

Abstract
We investigated N-adamantyl-N′-phenyl urea derivatives as simple sEH inhibitors. Salicylate ester derivatives have high inhibitory activities against human sEH, while the free benzoic acids are less active. The methyl salicylate derivative is a potent sEH inhibitor, which also has high metabolic and chemical stabilities; suggesting that such inhibitors are potential lead molecule for bioactive compounds acting in vivo.
Keywords: Soluble epoxide hydrolase, Urea, Salicylate, Intramolecular hydrogen bond, Metabolic and chemical stability, Hypertension, Anti-inflammation
Epoxide hydrolases (EH, EC 3.3.2.3) catalyze the hydrolysis of epoxides and arene oxides to their corresponding diols.1 The mammalian soluble EH (sEH) is involved in maintenance of homeostasis through hydration of endogenous lipid epoxides such as epoxyeicosatrienoic acids (EETs).1,2 EETs, derived from arachidonic acid by cytochrome P450 epoxygenation, have various biological activities. EETs have effect on vascular tone. In addition, it is known that the 11,12- and 14,15-regioisomers of EETs have anti-inflammatory activity.2 The EETs inhibit activation of nuclear factor kappa B (NF-κB) and its nuclear translocation. This means that, under certain conditions, EETs will reduce activation of a variety of pro-inflammatory peptides and proteins such as cyclooxygenase 2.3 Since sEH converts EETs to the less biological active corresponding diols (dihydroxyeicosatrienoic acids), sEH inhibition can result in anti-inflammatory and anti-hypertensive effects.
1,3-Disubstituted ureas, and the corresponding amides and carbamates are strong sEH inhibitors.4,5 Our previous findings suggested that ureas with two additional pharmacophores (P2 and P3, Fig. 1) have strong inhibition in vitro as well as good bioactivity in vivo.5 The primary pharmacophore is the urea group bearing a bulky and/or hydrophobic substituent such as the adamantyl, cyclohexyl, alkyl or aryl groups (R1, Fig. 1). The secondary pharmacophore (P2) is a polar group, such as ketone, ester, alcohol, sulfoxide, sulfonamide or ether located five or six atoms away from the carbonyl group of the urea. A hydrophobic linker (L) joins the urea or amide central pharmacophore to the secondary pharmacophore. Unlike straight alkyl chains, the presence of cyclic linker (L), such as a cyclohexane or benzene ring, between the primary (urea) and secondary (P2) pharmacophores, seemed to increase the bioavailability in a canine model.5 Such cyclic linkers also reduce flexibility and are thought to make compounds more “drug-like”.6 In addition, the introduction of polar group(s) on such linkers likely will improve solubility, ease of formulation, and often their bioavailability. The nature of group R2 situated between P2 and P3 is more open to variation in term of size and polarity as long as P3 that is a polar group is at least 12 Å away from the urea carbonyl.5
Figure 1.
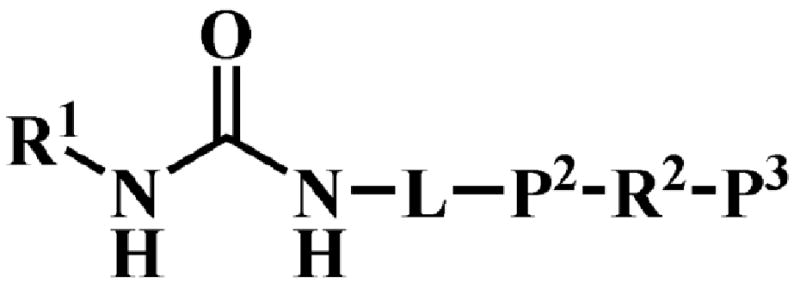
General schematic structure of urea-based human sEH inhibitors.
Therefore, in this study, we designed simple adamantyl ureas with a phenyl linker. These sEH inhibitors focus on the secondary pharmacophore. Usage of a phenyl moiety has several advantages for drug design, (1) the benzene ring, which is UV active, enables one to easily trace the target molecule during the synthesis or later scale-up and upon HPLC analysis; (2) as mentioned above, a benzene ring will play a role as a linker to keep a rigid distance between the primary and secondary pharmacophores; (3) a benzene ring, unlike a cycloalkane ring, has no stereoisomers, thus simplifying synthesis; and finally (4) numerous substituted anilines are commercially available simplifying the synthesis of ureas with functionalized phenyl rings, such as carboxylic acids that will increase the water solubility of the molecule. Many of these intermediates are inexpensive facilitating design of drugs for use in developing countries. Further, a carboxylic acid group can be connected with other functional group like alcohol or amine to design the tertiary pharmacophore. Herein, we report the sEH inhibitory activity as well as metabolic and chemical stabilities of simple N-adamantyl-N′-phenyl urea derivatives.
