

Abstract
Archaeal family-B DNA polymerases stall replication on encountering the pro-mutagenic bases uracil and hypoxanthine. This publication describes an X-ray crystal structure of Thermococcus gorgonarius polymerase in complex with a DNA containing hypoxanthine in the single-stranded region of the template, two bases ahead of the primer-template junction. Full details of the specific recognition of hypoxanthine are revealed, allowing a comparison with published data that describes uracil binding. The two bases are recognized by the same pocket, in the N-terminal domain, and make very similar protein-DNA interactions. Specificity for hypoxanthine (and uracil) arises from a combination of polymerase-base hydrogen bonds and shape fit between the deaminated bases and the pocket. The structure with hypoxanthine at the +2 position explains the stimulation of the polymerase 3′-5′ proof reading exonuclease, observed with deaminated bases at this location. A β hairpin element, involved in partitioning the primer strand between the polymerase and exonuclease active sites, inserts between the two template bases at the extreme end of the double stranded DNA. This denatures the two complementary primer bases and directs the resulting 3′ single-stranded extension towards the exonuclease active site. Finally the relative importance of hydrogen bonding and shape fit in determining selectivity for deaminated bases has been examined using non-polar isosteres. Affinity for both 2,4 difluorobenzene and fluorobenzimidazole, non-hydrogen bonding shape mimics of uracil and hypoxanthine respectively, is strongly diminished, suggesting polar protein-base contacts are important. However, residual interaction with 2,4 difluorobenzene is seen, confirming a role for shape recognition.
The family B DNA polymerases from the archaea are unusual in specifically recognizing uracil and hypoxanthine, the deamination products of cytidine and adenine, respectively. During replication these polymerases scan the template strand ahead of the replication fork and tightly bind such deaminated bases, should they be encountered four positions ahead of the primer-template junction (1-5). Following the capture of uracil, replication is aborted, thereby preventing the incorporation of adenine and the conversion of a C:G to a T:A base-pair. Similarly, cessation of polymerization in response to hypoxanthine results in the avoidance of an A:T to G:C transition mutation. Thus read-ahead recognition appears to be the first step in a DNA repair pathway that prevents mutations arising as a consequence of the deamination of cytidine or adenine. Additional surveillance is provided by strong stimulation of the 3′-5′ proof-reading exonuclease activity seen when the polymerase approaches nearer than four bases to uracil or hypoxanthine e.g. when these bases are at +2. This activity trims back the extending primer, re-setting the stalling position to four bases (6). Interaction with deaminated bases seems to be confined to the family-B DNA polymerases from the archaea. Family-B enzymes from bacteriophages (1) and eukaryotes (7), which have strong sequence and structural similarities to the archaeal enzymes, are unable to recognize these bases.
Read-ahead recognition requires exquisite selectivity for uracil and hypoxanthine. Any interaction with canonical DNA bases would result in aberrant termination of replication and premature cell death. Recently a crystal structure of the family B polymerase from Thermococcus gorgonarius (Tgo-Pol), in complex with a primer-template containing uracil at the optimal +4 position in the template, has elucidated the features responsible for specificity (4). Uracil was flipped into a specific binding pocket, located within the polymerase N-terminal domain, with the formation of two protein-base hydrogen bonds. Both interactions involve the peptide backbone with the amide nitrogens of Ile-114 and Tyr-37 interacting with the O2 and O4 atoms of uracil, respectively. The flipped uracil fits extremely snugly into the recognition pocket, with the side chains of Val-93 and Pro-36, and Ile-114 and Arg-119, packing above and below the base, respectively. Val-93 stacks on top of the uracil ring, with the isopropyl segment of the valine side chain lying flat and in the same plane as the uracil ring, mimicking the hydrophobic stacking seen in duplex DNA. The polymerase makes strong interactions with the phosphates flanking uracil; Tyr-7 forming a hydrogen bond with the 5′-phosphate and Arg-97 a salt bridge with the 3′-phosphate. Until now less information has been available for the mechanism responsible for specific recognition of hypoxanthine, which appears to interact with the polymerase about 1.5 – 4.5 fold less strongly than does uracil (5, 8). Nor has it been obvious how the polymerase is able to interact with both uracil (a pyrimidine) and hypoxanthine (a purine), while simultaneously rejecting the four standard DNA bases. It has been proposed that hypoxanthine may bind in the less favored syn conformation (5), in contrast to uracil which clearly binds as the preferred anti form (4). This publication presents a structure of Tgo-Pol with an oligodeoxynucleotide containing hypoxanthine two bases ahead of the primer-template junction, revealing fully how the deaminated purine is specifically recognized. Further, the ability of the polymerase to tightly bind both a purine and a pyrimidine is elucidated and clues are provided as to why locating these bases in the +2 position stimulates exonuclease activity.
