging from1.56μM to 50μM. (Panel B) [Lys7,Phe13(3,4-F2)]α-factor with yeast strain A3365 and ligand concentration ranging from1.56μM to 50μM. (Panel C) [Lys7,Phe13(2,3,4,5,6-F5)]α-factor with strain A3365 and ligand concentration ranging from 1.56μM to 50μM. (Panel D) [Lys7,Phe13(2,3,4,5,6-F5)]α-factor with strain A3365 and ligand concentration ranging from12.5μM to 400uM.
The halo size from the growth arrest assay was plotted versus log of the concentration of the pheromone (Fig. 5). All analogs showed a linear correlation between the halo diameter and the log of concentration of the pheromone over the range of concentrations that were examined. These plots were then used to determine the analog concentration required to give a halo diameter of 18 mm. The results showed that the [Lys7,Tyr13(OMe)]α-factor was the most potent agonist inducing an 18 mm halo at 1.66 μM concentration. It should be noted that the slope of the plot for this latter pheromone differed from that of all of the other analogs. This likely reflects differences in the diffusion of this compound in the agar. The wild-type, mono, di- and pentafluoro analogs gave activities of 5.0 μM, 5.4 μM, 11.7 μM and 655.6 μM, respectively, using this assay. Thus although all analogs are agonists, the pentafluorophenylalanine13 analog is about 130-fold less active than α-factor.
Figure 5.

Plot of halo diameter vs Log of pheromone concentration. (A) [Lys7,Tyr13]α-factor (WT), [Lys7,Phe13(4-F)]α-factor, [Lys7,Phe13(3,4-F2)]α-factor and [Lys7,Tyr13(OMe)]α-factor. (B) [Lys7,Phe13(2,3,4,5,6-F5)]α-factor. (C) [Lys7(NBD),Tyr13]α-factor, [Lys7(NBD),Phe13(4-F)]α-factor and [Lys7(NBD),Phe13(3,4-F2)]α-factor labeled α-factor analogs.
Four fluorescently labeled α-factor analogs ([Lys7(NBD),Tyr13]α-factor (WT), [Lys7(NBD),Phe13(4-F)]α-factor, [Lys7(NBD),Phe13(3,4-F2)]α-factor, [Lys7(NBD),Phe13(2,3,4,5,6-F5)]α-factor) were also studied to determine whether the fluorescent Lys7(NBD) analog used in the binding study were agonists (Fig. 5C). The overall halo diameters induced by NBD analogs were smaller compared to those of the unlabeled analogs. Since the overall diameters of the halos were smaller, the concentrations of NBD-labeled α-factor required to cause a 16mm halo, rather than a 18mm halo, were measured. Lys7(NBD)-labeled WT α-factor at 7.8μM produced a 16mm halo, whereas the labeled mono and difluoro analogs produced a similar halo diameter at 25.6μM and 66.8μM, respectively. The activity of [Lys7(NBD),Phe13(2,3,4,5,6-F5)]α-factor could not be measured by this method as the ligand started precipitating at concentrations above 25μM where this α-factor analog resulted in a halo which had a diameter of only 7mm.
DISCUSSION
Earlier studies carried out by our group employing photoaffinity labeling with [benzoylphenylalanine13]α-factor and oxidative cross linking experiments involving DOPA13 α-factor suggested that the Tyr13 position of α-factor interacts with the TM1 segment of Ste2p at residues R58 and Cys59, respectively (12, 13), and the presence of an aromatic ring at position 13 of α-factor was found to be necessary for efficient binding (11, 29). Arg58 on TM1 and His94 on TM2 in Ste2p are believed to be in the hydrophobic core of the membrane. Other members of the fungal pheromone GPCR subfamily conserve polar residues at similar positions (9). The conservation of positively charged residues in the first and second TM domains of these fungal GPCRs, and the critical nature of Tyr13 of α-factor and of other fungal α-factors suggested to us that ligand recognition by Ste2p might involve a cation-π type of interaction.