The urea compounds (1–4) were synthesized through the direct reaction with adamantyl isocyanate and the appropriate amine (Scheme 1). Although the isocyanate functional group can react with many nucleophiles such as alcohols, carboxylic acids and amines, the hydroxy group of aminophenol did not react significantly under our reaction conditions. However, for the synthesis of the carboxylic acid group-containing compound (14, 15, 19, 21, 23, 25 and 27), it was necessary to protect the acid function with a methyl ester that was removed following purification by alkaline hydrolysis (Scheme 1). The starting methyl esters (I–VII) were synthesized from the corresponding acids (i–vii) in a conventional manner. After the reaction with the isocyanate and appropriate amine followed by evaporation, the unreacted starting materials were removed by washing the crude product with a hexane:ethyl acetate mixture. This operation was simple but effective to remove not only the residual starting materials but also the N,N′-bisadamantyl urea which is not only present in the commercially available adamantyl isocyanate but can be formed as a side product during urea synthesis. This relatively insoluble high melting solid also shows strong inhibition towards sEH.4 After washing, the desired compound was purified through silica gel column chromatography and recrystallization as final purification stage. The compounds 8–11 were prepared from the corresponding phenol (3 or 4) and appropriate acyl chloride in a conventional manner.
Scheme 1.

Syntheses of the urea compounds used in this study. Reagents and conditions: (a) 1,2-dichloroethane, 45–50 °C, overnight; (b) THF, NaH, acetyl or benzoyl chloride, 45–50 °C, overnight; (c) methanol, concd H2SO4, overnight; (d) THF, 5% NaOH aq, reflux, 2–4 h; (e) dichloromethane, 4-dimethylaminophenol, 1-ethyl-3-[3-(dimethylamino)propyl] carbodiimide, appropriate amine, room temperature.
The inhibitory activity of the ureas with a benzene ring bearing a hydroxyl, ether, ester, carboxylic acid or amide functional group are given in Table 1. When a hydroxyl group was introduced in the meta (3) or para (4) position on the benzene ring, the inhibitory activity against human sEH was almost the same level as the non-functionalized compound 1. However, the introduction of hydroxy group as an ortho isomer (2) reduced the inhibitory potency by roughly 10-fold. It was found that the introduction of methoxy group in the ortho position (5) reduced the inhibitory activity dramatically although meta (6) and para (7) isomers possessed strong inhibition against sEH. These data indicated that the meta or para position on the benzene ring is a preferable position for the introduction of a functional group without adversely affecting sEH inhibition. We previously observed that only compounds bearing hydrogen or fluoride atoms on the ortho positions have sEH inhibition activity.4d The compounds bearing ester (8–13) as well as ether groups (6 and 7) were around 2- to 6-fold better sEH inhibitors than 3 and 4, and even better than 1, suggesting that an ether or ester group with hydrophobic properties helps bind to the active site of the human sEH enzyme. In previous studies,4,5 it was found that butyrate and caproate derivatives substituted in the 3 position of the urea were inactive as inhibitors of the sEH. However, their esters as secondary pharmacophores were highly active and could be used as soft drugs. In contrast, substitution of the 3 position of the urea with long chain acids such as dodecyl gave a tertiary pharmacophore as active as the ester, the acid or an acid mimic.5 In these cases, the ester is a pro-drug increasing ease of formulation and absorption. In the series of compounds described herein, most esters were very active (8–13). The dramatic decrease in the activity of the free acids is illustrated by compounds 14 and 15. Especially, compound 15, which had the acid group in the para position, is 83-fold less active towards sEH than the un-substituted phenyl 1 or the corresponding ester 13. Cyclic amides with a morpholine (16 and 17) or piperidine (18) were also synthesized. Unfortunately, up to 9-fold reduction in inhibitory potencies resulted from these amides compared with ester substitutions, suggesting that such amides are not suitable for yielding potent inhibitors. Interestingly, such heterocyclic groups when attached at the end of an alkyl chain yield ureas that have good pharmacokinetic properties in dogs while maintaining inhibitory potency.5f
Table 1.
