

Abstract
To overcome the limited availability of antibiotic resistance markers in filamentous fungi, we adapted the FLP/FRT recombination system from the yeast Saccharomyces cerevisiae for marker recycling. We tested this system in the penicillin producer Penicillium chrysogenum using different experimental approaches. In a two-step application, we first integrated ectopically a nourseothricin resistance cassette flanked by the FRT sequences in direct repeat orientation (FRT-nat1 cassette) into a P. chrysogenum recipient. In the second step, the gene for the native yeast FLP recombinase, and in parallel, a codon-optimized P. chrysogenum flp (Pcflp) recombinase gene, were transferred into the P. chrysogenum strain carrying the FRT-nat1 cassette. The corresponding transformants were analyzed by PCR, growth tests, and sequencing to verify successful recombination events. Our analysis of several single- and multicopy transformants showed that only when the codon-optimized recombinase was present could a fully functional recombination system be generated in P. chrysogenum. As a proof of application of this system, we constructed a ΔPcku70 knockout strain devoid of any heterologous genes. To further improve the FLP/FRT system, we produced a flipper cassette carrying the FRT sites as well as the Pcflp gene together with a resistance marker. This cassette allows the controlled expression of the recombinase gene for one-step marker excision. Moreover, the applicability of the optimized FLP/FRT recombination system in other fungi was further demonstrated by marker recycling in the ascomycete Sordaria macrospora. Here, we discuss the application of the optimized FLP/FRT recombination system as a molecular tool for the genetic manipulation of filamentous fungi.
Site-specific recombination is an important molecular tool for functional genetic studies in both prokaryotes and eukaryotes and is mediated by two major recombinase families, the resolvase/invertase family and the integrase family (9, 65). The resolvase/invertase family is characterized by the conserved catalytic amino acid serine, which allows intramolecular reactions, whereas the integrase family is able to mediate both intra- and intermolecular recombinations due to an autocatalytic activity of the conserved residue tyrosine (19, 35). The best-studied members of the integrase family are the λ integrase of bacteriophage λ, the recombinase Cre of Escherichia coli bacteriophage P1 (1, 62), the XerCD proteins of E. coli (6), and the eukaryotic FLP recombinase of the yeast Saccharomyces cerevisiae (9, 51). Common to all these systems are two different or identical recognition sites that serve as the DNA substrate for the recombinase.
In recent years, recombinases have become important tools to manipulate genetically prokaryotes as well as eukaryotes. In this study, we have modified the FLP/FRT recombination system from yeast for application in filamentous fungi. The FLP/FRT recombination system is encoded by the 2μm (6.4 kb) plasmid that is present in most isolates of S. cerevisiae. Due to its stable partitioning and amplification system, this plasmid is available with a copy number of 60 to 100 copies per cell (16, 63). If the copy number drops below a certain level, the 45-kDa FLP recombinase catalyzes recombination between two 599-bp sites. These sequences are present in inverted orientation and allow the 2μm plasmid to exist in two conformations, namely, the A conformation when it is silent and the B conformation when it can replicate itself and thus maintain a constant plasmid copy number per cell (64). Analysis of the 599-bp inverted repeat sequence revealed that only 34 bp, the FLP recognition targets (FRT), are necessary for successful recombination (54). For every FLP-mediated recombination, a total of four FLP recombinases and two FRT sequences are needed. Two of the four proteins bind to one FRT sequence because every FRT has two 13-bp FLP-binding sites which are interrupted by an 8-bp spacer region. In this spacer DNA strand breakage takes place, producing 8-bp overhanging ends. After strand breakage, the overhanging ends of the two FRT fragments come together by complementary base pairing so that a recombinant FRT sequence is generated (2, 51).
The FLP/FRT recombination system has been successfully applied in model organisms such as E. coli (10, 55) as well as in pathogenic and nonpathogenic Gram-negative and Gram-positive bacteria (53). In addition to prokaryotes, the FLP/FRT recombination system has been adapted for use in several eukaryotes (26). For example, it is well established in diverse yeasts (11, 41, 57, 60) and has been used successfully to modify the genomes of higher eukaryotes, including animal and human cell lines (5, 37, 58). Despite this tool's application in higher eukaryotes, it has not yet been used in filamentous fungi. We present the first application of the FLP/FRT recombination system in a filamentous fungus, the major penicillin producer Penicillium chrysogenum. A limiting factor to manipulate P. chrysogenum genetically is the small number of suitable resistance markers. To overcome this problem, we generated a codon-optimized FLP/FRT recombination system to establish an efficient marker recycling system in this fungus. Using the optimized FLP/FRT recombination system, we demonstrate in a two-step approach the functionality of this system by construction of a marker-free P. chrysogenum strain lacking the ku70 (Pcku70) gene for optimized homologous recombination. A further advancement was the construction of a novel inducible nat1 flipper cassette allowing marker recycling in a one-step approach. The broader applicability of this recombination system was finally shown by its use in the model ascomycete Sordaria macrospora (34).
MATERIALS AND METHODS
Strains, culture conditions, and transformation procedure.
E. coli strain K-12 XL1-blue (Stratagene) was used for plasmid construction and maintenance (7). All P. chrysogenum and S. macrospora strains used in this study are listed in Table 1. P. chrysogenum P2niaD18 and the ΔPcku70 strain lacking the Pcku70 gene (22) as well as the S. macrospora wild-type strain (34) served as recipients for the integration of different FRT-flanked resistance cassettes.
TABLE 1.
Fungal strains used in this study
| Organism | Strain | Genotype | Reference |
|---|---|---|---|
| P. chrysogenum | P2niaDi8 | Penicillin producer; niaD− | 23 |
| ΔPcku70 | ΔPcku70::trpC(p)::nat1 | 22 | |
| PcFRT1 | FRT::trpC(p)::nat1::FRT | This study | |
| PcFRT2Y | FRT::trpC(p)::nat1::FRT trpC(p)::flp ptrA(p)::ptrA | This study | |
| PcFRT2P | FRT trpC(p)::Pcflp ptrA(p)::ptrA | This study | |
| ΔPcku70FRT1 | ΔPcku70::FRT::trpC(p)::ble::FRT | This study | |
| ΔPcku70FRT2 | trpC(p)::Pcflp ptrA(p)::ptrA ΔPcku70::FRT | This study | |
| PcFLIP1 | FRT::xyl(p)::Pcflp::trpC(p)::nat1::FRT | This study | |
| PcFLIP2 | FRT | This study | |
| S. macrospora | SmFRT1 | FRT::trpC(p)::nat1::FRT trpC(p)::Pcflp trpC(p)::hph | This study |
| SmFRT2P | FRT trpC(p)::Pcflp trpC(p)::hph | This study |
All P. chrysogenum strains were grown at 27°C and 120 rpm for 3 days in liquid complex culture medium (CCM) or minimal medium (MM) or grown on solid CCM or MM (21, 39). To induce the P. chrysogenum flp (Pcflp)-mediated site-specific recombination, appropriate strains were grown on solid MM supplemented with 2% xylose (25). Liquid or solid Burkholder's minimal medium (BMM) was used for the cultivation of the S. macrospora strains (45).
Transformation of individual P. chrysogenum strains was performed as described by Janus et al. (25) and Hoff et al. (22). Transformation of S. macrospora was preformed according to Nowrousian et al. (45). Transformants were selected by growth on medium containing 150 μg ml−1 nourseothricin, 40 μg ml−1 phleomycin, or 7 μg ml−1 pyrithiamine.
Growth tests.
Recombination events in transgenic strains were detected by growth tests on MM and CCM containing 200 μg ml−1 nourseothricin, 40 μg ml−1 phleomycin, or 7 μg ml−1 pyrithiamine. Strains were grown for 5 days at 27°C. All transformants and the wild-type strains were grown on CCM without selection, as positive controls.
Construction of recombinant plasmids.
For construction of the FRT-nat1 cassette, the FRT sequences of the native FLP/FRT recombination system were used to flank the nourseothricin resistance gene (nat1) of Streptomyces noursei at the 5′ and 3′ ends. For this purpose the complementary oligonucleotides FRT1 and FRT2 were annealed to obtain the 5′ FRT sequence, and the primers FRT3 and FRT4 were used to generate the 3′ FRT sequence. The 5′ FRT was first inserted between the PstI and BamHI restriction sites of plasmid pD-NAT1 (32), generating plasmid pDNAT1-FRT1. This plasmid was then hydrolyzed with SacI and NotI and used for ligation with the 3′ FRT. The resulting plasmid carries the full FRT-nat1 cassette and was named pDNAT1-FRT1-2.
