

Abstract
The Holliday junction is a key intermediate of DNA repair, recombination, and replication. Branch migration of Holliday junctions is a process in which one DNA strand is progressively exchanged for another. Branch migration of Holliday junctions may serve several important functions such as affecting the length of genetic information transferred between homologous chromosomes during meiosis, restarting stalled replication forks, and ensuring the faithful repair of double strand DNA breaks by homologous recombination. Several proteins that promote branch migration of Holliday junctions have been recently identified. These proteins, which function during DNA replication and repair, possess the ability to bind Holliday junctions and other branched DNA structures and drive their branch migration by translocating along DNA in an ATPase-dependent manner. Here, we describe methods employing a wide range of DNA substrates for studying proteins that catalyze branch migration of Holliday junctions.
Keywords: branch migration, Holliday junction, ATPase, joint molecules
1. Introduction
The Holliday junction is a key intermediate in various genetic processes including homologous and site-specific recombination and DNA replication [1]. The Holliday junction is a four-stranded structure that, forms during exchange between two homologous DNA molecules. Robin Holliday first predicted this structure in his model of homologous recombination which provided the molecular basis for both gene conversion and crossing-over [2]. The remarkable feature of the Holliday junction is its ability to branch migrate along the DNA axis, in which one DNA strand is progressively exchanged for another.
Branch migration of Holliday junctions may serve several important functions in DNA repair, replication, and recombination. It may extend or shorten the heteroduplex DNA, formed during recombination, affecting the length of conversion tracks and thereby the amount of genetic information transferred between the two DNA molecules [3]. Branch migration may cause dissociation of recombination intermediates and thereby affect the choice between a crossover and non-crossover pathway by which recombination will proceed [4]. An increasing number of genetic and biochemical data indicate that formation and branch migration of Holliday junctions may also play a critical role in re-starting stalled replication forks during cell recovery after DNA damage [5-10]. It is thought that the major pathway of the DNA replication restart involves unwinding of the stalled replication fork to generate a Holliday junction [5]. Branch migration may also take a part in the control of frequency of recombination exchanges [11].
The Holliday junction is a substrate for proteins which promote its branch migration and for structure-specific nucleases which are responsible for its cleavage and formation of crossover products. Branch migration proteins have been identified in all domains of life [12-15] and have been classified as members of the helicase superfamilies [16]. These proteins rely on their DNA-dependent ATPase activity to translocate along DNA in a manner that drives the branch migration of Holliday junctions. The first branch migration enzymes identified were found also to possess classical helicase activity, i.e. the ability to separate strands of duplex DNA [1]. However, as more branch migration proteins are discovered [17] it appeared that not all of them have a helicase activity [16, 18]. Analysis of these proteins required the development of more specific approaches and DNA substrates. Here we describe the methods our laboratory used to characterize the branch migration activity of the homologous recombination protein Rad54. These assays are applicable for other branch migration proteins regardless of whether they have helicase activity or not.
2. Preparation of oligonucleotide substrates for analysis of branch migration proteins
2.1. Strategy for designing branched DNA oligonucleotide substrates
In earlier works, “non-movable” X-junctions (Holliday junctions), consisting of a central homologous “movable” core flanked by mutually heterologous terminal DNA branches [12] (Fig. 1A), were successfully used to detect the branch migration activities of prokaryotic enzymes (RuvAB, RecG) that have both branch migration and helicase activities [1]. However these substrates were unsuitable for detecting the branch migration activity of Rad54, which does not have canonical helicase activity [19]. We therefore constructed “movable” oligonucleotide substrates in which two of the four terminal DNA branches were mutually homologous [17]. This allowed the crossover point to move freely by branch migration to the end of the DNA molecule, resulting in complete separation of the two DNA duplexes without a need for helicase activity (Fig. 1B). Here we describe the protocols for constructing a series of branched DNA substrates including the X-junctions, PX-junctions (partial Holliday junctions), and movable replication forks.
Fig. 1.

Construction of oligonucleotide substrates to test the branch migration activity of proteins. (A) Both branch migration and DNA helicase activity are required to disrupt a non-movable X-junction. (B) The movable X-junctions contain a mismatch to block spontaneous branch migration and are formed by annealing of two forked DNA intermediates. (C) The branch migration of a PX-junction requires an additional step, the transition from a 3-stranded to 4-stranded structure. Shaded regions denote heterologous DNA terminal branches. The asterisk indicates 32P-label at the DNA 5′-end.
Oligonucleotide substrates can be prepared through the annealing of two simple DNA intermediates (forked, tailed, or ssDNAs) (Fig. 1B). When constructing substrates with four DNA arms (X-junctions, PX-junctions), it is important that two of the opposing arms of the predicted branched DNA molecule are non-homologous relative to each other, while the remaining two are homologous (Fig. 1B, C shaded). Without this orientation, it would be impossible to prepare forked DNA intermediates. To block spontaneous branch migration in a 4-stranded reaction [20, 21] at least one base pair heterology must be introduced in the two homologous duplexes involved in branch migration (Fig. 1B). The substrates used to study the 3-stranded branch migration reaction, where exchange occurs between one duplex DNA and one ssDNA, should have at least four bases of heterology. Regions of heterology shorter than four bases are not sufficient to block spontaneous 3-stranded branch migration, as this reaction proceeds more efficiently than the 4-stranded reaction [20, 21].
When constructing PX-junctions or replication forks a heterologous base should be placed at least three nucleotides away from the junction point. This provides room for the transition from 3-stranded to 4-stranded (Fig. 1C) to take place at the point in which a Holliday junction forms. We have observed that a single base heterology placed within three nucleotides of the junction will inhibit the rate of branch migration several fold over a heterology that is placed farther than three nucleotides from the junction (Mazin Lab, unpublished observation).
2.2. Gel purification of DNA oligonucleotides
To date, DNA oligonucleotides with a length up to 150 nucleotides are readily available from commercial sources. Because the quality of commercially-purified oligonucleotides varies, we usually purchase them in a dry desalted form and then purify them by electrophoresis in 6-10% denaturing polyacrylamide gels containing 50% urea.