Inhibitory activity of the ureas with a benzene ring containing a hydroxy, an ester, a carboxylic acid, amide or no functional group, against human sEH
| No. | 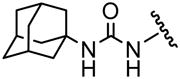 |
Human sEH IC50(nM)a |
|---|---|---|
| 1 | 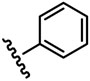 |
17 |
| 2 | 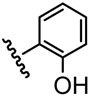 |
139 |
| 3 | 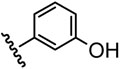 |
28 |
| 4 | 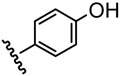 |
24 |
| 5 | 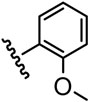 |
8544 |
| 6 | 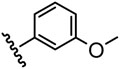 |
4.7 |
| 7 | 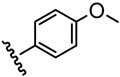 |
14 |
| 8 | 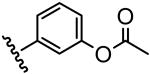 |
5.8 |
| 9 | 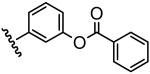 |
5.1 |
| 10 | 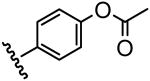 |
3.9 |
| 11 | 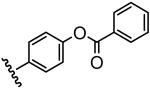 |
11 |
| 12 | 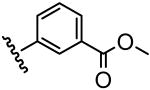 |
14 |
| 13 | 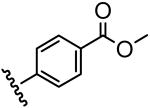 |
4.1 |
| 14 | 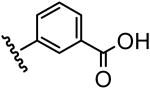 |
365 |
| 15 | 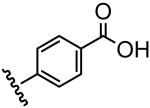 |
1411 |
| 16 | 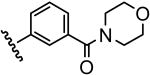 |
25 |
| 17 | 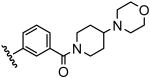 |
29 |
| 18 | 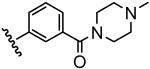 |
35 |
As determined via a kinetic fluorescent assay.13
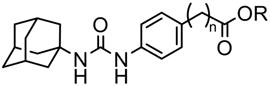
Compared to 15, inhibition potency on sEH is regained by moving the carboxylic acid away from the benzene ring (Table 2). A similar effect was observed with a series of alkyl-acids in place of the phenyl-acids.4d On the other hand, the corresponding methyl esters (13, 20, 22 and 24) showed strong inhibitory activities against sEH independently of the alkyl chain length between the ring and the methyl carboxylate. Recent X-ray analysis of the complex between the human sEH and urea inhibitors7 indicated that a basic amino acid residue(s) such as histidine and/or tryptophan, is present in the catalytic tunnel away from the active center of sEH. Such residues could make a salt bridge with an ionized carboxylate as the tertiary pharmacophore, and the salt formation would affect the orientation and affinity of such an ionizable acid molecule under physiological pH. Taken together, these data suggested that a free carboxylic acid or other ionizable polar group can be considered distal from the urea carbonyl group as the tertiary but not the secondary pharmacophore. When plotting the distance between the urea carbonyl and the acid carbonyl as a function of the inhibition potency for both a series of alkyl-acids and phenyl-acids (Fig. 2), we observed that both series of compounds followed a similar hyperbolic curve. These plots suggested that such acid functions as a tertiary pharmacophores should be roughly 12 Å form the urea carbonyl.
Table 2.
Inhibitory activity of the urea compounds with alkyl chains (C0–C3) between a benzene ring and a carboxylate functional group, against human sEH
 | ||||
|---|---|---|---|---|
| R=H | R = CH3 | |||
| Numbers of carbon (n) | Compound | IC50a (nM) | Compound | IC50a (nM) |
| 0 | 15 | 1411 | 13 | 4.1 |
| 1 | 19 | 392 | 20 | 4.0 |
| 2 | 21 | 126 | 22 | 2.2 |
| 3 | 23 | 19.9 | 24 | 3.1 |
As determined via a kinetic fluorescent assay.13
Figure 2.