Fluorine substitution has been utilized as an important tool to track cation-π interactions as fluorine is sterically similar to hydrogen but due to its extremely high electronegativity has a remarkable effect on the electronic structure of an aromatic ring. Progressive fluorination of the aromatic ring decreases the π-electron density and thereby decreases the ability of the π-face to interact with cations. Previously, we studied [Phe13]α-factor, [Phe13(3-F)]α-factor and [Phe13(4-F)]α-factor using a radioactive binding competition assay and found that these analogs competed strongly with α-factor binding and were good agonists (29). In the present investigation, using a direct fluorescent saturation binding assay and multiple fluorinated peptides we studied α-factor analogs with varying predicted cation-π binding energies and revealed that there is significant influence of the cation-π binding energies of the phenylalanine ring on the equilibrium binding constant Kd of the receptor. Phenylalanine has a similar cation-π binding energy as that of tyrosine (27) (Table 1) and ideally phenylalanine replacement should not adversely affect the binding affinity. In our studies, the Kd obtained for [Phe13]α-factor is similar to that of native [Tyr13]α-factor which appears to validate this contention.
Phe(3-F) (entry 3, Table 1) and Phe(4-F) (entry 4, Table 1) have similar calculated cation-π binding energies and α-factor analogs with these substitutions were found to have similar Kd values in our binding studies. This indicates that the position of the fluorine atom does not have a significant effect on the binding affinity and the trend that we begin to see is mainly due to the decrease in cation-π binding energy. Progressive fluorination of the phenylalanine ring resulted in an increase in Kd and the data fits well onto a linear correlation plot (Fig. 2A). Phe(2,3,4,5,6-F5) has the lowest cation-π binding energy among the fluorinated α-factor derivatives that were studied and this analog exhibited approximately a 10-fold decrease in binding affinity compared to α-factor. The associated decrease in binding affinity due to fluorination is not of the magnitude that was reported for ligand gated channels where up to a two log change in binding constants was observed. This suggests that the associated cation-π contribution to the α-factor–Ste2p binding may be weaker than the contribution to the ligand interactions in ion channels. In fact, we have recently found that position 13 of α-factor also interacts with Cys59 of Ste2p (13) so that contributions of H-bonding between the hydroxyl group of Tyr13 to Cys59 and a cation-π interaction between Tyr13 and Arg58 both contribute to the binding of α-factor to Ste2p.
An additional way to test for the cation-π interaction would be to carry out a similar binding analysis on ste2 mutants where Arg58 is replaced by other residues. We have generated 3 such mutants ((R58A); (R58D) and (R58E)). All of these are seriously deficient in binding, with Kd’s for α-factor from 10 to 50-fold higher. As shown in the present study the fluorescent fluorinated α-factor analogs had Kd’s for the wildtype Ste2p ranging from 26.2 nM to 177.3 nM (Table 1). To determine the latter value we needed to use 4μM peptide, saturation was barely achieved, and the non-specific fluorescence was quite high. Since the Ste2p containing the substitution R58A binds α-factor with nearly 10-fold lower affinity saturation binding experiments would require very high concentrations of [Lys7(NBD),Phe13(2,3,4,5,6-F5)]α-factor (~40 μM). As stated above this peptide precipitates at 25 μM and the experiment, therefore, could not be done. However, the marked decrease in affinity noted with the three ste2 mutants that we generated supports our conclusion that the Arg58 side chain contributes to the binding energy and would be consistent with the cation-π interaction.