Purify the individual oligonucleotides by electrophoresis in a denaturing polyacrylamide gel (acrylamide/bis-acrylamide ratio of 19:1) containing 50% urea. We use gels of 20 cm in width, 38 cm in length, and 0.8 mm in thickness. The optimal concentration of polyacrylamide gel depends on the length of the oligonucleotide to be purified (refer to Table 1). To prepare oligonucleotide samples for gel electrophoresis, dissolve lyophilized oligonucleotide in TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0) to a final concentration of 30 μg/ml, then mix 5 μl (150 μg) of the oligonucleotide stock with 25 μl of formamide containing 0.1% bromophenol blue. The 150 μg amount of oligonucleotide can be loaded on a gel well 25-30 mm wide. Prior to gel electrophoresis, heat the samples at 80°C for 3 min to remove any DNA secondary structure, and load them on the gel that was pre-run for at least 20 min. Before loading the samples, wash the wells with running buffer to remove urea that has defused from the gel. As a running buffer we use TBE (90 mM Tris borate, pH 8.3, and 1 mM EDTA). Run the gel at 1200 V (32 V/cm) for 2-3 h. After electrophoresis, remove one of the glass plates. Place the glass plate with the gel on top of a fluor-coated silica gel thin-layer chromatography plate. Then visualize the DNA bands by irradiating the gel from the top using a handheld short-wave (302 nM) ultraviolet lamp. This method, known as UV shadowing, allows for DNA visualization in the absence of ethidium bromide, which could inhibit subsequent branch migration reactions. Excise the DNA band with a blade, and transfer the gel slice to a microcentrifuge tube. Using a pipette tip break the gel slice up into small pieces, approximately 1 mm2. Add 500 μl (about 3 times of the gel slice volume) of an elution buffer containing 450 mM sodium acetate, pH 7.0 and 2 mM EDTA; then shake overnight on a vortex at room temperature. Pass the solution through a Spin-X centrifuge tube filter (Costar) and precipitate DNA in the flow-through by adding four volumes of ethanol followed by incubation for at least 1 h at -20°C. Centrifuge at >16,000 × g for 25 min at 4°C. Remove the ethanol, dry the pellet, and resuspend it in 40 μl of nanopure water. Determine concentrations of all oligonucleotides spectrophotometrically using known molar extinction coefficients (which can be obtained from the IDT website www.idtdna.com). Make concentration of oligonucleotides 0.5-1 mg/ml and store them at -20°C.
Table 1.
Percentage of polyacrylamide gel (acrylamide/bis-acrylamide ratio of 19:1) needed to purify oligonucleotides of indicated lengths.
| Length of Oligonucleotide | Percentage of polyacrylamide |
|---|---|
| < 40 nucleotides | 10% |
| 40 – 75 nucleotides | 8% |
| >75 nucleotides | 6% |
Label one of the oligonucleotides using [γ-32P] ATP and T4 polynucleotide kinase (NEB). For labeling, mix 2 μl of the oligonucleotide at concentration 0.5 mg/ml (1 μg), 2 μl of [γ-32P] ATP (10 mCi/ml; Perkin Elmer), 2 μl of 10 × T4 polynucleotide kinase buffer and 1 unit of T4 polynucleotide kinase in total volume of 20 μl. Incubate the reaction mixture for 1 h at 37 °C, then inactivate T4 polynucleotide kinase by heating the mixture for 10 min at 75 °C. Store labeled oligonucleotides at -20 °C.
2.3. Preparation of branched DNA substrates by annealing
Prepare tailed or forked DNA intermediates (Fig. 1 B,C) by mixing equal amounts of each complementary ssDNA strands (usually 3 μM molecules of each 100 nt long oligonucleotide) in 14 mM Tris-HCl, pH 7.5, 2 mM MgCl2, 1 mM dithiothreitol (DTT), 121 mM NaCl and 12.1 mM Na citrate. The reaction is heated for 2 min at 95°C, followed by annealing for 1 h at the optimal hybridization temperature (Th). Calculate Th using the formula: Th=1.24*Tm-43.8°C; where Tm is the melting temperature of the double-stranded portion of the DNA intermediate. Calculate the Tm using a tool from the Promega website (www.promega.com/biomath/calc11.htm). It is critically important when designing forked DNA that you make sure the single stranded regions do not contain even short fortuitous regions of homology; as they could decrease the efficiency of the subsequent annealing reaction.
2.3. 1. Preparation of branched DNA substrates for the branch migration assay
To prepare branched DNA substrates (Fig. 2), mix 32 nM of the 32P-labeled and 48 nM of the non-labeled annealed DNA intermediates in a reaction buffer containing magnesium (for DNA substrates to be used with RAD54, the following buffer was used: 25 mM Tris acetate, pH 7.5, 2 mM ATP, 5 mM magnesium acetate, 2 mM DTT, 100 μg/ml bovine serum albumin (NEB, nuclease and protease free), 15 mM phosphocreatine and 10 units/ml creatine phosphokinase). Anneal DNA intermediates for 10 min at 37°C, and then for another 10 min at the temperature of the branch migration assay. In order to inhibit spontaneous branch migration (especially on X-junctions), we usually use magnesium at concentrations of 3 mM or higher. If it is important to have magnesium concentration lower than 3 mM, the reaction temperature may need to be decreased to slow spontaneous branch migration.
Fig. 2.

An array of branched oligonucleotides substrates used to characterize the DNA binding specificity of branch migration proteins using the ATPase activity. Substrates are displayed in order of descending complexity. Shaded regions denote heterologous DNA terminal branches.
Note: For efficient annealing of DNA intermediates (Fig. 1B,C), complementary ssDNA regions should not contain a GC-content higher than 40-50%. If the DNA intermediates do not anneal efficiently, try to decrease GC-content or the length of the complementary DNA regions.
2.3.2. Preparation of branched DNA substrates for the ATPase assay
For the ATPase assay, it is more convenient to use “non-movable” branched DNA substrates with mutually heterologous terminal DNA branches, (e.g., Fig. 1A). To prepare these “non-movable” DNA substrates, mix equimolar amounts of non-labeled tailed and forked DNA intermediates (usually 3 μM molecules for DNA substrates of ∼ 60-100 nt/bp in size) in annealing buffer (14 mM Tris-HCl, pH 7.5, 2 mM MgCl2, 1 mM DTT, 121 mM NaCl and 12.1 mM Na citrate) followed by an 1 h incubation at the optimal hybridization temperature (Th). Preparation of X- and PX-junctions involves annealing of two ssDNA arms which may have different Th. In this case, calculate these two Th and perform annealing by incubating mixture subsequently 1 h at the higher Th, and then 1 h at the lower Th. These DNA substrates can be used in the ATPase assay without further purification.
3. Use of branched DNA substrates for characterization of ATPase activity of branch migration proteins
Because branch migration proteins translocate on DNA at the expense of ATP hydrolysis, their ATPase activity usually shows a characteristic increase with the increase in length of linear dsDNA or ssDNA [22-24]. Thus, the ATPase assay can be used to initially identify DNA translocating proteins, including branch migration proteins. Also, most branch migration proteins show high binding specificity to cruciform DNA substrates resembling the Holliday junction (illustrated in Fig. 2). Usually, this DNA binding specificity is determined by a band-shift assay [17, 25]. However, because the ATPase activities of all known branch migration proteins depend on DNA binding, ATPase can also be used as a readout of DNA-binding. Typically for branch migration proteins, high affinity to DNA substrates correlates with high rates of ATP hydrolysis. An exception to this rule may be an indicator of the mechanism of branch migration, e.g., the fact that binding of Rad54 to ssDNA poorly stimulates its ATPase activity indicates that this protein does not translocate on ssDNA.