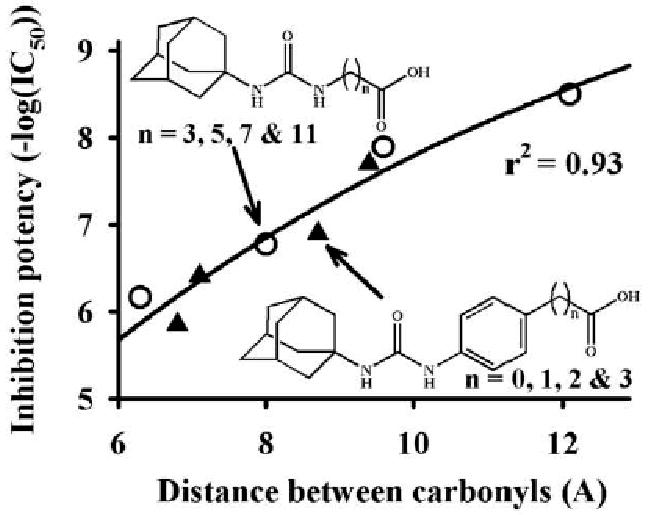
Effect of the distance between the urea carbonyl and the acid carbonyl on the inhibition potency. The distances were measured after minimizing the free energy of the molecule in gas phase, using the Chem3D® 8.0 software (Cambridge-Soft, Cambridge, MA).
The presence of a polar functional group in either the meta or para position on the phenyl group linker appeared beneficial for inhibition (Table 1). Thus, we designed and synthesized molecules containing both acid and hydroxyl functions (Table 3). The methyl salicylates 26 and 28 inhibited sEH strongly, with IC50s similar to those of 12 and 13, suggesting that for the methyl ester, the adjacent phenolic group did not negatively influence inhibitor binding to the enzyme. Surprisingly, the salicylic acids 25 and 27 showed 3- and 20-fold better inhibitory activity against sEH than 14 and 15, respectively. Infrared analysis of course shows strong internal hydrogen bonds of the salicylates (25 and 27) in contrast to the free carboxylic acids (14 and 15). Furthermore, the hydroxyl group by itself placed on meta 3 or para 4 position did not improved the inhibitor potency of 1, suggesting that the hydroxyl group plays an important role in binding with the active site of sEH only if a carboxylic function is placed on the adjacent carbon. In such cases, a hydrogen bond between the carboxylate and the hydroxyl is probably formed that certainly reduces the negative effect of the acid function on the inhibition potency.
Table 3.
Inhibitory activity of the benzoate- or the salicylate-based urea compounds against human sEH
| Compound | 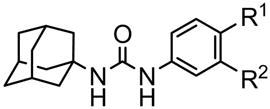 |
Human sEH IC50a (nM) | |
|---|---|---|---|
| R1 (para) | R2 (meta) | ||
| 25 | OH | COOH | 116 |
| 26 | OH | COOCH3 | 12 |
| 27 | COOH | OH | 71 |
| 28 | COOCH3 | OH | 2.8 |
As determined via a kinetic fluorescent assay.13
In general, the metabolic stability of a candidate compound will influence drug efficacy in vivo. Although all esters synthesized in this study showed strong inhibitory activities against the recombinant human sEH, it was anticipated that they would be hydrolyzed easily in the body. Thus, six compounds (10–13, 26 and 28) were tested for in vitro stability with human hepatic S9 fraction with or without NADPH, a cofactor necessary for cytochrome P450 activity.8 For all the compounds tested the results obtained were similar with or without NADPH, suggesting that the principal route of metabolism of these chemicals did not involve P450s. As expected, for most compounds only a small amount of the parent compound was left after 60 min of incubation (Table 4), and large amounts of the corresponding acids were detected. Unexpectedly, 90% or more of the methyl esters 13 and 28 remained after the reaction, and the levels of the corresponding acid metabolites (15 and 27, respectively) were below the limit of detection, indicating that, for this series of compounds, esters present on the para position are more metabolically stable than on a meta position. The quasi-absence of ester hydrolysis for the para compounds (13 and 28) is probably due to steric interactions that do not permit an optimal binding into the liver esterases for hydrolysis. Compared to 12, the presence of a hydroxyl group in para 26 increased the metabolic stability of the resulting compound 10-fold. Otherwise, 10 and 11, which give the phenol 4, were also decomposed easily under the same reaction conditions. The acetate 10 was hydrolyzed completely in an hour to give the corresponding phenol 4. Compound 11 was hydrolyzed slower than 10 to produce around 70% of the phenol 4, while 30% of the starting compound 11 was found. Imai et al.9 also reported that a phenyl acetate derivative was hydrolyzed more rapidly by liver microsomal carboxylesterases than methyl salicylate and benzoate derivatives. An earlier series of ether containing compounds with a 5 or 6 carbon alkyl spacer yielded potent inhibitors with excellent physical properties but poor stability.5a Compounds 29 and 30 indicate that the methyl ethers of 6 and 7 can be replaced successfully with polyethylene glycol chains.