In the case of plasmid pKOKU70_Phleo, a 5′ ku70-FRT fragment was amplified with primers 5′ku70FRT_s and 5′ku70FRT_a, and PCR with pKOKU70 as a template was conducted as published by Hoff et al. (22). The resulting 1.1-kb amplicon was ligated into plasmid pDrive (Qiagen, Hilden, Germany), and DNA sequencing was done by custom-provided services (MWG-Biotech, Germany). Plasmid pD5ku70FRT was hydrolyzed with KpnI-BamHI and the 5′ ku70-FRT fragment was ligated into plasmid pD-Phleo (22) using the corresponding restriction sites. The resulting recombinant plasmid was named p5ku70FRT_Phleo. In the next step, oligonucleotides 3′ku70FRT_s and 3′ku70FRT_a were annealed, and the resulting 3′ FRT site was ligated into p5ku70FRT_Phleo using EcoRI-HindIII restrictions sites to generate plasmid p53ku70FRT_Phleo. In a final step, plasmid pKOKU70 was hydrolyzed with NotI and HindIII, and the obtained 3′ ku70 fragment was ligated in p53ku70FRT_Phleo using the corresponding restriction sites. The resulting plasmid was designated pKOKU70_Phleo and carries the complete FRT-ku70 cassette together with the integrated ble gene under the control of the trpC promoter [trpC(p)] from Aspergillus nidulans.
For construction of plasmid pPTRII_FLP the recombinase gene was amplified with the primers 5-FLP and 3-FLP from plasmid pSFU1 (40) and further subcloned into pDrive (Qiagen, Germany) for DNA sequence analysis. In the resulting recombinant plasmid, the flp gene was flanked by the NcoI and SalI sites, which were used in a subsequent step to integrate flp behind the strong trpC promoter of plasmid pD-Phleo. In this plasmid a 1.7-kb KpnI fragment carries the fungal trpC promoter upstream of the flp gene. In order to generate a fungal transformation vector the 1.7-kb fragment was inserted in the free-replicating plasmid pPTRII (31). This new vector was named pPTRII_FLP.
The optimized Pcflp recombinase gene was cloned into plasmid pUC57PcFLP (GenScript). The sequence was further modified by amplification with primers P3, P4, and pUC57PcFLP as templates. Since primer P4 contains recognition sequences for KpnI and HindIII, the recombinant Pcflp sequence was flanked by these restriction sites. The resulting Pcflp gene, with a size of 1.3 kb, was ligated into pDrive, sequenced (MWG-Biotech, Germany), and subsequently isolated using NcoI-HindIII restriction sites. This fragment was ligated into plasmid pD-Phleo using restriction sites NcoI-HindIII. The resulting recombinant plasmid, carrying the Pcflp gene under the control of the trpC promoter, trpC(p), from A. nidulans, was designated pPtrpCPcFLP. In a final step, the trpC(p)-Pcflp fragment was digested with KpnI and cloned into the unique restriction site of plasmid pPTRII to generate plasmid pPTRII_PcFLP.
Plasmid pGPS2.1-Pcflp was designed as described by Dreyer et al. (12) with the following modifications: plasmid pPtrpCPcFLP was hydrolyzed with KpnI to isolate the Pcflp gene under the control of the trpC promoter, which was then integrated into pGPS2.1-hph.
The nat1 flipper cassette was designed by first annealing the complementary oligonucleotides 5FRTlong_s and 5FRTlong_a as well as 3FRTlong_s and 3FRTlong_a to produce either the 5′ FRT or 3′ FRT sequence. The 3′ FRT fragment was introduced into the NotI-ApaI restriction sites of plasmid pTOPO (Invitrogen, Germany). The resulting plasmid p3FRT was digested using HindIII and SacI for insertion of the 5′ FRT site to produce p53FRT. The next step involved eluting a 1.0-kb cassette containing the nat1 marker gene under the control of the trpC promoter from plasmid pD-NAT1 and inserting it into the BamHI-SacI restriction sites of plasmid p53FRT to generate p53FRTnat. Finally, p53FRTnat was hydrolyzed with NotI and BamHI to integrate the 3.0-kb xylP-Pcflp construct; the resulting plasmid was named pXPcFLPnatFRT and contained the complete nat1 flipper cassette. The sequences of all oligonucleotides and features of recombinant plasmids are given in Tables 2 and 3.
TABLE 2.
Sequences of oligonucleotides used in this work
| Oligonucleotide | Sequence (5′-3′) | Specificitya |
|---|---|---|
| FRT1 | GGAAGTTCCTATACTTTCTAGAGAATAGGAACTTCG | 5′ FRT sense + PstI + BamHI |
| FRT2 | GATCCGAAGTTCCTATTCTCTAGAAAGTATAGGAACTTCCTGCA | 5′ FRT antisense + PstI + BamHI |
| FRT3 | CGAAGTTCCTATACTTTCTAGAGAATAGGAACTTCGC | 3′ FRT sense + SacI + NotI |
| FRT4 | GGCCGCGAAGTTCCTATTCTCTAGAAAGTATAGGAACTTCGAGCT | 3′ FRT antisense + SacI + NotI |
| 5FRTlong_s | AGCTTGAAGTTCCTATACTTTCTAGAGAATAGGAACTTCGAGCT | 5′ FRT sense + HindIII + SacI |
| 5FRTlong_a | CGAAGTTCCTATTCTCTAGAAAGTATAGGAACTTCA | 5′ FRT antisense + HindIII + SacI |
| 3FRTlong_s | GGCCGCGAAGTTCCTATACTTTCTAGAGAATAGGAACTTCGGGCC | 3′ FRT sense NotI + ApaI |
| 3FRTlong_a | CGAAGTTCCTATTCTCTAGAAAGTATAGGAACTTCGC | 3′ FRT antisense NotI + ApaI |
| 5′ku70FRT_s | GGTACCATCCTCCATTTGCGCGCTTTCCCT | pKOKU70 (nt 285-308) + KpnI |
| 5′ku70FRT_a | GGATCCGAAGTTCCTATTCTCTAGAAAGTATAGGAACTTCTGTTAGCAAGTAGGTATCCTCGGGAGATTGGAAATATTAAAAGGTGTAAA | pKOKU70 (nt 1256-1305) + BamHI |
| 3′ku70FRT_s | AATTCGAAGTTCCTATACTTTCTAGAGAATAGGAACTTCA | 3′ FRT sense + EcoRI + HindIII |
| 3′ku70FRT_a | AGCTTGAAGTTCCTATTCTCTAGAAAGTATAGGAACTTCG | 3′ FRT antisense + EcoRI + HindIII |
| 5-FLP | CTCCATGGGAATGCCACAATTTGGTATATTATGTAA | flp gene of plasmid pSFU1 (nt 1-26) + NcoI |
| 3-FLP | GAGTCGACATAGGTACCTTATATGCGTCTATTTATGTAGGATGAAAGGT | flp gene of plasmid pSFU1 (nt 1250-1281) + KpnI + SalI |
| P1 | GCGGGCAGTGAGCGCAACGCAATTAA | pXPcFLPnat1FRT (nt 84-109) |
| P2 | GGCCTCTTCGCTATTACGCCAGCT | pXPcFLPnat1FRT (nt 4444-4467) |
| P3 | ATCTAGATACCATGGGTATGCCCCAGTT | pUC57PcFLP (nt 424-451) |
| P4 | TAAAGCTTAATGGTACCTAGGATCCTTAGATGCGGCGGT | pUC57PcFLP (nt 1699-1720) + KpnI + HindIII |
| P5 | CTCCTCTGGGTAGGTCTTAAGCTG | 5′ flanking region of Pcku70 (22) |
| P8 | CCCCCAGGTGGCCAAAGCATCTT | 3′ flanking region of Pcku70 (22) |
| FN_s | CCCAGGCTTTACACTTTATGCTT | pNATFRT1-2 (nt 135-157) |
| FN_a | CCAGTGAATTGTGCGGCCATTT | pNATFRT1-2 (nt 1420-1441) |
| recombinant_FRT | GTGAGCGGATAACAATTTCA | Recombinant FRT sequence (nt 182-201) |
nt, nucleotide.
TABLE 3.