The X-ray structure data published previously for uracil (4), along with that revealed for hypoxanthine in this paper, show binding of the two bases in an exquisitely tailored pocket, with formation of several protein-base hydrogen bonds. However, the role of these hydrogen bonds in the selective binding of deaminated bases has yet to be tested, for example using base analogues lacking hydrogen-bonding capability. Non-polar nucleoside isosteres were designed to be the same size and shape as natural bases (with benzenes and indoles/benzimidazoles replacing pyrimidines and purines, respectively), but to completely lack polar carbonyl, amino and imino functions. These bases have found extensive use as probes of the importance of polar functional groups and hydrogen bonding in protein-nucleic acid interactions (9-12). Perhaps the most surprising results were seen with A and B family DNA polymerases, which accepted isosteres (e.g. difluorotoluene, a deoxythymidine mimic), during replication with efficiency and specificity approaching those of natural bases (12-15). It was concluded that steric factors played a critical role during polymerase-catalyzed replication and the formation of base–pairs with appropriate size, shape and geometry was more important than the generation of Watson-Crick hydrogen bonds. However, polar interactions are important in other biological systems. For example family Y polymerases, involved in translesion synthesis and DNA repair, replicate poorly with difluorotoluene (15-18). The 3′-5′ proof reading exonuclease activity of family A DNA polymerases also depends on correct Watson-Crick base-pairing (19). With DNA repair proteins such as Fpg1 and MutY, isosteric analogues have shown that both Watson-Crick interactions between the two DNA strands and hydrogen bonds between the protein and the damaged base play important roles in the repair process (20). In this publication the non-polar isosteres difluorobenzene (diFBz) and fluorobenzimidazole (FBzIm), uracil and hypoxanthine analogues respectively (figure 1), have been used to shed light on the importance of hydrogen bonds in the selection of deaminated bases by archaeal DNA polymerase.
Figure 1.

The deaminated bases and isosteric base analogues used in this study. U, uracil; diFBz, difluorobenzene; H, hypoxanthine: FBzIm, fluorobenzimidazole. R = 2′-deoxyribose.
Methods
Polymerase purification
The purification of the family B DNA polymerases from Pyrococcus furiosus (Pfu-Pol) has been described previously (21). The polymerase from Thermococcus gorgonarius (Tgo-Pol) was produced in an identical manner. All polymerases used in this publication lacked the 3′-5′ proof reading exonuclease activity, by the incorporation of the D215A mutation (21) which abrogates exonucleolytic degradation of DNA but has no effect on the binding of the polymerase to deaminated bases (1).
Crystallization of Tgo polymerase
Complexes between Tgo-Pol and a number of oligodeoxynucleotides containing hypoxanthine were prepared for crystallization by mixing at a molar ratio of 1:1.2. Crystals with 5′-AAHGGAGACACGGCTTTTGCCGTGTCTC-3′ were obtained in 0.1M Bicine pH 9.0, 20% PEG 6000 from a robot screen where 100 nl of the complex (protein at 10 mg ml-1) was mixed with 100 nl of well solution. Crystals were flash frozen in liquid nitrogen using Paratone-N as a cryoprotectant. Diffraction data were collected at Diamond light source, U.K. on beamline I04. Data were processed and scaled using MOSFLM (22) and SCALA (23) from the CCP4 suite (24), respectively.
Structure determination and refinement
The co-crystal structure is in spacegroup P212121 with unit cell dimensions a = 79.5 Å, b = 98.5 Å, c = 116.9 Å and with one Tgo-pol:DNA complex per asymmetric unit. The structure was solved by molecular replacement using MOLREP (25) with the protein component of 2VWJ (4) as the search model. After rigid body fitting of the domains, clear density for a base was visible in the pocket. In addition, positive density, consistent with base stacking of DNA, was visible adjacent to the thumb and the sequence of the oligodeoxynucleotide used during crystallization fitted the density. In addition, density indicative of DNA was observed on the exterior surface of the polymerase, bridging the space between symmetry related molecules of the crystal. The positioning of the oligodeoxynucleotide used within this density was not possible and hence only a sugar phosphate backbone has been modeled, and all nucleotides labeled as deoxyadenosines within the PDB file. Manual rebuilding was carried out in COOT (26) interspersed with cycles of refinement using REFMAC (27) until convergence. Superimpositions were performed with LSQMAN (28).
Preparation of oligodeoxynucleotides containing difluorobenzene and fluorobenzimidazole
The diFBz and FBzIM nucleosides and their phosphoramidite derivatives were prepared as previously reported (29, 30). Both modified bases were incorporated into three oligodeoxynucleotides using standard phosphoramidite DNA-synthesis chemistry and characterized by MALDI-TOF mass spectroscopy. Two sequences were prepared: 5′-GGAGACAAGCXTGCTTGCCTGCAGGTCGACTCTAGAGGATCCCC-3′ (X = diFBz: calculated, 13,523; found, 13,527. X = FBzIm: calculated 13,544; found, 13,538); 5′-GGAGACAAGCTTGCXTGCCTGCAGGTCGACTCTAGAGGATCCCC-3′ (X = diFBz: calculated, 13,522; found, 13,523. X = FBzIm: calculated, 13,544; found, 13,537.