Table 4.
In vitro stability of the urea compounds containing an ester group on a phenyl ring with human liver S9 incubation
| Compound | 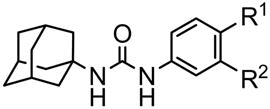 |
NADPH | Residual parent compound (%) | |||
|---|---|---|---|---|---|---|
| R1 (para) | R2 (meta) | − | + | |||
| 12 | H | COOCH3 | 3.5 | 6.9 | ||
| 26 | OH | COOCH3 | 46 | 51 | ||
| 13 | COOCH3 | H | 97 | 95 | ||
| 28 | COOCH3 | OH | 93 | 89 | ||
| 10 | OC(O)CH3 | H | <0.1 | <0.1 | ||
| 11 | OC(O)Ph | H | 32 | 31 | ||
| 29a | H | O(CH2CH2O)2C2H5 | N.D.b | 56 | ||
| 30a | O(CH2CH2O)2C2H5 H | N.D.b | 88 | |||
IC50s on the human sEH are 1.8 and 1.1 nM for 29 and 30, respectively.
Not determined. Since the polyethoxylates are ethers not esters, incubation in the absence of NADPH were not performed.
To investigate the surprising enzymatic stability of the benzoate esters (13 and 28), and because most esterases have a nucleophilic mechanism of action, we compared the chemical stability of these two compounds in alkaline solution (40 mM NaOH; 22 °C) with the para-phenolic esters (10 and 11) (Fig. 3). As observed with liver S9 (Table 4), 10 and 11 were hydrolyzed very fast with complete conversion to the corresponding phenol 4 in less than 15 min (data not shown). While 13 and 28 have similar metabolic stability with liver S9 (Table 4), they were hydrolyzed at a different rate in alkaline solution (Fig. 3). The methyl benzoate 13 was hydrolyzed gradually to the corresponding acid 17 with a 2-h half-life. By contrast, the methyl salicylate 28 was still more stable under the same reaction conditions, with ∼80% remaining after 7 h. These results suggest a different mechanism of hydrolysis for both compounds. In effect, the neighboring phenolate was shown to influence salicylate ester hydrolysis in alkaline conditions.10 This result suggested that introducing a hydroxyl group on the carbon adjacent to the benzoate ester improved not only metabolic but also chemical stability of a molecule by influencing its chemical reactivity. Furthermore, additional metabolic stability is obtained due to steric restrictions if the ester is placed on the para position.
Figure 3.
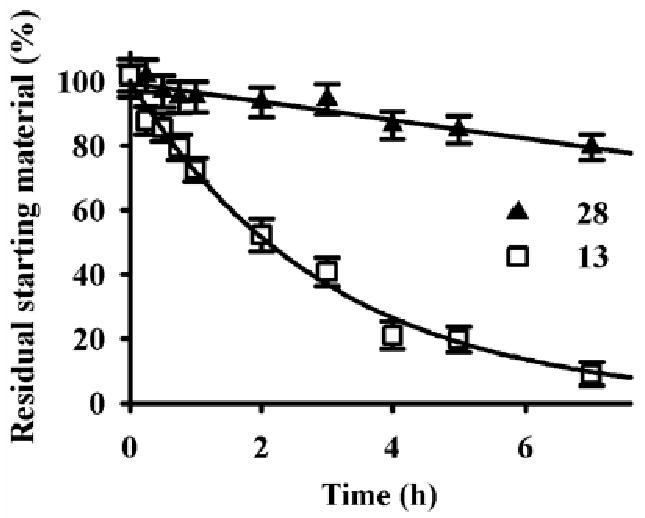
Time-dependent hydrolysis of the methyl benzoate- and salicylate-based urea compounds (13 and 28) under alkaline conditions (40 mM NaOH) at 22 °C.