There are several methods for measuring the rate of ATP hydrolysis; here we focus on two of them. In the first, ATP hydrolysis is coupled through additional enzymatic reactions that spectrophotometrically track the oxidation of NADH [26]. In the second, the products of ATP hydrolysis, ADP or inorganic phosphate, can be measured directly using thin-layer chromatography (TLC). The spectrophotometric assay has an important advantage of being rapid and very accurate. This assay can obtain important kinetic parameters of the ATPase activity of branch migration proteins, including Km and Vmax. The TLC-based asay is more laborious and therefore less suited for kinetic ananlysis, but can be used for initial characterization of the ATPase activity. TLC assay is especially useful, when i) the available amount of branch migration protein is small (less than a few μg), ii) the protein stock concentration is low (less than a few μM), or iii) the rate of ATP hydrolysis is low (less than 10-15 min-1). Also, in the spectrophotometric assay the enzymes that couple ATP regeneration with NADH oxidation, pyruvate kinase, and lactate dehydrogenase, may not be active under all experimental conditions, whereas the TLC assay is free from this limitation.
3.1. The TLC-based ATPase assay
We use polyethyleneimine-cellulose (Selecto Scientific) for separation of the products of ATP hydrolysis from ATP by TLC. As an eluent for separation of free phosphate from ATP we recommend 1 M formic acid with 0.5 M LiCl. A mixture of cold and radioactive [γ-32P] ATP can be used as a substrate. Typically, for a reaction mixture containing 1 mM ATP we add 20 μCi/ml [γ-32P] ATP (we prepare a 10 × ATP stock by mixing 1 μl of 10 mCi/ml stock with 50 μl of 10 mM ATP). Free phosphate produced during ATP hydrolysis migrates significantly faster that ATP substrate. After completion of TLC, we determine the amount of free phosphate and ATP using a PhosphorImager.
3.2. The spectrophotometric ATPase assay
We used the ATPase assay to characterize the DNA substrate specificity of RAD54 and to determine the minimal substrate size that supports the ATPase activity of RAD54 [27]. In this assay we use an Agilent 8453 diode array spectrophotometer equipped with an 8 cell motorized cuvette holder, UV-Visible ChemStation software, and a temperature controlled water bath (RC6 CS Lauda, Brinkmann). When an ATP molecule is hydrolyzed, pyruvate kinase converts the resultant ADP back to ATP by transfering the PO43-group from phosphoenol pyruvate (PEP) that is converted to pyruvate. Then, lactate dehydrogenase reduces the pyruvate to lactate while oxidizing NADH to NAD+. Per one ATP molecule hydrolysed, one molecule of NADH is oxidized; ATP hydrolysis and NADH oxidation are therefore coupled. The oxidation of NADH resulted in a decrease in absorbance at 340 nm that can be readily measured on a spectrophotometer. We use pyruvate kinase and lactate dehydrogenase (both type II from rabbit muscles, Sigma Chemical Co.,) that are supplied as a sulphate ammonium suspension. Prior to the experiment, pellet the enzymes by centrifugation for 3-5 min at 16,000 × g and resuspend them in the assay buffer. Mix together the components of the reaction mixture, except NADH, in a test-tube, and then transfer them into a spectrophometric cuvette. We use 50 μl microcuvettes (Agilent) with a 1 cm lightpass. Blank the mixture at 340 nM and then add NADH (200 μg/ml). The optimal A340 value should be 1.6-1.8. Incubate the reaction mixture at the reaction temperature for 5 min. Typically, ATP hydrolysis is initiated by addition of the branch migration enzyme; in some cases it may also be done by adding DNA, ATP, or another critical component of the reaction. Mix the enzyme well by flicking the cuvette and return it to the spectrophotometer holder. For human RAD54, the reactions were carried out in standard buffer containing 25 mM Tris acetate, pH 7.5, 10 mM magnesium acetate, 2 mM DTT, 2 mM ATP, 3 mM phosphoenolpyruvate, pyruvate kinase (20 units/ml), lactate dehydrogenase (20 units/ml), NADH (200 μg/ml), and different concentrations of RAD54 protein and DNA at 30 °C. In preliminary experiments, determine the saturating concentration of ATP for all DNA substrates used in the study.
Collect the absorbance data as a function of time. Calculate the rate of ATP hydrolysis using the following formula:
rate of ATP hydrolysis (μM/min) = rate of A340 decrease (s-1) × 9880 (μM).
We used GraphPad Prism 5 software to determine the kinetic parameters of the reactions.
3.3. The effect of dsDNA length on the ATPase activity of branch migration proteins
Previously, it was proposed by Peter von Hippel and co-workers that the ATPase activity of T4 gp41 helicase is proportional to DNA length when the rate of DNA binding is slow relative to the rate of translocation, and ATPase activity is coupled to translocation [22]. Based on this model the curve of ATPase activity versus DNA length is predicted to have three phases: (I) a sharp decrease in the Km(DNA) and concomitant increase in the ATPase Vmax when the size of DNA approaches the DNA binding site size of the protein; (II) an increase of ATPase activity proportional to DNA length when the protein translocates along DNA which length is below the limit of protein processivity; and (III) maximal ATPase velocity (Vmax) when DNA length reaches the processivity limit of the protein.
Measure the initial rate of ATP hydrolysis by the protein of interest as a function of dsDNA concentration for each dsDNA fragment from a set of linear DNA substrates with lengths varying from several base pairs (or nucleotides for ssDNA) to several kbp (or knt). In the range of short DNA fragments (less than 150 bs or nt) synthetic oligonucleotides can be used as substrates. Larger linear dsDNA (or ssDNA) fragments can be prepared from plasmid or phage DNA by cleavage with restriction endonucleases. After cleavage, the desired fragments can be purified by a 6% nondenaturing PAGE (if its size is less than 1 kb) or by 0.8% agarose gels and recovered as described [28].
From the obtained data, determine the Km (Km(DNA)) and Vmax for each dsDNA fragment. Then, plot Vmax of the ATPase as a function of DNA length. If the characteristic three-phase curve is observed, determine the average translocation distance for the protein of interest as the DNA length at half-plateau height.
By applying the model of Peter von Hippel and co-workers [22] described above we found that the apparent size of the RAD54 DNA binding site is between 63 and 90 bp; the increase in the velocity of RAD54 ATPase with the increase of dsDNA length observed for dsDNA fragments in a range from 63–90 bp to 1300 bp was consistent with translocation of RAD54 protein along the dsDNA; and the RAD54 processivity limit was found to be 280±30 bp under tested conditions [24].
3.4. The use of the spectrophotometric assay to study the DNA binding preferences of branch migration proteins
To examine the DNA substrate specificity of a branch migration protein, measure the initial rate of ATP hydrolysis in the presence of various branched oligonucleotide-derived DNA substrates (traditionally, these assays have been performed on substrates that were 61-63 bp long) (Fig. 2) over a range of DNA concentrations from 0.2 to 15 μM (nt). For instance, we demonstrated for RAD54 that branched DNA structures with one single-stranded arm (PX-junction, 3′-flap, 5′-flap) stimulate the maximal velocity (Vmax) of ATP hydrolysis 1.5–2.0-fold greater than branched DNA substrates with fully dsDNA arms (X-junction, replication fork) or with two ssDNA arms (Kappa and forked DNA) (25). The 3′-flap and 5′-flap structures containing two dsDNA arms and one ssDNA arm were nearly as efficient in stimulating the ATPase activity of hRad54 protein as the PX-junction, which was the best substrate among the previously examined structures. Overall, our results showed the following order of preference for DNA substrates according to their proficiency in inducing the RAD54 ATPase: PX-structure ≥3′-flap≥5′-flap≫Kappa≥X-structure=replication fork>forked DNA >dsDNA. These results were in agreement with those obtained in the band-shift assay [17].