Plasmids used to establish the native and optimized FLP/FRT recombination system in P. chrysogenum
| Plasmid | Characteristic(s)a | Reference or source |
|---|---|---|
| pD-NAT1 | trpC promoter of A. nidulans, nat1 gene of S. noursei | 32 |
| pDNAT1-FRT1 | trpC promoter of A. nidulans, 5′ FRT sequence, nat1 gene of S. noursei | This study |
| pDNAT1-FRT1-2 | 5′ FRT sequence, trpC promoter of A. nidulans, nat1 gene of S. noursei, 3′ FRT sequence | This study |
| pKOKU70 | 5′ ku70 fragment, trpC promoter of A. nidulans, nat1 gene of S. noursei, 3′ ku70 fragment | 22 |
| pD5ku70FRT | 5′ ku70 fragment | This study |
| p5ku70FRT_Phleo | 5′ ku70 fragment, 5′ FRT sequence, trpC promoter of A. nidulans, ble resistance gene of S. hindustanus | This study |
| p53ku70FRT_Phleo | 5′ ku70 fragment, 5′ FRT sequence, trpC promoter of A. nidulans, ble resistance gene of S. hindustanus, 3′ FRT sequence | This study |
| pKOKU70_Phleo | 5′ ku70 fragment, 5′ FRT sequence, trpC promoter of A. nidulans, ble resistance gene of S. hindustanus, 3′ FRT sequence, 3′ ku70 fragment | This study |
| p3FRT | 3′ FRT sequence | This study |
| p53FRT | 5′ FRT sequence, 3′ FRT sequence | This study |
| p53FRTnat | 5′ FRT sequence, trpC promoter of A. nidulans, nat1 gene of S. noursei, 3′ FRT sequence | |
| pXPcFLPnatFRT | 5′ FRT sequence, xyl promoter of P. chrysogenum, Pcflp gene, trpC promoter of A. nidulans, nat1 gene of S. noursei, 3′ FRT sequence | This study |
| pSFU1 | URA3 flipper cassette | 40 |
| pDrive | UA-based PCR cloning | Qiagen, Germany |
| pDFLP | flp gene | This study |
| pD-Phleo | trpC promoter of A. nidulans, ble resistance gene of S. hindustanus | 22 |
| pPtrpCFLP | trpC promoter of A. nidulans, Pcflp gene | This study |
| pPTRII | Self-replicating plasmid; ptrA resistance gene of A. oryzae, AMA1 sequences of A. nidulans | 31 |
| pPTRII_FLP | trpC promoter, flp gene, ptrA resistance gene of A. oryzae, AMA1 sequences of A. nidulans | This study |
| pUC57PcFLP | Pcflp gene | GenScript |
| pPtrpCPcFLP | trpC promoter of A. nidulans, Pcflp gene | This study |
| pPTRII_PcFLP | trpC promoter, Pcflp gene, ptrA resistance gene of A. oryzae, AMA1 sequences of A. nidulans | This study |
| pGPS2.1-hph | trpC promoter of A. nidulans, hph of K. pneumoniae, left and right borders from transposon Tn7 (New England Biolabs) | 12 |
| pGPS2.1-PcFLP | trpC promoter of A. nidulans, hph of K. pneumoniae, left and right borders from transposon Tn7 (New England Biolabs), Pcflp gene | This study |
S. hindustanus, Streptoalloteichus hindustanus; K. pneumoniae, Klebsiella pneumoniae.
Molecular analysis of fungal transformants.
Genomic DNA from both primary and secondary transformants was isolated as described previously (22, 48). For molecular analysis, genomic DNA of transformants from strain PcFRT1 was hydrolyzed with BamHI, separated by agarose gel electrophoresis, and blotted for Southern hybridization (52). A 32P-radiolabeled, 0.9-kb nat1 fragment was chosen as the probe. Bulk DNA was used for PCR analysis (23), followed by agarose gel electrophoresis to separate the amplicons. Template DNA from primary transformants or plasmid pDNAT1-FRT1-2 served as a positive control and generated a fragment of 1.3 kb with primers FN_s and FN_a (see Fig. 2). However, both primers amplified a 0.2-kb DNA molecule when DNA from secondary transformants PcFRT2Y (PcFRT2 carrying the yeast flp gene), PcFRT2P (PcFRT2 carrying the Penicillium-optimized codon usage), and SmFRT2 (S. macrospora secondary transformant) was used. The 0.2-kb fragment was sequenced (MWG, Germany) and further analyzed using the program LALIGN http://www.ch.embnet.org/software/LALIGN_form.html). In the case of strain PcFLIP1, the PCR analysis using genomic DNA generated fragments with sizes of 2.5 and 3.1 kb, whereas a 0.3-kb amplicon could be detected in the PcFLIP2 strain.
FIG. 2.

Analysis of secondary transformants. (A) Schematic map of the FRT-nat1 cassette before (top) and after (bottom) site-specific excision. Arrows indicate location of PCR primers and bars show the length of the predicted amplicons. (B) PCR analysis of secondary transformants as indicated. PcFRT2Y and PcFRT2P indicate fungal transformants carrying either the yeast (flp) or the codon-adapted recombinase (Pcflp) gene, respectively. T2.1, T2.2, T2.4, T2.5, T4.1, T4.2, T4.4, and T4.5 indicate single-copy primary transformants while T11.1, T11.2, T11.4, and T11.5 represent multicopy primary transformants. A 1.3-kb amplicon is generated when the complete FRT-nat1 cassette is present in the tested transformants, whereas a 0.2-kb fragment could be amplified only after an excision of the FRT-nat1 cassette by an FLP-mediated site-specific recombination event. DNA of plasmid pFRT served as a positive control.
Bioinformatics analysis.
In order to construct a codon-optimized flp gene, the codon usage of P. chrysogenum was analyzed using databases at http://www.kazusa.or.jp/codon/ and genome sequence data from van den Berg et al. (61). To decrease the GC content in the optimized recombinase sequence, not only the optimal codon triplets were chosen but also triplets in the second or third preference. After optimal codon selection, codon optimization was verified by using the program JCat (Java Codon Adaptation Tool [http://www.jcat.de/]) (18). The adapted recombinase sequence was extended at both ends using the restriction enzymes NcoI and BamHI and sent to GenScript (Piscataway, NJ) for custom synthesis and cloning into pUC57PcFLP. The resulting recombinant gene was named Pcflp.
RESULTS
Construction of the FRT-nat1 cassette and generation of primary transformants.
To generate a functional FLP/FRT recombination system, we constructed an FRT-nat1 cassette in which the nourseothricin resistance gene (nat1) of Streptomyces noursei is flanked by two FRT sites in direct-repeat orientation. In this construct, which is contained in plasmid pDNAT1_FRT1-2, the nat1 gene encoding an N-acetyltransferase is under the control of the strong and constitutive fungal trpC promoter from A. nidulans (32). For fungal DNA-mediated transformation, a 1.5-kb EarI restriction fragment from pDNAT1_FRT1-2, in which the FRT-nat1 cassette is flanked by short bacterial plasmid sequences, was used.
The ectopic integration of this linear DNA fragment into the P. chrysogenum recipient strain P2niaD18 generated a large number of primary transformants (PcFRT1). To confirm the presence of the FRT-nat1 cassette in PcFRT1 transformants, oligonucleotides FN_s and FN_a were used for PCR amplification from genomic DNA. The primers hybridize to the bacterial sequences of the linear transformation fragment, thus allowing amplification of the ectopically integrated cassette that otherwise is flanked by uncharacterized genomic DNA sequences. As expected, a 1.3-kb fragment was detected in all transformants (data not shown). Six recombinant strains, designated T2, T3, T4, T11, T12, and T18, were chosen for Southern hybridization analysis. The autoradiograph in Fig. 1B obtained from hybridization with a nat1 gene fragment as a radio-labeled probe identified a single band in strains T2 and T4, indicating that single-copy integration events of the FRT-nat1 cassette had taken place. In contrast, the several bands found in the other lanes indicate multicopy transformants. T11, which had four hybridizing bands, was chosen for further analysis as a representative example of a multicopy FRT-nat1 transformant. Both the single- and multicopy strains were used to generate secondary transformants.
FIG. 1.
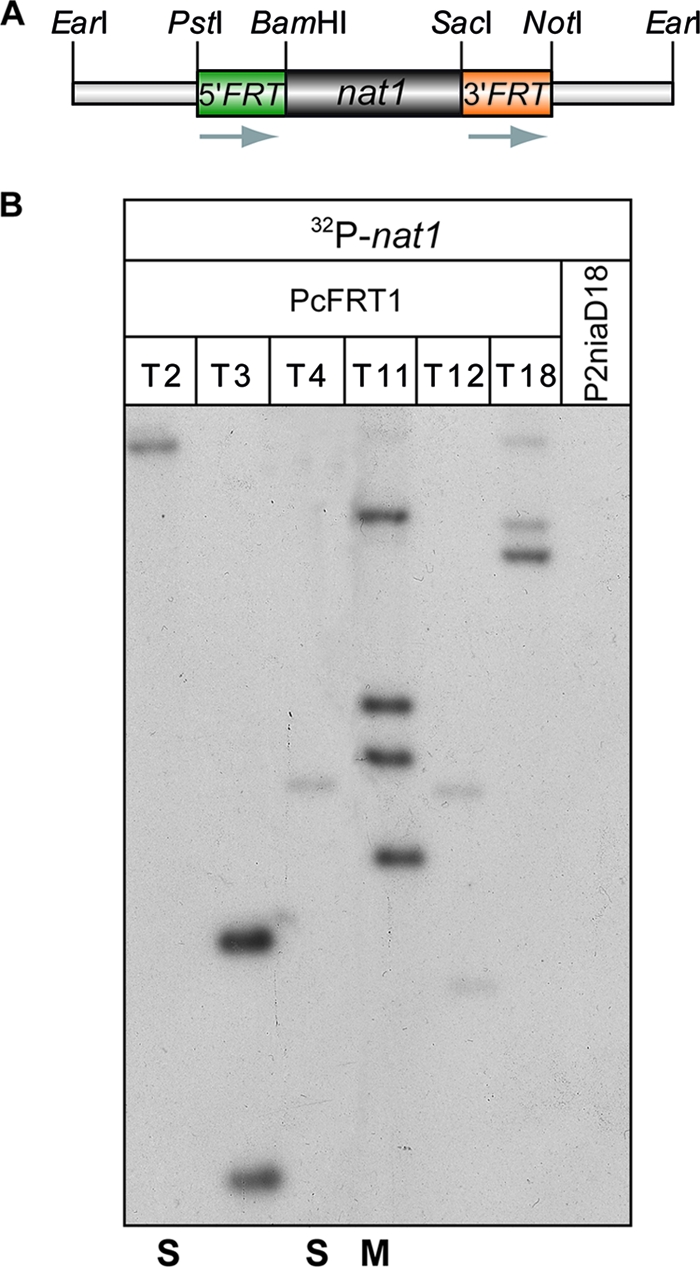
Characterization of primary transformants from P. chrysogenum. (A) Schematic representation of the FRT-nat1 cassette. Adjacent to the nat1 resistance gene are two FRT sites in direct repeat orientation, and both are flanked by vector sequences. (B) Southern hybridization analysis of the primary PcFRT1 transformants to identify the copy number of ectopically integrated FRT-nat1 cassettes. Genomic DNAs of the recipient strain P2niaD18 and the transformants were digested with BamHI and hybridized with a 32P-labeled nat1 probe. The single-copy transformants T2 and T4 and the multicopy transformant T11 were chosen for further experimental analyses. S, single copy; M, multicopy.