In addition to target potency and stability, a compound usually needs good solubility to give good exposure to the biochemical target in vivo. As shown in Table 5, the compounds with a free acid (15 and 27) have higher water solubility than the corresponding methyl esters (13 and 28). Furthermore, as expected the addition of a hydroxyl group on 27 led to a compound 20-fold more soluble in water than 17. Interestingly, no such effect was observed for the methyl esters 13 and 28. On the other hand, the hydroxyl group increased solubility in octanol, which is a model of solubility in biological membranes,11 but only for the methyl ester 28 not its corresponding acid 27. This result suggests that for 28 there is probably an intramolecular bond between the hydroxyl group and the ester carbonyl leading to increase miscibility with oil, but not in water. These properties could aid absorption in the gut. Finally, for the methyl esters (13 and 28), the measured logP are very similar to the one calculated ones (clogP); however, for the acids (17 and 27), the measured logP are between the clogP of the acids and corresponding base forms. It suggests that both forms of 17 and 27 are present at pH 7.4.
Table 5.
Physicochemical properties of the benzoate- or the salicylate-based urea compounds
| Compound | 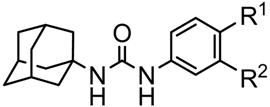 |
Solubility in watera (mg/mL) | Mp (°C) | Experimental parameters and logP | ||||
|---|---|---|---|---|---|---|---|---|
| R1 (para) | R2 (meta) | Concentrationb (μM) | logPc | clogPd | ||||
| Octanol | Buffer | |||||||
| 13 | COOCH3 | H | 0.4 | 219–220 | 2445 | <0.3 | >3.9 | 4.3 |
| 17 | COOH | H | 10 | >300 | 2720 | 17 | 2.2 | 3.8/−0.5e |
| 27 | COOH | OH | 199 | 205–206 | 1641 | 39 | 1.6 | 3.9/−1.1e |
| 28 | COOCH3 | OH | 0.2 | 212–213 | 3886 | <0.3 | >4.1 | 4.1 |
Solubility in 0.1 M sodium phosphate buffer (pH 7.4) at 23 ± 1.5 °C.
Buffer saturated 1-octanol and 0.1 M sodium phosphate buffer (pH 7.4) were used. See Supplementary information in detail.
logP value was calculated following the equation; logP = log10 ([conc. of compound]octanol/[conc. of compound]buffer).
clogPwas calculated using ChemDraw® software (CambridgeSoft, Cambridge, MA).
The clogP of both the acid and base forms of the carboxylic acid are given.
Finally, to test their bioavailability, the methyl salicylate 28 that had good in vitro metabolic and chemical stability in addition to potent sEH inhibitory activity, and its corresponding acid 27 were administered orally to a dog. Blood levels of 28 and 27 were determined by LC/MS–MS (Fig. 4). We used AUDA, which has been utilized in various biological studies,3,12 for comparison. As shown in Figure 4, the blood concentration of 28 was higher than that of the classical sEH inhibitor AUDA, indicating a good bioavailability. While 28 and AUDA have similar IC50s (∼3 nM) for the human sEH, 28 is more water soluble and metabolically stable than AUDA.5d Thus, put together, 28 should be more efficient than AUDA to inhibit sEH in vivo. Surprisingly, no 27 was detected in the blood at all, indicating poor absorption.
Figure 4.
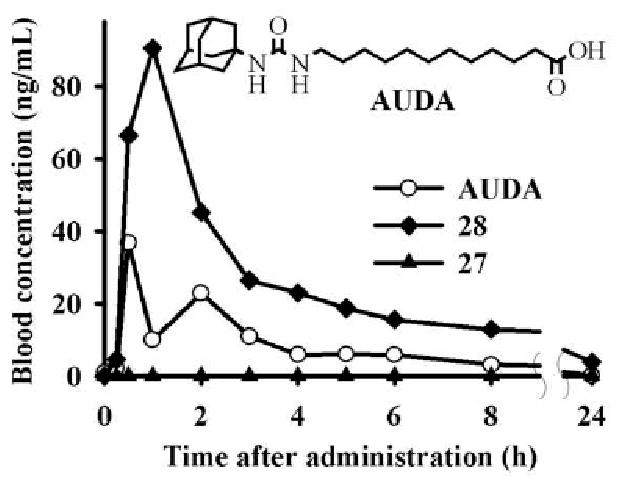
Blood concentration–time profiles of sEH inhibitors (27, 28 and AUDA) in dogs following oral gavage at a dose of 0.3 mg/kg in 6 mL of tri-olein rich oil. Pharmacokinetic parameters are given in Supplementary materials.