Primer-template extensions
The ability of Pfu-Pol to copy beyond modified bases was measured using primer-template extension assays (1-5). A Cy-5 labeled primer 24 bases in length (5′-Cy5-GGGGATCCTCTAGAGTCGACCTGC-3′) was used with two related 44-mer templates (5′-GGAGACAAGCX1TGCX2TGCCTGCAGGTCGACTCTAGAGGATCCCC-3′). With X1 = T and X2 = either uracil, hypoxanthine, stable abasic site (tetrahydrofuran; sold as dSpacer phosphoramidite, Glenn Research, Stirling, VA), diFBz or FBzIm, the modified bases are positioned 6 bases ahead of the primer-template junction. With X1 = either uracil, hypoxanthine, stable abasic site, diFBz, or FBzIm, and X2 = T, the modified bases are positioned 10 bases ahead of the primer-template junction. Reactions were carried out, at 37 °C, in 150 μl volumes containing 20 mM Tris pH 8.5, 10 mM (NH4)2SO4, 2 mM MgSO4, 15 μg acetylated bovine serum albumin, 0.1 mM each of the four dNTPs, 20 nM primer-template and 100 nM Pfu-Pol. After 2, 7, 15 and 30 minutes a 25 μl aliquot was withdrawn and the reaction quenched by adding an equal volume of 80 % formamide, 10 % glycerol, 0.1 M EDTA and orange G. An excess of “competitor” DNA, corresponding to the sequence of a fully extended primer (but lacking Cy-5) was added and the samples heated at 90 °C for 10 minutes, then rapidly cooled on ice (the competitor sequesters the template and so prevents any re-annealing of the extended Cy-5 primer, giving cleaner electrophoresis data) (6). Extension products were detected by denaturing polyacrylamide (15 %) gel electrophoresis followed by fluorescence detection using a Typhoon scanner (GE Healthcare).
Binding of Pfu-Pol to DNA containing modified bases
The interaction of Pfu-Pol with DNA containing modified bases was measured using fluorescence anisotropy with hexachlorofluorescein (Hex)-labeled DNA (3, 4). A 5′-Hex-labelled primer (5′-Hex-GGGGATCCTCTAGAGTCGACCTGCAG-3′), 26 bases in length, was annealed to a 44-mer template, (5′-GGAGACAAGCTTGCXTGCCTGCAGGTCGACTCTAGAGGATCCCC-3′; X = either T, uracil, hypoxanthine, diFBz or FBzIm). This combination locates the modified base four positions ahead of the primer-template junction, resulting in tightest possible binding (3). Binding was measured in volumes of 1 ml containing 25 mM Hepes, pH 7.5, 100 mM NaCl, 1 mM EDTA and 5 nM primer-template. Pfu-Pol was added in aliquots and the anisotropy measured after each addition. Data was fitted to binding isotherms, assuming a 1:1 stoichiomety, using Grafit (Erithacus Software, London).
Incorporation of a single dNTP into primer-templates containing modified bases
Single turnover incorporations (8) used essentially the same volumes, buffer conditions and temperatures described for primer-template extensions, save the primer-template concentration was 10 nM. The primer-template consisted of 5′-Cy5-GGGGATCCTCTAGAGTCGACCTGCAG-3′ (primer) and (5′-GGAGACAAGCTTGCXTGCCTGCAGGTCGACTCTAGAGGATCCCC-3′, X = either T, diFBz or FBzIm). This combination places the modified bases 4 positions ahead of the primer-template junction. In addition Pfu-PCNA (300 nM) was added to ensure complete binding of the polymerase (100 nM) to the DNA and, hence, single turnover conditions (8). dGTP, complementary to the first single stranded base in the template, was the only dNTP added. The reaction was quenched and analyzed by gel electrophoresis as above. Data obtained at each dGTP concentration were fitted to single exponentials to obtain kobs values, which were replotted against dGTP concentration using kobs = kpol[dGTP]/(KD + [dGTP]). Grafit (Erithacus Software, London) was used for both fitting procedures.
Results
Structure of Tgo-Pol with a hypoxanthine-containing oligodeoxynucleotide
The crystal structure of Tgo-Pol with a primer-template containing uracil at the +4 position has been described previously (4). Comparison with bacteriophage RB69 Pol (a family B member) containing DNA bound in either the polymerization (31) or editing (32) mode, indicated that the overall localization of the U+4 primer-template, was reminiscent of that observed for the DNA in the RB69 editing structure. However, no bases at the 3′-end of the U+4 primer were unwound to give single strands and primer-templates with uracil at this position are only slowly subject to proof reading exonuclease activity (6). In the light of results showing that deaminated bases at 0, +1, +2 and +3 lead to more rapid exonucleolytic degradation of the primer than when at the +4 position (6), we were curious to investigate such a complex. Additionally, the absence of data for the binding of hypoxanthine precluded a full understanding of the selectivity of the pocket, which is able to recognize both a deaminated pyrimidine and a purine (5). We thus determined the structure of a DNA-polymerase complex, using AAHGGAGACACGGCTTTTGCCGTGTCTC, an oligodeoxynucleotide that folds to a stem-loop structure to produce a primer template mimic, which places hypoxanthine at the +2 position (figure 2a). An analogous oligodeoxynucleotide was used in our previous study of the binding of U+4 (4). Data collection and refinement statistics for the final model are summarized in table 1.
Figure 2.