Synthesis of a codon-optimized flp recombinase gene for construction of an expression plasmid.
When we compared the codon bias of the native flp recombinase gene from yeast with the general codon usage in P. chrysogenum using the online databases at http://www.kazusa.or.jp/codon/ and the genome sequence of P. chrysogenum (61), we found distinct differences. For example, in the native flp gene, the amino acid arginine is mostly encoded by the triplets AGA and AGG but never by codon CGC. In contrast, CGC is the favored codon for arginine in P. chrysogenum, with AGA and AGG being used only in <10% of cases (data not shown). Furthermore, the S. cerevisiae flp gene has a GC content of 37%. In contrast, the nuclear genome of P. chrysogenum has an overall GC content of 48.9%, with exon regions having a GC content of 52.8% and intron sequences having a lower GC content of 45.3% (61). Due to these differences between S. cerevisiae and P. chrysogenum, the codon usage bias for each amino acid in the flp recombinase gene was determined. Based on these properties, we decided to synthesize an flp recombinase gene with a bias for preferred P. chrysogenum codons.
In a first attempt, we generated a codon-optimized recombinase gene in silico with a GC content of 61%. Since P. chrysogenum exons have an average GC content of 52.8%, we generated in silico a second version with a mixture of codons used in the first, second, and third preferences. The resulting sequence had a GC content of 57% with 73% DNA homology to the native flp recombinase gene, despite modification of over 66% of all triplets. To determine whether the modified Pcflp sequence was mostly compatible for P. chrysogenum, we used the program JCat (18) for codon adaptation. This program compares the sequence of any gene with the codon usage database of a chosen organism. As a database for P. chrysogenum is not available in JCat, the database of the next most closely related filamentous fungus, Aspergillus niger, was used to test the quality of the codon adaptation as the species have a very similar codon usage (47, 61). The codon adaptation index (CAI) was calculated for both the native flp gene and the codon-optimized Pcflp sequence. The CAI can have a value between 0 and 1; the more the CAI tends to 1, the less optimization is needed. The native flp recombinase gene has a CAI of 0.17, indicating that this gene is not fully suitable for optimal expression in P. chrysogenum. In contrast, the CAI of 0.81 of the codon-adapted Pcflp recombinase gene (see Fig. S1 in the supplemental material) is a strong indicator that this gene can be used for optimal expression in filamentous fungi.
Expression of the native and codon-adapted FLP recombinase genes in P. chrysogenum.
Using the primary PcFRT1 transformants T2, T4, and T11, we generated secondary transformants that carry either the yeast gene (flp) or the codon-optimized recombinase gene (Pcflp) by transformation with the self-replicating plasmids pPTRII_FLP and pPTRII_PcFLP, respectively. The resulting transformants, carrying the yeast gene (PcFRT2Y) or the synthetic recombinase gene with the Penicillium-optimized codon usage (PcFRT2P), were characterized by PCR analysis, growth tests, and sequencing to verify the functionality and efficiency of the FLP/FRT systems in P. chrysogenum. We expected that after FLP- or Pcflp-mediated site-specific recombination, a single, recombinant 34-bp FRT site remains as a remnant of the complete FRT-nat1 in the genome of the resulting strain. As a consequence, the secondary transformants should be sensitive to nourseothricin.
In the case of strains transformed with flp, growth tests showed that none of 30 secondary PcFRT2Y transformants was nourseothricin sensitive when grown on the selection medium. Growth tests with strains transformed with flp showed that none of 40 secondary transformants had lost the nourseothricin resistance gene. To exclude the possibility that heterokaryons are responsible for nourseothricin resistance, a total of 300 single-spore isolates from two single-copy and two multicopy PcFRT2Y transformants were tested on nourseothricin-containing medium (data not shown). None had lost the resistance marker, indicating that only a very minor fraction of nuclei is carrying the FRT sequence without the resistance marker gene.
A completely different result was obtained with transformants PcFRT2P carrying Pcflp. All 34 tested transformants derived either from the single-copy PcFRT1 T2 or T4 transformant showed sensitivity to the antibiotic nourseothricin. Similarly, 16 out of 20 analyzed transformants received from the multicopy transformant PcFRT1 T11 showed no growth on nourseothricin-containing medium (see Fig. S2 in the supplemental material). These data clearly indicate that the codon-adapted FLP/FRT recombination system can efficiently work in different P. chrysogenum transformants.
These results from the growth tests correlate perfectly with our PCR analyses (Fig. 2). As indicated in Fig. 2A, oligonucleotides FN_s and FN_a hybridize to sequences flanking the FRT-nat1 cassette. Amplification of the cassette with these two primers generated a 1.3-kb amplicon, whereas a 0.2-kb fragment was generated after excision of the FRT-nat1 cassette by an FLP-mediated site-specific recombination event. For amplification, we used genomic DNA from the above-described fungal transformants carrying one of the two recombinase genes. All recombinant strains (PcFRT2Y) carrying the yeast flp gene delivered both the large and the small amplicons, thus indicating that the recombinase-mediated recombination was highly inefficient in these transformants (Fig. 2B). The large fragment of 1.3 kb, however, was completely missing in all transformants (PcFRT2P) carrying the codon-optimized Pcflp recombinase gene. From the results of the PCR analyses together with data from growth tests, we conclude that all FRT-nat1 cassettes were excised from the chromosomal DNA.
That successful recombination had occurred was confirmed by sequencing a recombinant FRT sequence from the secondary transformants PcFRT2P. This site consists of the left and right halves from both FRT sequences of the FRT-nat1 cassette (Fig. 3). Both FRT sequences are flanked by different restriction sites, namely, by the PstI and BamHI and by the pair SacI and NotI. After excision of the FRT-nat1 cassette, the recombinant FRT should be flanked by restriction sites PstI and NotI. Sequencing of the 0.2-kb fragment from the secondary PcFRT2P transformants confirmed this prediction (Fig. 3). Taken together, our data clearly demonstrate that the optimized FLP/FRT recombination system is functional in P. chrysogenum and can be used as an efficient marker recycling system to generate multiple deletion strains with a limited number of resistant markers.
FIG. 3.

Structure and sequence of a recombinant FRT sequence in the genomic DNA. FRT sequences before (top) and after (middle) recombination are flanked by different restriction sites, as indicated. The recombinant DNA sequence obtained from PcFRT2P is shown at the bottom.
Construction of a marker-free recipient for the generation of knockout strains.