4. Use of oligonucleotide substrates for analysis of branch migration activity
First, test for branch migration activity across a range of protein concentrations by adding increasing amounts of your protein to the annealed branched DNA substrate (e.g. branch migration activity of RAD54 can be detected in 5-1000 nM range) and incubating the mixture for a set period of time and temperature. For RAD54, the following buffer was used: 25 mM Tris acetate, pH 7.5, 2 mM ATP, 5 mM magnesium acetate, 2 mM DTT, 100 μg/ml bovine serum albumin (NEB, nuclease and protease free), 15 mM phosphocreatine and 10 units/ml creatine phosphokinase. However, higher RAD54 concentrations (> 100 nM) require higher concentrations of the ATP regeneration system (up to 20 mM phosphocreatine and 30 units/ml creatine phosphokinase). For each particular enzyme, concentrations of ATP and Mg2+ need to be optimized. The concentration of free divalent metal ions (in this case Mg2+) determines whether the branched DNA molecule will exist predominately in an open or stacked conformation. Since branch migration only occurs during the times when the DNA substrate exists in the open conformation, the equilibrium between these two states affects the rate of branch migration [27]. Once a suitable protein concentration is found, measure the time course of the branch migration reaction. As a control, always determine the extent of spontaneous (protein independent) branch migration in a control mixture by using storage buffer instead of your branch migration protein.
4.1 Visualizing branch migration products
Terminate the reaction by adding SDS (to 1.5%) and mix with a 1/10 volume of loading buffer (70% glycerol, 0.1% bromophenol blue). Analyze the sample by electrophoresis on a native 5-10% polyacrylamide gel in TBE (90 mM Tris borate, pH 8.3, and 1 mM EDTA). Run the gel at 11 V/cm for 1.5-2 h. Some proteins form very stable complexes with DNA, and therefore require treatment with SDS (1.5%) and proteinase K (800 μg/ml) for 1-15 min at room temperature before electrophoresis to disrupt. Alternatively, the reaction can be stopped by the addition of 1/10 volume of poly dT (1 mg/ml, GE Healthcare). For this method, incubate the mixture for 1-5 min at room temperature then perform deproteinization with SDS and proteinase K as described above. The concentration and acrylamide/bis-acrylamide ratio of the polyacrylamide gels may vary depending on the size and structure of branch migration substrates. (e.g. we use an 8% polyacrylamide gel with a 29:1 acrylamide/bis-acrylmaide ratio to separate branch migration products of our standard branched DNA molecules constructed from 94-mer oligonucleotides [17]). Some structures (e.g. X-junctions) become unstable during electrophoresis at room temperature and should be resolved at 4°C instead.
Dry the gel on DE81 chromatography paper (Whatman), visualize, and quantify the products of branch migration using a PhosphorImager system (GE Healthcare), or another appropriate device.
5. Branch migration on plasmid-based substrates
Oligonucleotides are invaluable tools for dissecting the properties of DNA helicase family proteins. They provide an efficient, flexible, rapid, and affordable method for generating a wide range of DNA substrates to analyze your protein with. But oligonucleotide-based DNA substrates also have their drawbacks. By their very nature, these substrates provide very little room for DNA translocation, which may bias the results based on the DNA binding properties of the protein. The DNA ends of oligonucleotides also undergo free rotation and lack any of the topological characteristics of long DNA molecules. Therefore, these substrates differ substantially from in vivo recombination intermediates. For these reasons, we also use plasmid-based substrates to study the branch migration activity of DNA helicase proteins.
Plasmid-based substrates serve as better chromosomal analogs, but they are also far more laborious to produce, requiring the purification of several DNA fragments followed by an enzymatic reaction to produce recombination intermediates that are suitable for branch migration.
To produce plasmid-based recombination intermediates (known as joint molecules or JM) DNA strand exchange reaction is carried out between gapped circular dsDNA and homologous linear DNA substrates. The joint molecules are deproteinized and used as a substrate for branch migration proteins (Fig. 6C).
Fig. 6.
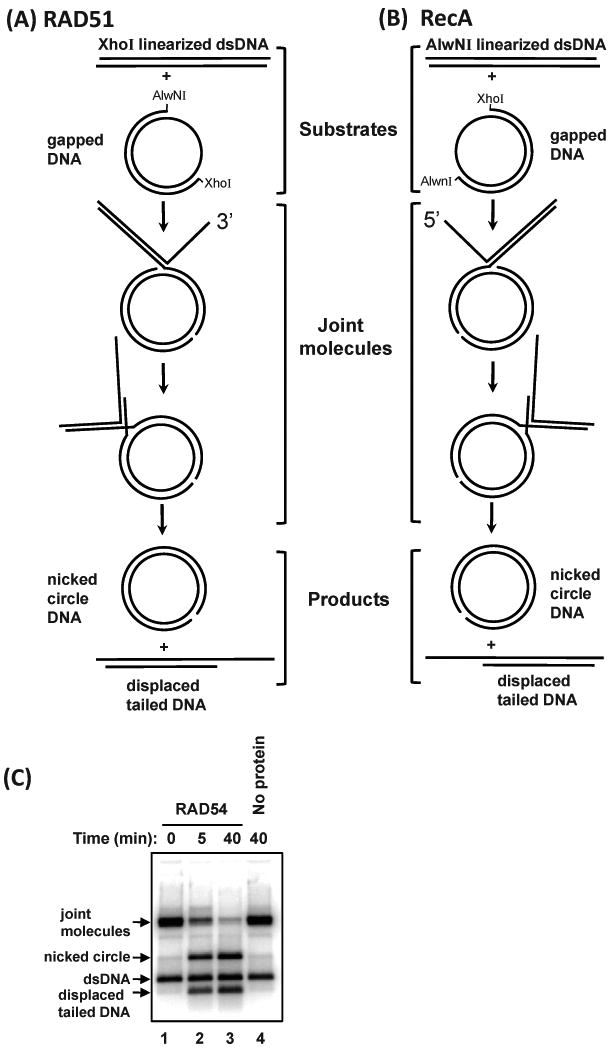
Production of joint molecules used to test the branch migration activity of proteins. (A) Joint molecules with a 3′ displaced ssDNA tail are produced by RAD51 using gapped DNA and pBS II K (+) dsDNA linearized with XhoI. (B) Joint molecules with a 5′ displaced ssDNA tail are produced by RecA using gapped DNA and pBS II K (+) dsDNA linearized with AlwNI. Joint molecules are deproteinized and used as substrates to test the branch migration activity of proteins of interest. (C) The branch migration of joint molecules with a 3′ displaced ssDNA tail (0.5 nM, molecules) by RAD54 (200 nM) at 30 °C. In the control reaction (Lane 4), RAD54 was replaced with storage buffer and carried out on for 40 min. Over the time course of the experiment, the amount of joint molecules decreases, while nicked circle and displaced tailed DNA increase.