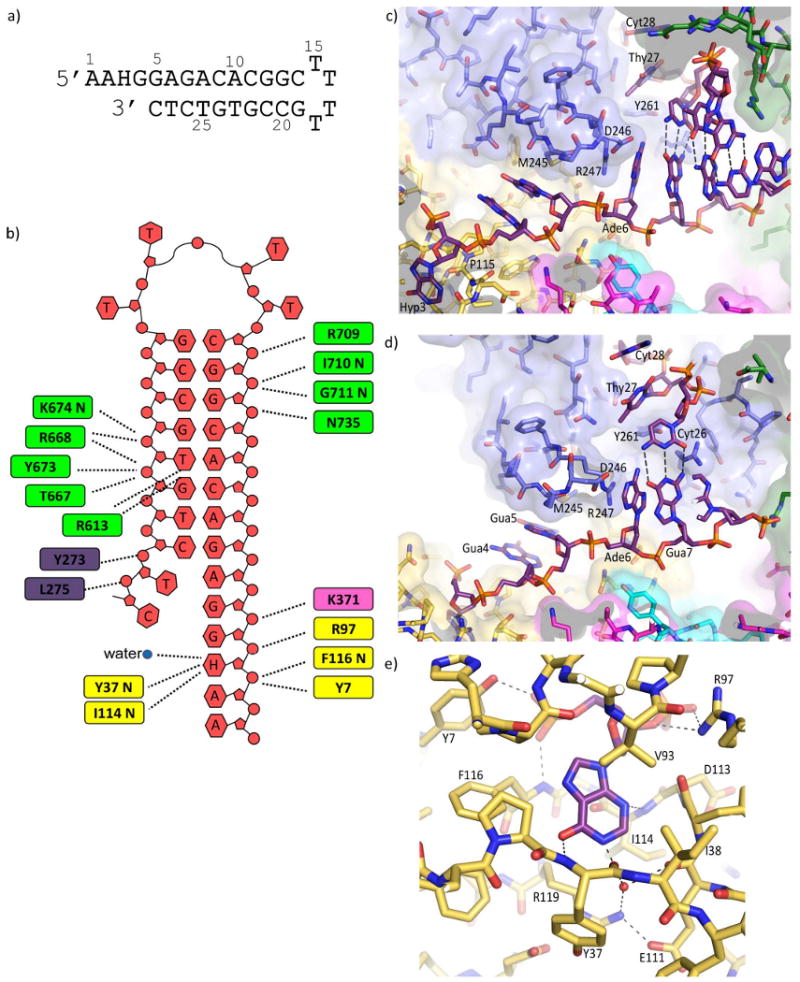
Structure of Tgo-Pol DNA complex. (a) The hypoxanthine-containing DNA used to obtain structural data. The oligodeoxynucleotide folds to produce a primer-template mimic that places hypoxanthine two bases (+2) ahead of the primer-template junction. (b) Summary of the interactions seen between Tgo-Pol and the H+2 primer-template mimic. Although all the bases are shown, A1, A2, and C14 to G19 are not visible in the structure. The extreme 3′ bases (T27 and C28) become single stranded when bound to the polymerase. The amino acids contacting the DNA are color coded according to polymerase sub-domain: thumb, green; palm, pink; amino-terminal, yellow; exonuclease, blue. The interactions made by the β-hairpin to G5 and A6 are not shown. (c and d) Two views illustrating key features of the interactions of the H+2 primer-template mimic with Tgo-Pol. Hypoxanthine (Hyp 3) is bound in the anti-conformation. G4 and G5 are sandwiched between Pro 115 and M245, R247 stacks against A6 and D246 appears to hydrogen bond with this base. As a consequence DNA denaturation occurs with the primer bases T27 and C28 becoming single stranded and well separated from their complementary template bases, G5 and A6. T27 and C28 are located in the editing channel and Y261, which is positioned between T27 and A6, hinders re-annealing. Polymerase amino acids are color coded by sub-domain as for figure 2b. (e) Contacts between Tgo-Pol and hypoxanthine. Three protein-base hydrogen bonds are seen: Tyr 37 (backbone)-O6; Ile 116 (backbone)-N3; Glu 111/R119-N1 (water-mediated). The side chains of E111 and Arg 119 form a salt bridge. The interactions of Tyr 7 and Arg 97 with the flanking phosphates (see also figure 3a) and the stacking of Val 93 with hypoxanthine are also shown.
Table 1.
X-ray diffraction data collection and refinement statistics
| Data Collection | |
| Space group | P212121 |
| Cell dimensions (Å) | a=79.45 b=98.37 c=116.63 |
| Resolution Range (Å) | 47.01-2.72 (2.87-2.72) |
| No. of unique reflections | 23740 (3473) |
| Completeness (%) | 95.1 (97.0) |
| Multiplicity | 3.6 (3.7) |
| Rmerge (%) | 10.9 (25.0) |
| Mean (I/σI) | 8.8 (4.2) |
| Refinement | |
| Resolution | 47.01-2.72 (2.79-2.72) |
| Rwork | 23.3 (31.5) |
| Rfree | 29.4 (39.4) |
| No. non-H atoms | 6484 |
| Protein | 5949 |
| DNA | 512 |
| Water | 22 |
| Mean B, all atoms (Å2) | 34 |
| No. Ramachandran outliers | 0 |
| Ramachandran favored (%) | 97.82 |
| Rmsd bond lengths (Å) | 0.006 |
| Rmsd bond angles (°) | 0.944 |
Values in parentheses refer to values in the highest resolution shell
The protein-DNA interactions seen with H+2 are summarized in figure 2b and the DNA largely makes the same contacts with the thumb, palm and amino terminal domains as observed in the uracil structure (4). However, with H+2 the thumb domain of the polymerase is moved even further, in comparison to U+4, from the position it occupies in the apo enzyme form (33). The two bases immediately 3′ of the hypoxanthine (G4 and G5) are found stacking within the single-strand template channel T of the polymerase between Pro 115 and Met 244 (figure 2c-d). The majority of the remaining DNA exhibits approximate double-stranded B form within the main polymerase cavity, although six bases (T15-C20) at the hairpin bend and the two extreme 5′ bases (A1, A2) are not visible in the electron density maps. The sequence should allow G5 and A6 to basepair with T27 and C28 and, indeed, with the U+4 structure all bases in the double-stranded region adjacent to the primer-template junction are paired. However, in the H+2 structure T27 and C28 have peeled away from their complementary bases in the template strand to give a two base single-stranded extension, with T27 positioned approximately 6 Å from A6. This denaturation appears to be driven by the β-hairpin motif, two anti-parallel β-strands joined by a loop and comprising amino acids 240-251. Arg 247, found in the loop region of the β-hairpin, inserts between G5 and A6, stacking against A6 and acting as a wedge to force these two bases apart (figure 2c-d). The separation between the C1′ atoms of G5 and A6 is 8.4 Å; for the N9 atoms a distance of 9.8 Å is observed. Additionally, D246, also present in the loop region, appears to form a hydrogen bond with A6, and may contribute to strand separation. As a consequence T27 and C28 are displaced into the editing channel (figure 2c-d) and additional hydrogen bonds between the phosphates of T27 and C28 and the backbone amide nitrogens of Y273 and L275 (amino acids in the exonuclease domain) are observed (not shown). Y261 helps maintain the primer strand in the editing channel by holding T27 away from its template partner, A6 (figure 2c-d). Although T27 and C28 are directed towards the 3′-5′ exonuclease site, the extreme 3′ base, C28, does not actually enter the active site and so is not correctly positioned for excision. Previously (6) it was predicted that location of a deaminated base at +2 resulted in the formation of a single stranded region in the primer, with the 3′ base inserting into the 3′-5′ exonuclease active site for rapid removal. While the H+2 structure confirms the formation of a single stranded region, entry into the exonuclease site is not seen. The latter may arise through the use of D215A (an exo- mutant) and the absence of divalent metal ions essential for exonuclease activity (34). It is presumed that in the complete system a further conformational change places the 3′ base in a suitable position for catalysis.
Hypoxanthine is observed in the same binding pocket used to accommodate uracil and is clearly bound in the anti conformation (figure 2c), contrary to an earlier prediction that the purine may be recognized in the syn conformation (5). Thus both uracil and hypoxanthine interact with the polymerase as anti conformers. Comparison of the structure of the uracil complex (4) with the apo-enzyme (33) revealed a pre-formed binding pocket, with no conformational change taking place following interaction with the deaminated base. Similarly the binding of hypoxanthine does not alter the structure of the pocket and the amino acids involved in selective binding of uracil are again observed to be critical for recognition of hypoxanthine (figure 2e). The same two backbone amide nitrogens position the hypoxanthine within the pocket; Tyr 37 and Ile 114 hydrogen bond with the exocyclic O6 group and the ring N3, respectively (figure 2e) (with uracil these amino acids recognize the spatially near-equivalent O4 and O2 functions). It is clear that hypoxanthine alone is not sufficient to account for all the observed density in the pocket, as an additional feature is visible at a distance of about 2.8 Å from the N1 of the purine. This density, which is situated within hydrogen bonding distance of NH1 of Arg 119, and the main-chain O of Glu 111, has been modeled as a water molecule (figure 2e). A water-mediated hydrogen bond network linking Arg 119 and Glu 111 with the N1 of hypoxanthine would further increase selectivity for this deaminated base. In addition the side chains of Arg 119 and Glu 111 form a salt bridge, (figure 2e) as they do in the U+4 (4) and apo enzyme (33) structures. While a corresponding water molecule is not visible in the model of the U+4 structure, the modest resolution does not allow the presence of such a water to be unequivocally excluded. Indeed, it is possible to model a water molecule in the pocket in a manner that makes chemical sense, albeit at a slightly shifted position due to the differences between the purine and pyrimidine rings. The hydrophobic contributions seen with uracil are preserved with hypoxanthine. Again of particular note is Val 93, whose isopropyl group effectively stacks with the plane of the base. The N7 side of the imidazole ring is flanked by Pro 90, Pro 36, Phe 116 and the side chains of Arg 119 and Ile 114 also contribute to the shape of the pocket (figure 2e). Overlay of the U+4 and H+2 structures (figure 3a-b) demonstrates that the base, sugar and flanking phosphates of the two deaminated bases occupy virtually identical positions, as do the side chains of Tyr 7 and Arg 97, which tether the flanking phosphates at the mouth of the pocket. The three amino acids, Pro 90, Pro36 and Phe116, on one side of the pocket (the left side in figure 3b) are close to the C5-C6 double bond of uracil, such that binding of thymine would give rise to a severe steric clash with the 5-CH3 group (4). The main chain oxygen of Glu 111, on the other side of the pocket (right hand side in figure 3b) plays a similar role with purines. This backbone atom is located near the C2 of hypoxanthine, such that the exocyclic amino group present in guanine would lead to a steric clash ensuring its exclusion from the pocket (figure 3b).
Figure 3.