The use of NBD derivatives of α-factor might lead to a change in the interaction of position-13 of the pheromone and its receptor. However all of the analogs that we tested were agonists indicating productive binding to the receptor, and the potencies of the fluorinated α-factor analogs were within a factor of 3 to 4 of that of α-factor with the exception of the pentafluorinated analog. Although the trends in the binding affinities were not directly parallel to the trends in the agonist potencies, we have observed a similar lack of correlation in our previous study with α-factor analogs (29). Moreover, the weakest binding fluorinated analog ([Phe13(2,3,4,5,6-F5)]α-factor) was by far the weakest agonist. Finally, α-factor efficiently competed with the binding of the Lys7(NBD) analogs to Ste2p. Based on these observations and previous studies on α-factor derivatized at the Lys7 side chain (30–33) we conclude that the fluorescent labeling does not result in a significant perturbation of the interactions of the position-13 side chain and the receptor. Interestingly, two non-fluorinated analogs, [Phe13(4-Me)]α-factor and [Phe13(4-OMe)]α-factor each showed a marked lack of correlation between the Kd values and cation-π binding energies (Fig. 2B). This finding contrasts with the correlation observed with fluorinated phenylalanine analogs (Fig. 2A) and indicates that while the insertion of fluorine in place of a hydrogen results in primarily an electronic effect on the benzene ring, other groups can have both electronic and steric effects on binding. These analogs have marginally higher cation-π binding energies as compared to phenylalanine and tyrosine (Table 1) and would be predicted to result in stronger binding. We observed a 3-fold decrease in the binding ability for [Phe13(4-Me)]α-factor and the difference reduces to 2-fold upon re-introduction of phenolic oxygen which should be a good electron donor. It is possible that the effect of re-introduction of the donor-oxygen atom is at least partially nullified by the steric bulk of the methoxy group. Despite the fact that it cannot participate in a cation-π interaction, the [Lys7(NBD),Ala13]α-factor does bind weakly to Ste2p. Thus, it is evident that additional effects involving steric, electrostatic and perhaps Van der Waals forces may also play a role in α-factor-Ste2p recognition at position-13 of the pheromone.
Biological Implications
The results of this paper support the existence of a cation-π interaction between the carboxyl terminal Tyr13 residue of the α-factor peptide and the Arg58 residue in the first transmembrane domain of Ste2p. This interaction contributes to the favorable energetics of binding of ligand to receptor, since removal of Tyr from the carboxyl terminus drastically reduces the peptide affinity and leads to an inactive pheromone (34). However this interaction involving the C-terminal of α-factor is not, by itself, sufficient for receptor activation because N-terminally truncated analogs of α-factor bind strongly to the receptor but do not lead to signal transduction (34).
Detection of the interaction of Arg58 of the receptor with Tyr13 of α-factor raises the question of how the positively charged Arg58 is accommodated when there is no ligand bound to receptor, since the existence of an isolated arginine sidechain within the hydrophobic transmembrane region of Ste2p is expected to be energetically unfavorable. One possibility is that, in the absence of ligand, Arg58 participates in an intramolecular cation-π interaction with an aromatic residue elsewhere in the receptor. Each of the seven phenylalanine and two tryptophan residues within the predicted transmembrane regions of Ste2p can be mutated to non-aromatic residues without causing loss of function (35). Similarly, aromatic residues do not appear to be required at the positions of two of the three tyrosine residues in these regions. The remaining tyrosine, Tyr266 can only be mutated to Trp, Phe or His (35, 36). However current models of Ste2p (9), place Tyr266 too far from Arg58 for any direct interaction.
This suggests that the any aromatic side chain involved in an intramolecular cation-π interaction with Arg58 in the absence of ligand would reside in an extracellular tail or loop. The most likely site for the interacting side chains would be the first extracellular loop EL1, based on its expected proximity to Arg58 and on the following: 1) The results of cysteine scanning accessibility measurements on Ste2p indicate that burial of this loop in the transmembrane core of the receptor is involved in stabilizing the inactive state (37); 2) Mutation of Phe119, also in EL1, has been reported to lead to constitutive activity, as would be expected if EL1 is involved in stabilizing the inactive state (38). 3) Removal of residues 114 to 129 from EL1 of Ste2p yields a receptor that signals poorly but is constitutively active (Bhuiyan, Cohen and Hauser unpublished results); and 4) The substitution Tyr111C in EL1 of Ste2p results in a receptor that is incapable of signaling while retaining high affinity α-factor binding, suggesting that Tyr111 is involved in the activation pathway.
Thus, we hypothesize that either Tyr111 or F119 in EL1 may interact with Arg58 in the ligand-free receptor via a π-cation interaction, stabilizing the ground state (see Fig. 6 for a model of the Tyr111-Arg58 interaction in Ste2p). Upon ligand binding, the intramolecular interaction is replaced by the cation-π interaction with Tyr13 of α-factor, anchoring the carboxyl terminal of the ligand to the receptor and allowing the amino terminal of the pheromone to establish additional interactions that induce a conformational change that is propagated through other transmembrane domains, perhaps through the sequential actions of a series of “micro-switches” (39), to sites interacting with the G protein. A role for EL1 in signal transduction by other GPCRs has been proposed based on studies of rhodopsin, the dopamine receptor and the C5a receptor (40–43). In addition, recent solid state and solution NMR studies have provided evidence for significant conformational changes in other extracellular loops of Class A GPCRs (44, 45).