In conclusion, benzoate–urea-based sEH inhibitors used in this study had simple chemical structures and were easily synthesized. The aromatic group made them easy to follow on thin layer chromatography. Furthermore, among them, the methyl salicylate 28 which is quite a potent sEH inhibitor has high metabolic and chemical stabilities, as well as a good bioavailability. These data suggest that such inhibitor is a potential lead molecule for a bioactive compound in vivo. Finally, Schmelzer et al. reported recently that co-administration of non-steroidal anti-inflammatory drugs and sEH inhibitors enhanced anti-inflammatory and anti-nociceptional effects through dramatic suppression of prostaglandin E2 production.3 Since the salicylate–urea compound in this study is also a prospective cyclooxygenase inhibitor, this or related molecules may have additional in vivo benefits with biological activities as dual sEH-COX inhibitors. However, using a fluorescent inhibitor screening assay kit (#700100, Cayman Chemicals, Ann Arbor, MI), we were not able to show any inhibition of COX-1 and -2 with compound 28 at 10 μM.
Supplementary Material
Acknowledgments
The authors thank Dr. Jun-Yan Liu for his assistance on MS analyses. This study was supported in part by NIEHS Grant R37 ES02710, NIEHS Superfund Grant P42 ES004699, and NIH/NHLBI R01 HL059699. H.J.T. is a recipient of a Howard Hughes fellowship.
Footnotes
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.bmcl.2009.01.069.
References and Notes
- 1.Morisseau C, Hammock BD. Annu Rev Pharmacol Toxicol. 2005;45:311. doi: 10.1146/annurev.pharmtox.45.120403.095920. [DOI] [PubMed] [Google Scholar]
- 2.(a) Harder DR, Campbell WB, Roman RJ. J Vasc Res. 1995;32:79. doi: 10.1159/000159080. [DOI] [PubMed] [Google Scholar]; (b) Campbell WB, Gebremedhin D, Pratt PF, Harder DR. Circ Res. 1996;78:415. doi: 10.1161/01.res.78.3.415. [DOI] [PubMed] [Google Scholar]; (c) Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, Zeldin DC, Liao JK. Science. 1999;285:1276. doi: 10.1126/science.285.5431.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Newman JW, Morisseau C, Hammock BD. Prog Lipid Res. 2005;44:1. doi: 10.1016/j.plipres.2004.10.001. [DOI] [PubMed] [Google Scholar]; (e) Spiecker M, Liao JK. Arch Biochem Biophys. 2005;433:413. doi: 10.1016/j.abb.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 3.(a) Schmelzer KR, Kubala L, Newman JW, Kim IH, Eiserich JP, Hammock BD. Proc Natl Acad Sci USA. 2005;102:9772. doi: 10.1073/pnas.0503279102. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Schmelzer KR, Inceoglu B, Kubala L, Kim IH, Links SL, Eiserich JP, Hammock BD. Proc Natl Acad Sci USA. 2006;103:13646. doi: 10.1073/pnas.0605908103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Morisseau C, Goodrow MH, Dowdy D, Zheng J, Greene JF, Sanborn JR, Hammock BD. Proc Natl Acad Sci USA. 1999;96:8849. doi: 10.1073/pnas.96.16.8849. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nakagawa Y, Wheelock CE, Morisseau C, Goodrow MH, Hammock BG, Hammock BD. Bioorg Med Chem. 2000;8:2663. doi: 10.1016/s0968-0896(00)00198-x. [DOI] [PubMed] [Google Scholar]; (c) Morisseau C, Newman JW, Dowdy DL, Goodrow MH, Hammock BD. Chem Res Toxicol. 2001;14:409. doi: 10.1021/tx0001732. [DOI] [PubMed] [Google Scholar]; (d) Morisseau C, Goodrow MH, Newman JW, Wheelock CE, Dowdy DL, Hammock BD. Biochem Pharmacol. 2002;63:1599. doi: 10.1016/s0006-2952(02)00952-8. [DOI] [PubMed] [Google Scholar]; (e) Morisseau C, Newman JW, Tsai HJ, Baecker PA, Hammock BD. Bioorg Med Chem Lett. 2006;16:5439. doi: 10.1016/j.bmcl.2006.07.073. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hwang SH, Morisseau C, Do Z, Hammock BD. Bioorg Med Chem Lett. 2006;16:5773. doi: 10.1016/j.bmcl.2006.08.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Kim IH, Morisseau C, Watanabe T, Hammock BD. J Med Chem. 2004;47:2110. doi: 10.1021/jm030514j. [DOI] [PubMed] [Google Scholar]; (b) Kim IH, Heirtzler FR, Morisseau C, Nishi K, Tsai HJ, Hammock BD. J Med Chem. 2005;48:3621. doi: 10.1021/jm0500929. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Jones PD, Tsai HJ, Do ZN, Morisseau C, Hammock BD. Bioorg Med Chem Lett. 2006;16:5216. doi: 10.1016/j.bmcl.2006.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kim IH, Nishi K, Tsai HJ, Bradford T, Koda Y, Watanabe T, Morisseau C, Blanchfield J, Toth I, Hammock BD. Bioorg Med Chem. 2007;15:312. doi: 10.1016/j.bmc.2006.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Hwang SH, Tsai HJ, Liu JY, Morisseau C, Hammock BD. J Med Chem. 2007;50:3825. doi: 10.1021/jm070270t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Kim IH, Tsai HJ, Nishi K, Kasagami T, Morisseau C, Hammock BD. J Med Chem. 2007;50:5217. doi: 10.1021/jm070705c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Adv Drug Deliv Rev. 2001;46:3. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 7.Gomez GA, Morisseau C, Hammock BD, Christianson DW. Protein Sci. 2006;15:58. doi: 10.1110/ps.051720206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Watanabe T, Morisseau C, Newman JW, Hammock BD. Drug Metab Dispos. 2003;31:846. doi: 10.1124/dmd.31.7.846. [DOI] [PubMed] [Google Scholar]
- 9.Imai T, Taketani M, Shii M, Hosokawa M, Chiba K. Drug Metab Dispos. 2006;34:1734. doi: 10.1124/dmd.106.009381. [DOI] [PubMed] [Google Scholar]
- 10.Khan MN, Fatope IL, Isaac KI, Zubair MO. J Chem Soc Perkin Trans 2. 1986;5:655. [Google Scholar]
- 11.Dennis AS, van de Waterbeemd H, Walker DK. In: Pharmacokinetics and Metabolism in Drug Design. Mannhold R, Kubinyi H, Timmerman H, editors. Vol. 31. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim: 2006. pp. 1–15. [Google Scholar]
- 12.(a) Jung O, Brandes RP, Kim IH, Schweda F, Schmidt R, Hammock BD, Busse R, Fleming I. Hypertension. 2005;45:759. doi: 10.1161/01.HYP.0000153792.29478.1d. [DOI] [PubMed] [Google Scholar]; (b) Dorrance AM, Rupp N, Pollock DM, Newman JW, Hammock BD, Imig JD. J Cardiovasc Pharmacol. 2005;46:842. doi: 10.1097/01.fjc.0000189600.74157.6d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Imig JD, Zhao X, Zaharis CZ, Olearczyk JJ, Pollock DM, Newman JW, Kim IH, Watanabe T, Hammock BD. Hypertension. 2005;46:975. doi: 10.1161/01.HYP.0000176237.74820.75. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Olearczyk JJ, Field MB, Kim IH, Morisseau C, Hammock BD, Imig JD. J Pharmacol Exp Ther. 2006;318:1307. doi: 10.1124/jpet.106.103556. [DOI] [PubMed] [Google Scholar]; (e) Huang H, Morisseau C, Wang J, Yang T, Falck JR, Hammock BD, Wang MH. Am J Physiol Renal Physiol. 2007;293:F342. doi: 10.1152/ajprenal.00004.2007. [DOI] [PubMed] [Google Scholar]
- 13.(a) Jones PD, Wolf NM, Morisseau C, Whetstone P, Hock B, Hammock BD. Anal Biochem. 2005;343:66. doi: 10.1016/j.ab.2005.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Morisseau C, Hammock BD. In: Techniques for analysis of chemical biotransformation, Current Protocols in Toxicology. Bus JS, Costa LG, Hodgson E, Lawrence DA, Reed DJ, editors. John Wiley & Sons; New Jersey: 2007. pp. 4.23.1–4.23.18. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.