5.1. Preparation of gapped DNA substrate
The preparation of gapped DNA takes several days to complete. First plasmid DNA is digested with two restriction enzymes to produce a linear dsDNA that is shorter than the original plasmid DNA by several hundred nucleotides (the size of the future gap). The dsDNA fragment of interest is isolated via agarose gel electrophoresis, excised from the gel, and then electroeluted. Next, the purified dsDNA fragment is annealed to circular ssDNA to create gapped DNA. Finally, the gapped DNA is purified via agarose gel electrophoresis, followed by gel excision and electroelution (Fig. 3).
Fig. 3.

The scheme used to produce gapped DNA. i) pBS II K (+) plasmid DNA is digested with XhoI and AlwNI. ii) The large DNA fragment from this digest is purified by gel electrophoresis in agarose gels, and then iii) annealed to circular ssDNA to generate gapped DNA. iv) Finally, the gapped DNA is purified by electrophoresis in agarose gels.
5.1.1. Preparation of linear dsDNA fragment
To prepare the linear dsDNA fragment, perform a digest of pBS II K (+) plasmid DNA first with the restriction endonuclease XhoI, and then with AlwNI (New England Biolabs). This digest generates two DNA fragments of 2065 bp and 896 bp (Fig. 3i). Although, both restriction endonucleases have approximately 100% activity in 1× NEBuffer 2, we do not perform double digest, because in agarose gels the electrophoretic mobility of pBS II K (+) supercoiled plasmid DNA is very close to the mobility of 2065 bp dsDNA fragment that makes it difficult to judge completeness of the digestion. As a starting material, we use 60 μg of pBS II K (+) plasmid DNA per an agarose gel with the dimensions: 10 × 15 × 0.5 cm. For larger scale dsDNA fragment preparation use larger agarose gels or multiple gels; do not overload the gel. Digest 60 μg of pBS II K (+) with 450 units of XhoI in 1× NEBuffer 2 with100 μg/ml BSA, in a total volume of 0.6 ml. After 1 hr, test completeness of XhoI digestion by analytical electrophoresis, e.g., in a 6 cm-long 0.8 % agarose gel. When XhoI-digest is complete add 450 units AlwNI and continue digestion at 37 °C. The reaction is performed with a 3-fold excess of both restriction endonucleases to ensure completeness of digestion. Incubate the reaction mixture for 2 h at 37°C, and then test completeness of digestion by analytical electrophoresis in a 6 cm-long 0.8 % agarose gel. To rectify incomplete digestion, the incubation times and/or concentration of restriction endonucleases may be increased. Otherwise stop the digest by heating the reaction mixture for 20 min at 75°C. Then add NaCl to 300 mM and precipitate the DNA products by adding 2.5 volumes of ethanol and incubation at -20°C for at least 1 h (or overnight). Centrifuge at >16,000 g for 30 min, dry the pellet, resuspend it in 300 μl of TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0) and proceed to purification of the 2065 bp DNA fragment. Add 45 μl of loading buffer containing 50% glyercol, 0.25% bromophenol blue, and 1 mM EDTA and load the samples on a 1.1 % agarose gel with the dimensions: 10 × 15 × 0.5 cm (total volume 76 ml). A standard 20-well comb was modified with tape to create a 5-well comb with 2 preparative wells (∼50 mm wide) and 3 analytical wells (5 mm wide) as shown in Figure 4. Volumes of 10 μl were loaded in each of analytical wells (Lanes A, C, and E), with the remaining 315 μl being split between two preparative wells (Lanes B and D). The gel is run in TAE buffer (40 mM Tris acetate, pH 8.3, and 1 mM EDTA) at 1.7 V/cm for 13-14 h.
Fig. 4.

Illustration of the 0.8% agarose gel used to purify the large (2065 bp) dsDNA fragment of pBS II K (+) following digestion with XhoI and AlwNI. After electrophoresis, lanes A, C, and E are excised from the gel and stained with ethidium bromide (dashed black lines). These lanes contain two bands (white bands). Using these bands as markers, the area that contains the 2065 bp dsDNA fragment is cut out of lanes B and D (dashed red boxes), and then purified by electroelution.
Following electrophoresis, excise Lanes A, C, and E from the gel and stain them with ethidium bromide. Reassemble the gel and visualize it under a UV-lamp, revealing two bands. Using Lanes A, C, and E as markers, excise the sections of agarose containing the upper band (the 2065 fragment of DNA) from Lanes B and D (Red boxes in Fig. 4). The DNA is extracted from the gel by electroellution; we use a Schleicher & Schuell Elutrap. Place two agarose gel slices into an Elutrap following the manufacturer's instructions. Carry out electroelution at 150 mA for 4 hr in TAE buffer (see above). To increase the amount of extracted DNA we carry out second elution at 150 V for 4 hr or at 75 V overnight.
Note: During the two gel excision steps, be sure to minimize the width of the gel slices, as excess agarose will decrease the yield of DNA recovered. After removing the bands, you can stain the remainder of the gel with ethidium bromide to determine how successful the band excision was.
Concentrate DNA recovered from the first and the second eluate (500-930 μl) to ∼200 μl and 100 μl, respectively, by extraction with 1-butanol (after extraction discard the upper phase). Precipitate the DNA from each eluate by adding 1/10 volume of 3 M sodium acetate, pH 7.0 and 2.5 volumes of ethanol and incubation for at least 1 h (or overnight) at -20°C. Centrifuge at >16,000 g for 30 min, dry the pellets and resuspend each in 20 μl of TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0). Before proceed further, ensure that DNA electroelution was successful by determining the DNA concentration recovered from first eluate (usually 1:30 – 1:60 is required). Butanol extraction of DNA eluates increases salt concentration so that salt may precipitate during ethanol precipitation. To desalt the DNA solution combine the DNA from recovered from both eluates (∼40 μl) and pass the DNA solution through a Micro-BioSpin 6 column (Bio-Rad). Determine the DNA concentration by measuring the absorbance at 260 nm on a spectrophotometer using 50 μl cuvettes (Agilent). Good yields are in the range of 20-27 μg of dsDNA (∼50% of theoretical yield, Fig. 3ii).
5.1.3. Preparation of gapped DNA by annealing of circular ssDNA with linear dsDNA
Circular ssDNA of pBS II K (+) was prepared as previously described in the Sambrook and Russell Molecular Cloning Laboratory Manual “Producing Single-stranded DNA with Phagemid Vectors” protocol [28]. Perform a trial small-scale annealing reaction (22 μl) to determine the optimum ratio between ssDNA circular and dsDNA fragment for generating gapped DNA. Typically, 1 μg of linear duplex fragment is mixed with 1, 1.5, and 2 μg of circular pBS II K (+) ssDNA in annealing buffer, containing 25 mM Tris-HCl, pH 7.5, 50 mM NaCl, and 50% formamide. The tube is incubated for 10 min at 75°C to denature the large fragment of dsDNA, and then for at least 3 h at room temperature to allow annealing to occur (reaction can be left overnight at room temperature). Analyze the DNA before and after annealing on a 1.4% agarose gel. Optimum ratio can be different with different preparation of circular ssDNA.