We have recently reported the generation of a ΔPcku70 strain as a recipient for the construction of knockout strains in P. chrysogenum (22). As shown in Fig. 4A, the Pcku70 gene of P2niaD18 was replaced in the ΔPcku70 strain by homologous recombination using the nat1 resistance marker. This strain was used as recipient for a further transformation experiment with a vector molecule carrying the phleomycin (ble) resistance marker. The ble gene is flanked by two FRT sites as well as sequences homologous to the 5′ and 3′ regions of the Pcku70 gene. After selection of transformants on phleomycin-containing medium, we obtained a ΔPcku70FRT1 strain where the nat1 cassette was replaced by the ble marker. Due to efficient homologous recombination, 9 out of 18 tested transformants contained the expected substitution. These strains have lost the ability to grow on nourseothricin-containing medium. For the next step, we used two randomly selected transformants as recipients to introduce the self-replicating plasmid pPTRII_PcFLP carrying the Pcflp recombinase gene. After selection on pyrithiamine, a total of 18 transformants were tested for their capability to still grow on phleomycin. Two transformants (ΔPcku70FRT2) had lost the phleomycin resistance phenotype, thus suggesting that excision of the phleomycin resistance gene had occurred (see Fig. S3 in the supplemental material). The above described substitutions and excisions in the diverse strains shown in Fig. 4 were further verified by PCR analyses. Amplification was performed with primer pair P5 and P8 flanking the Pcku70 region and located outside the recombination sites (Fig. 4A). As indicated in Fig. 4B, the different events result in amplicons that can clearly be distinguished by their sizes. The smallest amplicon of 2.4 kb, indicating a Pcflp-mediated site-specific recombination event, was found in the ΔPcku70FRT2 strain. A further loss of the recombinase gene carrying the plasmid was achieved by growth of the transformants on pyrithiamine-free medium. In conclusion, we have constructed a recipient strain for efficient homologous recombination that is free of any heterologous gene sequences. Only the 34 bp of a single FRT site reside within the fungal genome. This experimental approach and the generated transformants provide the basis for both the nat1 and the ble marker to be used for further construction of double or triple knockout mutants.
FIG. 4.

Generation of a marker-free ΔPcku70 strain. (A) Strategy for construction of different strains. Location of primers for the PCR analysis in panel B is shown, and the predicted sizes of amplicons are given. (B) PCR analysis with DNA from different strains as indicated. HR, homologous recombination. P5 × P8 indicates the pair of primers used for PCR amplification.
Construction of an nat1 flipper cassette for one-step marker excision.
All above-described experiments require a two-step strategy for successful excision of the FRT-nat1 cassette. We therefore constructed an nat1 flipper cassette for one-step marker recycling. The cassette contains the nat1 resistance marker as well as the gene for the Pcflp recombinase, which is under the control of the xylose-inducible xyl promoter (70). As shown in Fig. 5A, both genes are flanked by the two FRT sites on the nat1 flipper cassette. For transformation of P2niaD18, a linear restriction fragment was used carrying the nat1 flipper cassette as well as flanking plasmid sequences. The corresponding transformants (PcFLIP1) were selected on nourseothricin-containing medium with glucose as a carbon source. To induce the Pcflp-mediated recombination, 12 transformants were further grown for 72 h in liquid medium with xylose as a carbon source but lacking any antibiotics. Subsequently, these transformants were transferred onto solid medium with or without nourseothricin but lacking xylose in both cases. We found that 2 out of 12 transformants had lost nourseothricin resistance, and these were designated PcFLIP2 T1 and PcFLIP2 T3 (Fig. 5A). In the following PCR analyses (Fig. 5B), the lack of the nat1 flipper cassette was confirmed with the primer pairs as indicated. The whole flipper cassette is still present in strains (PcFLIP1) that grow on glucose as a noninducible carbon source. After induction on xylose, the 0.3-kb amplicon, carrying only the recombinant FRT sequence, can be detected in the PcFLIP2 transformants. Thus, with the optimized FLP/FRT recombination system described here, it is possible to eliminate and to recycle any marker using a one-step approach.
FIG. 5.

One-step marker excision. (A) Map of the nat1 flipper cassette together with the location of primers for PCR analysis before (top) and after (bottom) excision. The location of primers for panel B is given. (B) PCR analysis with DNA from strains as indicated. xyl(p), xyl promoter of P. chrysogenum P1 × P3, P2 × P4, and P1 × P2 indicate primer pairs used for PCR amplification.
Establishment of the FLP/FRT recombination system in S. macrospora.
To extend the application of the optimized FLP/FRT recombination system to a further filamentous fungus, we chose the model ascomycete S. macrospora (46), which belongs to the Sordariomycetes and is distantly related to P. chrysogenum, a member of the Eurotiomycetes. For our analysis, we used plasmids which were successfully tested in P. chrysogenum as described above. First, the 1.3-kb FRT-nat1 cassette was ectopically integrated into the wild-type strain to generate primary SmFRT1 transformants. The codon-adapted Pcflp recombinase gene was then introduced into three of the primary SmFRT1 transformants using plasmid pGPS2.1-Pcflp conferring hygromycin resistance. Characterization of the 53 resulting secondary SmFRT2P transformants revealed that 26 were unable to grow on selection medium containing nourseothricin; thus, excision of the FRT-nat1 cassette by the Pcflp recombinase resulted in the loss of the nat1 gene. To verify this recombination event, a PCR analysis with the primer pair FN_s and FN_a was performed. As shown in Fig. 6, the 1.3-kb FRT-nat1 cassette was detectable in only the primary SmFRT1 transformants, while all nourseothricin-sensitive secondary SmFRT2P transformants carry only the 0.2-kb amplicon, indicating a successful recombination event in S. macrospora. It can be foreseen that the optimized FLP/FRT recombination system will be applied to fungi unrelated to the two species investigated in this study.
FIG. 6.
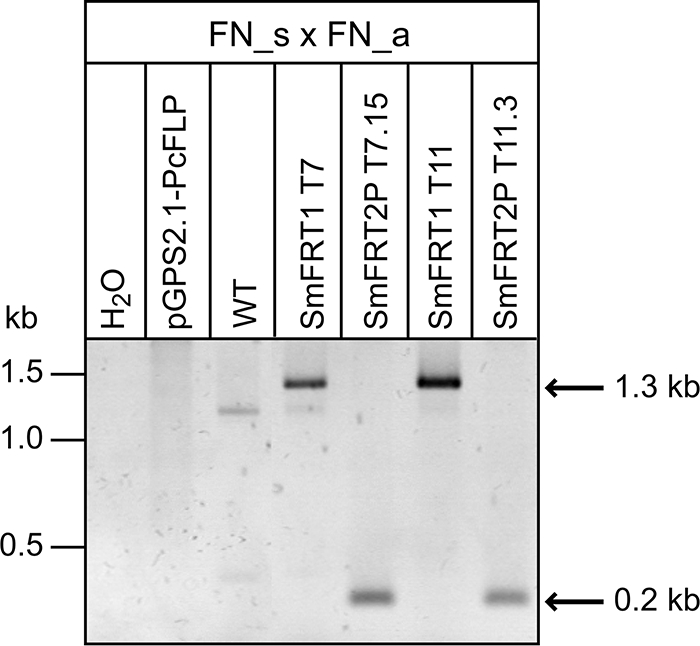
PCR analysis of S. macrospora transformants with primers as indicated. The location of both primers is shown in Fig. 2A. WT, wild type. FN_s × FN_a indicates the primer pair used for PCR amplification.
DISCUSSION
The complete genome sequences of a large number of filamentous fungi are already available, thereby enabling the application of fundamental functional genetic studies. One approach will be the analysis of candidate genes, e.g., involved in the regulation of metabolism and/or morphogenesis. A common and frequently used strategy in fungal genetics is the disruption of the primary gene structure by insertion or substitution of marker genes using homologous recombination systems (33). Usually, the number of gene disruptions followed by complementation experiments within a single strain is dependent on the available marker genes applicable to a given fungus. One option to generate strains with multiple gene substitutions is sexual recombination; however, this is not possible for most biotechnically relevant fungi since they usually propagate exclusively by asexual means. Note that the FLP/FRT recombination system established here should be applicable to any given filamentous fungus.
Efficient expression of codon-adapted genes in filamentous fungi.
Filamentous fungi are often used as producers for homo- and heterologous proteins because of their capability to synthesize and secrete huge amounts of diverse products (8, 27, 42, 59). Heterologous gene expression is especially important for increased production of pharmaceuticals and food additives because often the extraction of these compounds from their natural sources is not cost-effective or production in the corresponding hosts is impossible (4, 67). Unfortunately, however, the yield of most heterologous proteins is generally far lower than that of homologous proteins (8). To solve this problem, different approaches have been developed, including multicopy integration of the coding gene, use of strong regulatory promoters, and fusion with carrier proteins (3, 38). Moreover, codon usage has been identified as an important factor for efficient gene expression, indicating that differences in the codon usage between two species, in general, and between prokaryotes and eukaryotes, in particular, can result in translation inefficiency (20, 27, 36, 59). Because of this, strategies to remove codons from the native gene that are rare in the new host or the introduction of tRNAs for rare codons into the genome of a given organism have been developed (4, 68, 71). Another alternative is the complete synthesis of recombinant genes, an option which has become affordable recently, making the adaption of a complete heterologous gene to the codon usage or codon bias of the organism of choice possible.