Figure 6.
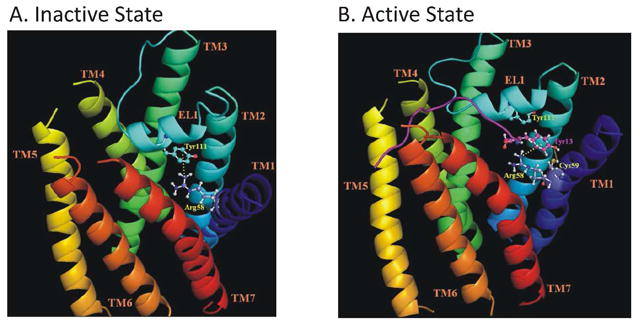
Model for change in EL1 loop receptor interactions in presence and absence of α-factor. The transmembrane helices (TM) of Ste2p are shown in different colors and labeled. The structure of EL1 with a short 310-helix (106YSSVTYALT114) predicted by (46) is also shown. The α-factor in panel B is shown as a purple ribbon. Panel A: Cation-p interaction between the Arg58 guanidinium moiety in TM1 and the phenyl ring of Tyr111 (EL1) in the inactive state of Ste2p. Panel B: This interaction is disturbed upon α-factor binding and replaced by a similar cation-p interaction between Arg58 and Tyr13 of α-factor. The phenolic hydroxyl group of Tyr13 of α-factor is also suggested to form a hydrogen bond with the thiol group of Cys59. The interactions are shown as broken yellow lines.
CONCLUSION
We synthesized a series of position-13 α-factor analogs with varying predicted cation-p binding energies and applied a flow cytometry-based assay to investigate the binding of these peptide ligands to their cognate GPCR. The trends in Kd obtained by the fluorescence-based saturation binding experiments are well-correlated with the predicted energies of a cation-π interaction for fluorinated phenylalanines at position 13 of α-factor. All analogs function as agonists and appear to bind to the α-factor pocket of Ste2p. The experimental results suggest that there is a cation-π interaction that facilitates binding of α-factor to Ste2p. However, electrostatic contributions associated with the phenolic oxygen of tyrosine and steric effects also appear to be involved in the interaction of position-13 and the receptor.
Acknowledgments
We thank Sara Connelly and Elizabeth Matthew for help in the binding and bioassays. We also thank Boris Arshava and Leah Cohen for help with the preparation of the manuscript and George Umanah for preparation of Figure 6.
This work was supported by NIH grants GM22087 (JMB and FN) and GM059357 (MED).