Based on this result, prepare a large-scale annealing reaction. Usually, we mix 30 μg of the linear dsDNA fragment and 30 μg of circular ssDNA (∼ 1.4 fold excess of circular pBS II K (+) ssDNA in terms of DNA molecules) and anneal as described above in 660 μl of a total volume (Fig. 3iii). Precipitate DNA from the annealing mixture by adding 1/10 volume of 3 M sodium acetate, pH 7.0 and 2.5 volumes of ethanol. Centrifuge at >16,000 g for 30 min, dry the pellet and resuspend in 300 μl TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0). Add 45 μl of loading buffer containing 50% glycerol, 0.25% bromophenol blue, and 1 mM EDTA, and then load the sample on a 5-well agarose gel (1.4%). (The gel is made with a higher percentage of agarose to provide better separation of DNA products). Run the gel at 2 V/cm for 14 h. Following electrophoresis, cut off Lanes A, C, and E and stain with ethidium bromide (Fig. 5). Reassemble the gel and visualize it under a UV-lamp, to reveal four major bands. Using Lanes A, C, and E as markers, excise the section of agarose containing the upper-most band (containing the gapped DNA) from Lanes B and D. (Red boxes in Fig. 5) You must take care to be more precise when cutting out this band, otherwise you will pick up contaminates that are similar in size to the gapped DNA. Note: You may see other minor bands in the annealing reaction.
Fig. 5.
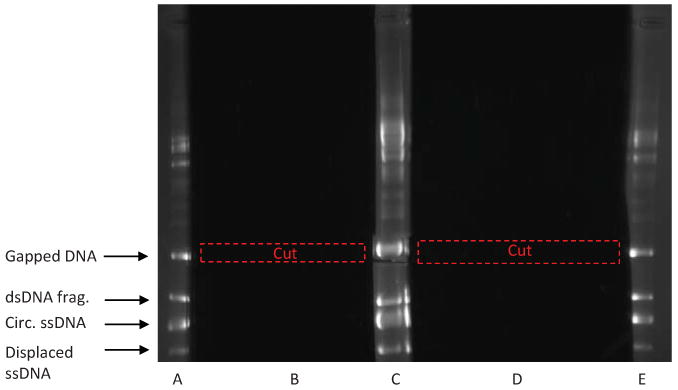
Purification of gapped DNA by electrophoresis in a 1.4% agarose gel. Following electrophoresis, lanes A, C, and E are cut out and stained with ethidium bromide. These lanes should contain four major bands: gapped DNA, circular ssDNA, large dsDNA fragment, and displaced ssDNA. Other higher order bands correspond to VCS-M13 helper phage DNA (∼ 6 kb) present in the circular ssDNA preparation and also to oligomeric forms of plasmid ssDNA. Using these bands as markers, the area that contains the gapped DNA is cut out of lanes B and D (dashed red boxes), and then purified by electroelution.
Electroelute the DNA, as described above for 2065 bp fragment, concentrate with 1-butanol to 100-150 μl, add 1/10 volume of 3 M sodium acetate, pH 7.0, precipitate with 2.5 volumes of ethanol by incubating for at least 1 hr at -20 °C, centrifuge at >16,000 g for 30 min, dry the pellet, and resuspend the DNA pellet in a volume of ∼20 μl of 10 mM Tris-HCl, pH 7.5 in the same manner as described in the previous section. Finally, purify the resuspended gapped DNA using a Bio-Rad Micro Bio-Spin 6 Chromatography Column (Catalog # 732-6221) to remove any large particles and salt. Determine the final concentration of gapped DNA by measuring the optical density (OD) at 260 nm; 1 OD260 Unit corresponds to gapped DNA concentration of 45 μg/ml. Good yields are in the range of 15 – 19 μg of gapped DNA (∼10% of theoretical yield for the entire protocol, Fig. 3iv).
5.2. Labeling of linear dsDNA
When constructing plasmid-based recombination intermediates it is important to consider the desired polarity of the substrates. If the polarity of the branch migration protein in question is unknown, then it may useful to construct substrates suitable for branch migration with the 5′ to 3′ and the 3′ to 5′ polarity in order to test both. Described below are protocols for constructing plasmid-based substrates with either polarity.
5.2.1. 32P-labeling of XhoI-linearized pBSK(+) dsDNA at the 5′-end for Rad51 promoted-joint molecule formation
The ssDNA portion of gapped DNA is defined by the AlwNI and XhoI restriction sites of pBS II K (+). From the vector map, you can determine that the ssDNA of the gap runs in a 5′ to 3′ direction from the AlwNI site to the XhoI site. To produce joint molecules with a 3′ displaced ssDNA, pBS II K (+) plasmid DNA must be digested with XhoI (Fig. 6A). And since XhoI cleavage creates a DNA end with a 5′ overhang, it can readily be radiolabeled by T4 polynucleotide kinase.
Begin by digesting 10 μg of pBS II K (+) plasmid DNA with 40 units of XhoI (NEB) in the presence of 2 units of Shrimp Alkaline Phosphatase (USB) for 1 h at 37°C in a 100 μl volume. The phosphatase removes the 5′ terminal phosphate group following digestion, releasing the end to be labeled in the next step. Test completeness of digestion by electrophoresis on a 0.8% agarose gel. If digestion is full, inactivate both enzymes for 20 min at 75°C. Precipitate the DNA with ethanol and resuspend the pellet in 20 μl of 10 mM Tris-HCl, pH 8.0 to give a DNA concentration of ∼0.5 μg/μl.
Radiolabel 5 μg of XhoI–linearized pBS II K (+) plasmid DNA in a 40 μl reaction volume containing 100 μCi of [γ-32P]ATP (10 mCi/ml; Perkin Elmer), 1 × T4 polynucleotide kinase buffer and 2 units of T4 polynucleotide kinase (NEB). Incubate the reaction for 1 h at 37°C, and then inactivate the enzyme for 20 min at 75°C. Purify the DNA through a Micro-BioSpin 6 column (Bio-Rad) and determine the DNA concentration again.
5.2.2. 32P-labeling of AlwNI-linearized pBSK(+) dsDNA at the 3′-end for RecA promoted joint molecule formation
Joint molecules can also be produced with a 5′-end displaced ssDNA strand if pBS II K (+) is digested with AlwNI (Fig. 6B). AlwNI digestion produces a DNA end with a 3′ overhang, thus this linear DNA should be 3′ radiolabeled with terminal deoxynucleotidyl transferase (TdT).
Digest 10 μg of pBS II K (+) plasmid DNA with 40 units of AlwNI (NEB) in a 100 μl volume. Incubate the reaction for 1 h at 37°C, and then test digestion on a 0.8% agarose gel. If digestion is incomplete, increase the time of incubation or concentration of the enzyme, otherwise inactivate restriction endonuclease for 20 min at 75°C. Precipitate the cleavage DNA product with ethanol and resuspend the pellet in 20 μl of Tris-HCl, pH 8.0 to give a DNA concentration ∼0.5 μg/μl.