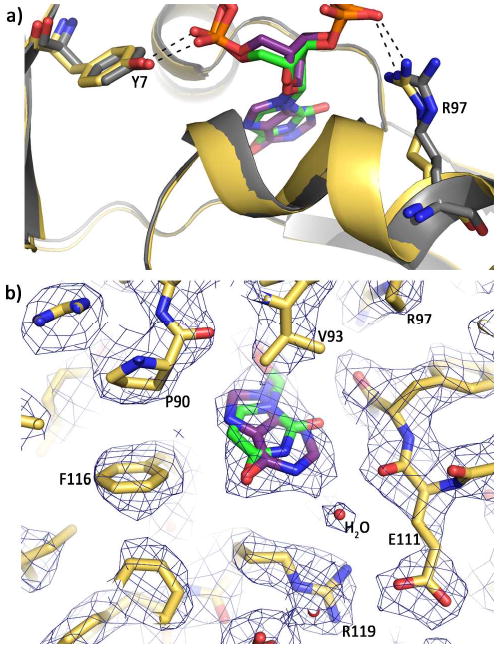
Comparison of hypoxanthine and uracil recognition. (a) Overlay of the hypoxanthine (purple) (this publication) and uracil (green) (4) structures with Tgo-Pol. The base, sugar and phosphates of the two deaminated bases are nearly super-imposable, as are the amino-terminal domains (yellow and grey for the hypoxanthine and uracil structures respectively). Side chain overlap is also very high, as shown for Tyr 7 and Arg 97, which bind the phosphates flanking the deaminated bases. (b) Superimposition of uracil (green) onto the structure of hypoxanthine (purple) bound to Tgo-Pol. The C5-C6 edge of uracil is near Pro 90 and Phe 116 (and also Pro 36, which is not visible in this view) and the C2 of hypoxanthine is close to the main chain carbonyl oxygen of Glu 111. Such tight packing of the deaminated bases results in steric exclusion of the larger thymine (5-CH3) and guanine (2-NH2) bases. The water molecule involved in a water-mediated hydrogen bond with hypoxanthine is also shown.
Replication of modified-base containing primer-templates
The inability of Pfu-Pol to copy beyond deaminated bases is most simply observed using primer extension assays, where the base is located at a defined position in the template strand, and its ability to impede polymerization noted (1-8). This method has been applied as an initial test of the interaction of both diFBz and FBzIm with Pfu-Pol. Although different Pols were used for crystallization and biochemical experiments, Tgo-Pol and Pfu-Pol have ∼80% amino acid identity and their crystal structures are essentially identical (33, 35). The two polymerases show indistinguishable behavior with deaminated bases (4). The results observed for difluorobenzene (diFBz), after 15 minutes incubation time, are shown in figure 4a. The control template, comprising only the four natural DNA bases, was completely extended (lane 2). Templates containing uracil (U), either +6 or +10 steps ahead of the primer-template junction, gave truncated products, consistent with stalling of replication 3-4 bases prior to its encounter (2, 3, 6) (lanes 3 and 6, products resulting from stalling marked with an arrow). With a stable abasic site (Ab), shortened products were also observed (panel lanes 4 and 7) as the polymerase cannot effectively add a base opposite the non-coding site. Note that the polymerase pauses at the abasic site itself, rather than stalling 3-4 bases upstream and, therefore, any truncated product seen with abasic sites are longer than those observed with uracil. With diFBz, some evidence of stalling is seen for the +10 position, with an extended product, corresponding to that found with uracil, being produced (lane 8, key product marked with an arrow). However, a band equivalent to that seen with the abasic site (arising as the polymerase is poor at inserting a base opposite the template strand isostere), along with full length product are also seen. Further, there is no evidence of stalling with diFBz at +6, the observed band running with that produced by an abasic site (lane 5). A shorter time (2 minutes) was used to better visualize any transient stalling caused by diFBz (figure 4b). With diFBz at +10 most of the product now corresponds to the stalled band seen for U+10 (lane 8, key band indicated with an arrow). Even with diFBz+6 traces of pocket mediated stalling are now visible as a minor band (lane 5, critical band arrowed) running just below the main truncated product, which co-migrates at the Ab+6 location. Overall it is clear that any pausing of replication due to the specific capture of diFBz in the deaminated base binding pocket is significantly weaker than seen with uracil. Figure 4c shows the gel patterns observed with fluorobenzimidazole (FBzIm) after 15 minutes. This gel additionally contains lanes corresponding to hypoxanthine (H) at +6 and +10, which give stalled products similar to those seen with uracil at the same location (lanes 3, 4, 7 and 8, bands arising from stalling arrowed). Wth FBzIm the gel shows no evidence of pocket-mediated capture (lanes 6 and 10), rather all extended products correspond to those seen with an abasic site, consistent with the enzyme's active site being inefficient at using FBzIm as a coding base. Shorter incubation times, comparable to the two minutes used with uracil, gave identical results to those seen after 15 minutes (data not shown). Thus FBzIm does not interact with the deaminated base binding pocket, resulting in stalling of replication.
Figure 4.