References
- 1.Filmore D. It’s a GPCR World. Modern Drug Discovery. 2004;7:24–28. [Google Scholar]
- 2.Marshall GR. Peptide interactions with G-protein coupled receptors. Biopolymers. 2001;60:246–277. doi: 10.1002/1097-0282(2001)60:3<246::AID-BIP10044>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 3.Kristiansen K. Molecular mechanisms of ligand binding, signaling, and regulation within the superfamily of G-protein-coupled receptors: molecular modeling and mutagenesis approaches to receptor structure and function. Pharmacology & therapeutics. 2004;103:21–80. doi: 10.1016/j.pharmthera.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 4.Schyler S, Horuk R. I want a new drug: G-protein-coupled receptors in drug development. Drug discovery today. 2006;11:481–493. doi: 10.1016/j.drudis.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 5.Fredriksson R, Schioth HB. The repertoire of G-protein-coupled receptors in fully sequenced genomes. Molecular pharmacology. 2005;67:1414–1425. doi: 10.1124/mol.104.009001. [DOI] [PubMed] [Google Scholar]
- 6.Crowe ML, Perry BN, Connerton IF. Golf complements a GPA1 null mutation in Saccharomyces cerevisiae and functionally couples to the STE2 pheromone receptor. Journal of receptor and signal transduction research. 2000;20:61–73. doi: 10.3109/10799890009150037. [DOI] [PubMed] [Google Scholar]
- 7.King K, Dohlman HG, Thorner J, Caron MG, Lefkowitz RJ. Control of yeast mating signal transduction by a mammalian beta 2-adrenergic receptor and Gs alpha subunit. Science (New York, NY. 1990;250:121–123. doi: 10.1126/science.2171146. [DOI] [PubMed] [Google Scholar]
- 8.Pausch MH. G-protein-coupled receptors in Saccharomyces cerevisiae: high-throughput screening assays for drug discovery. Trends in biotechnology. 1997;15:487–494. doi: 10.1016/S0167-7799(97)01119-0. [DOI] [PubMed] [Google Scholar]
- 9.Eilers M, Hornak V, Smith SO, Konopka JB. Comparison of class A and D G protein-coupled receptors: common features in structure and activation. Biochemistry. 2005;44:8959–8975. doi: 10.1021/bi047316u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neumoin A, Cohen LS, Arshava B, Tantry S, Becker JM, Zerbe O, Naider F. Structure of a double transmembrane fragment of a G-protein-coupled receptor in micelles. Biophysical journal. 2009;96:3187–3196. doi: 10.1016/j.bpj.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abel MG, Zhang YL, Lu HF, Naider F, Becker JM. Structure-function analysis of the Saccharomyces cerevisiae tridecapeptide pheromone using alanine-scanned analogs. J Pept Res. 1998;52:95–106. doi: 10.1111/j.1399-3011.1998.tb01363.x. [DOI] [PubMed] [Google Scholar]
- 12.Son CD, Sargsyan H, Naider F, Becker JM. Identification of ligand binding regions of the Saccharomyces cerevisiae alpha-factor pheromone receptor by photoaffinity cross-linking. Biochemistry. 2004;43:13193–13203. doi: 10.1021/bi0496889. [DOI] [PubMed] [Google Scholar]
- 13.Umanah GK, Son C, Ding F, Naider F, Becker JM. Cross-linking of a DOPA-containing peptide ligand into its G protein-coupled receptor. Biochemistry. 2009;48:2033–2044. doi: 10.1021/bi802061z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma JC, Dougherty DA. The Cationminus signpi Interaction. Chemical reviews. 1997;97:1303–1324. doi: 10.1021/cr9603744. [DOI] [PubMed] [Google Scholar]
- 15.Gallivan JP, Dougherty DA. Cation-pi interactions in structural biology. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:9459–9464. doi: 10.1073/pnas.96.17.9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crowley PB, Golovin A. Cation-pi interactions in protein-protein interfaces. Proteins. 2005;59:231–239. doi: 10.1002/prot.20417. [DOI] [PubMed] [Google Scholar]
- 17.Zacharias N, Dougherty DA. Cation-pi interactions in ligand recognition and catalysis. Trends in pharmacological sciences. 2002;23:281–287. doi: 10.1016/s0165-6147(02)02027-8. [DOI] [PubMed] [Google Scholar]