Radiolabel 5 μg of AlwNI –linearized pBS II K (+) plasmid DNA in a 40 μl reaction volume containing 16 μCi of [α-32P]dCTP (10 mCi/ml; Perkin Elmer), 1× TdT buffer supplemented with 0.25 mM CoCl2 and 10 units of TdT (NEB), as specified by the manufacturer. Incubate the reaction for 15 min at 37°C, and then inactivate the enzyme for 10 min at 75°C. Purify the DNA through a Micro-BioSpin 6 column (Bio-Rad) and determine the DNA concentration again. The number of incorporated nucleotide residues depends on the ratio of 3′ DNA ends: dNTP and also on the nature of dNTP. For [α-32P]dCTP we used the DNA ends: dNTP ratio 100: 1. Under these conditions dCTP supports smaller number of incorporated nucleotide residues on a single DNA molecule (1-3 nts) than dATP or dTTP (1-5 nts), and therefore is preferable. [α-32P]ddNTP that allows only a single nucleotide incorporation is even better, though is more expensive.
5.3. Joint molecule formation
5.3.1. RAD51-promoted formation of joint molecules with a 3′-ssDNA displaced strand
RAD51 polymerizes on ssDNA in a 3′ to 5′ direction and will promote strand exchange in the same direction between gapped DNA and linear dsDNA. To prepare joint molecules with a 3′-displaced ssDNA strand, mix 20 μM (nt) of gapped DNA with 5 μM of RAD51 in a reaction buffer containing 25 mM Tris acetate, pH 7.5, 275 mM NaCl, 2 mM ATP, 1 mM MgCl2, 1 mM CaCl2, 1 mM DTT, 100 μg/ml acetylated bovine serum albumin (NEB, nuclease and protease free) in an 90 μl volume. Incubate for 5 min at 37°C, then add 0.4 μM of Replication Protein A (RPA), incubate for another 1 min, and finally add 20 μM (nt) of 32P-labeled XhoI–linearized dsDNA to initiate joint molecule formation (Fig. 6A). Incubate the mixture for 2 h at 37°C. Take a 10 μl aliquot and estimate the extent of joint molecule formation on a 1.5 % agarose gel. If the yield is in the range of 50-60%, the joint molecules can be purified (see section 5.3.3) and used for testing branch migration activities. If the yield is lower, increase time of joint molecule formation up to 22 h.
5.3.2. RecA-generated formation of joint molecules with a 5′-ssDNA displaced strand
RecA has a 5′ to 3′ polarity of polymerization on DNA (opposite of RAD51), and will promote strand exchange in a 5′ to 3′ direction. To prepare joint molecules with a 5′-ssDNA displaced strand, mix 20 μM (nt) of gapped DNA with 4 μM of RecA in a reaction buffer containing 35 mM Tris-HCl, pH 7.5, 3 mM ATP, 15 mM MgCl2, 2 mM DTT, 100 μg/ml bovine serum albumin (NEB, nuclease and protease free), 10 mM phosphocreatine and 10 units/ml creatine phosphokinase) in a 90 μl volume. Incubate for 5 min at 37°C, then add 0.33 μM of SSB, incubate for another 1 min, and finally add 20 μM (nt) of 32P-labeled AlwNI –linearized dsDNA to initiate joint molecule formation (Fig. 6B). Incubate at 37°C for 10 min. Take 10 μl aliquot and estimate the extent of joint molecule formation on a 1.5 % agarose gel. If the yield is in the range of 50-60%, the joint molecules can be purified (see section 5.3.3) and used for testing branch migration activities.
Note: In the presence of calcium ion, RAD51 forms a stable filament that will produce a relatively homogeneous population of joint molecules through its DNA strand exchange activity. On the other hand, RecA forms a far more dynamic filament that will promote both strand exchange and branch migration, resulting in a more heterogeneous population of joint molecules. The 10 min reaction time for the RecA-promoted joint molecule formation reaction is a compromise. This time point provides for the largest accumulation of joint molecules before branch migration products become visible. To produce a more homogenous population with RecA, 3 mM ATP can be replaced with 1 mM ATPγS. In the presence of ATPγS, the ATP regeneration system of phosphocreatine and creatine phosphokinase is removed, RecA concentration is increased to 12 μM, MgCl2 concentration is decreased to 4 mM and the reaction time is increased from 10 min to 24 h. Besides taking over 100 times longer, RecA-promoted joint molecule formation in the presence of ATPγS is half as efficient as in the presence of ATP (30 and 60%, respectively).
5.3.3. Deproteinization and purification of joint molecules
Following joint molecule formation (regardless of whether they were produced by RAD51 or RecA), stop the reaction and deproteinize DNA by the addition of SDS to 0.8%, EDTA to 2 mM, and proteinase K to 1.6 mg/ml. Incubate for 15 min at 37°C. Pass deproteinized joint molecules twice through S-400 Spin column (GE Healthcare) equilibrated with 30 mM Tris-HCl, pH 7.5. Add 2-10 mM of MgCl2 to inhibit spontaneous branch migration. Store purified joint molecules at -20 °C. The concentration of the purified joint molecules can be calculated by comparing their radioactivity to a known amount of labeled dsDNA after electrophoresis on the same gel.
5.4. Testing for branch migration activity on plasmid-based substrates
Using purified joint molecules as a DNA substrate, test the branch migration activity of the protein of interest. For RAD54, we used 0.5 nM joint molecules in buffer containing 25 mM Tris acetate, pH 7.5, 2 mM ATP, 3 mM magnesium acetate, 2 mM DTT, 100 μg/ml bovine serum albumin (NEB, nuclease and protease free), 20 mM phosphocreatine and 30 units/ml creatine phosphokinase. Add increasing amounts of the branch migration protein (e.g., branch migration activity of RAD54 can be detected in 5-1000 nM range) and incubate the reaction mixture for a set period of time and temperature. Deproteinize the DNA products by treatment with 1.6 mg/ml proteinase K, 0.8 % SDS, 6 % glycerol, 0.01 % bromphenol blue for 15 min at 37 °C. Analyze the DNA products by electrophoresis on a 1.5% agarose gel in TAE (40 mM Tris acetate, pH 8.3, and 1 mM EDTA). Run the gel at 5 V/cm for 3 h. Dry the gel on DE81 chromatography paper, visualize, and quantify the products of branch migration using a PhosphorImager system (GE Healthcare), or another appropriate device. If branch migration has occurred, the gel will reveal a time-dependent disappearance of joint molecules and a concurrent appearance of nicked circle and displaced tailed DNA (Fig.6C). Once a suitable protein concentration is found, measure the time course of the branch migration reaction.
As a control, always determine the extent of spontaneous (protein independent) branch migration in a control mixture by using storage buffer instead of your branch migration protein.
6. Concluding Remarks
Helicases and DNA/RNA translocases are a ubiquitous, highly diverse group of proteins that perform an extraordinary variety of functions in cells [16, 30]. Recently, it was discovered that several members of different helicase families promote branch migration of Holliday junctions. These proteins that function during DNA replication, recombination, and repair include (1) RecG, a member of the RecG family; (2) several members of the RecQ helicase family; (3) FANCM, a member of the DEAH helicase family; (4) Rad54, a member of the Snf2 family of DNA translocases; (5) RuvB, a member of the ATPases associated with diverse cellular activities (AAA+) family [1, 25, 31-35].