Recent examples from fungal systems include the complete reconstruction of the firefly luciferase gene, resulting in a significant increase in gene expression in Neurospora crassa (17). Another example from the fungal field is the novel synthesis of the Der f 7 gene encoding a mite allergen. This synthetic gene was fused with the glucoamylase gene for optimal expression in Aspergillus oryzae, offering the possibility to produce recombinant allergens for immunotherapy (59). Both studies demonstrate that codon optimization is a powerful strategy to improve the level of gene expression to allow successful protein synthesis in a given host. To establish the FLP/FRT recombination system in P. chrysogenum, we have optimized the codon usage of the native flp recombinase gene from S. cerevisiae, an approach which was previously done for optimal expression in the prokaryote Mycobacterium bovis (56). This was necessary since expression of the yeast flp gene was not sufficient to generate any nourseothricin-sensitive single-spore isolates out of 300 tested spores. A more practical system was obtained with the codon-optimized Pcflp version, resulting in expression levels for efficient excision of the FRT-nat1 cassette in P. chrysogenum. Thus, the adapted codon usage most likely allows the translational system to be optimized. Preferred codons usually correlate with the abundance of cognate tRNAs within a cell, a prerequisite to balance codon with isoacceptor tRNA concentrations. Differences in codon usage were shown previously to be significant obstacles for efficient heterologous gene expression (20). We observed variable frequencies of excision between individual experimental approaches. Different promoters in front of the Pcflp gene may result in different expression levels, which are responsible for the variable excision frequency. For example the xyl promoter was activated for only 72 h, and longer incubation times might result in higher excision frequencies. Another explanation might be the different host strains, e.g., P2niaD18 versus ΔPcku70, used as recipients in different experiments.
Generation of marker-free transgenic strains.
The application of the marker recycling system in the two filamentous fungi P. chrysogenum and S. macrospora opens up the possibility of wider usage in diverse fungal systems. Such an application will be an attractive option for applied microbiologists when genetically engineered fungal strains are needed in biotechnical production processes. Moreover, as the recombinant strains will be marker free, there should be no problem in fulfilling all essential safety requirements needed to obtain marketing approval. We have previously shown that homologous resistance markers are suitable tools to construct recombinant strains devoid of foreign DNA sequences. Using the homologous β-tubulin gene from the cephalosporin C producer Acremonium chrysogenum, we generated a mutated gene version that confers resistance against the fungicide benomyl (44). Using a linear DNA fragment carrying the mutated β-tubulin gene, we were able to generate transgenic strains lacking any heterologous DNA. Similarly, pyrithiamine resistance-conferring riboswitches from fungi can be used as homologous selection markers (30). In this case, the resistance marker resides within the fungal genome and can be used only once. Alternative systems that allow elimination of homologous as well as heterologous marker genes have been developed for microorganisms and plants. For example, cotransformation of two separate DNAs can be conducted; one incorporates a gene of interest, and the other is a self-replicating plasmid carrying the marker.
We have recently found for a filamentous fungus that self-replicating plasmids can be eliminated from a host strain when it is kept on nonselective growth medium (Hoff, unpublished data). In another approach, intragenomic relocation of transgenes via transposable elements was used to eliminate selectable marker genes in filamentous fungi (69). A similar study used a counter-selectable marker flanked by direct repeats to reestablish a functional allele in a transgenic strain (43). The most elegant systems use site-specific recombinases for efficient marker recycling. As demonstrated here and shown by others, marker-free knockout strains can be generated carrying only small sequences of foreign DNA (14-15, 29). Here, we clearly demonstrated how highly applicable this system is by generating a ΔPcku70FRT2 strain devoid of any heterologous gene. Ku70 is an important component of the nonhomologous end-joining (NHEJ) pathway, a mechanism employed by filamentous fungi to repair double-strand breaks. The deletion of the ku70 gene, a component of the NHEJ pathway, leads to much higher integration frequencies of foreign DNA by homologous recombination, making the construction of knockout strains a relatively straightforward approach (33). Moreover, the optimized recombination system offers recycling of the integrated resistance marker in a Δku70 background, thus allowing the possibility for fast and efficient production of multiple deletion strains as the availability of suitable marker genes is no longer the limiting factor. All comparable approaches with filamentous fungi have used the Cre/loxP recombination system of bacteriophage P1. For example, Krappmann et al. (29) were able to generate a double knockout mutant in Aspergillus fumigatus by integrating the Cre/loxP system in a two-step approach. A resistance cassette flanked by loxP sequences and a self-replicating plasmid carrying the cre gene were used to delete the pabaA gene encoding the para-aminobenzoic acid (PABA) synthetase, which is involved in virulence. After marker rescue by Cre-mediated recombination, these investigators used the system a second time to remove the veA gene encoding a developmental protein.
Application potential of the recombination systems.
The FLP/FRT recombination system provides multiple options to manipulate the genome of a chosen organism. In addition to the efficient deletion of genes followed by marker recycling as described in this report, integration of genes at defined loci (24), gene tagging at the endogenous locus (49), and transient gene inactivation (60) are useful applications to manipulate organisms genetically.
To establish an efficient and safe recombination system, it is important to control the time point of marker gene excision mediated by the recombinase. To achieve this, different approaches have been developed. In mouse, for example, two independent strains were generated, one containing the marker gene flanked by the recombination sites and the other one harboring the recombinase gene under the control of a tissue- or cell type-specific promoter. After crossing of the two strains, introduction of the recombination system results in site-specific recombination of the marker gene in a specific tissue (28). In addition to this application, an inducible Cre/loxP system has been reported for A. nidulans that allows consecutive deletions to be introduced (15). In this system, the cre gene was placed under the control of the xylose-inducible xlnA promoter, which can be repressed on medium containing glucose. Development of an appropriate protocol enables the efficient generation of sequential deletions in this fungus. However, this system uses a two-step strategy since the available components are located on two different DNA constructs. An alternative and efficient one-step strategy was recently applied for Candida species. Different versions of so-called flipper cassettes flanked by the FRT sequences and containing both the resistance gene and the inducible recombinase gene were generated (13, 40, 50, 57, 60, 66). Using a similar strategy, we generated the nat1 flipper cassette for P. chrysogenum. To prevent a spontaneous loss of the flipper, we used the inducible xyl promoter from P. chrysogenum, which was shown recently to be a valuable tool for the controlled off/on regulation of gene expression (25). After only 72 h of induction we obtained P. chrysogenum strains that had lost the flipper cassette. However, it remains to be investigated whether longer induction times lead to higher recombination efficiencies.
In conclusion, we have demonstrated by a two-step strategy that the established FLP/FRT system enables the production of marker-free deletion strains. This provides the opportunity in future experiments to use the nat1 flipper in a fast and efficient one-step approach for targeted gene excision.
Supplementary Material
Acknowledgments
We thank Ingeborg Godehardt, Ivonne Schelberg, and Susanne Schlewinski for their excellent technical assistance, Gabriele Frenßen-Schenkel for preparing the figures, N. Frankenberg-Dinkel for suggesting the use of JCat for codon adaptation, and Hubert Kürnsteiner, Ernst Friedlin, Ivo Zadra, Heiko Eichhorn, Rudolf Mitterbauer, and Thomas Specht (Sandoz GmbH) for their interest and support. We acknowledge receipt of the FRT and flp sequences from J. Morschhäuser (Würzburg, Germany).
This work was funded by Sandoz GmbH (Kundl, Austria) and the Christian Doppler Society (Vienna, Austria).
Footnotes
Published ahead of print on 14 May 2010.