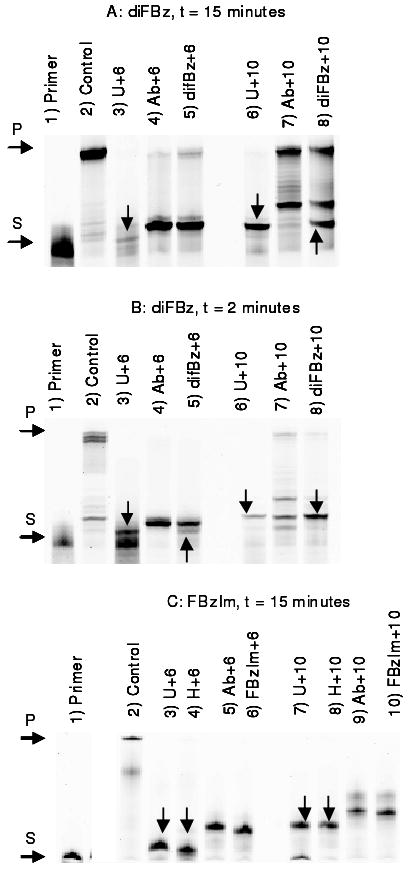
Primer extension reactions. Pfu-Pol was used to extend a Cy5-labelled primer (24 bases in length) annealed to different templates (all 44 bases long) that place uracil (U), hypoxanthine (H), a stable abasic site (Ab), difuorobenzene (diFBz) and fluorobenzimidazole (FBzIm) either 6 (+6) or 10 (+10) bases ahead of the primer-template junction). Controls use a template that contains only canonical DNA bases, resulting in full extension of the primer. U and H stall replication 3-4 bases prior to encounter, resulting in truncated products (such stalling products are highlighted with a vertical arrow). The abasic site also gives shortened products but these are longer than those seen with U and H as, here, the polymerase stops directly at the abasic site. The positions of the starting primer and fully extended primer are shown with the horizontal arrows at the sides of the gel, labeled S and P, respectively. (A) Results seen with diFBz after 15 minutes; a stalled product (arrowed) is seen with diFBz+10 but not with diFBz+6. (B) Results found with diFBz after 2 minutes; here a stalled product (arrowed) can be seen with diFBz+6, although the main product runs with that produced by the abasic site. (C) Results seen with FBzIm after 15 minutes; no evidence of stalling is visible, rather extended products seen with this isostere correspond to those produced by the abasic site. Similar results were found at shorter times.
Binding of Pfu-Pol to primer-templates containing isosteric bases
Results obtained using primer-extension assays only give a qualitative indication of recognition by the deaminated base binding pocket. To obtain a more quantitative measure of any affinity for diFBz and FBzIm, binding titrations using fluorescence anisotropy have been performed (2-5). These studies have been carried out with primer-templates that position the modified base at the +4, a location that results in the tightest binding (3). As shown in figure 5 and summarized in table 2 a primer-template with uracil at the +4 location bound strongly to the polymerase, giving a KD value of ∼ 5 nM. The binding of diFBz and FBzIm, located in an identical sequence, is clearly much weaker. In fact no differences in affinity between FbzIm and a control, containing T at +4, could be discerned; in both cases a KD of around 175 nM was found. Slightly better binding was seen with diFbz (KD ∼ 50 nM), but the U+4 primer-template still interacts some 10-fold more strongly with the polymerase. The KD values summarised in table 2 agree reasonably well with those obtained previously for primer-templates using competitive fluorescence titrations (3), also quoted in table 2. Unfortunately it proved impossible to apply the competitive approach, where a non-labeled primer-template is used to displace a hexachlorofluorescein-labeled uracil-containing oligodeoxynucleotide from the polymerase. The poor affinities of both diFBz and FBzIm necessitate the use of high concentrations to obtain full or near-full displacement and the amount of material required was not available.
Figure 5.

Binding of Pfu-Pol to hex-labeled primer-templates (sequences given in materials and methods) containing varying bases at the +4 position in the template strand. A: diFBz+4 (▲), FBzIM +4 (■), T+4 (control) (●). B: U+4 (●). The KD values found for these titrations are summarized in table 2.
Table 2.
Binding of primer-templates to P. furiosus DNA polymerase
| Primer-template | KD (nM)a |
|---|---|
| Control (T) | 182 ± 23 (270) |
| U | 4.4 ± 1.3 (1.5) |
| diFBz | 52 ± 8 |
| FBzIm | 176 ± 19 |
The binding constants for the interaction of primer-templates (sequences given in the materials and methods) containing the bases indicated at the + 4 position were measured by direct titration (figure 5). The values (± standard deviation) are the averages found from four determinations. The values given in brackets for T and U were measured previously for analogous primer-templates using competitive titration (3).
Single dNTP incorporation into primer-templates
A final approach to determine the barrier to polymerization that arises from the presence of a deaminated base involves measuring the kinetic constants for the incorporation of one dNTP, under single turnover conditions (8). In order to ensure complete binding of Pfu-Pol to primer-templates, particularly those lacking deaminated bases, it is necessary to include an excess of PCNA. As found previously PCNA increases the affinity of the polymerase for DNA, allowing saturation of the DNA (measured by attainment of the ceiling reaction velocity), at achievable concentrations of the enzyme (8). It is not necessary to add the PCNA loader (RF-C in archaea) or “block” the ends of the DNA, as used in studies of other polymerases, to realize this outcome (36). Values for kpol (reaction rate under single turnover conditions) and KD (binding affinity) have been determined for primer-templates containing thymine (control), diFBz and FBzIm at the +4 position, as shown in figure 6 and summarized in table 3. Very similar values of kpol and KD were observed with T and FBzIm, showing that the purine-based isostere does not result in any significant block to dNTP incorporation and the progression of the polymerase. With diFBz the reaction was slowed by about an order of magnitude, as a result of a 10-fold drop in kpol and, consequently, a similar decrease in the specificity constant kpol/KD. Therefore, diFBz shows some degree of interaction with the deaminated base binding pocket, resulting in reduced incorporation of incoming dNTPs and abatement in the rate of polymerization. With primer-templates containing U or H at + 4, incorporation of the single dNTP was too slow (∼ 103 – 104 fold reduced when compared to T) to allow kinetic constant determination. However, table 3 also gives results obtained in an earlier investigation with T, U and H (8). The two studies used different temperatures; 37 °C here, 50 °C previously, accounting for the lower kpol observed for T controls in this investigation. Additionally, the changes in both primer-templates and incoming dNTP (dGTP here, dATP earlier), can account for the small changes in KD. Nevertheless, it is abundantly clear that the decreases of ∼ 4 × 103 in kpol and (kpol/KD) seen with U and H, relative to the values seen for the T control, are much greater than the 10 fold reduction observed with diFBz. The results presented in this section are in agreement with those found above, confirming diFBz interacts with the deaminated base binding pocket a little better than controls, but much more weakly than either U or H. With FBzIm, identical kinetic parameters to that observed with the control primer-template, containing T+4, were found. Thus, in agreement with the primer-template extensions and binding assays, FBzIm does not appear to interact with the deaminated base binding pocket.