The wide range of experimental approaches presented in this paper have been used to investigate the branch migration activities of different proteins. These approaches provide a basis for initial investigation of proteins suspected to possess branch migration activity. Furthermore, the DNA substrates described in this paper can also be applied in a wide array of biochemical experiments (i.e. x-ray crystallography, footprinting assays, protection assays, and crosslinking) to determine the molecular mechanisms of branch migration proteins and the roles these proteins play in cellular processes.
Acknowledgments
This work was supported by the NIH Grant CA100839, MH084119, and the Leukemia and Lymphoma Society Scholar Award 1054-09 (to AVM) and NIH Grant F31 AG033484-01 (to MJR).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Liu Y, West SC. Nat Rev Mol Cell Biol. 2004;5:937–944. doi: 10.1038/nrm1502. [DOI] [PubMed] [Google Scholar]
- 2.Holliday R. Genet Res. 1964;5:282–304. [Google Scholar]
- 3.Pâques F, Haber JE. Microbiology and Molecular Biology Reviews. 1999;63:349–404. doi: 10.1128/mmbr.63.2.349-404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bugreev DV, Hanaoka F, Mazin AV. Nat Struct Mol Biol. 2007;14:746–753. doi: 10.1038/nsmb1268. [DOI] [PubMed] [Google Scholar]
- 5.McGlynn P, Lloyd RG, Marians KJ. Proc Natl Acad Sci U S A. 2001;98:8235–8240. doi: 10.1073/pnas.121007798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Postow L, Ullsperger C, Keller RW, Bustamante C, Vologodskii AV, Cozzarelli NR. J Biol Chem. 2001;276:2790–2796. doi: 10.1074/jbc.M006736200. [DOI] [PubMed] [Google Scholar]
- 7.Michel B, Grompone G, Flores MJ, Bidnenko V. Proc Natl Acad Sci U S A. 2004;101:12783–12788. doi: 10.1073/pnas.0401586101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grompone G, Seigneur M, Ehrlich SD, Michel B. Mol Microbiol. 2002;44:1331–1339. doi: 10.1046/j.1365-2958.2002.02962.x. [DOI] [PubMed] [Google Scholar]
- 9.Rothstein R, Michel B, Gangloff S. Genes Dev. 2000;14:1–10. [PubMed] [Google Scholar]
- 10.Seigneur M, Bidnenko V, Ehrlich SD, Michel B. Cell. 1998;95:419–430. doi: 10.1016/s0092-8674(00)81772-9. [DOI] [PubMed] [Google Scholar]
- 11.Lanzov V, Stepanova I, Vinogradskaja G. Biochimie. 1991;73:305–312. doi: 10.1016/0300-9084(91)90217-o. [DOI] [PubMed] [Google Scholar]
- 12.West SC. Annual Review of Genetics. 1997;31:213–244. doi: 10.1146/annurev.genet.31.1.213. [DOI] [PubMed] [Google Scholar]
- 13.Sharples GJ, Ingleston SM, Lloyd RG. J Bacteriol. 1999;181:5543–5550. doi: 10.1128/jb.181.18.5543-5550.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seitz EM, Haseltine CA, Kowalczykowski SC. Adv Appl Microbiol. 2001;50:101–169. doi: 10.1016/s0065-2164(01)50005-2. [DOI] [PubMed] [Google Scholar]
- 15.Karow JK, Wu L, Hickson ID. Curr Opin Genet Dev. 2000;10:32–38. doi: 10.1016/s0959-437x(99)00039-8. [DOI] [PubMed] [Google Scholar]
- 16.Singleton MR, Dillingham MS, Wigley DB. Annu Rev Biochem. 2007;76:23–50. doi: 10.1146/annurev.biochem.76.052305.115300. [DOI] [PubMed] [Google Scholar]
- 17.Bugreev DV, Mazina OM, Mazin AV. Nature. 2006;442:590–593. doi: 10.1038/nature04889. [DOI] [PubMed] [Google Scholar]
- 18.Swagemakers SM, Essers J, de Wit J, Hoeijmakers JH, Kanaar R. J Biol Chem. 1998;273:28292–28297. doi: 10.1074/jbc.273.43.28292. [DOI] [PubMed] [Google Scholar]
- 19.Petukhova G, Stratton S, Sung P. Nature. 1998;393:91–94. doi: 10.1038/30037. [DOI] [PubMed] [Google Scholar]
- 20.Panyutin IG, Hsieh P. J Mol Biol. 1993;230:413–424. doi: 10.1006/jmbi.1993.1159. [DOI] [PubMed] [Google Scholar]
- 21.Panyutin IG, Hsieh P. Proc Natl Acad Sci U S A. 1994;91:2021–2025. doi: 10.1073/pnas.91.6.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Young MC, Kuhl SB, von Hippel PH. J Mol Biol. 1994;235:1436–1446. doi: 10.1006/jmbi.1994.1099. [DOI] [PubMed] [Google Scholar]
- 23.Saha A, Wittmeyer J, Cairns BR. Genes Dev. 2002;16:2120–2134. doi: 10.1101/gad.995002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mazina OM, Mazin AV. J Biol Chem. 2004;279:52042–52051. doi: 10.1074/jbc.M410244200. [DOI] [PubMed] [Google Scholar]
- 25.Karow JK, Constantinou A, Li JL, West SC, Hickson ID. Proc Natl Acad Sci U S A. 2000;97:6504–6508. doi: 10.1073/pnas.100448097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kowalczykowski SC, Clow J, Somani R, Varghese A. J Mol Biol. 1987;193:81–95. doi: 10.1016/0022-2836(87)90629-2. [DOI] [PubMed] [Google Scholar]
- 27.Mazina OM, Rossi MJ, Thomaa NH, Mazin AV. J Biol Chem. 2007;282:21068–21080. doi: 10.1074/jbc.M701992200. [DOI] [PubMed] [Google Scholar]
- 28.Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. Third. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 2001. [Google Scholar]
- 29.Gaillard C, Strauss F. Nucleic Acids Res. 1990;18:378. doi: 10.1093/nar/18.2.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brosh RM, Jr, Bohr VA. Nucleic Acids Res. 2007;35:7527–7544. doi: 10.1093/nar/gkm1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Constantinou A, Tarsounas M, Karow JK, Brosh RM, Bohr VA, Hickson ID, West SC. EMBO Rep. 2000;1:80–84. doi: 10.1093/embo-reports/kvd004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kanagaraj R, Saydam N, Garcia PL, Zheng L, Janscak P. Nucleic Acids Res. 2006;34:5217–5231. doi: 10.1093/nar/gkl677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bugreev DV, Brosh RM, Jr, Mazin AV. J Biol Chem. 2008;283:20231–20242. doi: 10.1074/jbc.M801582200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu L, Bachrati CZ, Ou J, Xu C, Yin J, Chang M, Wang W, Li L, Brown GW, Hickson ID. Proc Natl Acad Sci U S A. 2006;103:4068–4073. doi: 10.1073/pnas.0508295103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lloyd RG, Sharples GJ. EMBO J. 1993;12:17–22. doi: 10.1002/j.1460-2075.1993.tb05627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]