- 18.Zhong W, Gallivan JP, Zhang Y, Li L, Lester HA, Dougherty DA. From ab initio quantum mechanics to molecular neurobiology: a cation-pi binding site in the nicotinic receptor. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:12088–12093. doi: 10.1073/pnas.95.21.12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beene DL, Brandt GS, Zhong W, Zacharias NM, Lester HA, Dougherty DA. Cation-pi interactions in ligand recognition by serotonergic (5-HT3A) and nicotinic acetylcholine receptors: the anomalous binding properties of nicotine. Biochemistry. 2002;41:10262–10269. doi: 10.1021/bi020266d. [DOI] [PubMed] [Google Scholar]
- 20.Lummis SCDLB, Harrison NJ, Lester HA, Dougherty DA. A cation-pi binding interaction with a tyrosine in the binding site of the GABAC receptor. Chemistry & biology. 2005;12:993–997. doi: 10.1016/j.chembiol.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 21.Ward SD, Curtis CA, Hulme EC. Alanine-scanning mutagenesis of transmembrane domain 6 of the M(1) muscarinic acetylcholine receptor suggests that Tyr381 plays key roles in receptor function. Molecular pharmacology. 1999;56:1031–1041. doi: 10.1124/mol.56.5.1031. [DOI] [PubMed] [Google Scholar]
- 22.Ding FX, Lee BK, Hauser M, Davenport L, Becker JM, Naider F. Probing the binding domain of the Saccharomyces cerevisiae alpha-mating factor receptor with rluorescent ligands. Biochemistry. 2001;40:1102–1108. doi: 10.1021/bi0021535. [DOI] [PubMed] [Google Scholar]
- 23.Bajaj A, Celic A, Ding FX, Naider F, Becker JM, Dumont ME. A fluorescent alpha-factor analogue exhibits multiple steps on binding to its G protein coupled receptor in yeast. Biochemistry. 2004;43:13564–13578. doi: 10.1021/bi0494018. [DOI] [PubMed] [Google Scholar]
- 24.Leavitt LM, Macaluso CR, Kim KS, Martin NP, Dumont ME. Dominant negative mutations in the alpha-factor receptor, a G protein-coupled receptor encoded by the STE2 gene of the yeast Saccharomyces cerevisiae. Mol Gen Genet. 1999;261:917–932. doi: 10.1007/s004380051039. [DOI] [PubMed] [Google Scholar]
- 25.Gehret AU, Bajaj A, Naider F, Dumont ME. Oligomerization of the yeast alpha-factor receptor: implications for dominant negative effects of mutant receptors. The Journal of biological chemistry. 2006;281:20698–20714. doi: 10.1074/jbc.M513642200. [DOI] [PubMed] [Google Scholar]
- 26.Rose M, Winston F, Hieter P. Methods in Yeast Genetics. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1990. [Google Scholar]
- 27.Mecozzi S, West AP, Jr, Dougherty DA. Cation-pi interactions in aromatics of biological and medicinal interest: electrostatic potential surfaces as a useful qualitative guide. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:10566–10571. doi: 10.1073/pnas.93.20.10566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raines DE, Gioia F, Claycomb RJ, Stevens RJ. The N-methyl-D-aspartate receptor inhibitory potencies of aromatic inhaled drugs of abuse: evidence for modulation by cation-pi interactions. The Journal of pharmacology and experimental therapeutics. 2004;311:14–21. doi: 10.1124/jpet.104.069930. [DOI] [PubMed] [Google Scholar]
- 29.Liu S, Henry LK, Lee BK, Wang SH, Arshava B, Becker JM, Naider F. Position 13 analogs of the tridecapeptide mating pheromone from Saccharomyces cerevisiae: design of an iodinatable ligand for receptor binding. J Pept Res. 2000;56:24–34. doi: 10.1034/j.1399-3011.2000.00730.x. [DOI] [PubMed] [Google Scholar]
- 30.Shenbagamurthi P, Baffi R, Khan SA, Lipke P, Pousman C, Becker JM, Naider F. Structure-activity relationships in the dodecapeptide alpha factor of Saccharomyces cerevisiae. Biochemistry. 1983;22:1298–1304. doi: 10.1021/bi00274a047. [DOI] [PubMed] [Google Scholar]
- 31.Raths SK, Naider F, Becker JM. Peptide analogues compete with the binding of alpha-factor to its receptor in Saccharomyces cerevisiae. The Journal of biological chemistry. 1988;263:17333–17341. [PubMed] [Google Scholar]
- 32.Naider F, Yaron A, Ewenson A, Tallon M, Xue CB, Srinivasan JV, Eriotou-Bargiota E, Becker JM. Synthetic probes for the alpha-factor receptor. Biopolymers. 1990;29:237–245. doi: 10.1002/bip.360290128. [DOI] [PubMed] [Google Scholar]