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Abremski, K., and R. Hoess. 1984. Bacteriophage P1 site-specific recombination. Purification and properties of the Cre recombinase protein. J. Biol. Chem. 259:1509-1514. [PubMed] [Google Scholar]
- 2.Amin, A., H. Roca, K. Luetke, and P. D. Sadowski. 1991. Synapsis, strand scission, and strand exchange induced by the FLP recombinase: analysis with half-FRT sites. Mol. Cell. Biol. 11:4497-4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Archer, D. B., I. F. Connerton, and D. A. MacKenzie. 2008. Filamentous fungi for production of food additives and processing aids. Adv. Biochem. Eng. Biotechnol. 111:99-147. [DOI] [PubMed] [Google Scholar]
- 4.Berlec, A., Z. Jevnikar, A. C. Majhenic, I. Rogelj, and B. Strukelj. 2006. Expression of the sweet-tasting plant protein brazzein in Escherichia coli and Lactococcus lactis: a path toward sweet lactic acid bacteria. Appl. Microbiol. Biotechnol. 73:158-165. [DOI] [PubMed] [Google Scholar]
- 5.Birling, M. C., F. Gofflot, and X. Warot. 2009. Site-specific recombinases for manipulation of the mouse genome. Methods Mol. Biol. 561:245-263. [DOI] [PubMed] [Google Scholar]
- 6.Blakely, G. W., A. O. Davidson, and D. J. Sherratt. 2000. Sequential strand exchange by XerC and XerD during site-specific recombination at dif. J. Biol. Chem. 275:9930-9936. [DOI] [PubMed] [Google Scholar]
- 7.Bullock, W. O., J. M. Fernandez, and J. M. Short. 1987. XL1-Blue—a high-efficiency plasmid transforming recA Escherichia coli strain with β-galactosidase selection. Biotechniques 5:376-378. [Google Scholar]
- 8.Cardoza, R. E., S. Gutierrez, N. Ortega, A. Colina, J. Casqueiro, and J. F. Martin. 2003. Expression of a synthetic copy of the bovine chymosin gene in Aspergillus awamori from constitutive and pH-regulated promoters and secretion using two different pre-pro sequences. Biotechnol. Bioeng. 83:249-259. [DOI] [PubMed] [Google Scholar]
- 9.Chen, Y., and P. A. Rice. 2003. New insight into site-specific recombination from Flp recombinase-DNA structures. Annu. Rev. Biophys. Biomol. Struct. 32:135-159. [DOI] [PubMed] [Google Scholar]
- 10.Chiang, C. J., P. T. Chen, and Y. P. Chao. 2008. Replicon-free and markerless methods for genomic insertion of DNAs in phage attachment sites and controlled expression of chromosomal genes in Escherichia coli. Biotechnol. Bioeng. 101:985-995. [DOI] [PubMed] [Google Scholar]
- 11.Ding, C., and G. Butler. 2007. Development of a gene knockout system in Candida parapsilosis reveals a conserved role for BCR1 in biofilm formation. Eukaryot. Cell 6:1310-1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dreyer, J., H. Eichhorn, E. Friedlin, H. Kürnsteiner, and U. Kück. 2007. A homologue of the Aspergillus velvet gene regulates both cephalosporin C biosynthesis and hyphal fragmentation in Acremonium chrysogenum. Appl. Environ. Microbiol. 73:3412-3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dunkel, N., J. Blass, P. D. Rogers, and J. Morschhäuser. 2008. Mutations in the multi-drug resistance regulator MRR1, followed by loss of heterozygosity, are the main cause of MDR1 overexpression in fluconazole-resistant Candida albicans strains. Mol. Microbiol. 69:827-840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Florea, S., K. Andreeva, C. Machado, P. M. Mirabito, and C. L. Schardl. 2009. Elimination of marker genes from transformed filamentous fungi by unselected transient transfection with a Cre-expressing plasmid. Fungal Genet. Biol. 46:721-730. [DOI] [PubMed] [Google Scholar]
- 15.Forment, J. V., D. Ramon, and A. P. MacCabe. 2006. Consecutive gene deletions in Aspergillus nidulans: application of the Cre/loxP system. Curr. Genet. 50:217-224. [DOI] [PubMed] [Google Scholar]
- 16.Futcher, A. B. 1986. Copy number amplification of the 2 μm circle plasmid of Saccharomyces cerevisiae. J. Theor. Biol. 119:197-204. [DOI] [PubMed] [Google Scholar]
- 17.Gooch, V. D., A. Mehra, L. F. Larrondo, J. Fox, M. Touroutoutoudis, J. J. Loros, and J. C. Dunlap. 2008. Fully codon-optimized luciferase uncovers novel temperature characteristics of the Neurospora clock. Eukaryot. Cell 7:28-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grote, A., K. Hiller, M. Scheer, R. Munch, B. Nortemann, D. C. Hempel, and D. Jahn. 2005. JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 33:W526-W531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Groth, A. C., and M. P. Calos. 2004. Phage integrases: biology and applications. J. Mol. Biol. 335:667-678. [DOI] [PubMed] [Google Scholar]
- 20.Gustafsson, C., S. Govindarajan, and J. Minshull. 2004. Codon bias and heterologous protein expression. Trends Biotechnol. 22:346-353. [DOI] [PubMed] [Google Scholar]
- 21.Haas, H., K. Angermayr, I. Zadra, and G. Stoffler. 1997. Overexpression of nreB, a new GATA factor-encoding gene of Penicillium chrysogenum, leads to repression of the nitrate assimilatory gene cluster. J. Biol. Chem. 272:22576-22582. [DOI] [PubMed] [Google Scholar]
- 22.Hoff, B., J. Kamerewerd, C. Sigl, I. Zadra, and U. Kück. 2010. Homologous recombination in the antibiotic producer Penicillium chrysogenum: strain ΔPcku70 shows up-regulation of genes from the HOG pathway. Appl. Microbiol. Biotechnol. 85:1081-1094. [DOI] [PubMed] [Google Scholar]
- 23.Hoff, B., S. Pöggeler, and U. Kück. 2008. Eighty years after its discovery, Fleming's Penicillium strain discloses the secret of its sex. Eukaryot. Cell 7:465-470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang, L. C., E. A. Wood, and M. M. Cox. 1997. Convenient and reversible site-specific targeting of exogenous DNA into a bacterial chromosome by use of the FLP recombinase: the FLIRT system. J. Bacteriol. 179:6076-6083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Janus, D., B. Hoff, and U. Kück. 2009. Evidence for Dicer-dependent RNA interference in the industrial penicillin producer Penicillium chrysogenum. Microbiology 155:3946-3956. [DOI] [PubMed] [Google Scholar]
- 26.Kilby, N. J., M. R. Snaith, and J. A. Murray. 1993. Site-specific recombinases: tools for genome engineering. Trends Genet. 9:413-421. [DOI] [PubMed] [Google Scholar]
- 27.Koda, A., T. Bogaki, T. Minetoki, and M. Hirotsune. 2005. High expression of a synthetic gene encoding potato alpha-glucan phosphorylase in Aspergillus niger. J. Biosci. Bioeng. 100:531-537. [DOI] [PubMed] [Google Scholar]
- 28.Kos, C. H. 2004. Cre/loxP system for generating tissue-specific knockout mouse models. Nutr. Rev. 62:243-246. [DOI] [PubMed] [Google Scholar]
- 29.Krappmann, S., O. Bayram, and G. H. Braus. 2005. Deletion and allelic exchange of the Aspergillus fumigatus veA locus via a novel recyclable marker module. Eukaryot. Cell 4:1298-1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kubodera, T., N. Yamashita, and A. Nishimura. 2000. Pyrithiamine resistance gene (ptrA) of Aspergillus oryzae: cloning, characterization and application as a dominant selectable marker for transformation. Biosci. Biotechnol. Biochem. 64:1416-1421. [DOI] [PubMed] [Google Scholar]
- 31.Kubodera, T., N. Yamashita, and A. Nishimura. 2002. Transformation of Aspergillus sp. and Trichoderma reesei using the pyrithiamine resistance gene (ptrA) of Aspergillus oryzae. Biosci. Biotechnol. Biochem. 66:404-406. [DOI] [PubMed] [Google Scholar]
- 32.Kück, U., and B. Hoff. 2006. Application of the nourseothricin acetyltransferase (nat1) as dominant marker for the transformation of filamentous fungi. Fungal Genet. Newsl. 53:9-11. [Google Scholar]
- 33.Kück, U., and B. Hoff. 2010. New tools for the genetic manipulation of filamentous fungi. Appl. Microbiol. Biotechnol. 86:51-62. [DOI] [PubMed] [Google Scholar]
- 34.Kück, U., S. Pöggeler, M. Nowrousian, N. Nolting, and I. Engh. 2009. Sordaria macrospora, a model system for fungal development, p. 17-39. In T. Anke and D. Weber (ed.), The Mycota XV: physiology and genetics. Springer-Verlag, Berlin, Germany.