- 33.Naider F, Becker JM. The alpha-factor mating pheromone of Saccharomyces cerevisiae: a model for studying the interaction of peptide hormones and G protein-coupled receptors. Peptides. 2004;25:1441–1463. doi: 10.1016/j.peptides.2003.11.028. [DOI] [PubMed] [Google Scholar]
- 34.Eriotou-Bargiota E, Xue CB, Naider F, Becker JM. Antagonistic and synergistic peptide analogues of the tridecapeptide mating pheromone of Saccharomyces cerevisiae. Biochemistry. 1992;31:551–557. doi: 10.1021/bi00117a036. [DOI] [PubMed] [Google Scholar]
- 35.Martin NP, Celic A, Dumont ME. Mutagenic mapping of helical structures in the transmembrane segments of the yeast alpha-factor receptor. Journal of molecular biology. 2002;317:765–788. doi: 10.1006/jmbi.2002.5444. [DOI] [PubMed] [Google Scholar]
- 36.Lee BK, Lee YH, Hauser M, Son CD, Khare S, Naider F, Becker JM. Tyr266 in the sixth transmembrane domain of the yeast alpha-factor receptor plays key roles in receptor activation and ligand specificity. Biochemistry. 2002;41:13681–13689. doi: 10.1021/bi026100u. [DOI] [PubMed] [Google Scholar]
- 37.Hauser M, Kauffman S, Lee BK, Naider F, Becker JM. The first extracellular loop of the Saccharomyces cerevisiae G protein-coupled receptor Ste2p undergoes a conformational change upon ligand binding. The Journal of biological chemistry. 2007;282:10387–10397. doi: 10.1074/jbc.M608903200. [DOI] [PubMed] [Google Scholar]
- 38.Parrish W, Eilers M, Ying W, Konopka JB. The cytoplasmic end of transmembrane domain 3 regulates the activity of the Saccharomyces cerevisiae G-protein-coupled alpha-factor receptor. Genetics. 2002;160:429–443. doi: 10.1093/genetics/160.2.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nygaard R, Frimurer TM, Holst B, Rosenkilde MM, Schwartz TW. Ligand binding and micro-switches in 7TM receptor structures. Trends in pharmacological sciences. 2009;30:249–259. doi: 10.1016/j.tips.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 40.Lawson Z, Wheatley M. The third extracellular loop of G-protein-coupled receptors: more than just a linker between two important transmembrane helices. Biochemical Society transactions. 2004;32:1048–1050. doi: 10.1042/BST0321048. [DOI] [PubMed] [Google Scholar]
- 41.Shi L, Javitch JA. The second extracellular loop of the dopamine D2 receptor lines the binding-site crevice. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:440–445. doi: 10.1073/pnas.2237265100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Klco JM, Wiegand CB, Narzinski K, Baranski TJ. Essential role for the second extracellular loop in C5a receptor activation. Nature structural & molecular biology. 2005;12:320–326. doi: 10.1038/nsmb913. [DOI] [PubMed] [Google Scholar]
- 43.Klco JM, Nikiforovich GV, Baranski TJ. Genetic analysis of the first and third extracellular loops of the C5a receptor reveals an essential WXFG motif in the first loop. The Journal of biological chemistry. 2006;281:12010–12019. doi: 10.1074/jbc.M600548200. [DOI] [PubMed] [Google Scholar]
- 44.Ahuja S, Hornak V, Yan EC, Syrett N, Goncalves JA, Hirshfeld A, Ziliox M, Sakmar TP, Sheves M, Reeves PJ, Smith SO, Eilers M. Helix movement is coupled to displacement of the second extracellular loop in rhodopsin activation. Nature structural & molecular biology. 2009;16:168–175. doi: 10.1038/nsmb.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bokoch MP, Zou Y, Rasmussen SG, Liu CW, Nygaard R, Rosenbaum DM, Fung JJ, Choi HJ, Thian FS, Kobilka TS, Puglisi JD, Weis WI, Pardo L, Prosser RS, Mueller L, Kobilka BK. Ligand-specific regulation of the extracellular surface of a G-protein-coupled receptor. Nature. 2010;463:108–112. doi: 10.1038/nature08650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Akal-Strader A, Khare S, Xu D, Naider F, Becker JM. Residues in the first extracellular loop of a G protein-coupled receptor play a role in signal transduction. The Journal of biological chemistry. 2002;277:30581–30590. doi: 10.1074/jbc.M204089200. [DOI] [PubMed] [Google Scholar]