- 35.Landy, A. 1989. Dynamic, structural, and regulatory aspects of lambda site-specific recombination. Annu. Rev. Biochem. 58:913-949. [DOI] [PubMed] [Google Scholar]
- 36.Lithwick, G., and H. Margalit. 2003. Hierarchy of sequence-dependent features associated with prokaryotic translation. Genome Res. 13:2665-2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luo, H., and A. P. Kausch. 2002. Application of FLP/FRT site-specific DNA recombination system in plants. Genet. Eng. (N. Y.) 24:1-16. [DOI] [PubMed] [Google Scholar]
- 38.Meyer, V. 2008. Genetic engineering of filamentous fungi-progress, obstacles and future trends. Biotechnol. Adv. 26:177-185. [DOI] [PubMed] [Google Scholar]
- 39.Minuth, W., P. Tudzynski, and K. Esser. 1982. Extrachromosomal genetics of Cephalosporium acremonium. 1. Characterization and mapping of mitochondrial-DNA. Curr. Genet. 5:227-231. [DOI] [PubMed] [Google Scholar]
- 40.Morschhäuser, J., S. Michel, and P. Staib. 1999. Sequential gene disruption in Candida albicans by FLP-mediated site-specific recombination. Mol. Microbiol. 32:547-556. [DOI] [PubMed] [Google Scholar]
- 41.Morschhäuser, J., P. Staib, and G. Köhler. 2005. Targeted gene deletion in Candida albicans wild-type strains by MPAR flipping. Methods Mol. Med. 118:35-44. [DOI] [PubMed] [Google Scholar]
- 42.Nelson, G., O. Kozlova-Zwinderman, A. J. Collis, M. R. Knight, J. R. Fincham, C. P. Stanger, A. Renwick, J. G. Hessing, P. J. Punt, C. A. van den Hondel, and N. D. Read. 2004. Calcium measurement in living filamentous fungi expressing codon-optimized aequorin. Mol. Microbiol. 52:1437-1450. [DOI] [PubMed] [Google Scholar]
- 43.Nielsen, J. B., M. L. Nielsen, and U. H. Mortensen. 2008. Transient disruption of non-homologous end-joining facilitates targeted genome manipulations in the filamentous fungus Aspergillus nidulans. Fungal Genet. Biol. 45:165-170. [DOI] [PubMed] [Google Scholar]
- 44.Nowak, C., R. Radzio, and U. Kück. 1995. DNA-mediated transformation of a fungus employing a vector devoid of bacterial DNA sequences. Appl. Microbiol. Biotechnol. 43:1077-1081. [DOI] [PubMed] [Google Scholar]
- 45.Nowrousian, M., S. Masloff, S. Pöggeler, and U. Kück. 1999. Cell differentiation during sexual development of the fungus Sordaria macrospora requires ATP citrate lyase activity. Mol. Cell. Biol. 19:450-460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nowrousian, M., J. E. Stajich, M. Chu, I. Engh, E. Espagne, K. Halliday, J. Kamerewerd, F. Kempken, B. Knab, H. C. Kuo, H. D. Osiewacz, S. Poggeler, N. D. Read, S. Seiler, K. M. Smith, D. Zickler, U. Kück, and M. Freitag. 2010. De novo assembly of a 40 Mb eukaryotic genome from short sequence reads: Sordaria macrospora, a model organism for fungal morphogenesis. PLoS Genet. 6:e1000891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pel, H. J., J. H. de Winde, D. B. Archer, P. S. Dyer, G. Hofmann, P. J. Schaap, G. Turner, R. P. de Vries, R. Albang, K. Albermann, M. R. Andersen, J. D. Bendtsen, J. A. Benen, M. van den Berg, S. Breestraat, M. X. Caddick, R. Contreras, M. Cornell, P. M. Coutinho, E. G. Danchin, A. J. Debets, P. Dekker, P. W. van Dijck, A. van Dijk, L. Dijkhuizen, A. J. Driessen, C. d'Enfert, S. Geysens, C. Goosen, G. S. Groot, P. W. de Groot, T. Guillemette, B. Henrissat, M. Herweijer, J. P. van den Hombergh, C. A. van den Hondel, R. T. van der Heijden, R. M. van der Kaaij, F. M. Klis, H. J. Kools, C. P. Kubicek, P. A. van Kuyk, J. Lauber, X. Lu, M. J. van der Maarel, R. Meulenberg, H. Menke, M. A. Mortimer, J. Nielsen, S. G. Oliver, M. Olsthoorn, K. Pal, N. N. van Peij, A. F. Ram, U. Rinas, J. A. Roubos, C. M. Sagt, M. Schmoll, J. Sun, D. Ussery, J. Varga, W. Vervecken, P. J. van de Vondervoort, H. Wedler, H. A. Wösten, A. P. Zeng, A. J. van Ooyen, J. Visser, and H. Stam. 2007. Genome sequencing and analysis of the versatile cell factory Aspergillus niger CBS 513.88. Nat. Biotechnol. 25:221-231. [DOI] [PubMed] [Google Scholar]
- 48.Pöggeler, S., M. Nowrousian, S. Jacobsen, and U. Kück. 1997. An efficient procedure to isolate fungal genes from an indexed cosmid library. J. Microbiol. Methods 29:49-61. [Google Scholar]
- 49.Prein, B., K. Natter, and S. D. Kohlwein. 2000. A novel strategy for constructing N-terminal chromosomal fusions to green fluorescent protein in the yeast Saccharomyces cerevisiae. FEBS Lett. 485:29-34. [DOI] [PubMed] [Google Scholar]
- 50.Reuss, O., A. Vik, R. Kolter, and J. Morschhäuser. 2004. The SAT1 flipper, an optimized tool for gene disruption in Candida albicans. Gene 341:119-127. [DOI] [PubMed] [Google Scholar]
- 51.Sadowski, P. 1986. Site-specific recombinases: changing partners and doing the twist. J. Bacteriol. 165:341-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 53.Schweizer, H. P. 2003. Applications of the Saccharomyces cerevisiae Flp-FRT system in bacterial genetics. J. Mol. Microbiol. Biotechnol. 5:67-77. [DOI] [PubMed] [Google Scholar]
- 54.Senecoff, J. F., R. C. Bruckner, and M. M. Cox. 1985. The FLP recombinase of the yeast 2-μm plasmid: characterization of its recombination site. Proc. Natl. Acad. Sci. U. S. A. 82:7270-7274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sharan, S. K., L. C. Thomason, S. G. Kuznetsov, and D. L. Court. 2009. Recombineering: a homologous recombination-based method of genetic engineering. Nat. Protoc. 4:206-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Song, H., and M. Niederweis. 2007. Functional expression of the Flp recombinase in Mycobacterium bovis BCG. Gene 399:112-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Staib, P., M. Kretschmar, T. Nichterlein, H. Hof, and J. Morschhäuser. 2000. Differential activation of a Candida albicans virulence gene family during infection. Proc. Natl. Acad. Sci. U. S. A. 97:6102-6107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Theodosiou, N. A., and T. Xu. 1998. Use of FLP/FRT system to study Drosophila development. Methods 14:355-365. [DOI] [PubMed] [Google Scholar]
- 59.Tokuoka, M., M. Tanaka, K. Ono, S. Takagi, T. Shintani, and K. Gomi. 2008. Codon optimization increases steady-state mRNA levels in Aspergillus oryzae heterologous gene expression. Appl. Environ. Microbiol. 74:6538-6546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ueno, K., J. Uno, H. Nakayama, K. Sasamoto, Y. Mikami, and H. Chibana. 2007. Development of a highly efficient gene targeting system induced by transient repression of YKU80 expression in Candida glabrata. Eukaryot. Cell 6:1239-1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van den Berg, M. A., R. Albang, K. Albermann, J. H. Badger, J. M. Daran, A. J. Driessen, C. Garcia-Estrada, N. D. Fedorova, D. M. Harris, W. H. Heijne, V. Joardar, J. A. Kiel, A. Kovalchuk, J. F. Martin, W. C. Nierman, J. G. Nijland, J. T. Pronk, J. A. Roubos, I. J. van der Klei, N. N. van Peij, M. Veenhuis, H. von Döhren, C. Wagner, J. Wortman, and R. A. Bovenberg. 2008. Genome sequencing and analysis of the filamentous fungus Penicillium chrysogenum. Nat. Biotechnol. 26:1161-1168. [DOI] [PubMed] [Google Scholar]
- 62.Van Duyne, G. D. 2001. A structural view of cre-loxp site-specific recombination. Annu. Rev. Biophys. Biomol. Struct. 30:87-104. [DOI] [PubMed] [Google Scholar]
- 63.Veit, B. E., and W. L. Fangman. 1988. Copy number and partition of the Saccharomyces cerevisiae 2μm plasmid controlled by transcription regulators. Mol. Cell. Biol. 8:4949-4957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vetter, D., B. J. Andrews, L. Roberts-Beatty, and P. D. Sadowski. 1983. Site-specific recombination of yeast 2-μm DNA in vitro. Proc. Natl. Acad. Sci. U. S. A. 80:7284-7288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Voziyanov, Y., S. Pathania, and M. Jayaram. 1999. A general model for site-specific recombination by the integrase family recombinases. Nucleic Acids Res. 27:930-941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wirsching, S., S. Michel, and J. Morschhäuser. 2000. Targeted gene disruption in Candida albicans wild-type strains: the role of the MDR1 gene in fluconazole resistance of clinical Candida albicans isolates. Mol. Microbiol. 36:856-865. [DOI] [PubMed] [Google Scholar]
- 67.Yang, B., Z. Guo, Y. Huang, and S. Zhu. 2004. Codon optimization of MTS1 and its expression in Escherichia coli. Protein Expr. Purif. 36:307-311. [DOI] [PubMed] [Google Scholar]
- 68.Yang, Y., M. Malten, A. Grote, D. Jahn, and W. D. Deckwer. 2007. Codon optimized Thermobifida fusca hydrolase secreted by Bacillus megaterium. Biotechnol. Bioeng. 96:780-794. [DOI] [PubMed] [Google Scholar]
- 69.Yoder, J. I., and A. P. Goldsbrough. 1994. Transformation systems for generating marker-free transgenic plants. Nat. Biotechnol. 12:263-267. [Google Scholar]
- 70.Zadra, I., B. Abt, W. Parson, and H. Haas. 2000. xylP promoter-based expression system and its use for antisense downregulation of the Penicillium chrysogenum nitrogen regulator NRE. Appl. Environ. Microbiol. 66:4810-4816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhou, Z., P. Schnake, L. Xiao, and A. A. Lal. 2004. Enhanced expression of a recombinant malaria candidate vaccine in Escherichia coli by codon optimization. Protein Expr. Purif. 34:87-94. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.