Figure 6.

Incorporation of a single dGTP by Pfu-Pol into primer-templates containing varying bases in the template strand at +4, under single turnover conditions. Initially kobs was determined at various dGTP concentrations (data not shown) and used to generate secondary plots of kobs against [dGTP] for determination of kinetic parameters. A: diFBz+4 (●). B: T+4 (control) (■), FbzIm+4 (●). Kinetic parameters are summarized in table 3.
Table 3.
Kinetic parameters for the interaction of dNTP with P. furiosus polymerase/primer-template complexes.
| This studya | Published work (8)b | |||||
|---|---|---|---|---|---|---|
| Primer-template | kpolc (s-1) |
KDc (μM) |
kpol/KDc (M-1 s-1) |
kpolc (s-1) |
KDc (μM) |
kpol/KDc (M-1 s-1) |
| Thymidine | 2.1 | 9.7 | 1.7 × 105 | 7.7 | 45 | 1.5 × 105 |
| diFBz | 0.2 | 10.5 | 2.0 × 104 | not determined | ||
| FBzIm | 1.6 | 10 | 1.6 × 105 | not determined | ||
| Uracil | too slow for kpol/KD determination | 1.8 × 10-3 | 52 | 35 | ||
| Hypoxanthine | too slow for kpol/KD determination | 2.5 × 10-3 | 51 | 50 | ||
Experiments carried out at 37 °C with insertion of a single dGTP
Experiments carried out at 50 °C with insertion of a single dATP.
The kinetic parameters for the incorporation of a single dNTP into primer-templates (sequences given in the materials and methods) containing the base indicated at +4 were measured under single turnover conditions (figure 6). Values are the averages of three determinations and are accurate to ± 25%.
Discussion
Comparison of uracil and hypoxanthine recognition
The X-ray structure of Tgo-Pol bound to a primer-template containing hypoxanthine elucidates how a deaminated purine is recognized and reveals the mechanism for specific binding of both a purine and a pyrimidine deaminated base. Comparison of the structure of apo Tgo-Pol (33) with a uracil-bound form (4), showed no conformational changes in the amino-terminal domain, which houses the deaminated base binding pocket. The hypoxanthine structure confirms the rigidity of this domain, with a pre-formed pocket poised to accept deaminated bases as they are encountered during replication. Comparing the uracil and hypoxanthine structures reveals remarkable superposition between atoms involved in both polar and hydrophobic protein-DNA contacts, with profound overlap of the two bases (figure 3a). Uracil and hypoxanthine are discriminated from the four canonical DNA bases by a combination of shape match to the pocket and protein-base hydrogen bonds. The backbone amide nitrogen of Tyr 37 is critical for identifying an oxygen at the deaminated position (O4 in uracil, O6 in hypoxanthine), with the backbone amide of Ile 114 stabilizing binding by selecting a hydrogen bond acceptor (O2 in uracil, N3 in hypoxanthine) on the opposite side of the ring. With hypoxanthine an additional, water-mediated hydrogen bond, is observed between the ring NH1 and Glu 111/Arg 119. As mentioned in the results section, higher resolution structures will be needed to unequivocally confirm if such an interaction is also formed to the equivalent NH3 position of uracil. Forming two hydrogen bonds to the C(6)O-N(1)H system of hypoxanthine (and possibly to the analogous C(4)O-N(3)H group of uracil) provides excellent discrimination against adenine (and potentially cytosine), as in the latter two bases the hydrogen bonding pattern is reversed. This recognition pattern is analogous to that used by uracil-DNA-glycosylases to distinguish between uracil and cytosine (37, 38). The importance of Glu 111 and Arg 119 are confirmed by their high degree of conservation within the euryarchaeal family-B DNA polymerases and the observation that the mutation R119A no longer interacts with deaminated bases (4). While hydrogen bonding accounts for preferential binding of uracil and hypoxanthine, relative to their precursors, cytosine and adenine, rejection of thymine and guanine relies on shape matching and steric features. The additional bulk of thymine (5-CH3 group) and guanine (2-NH2 group) would not occupy the same spatial positions, should these two bases interact with the pocket in the same manner as uracil and hypoxanthine (figure 3b). Therefore, different groups in the pocket are used for rejecting the thymine 5-methyl and guanine 2-amino functions. As described previously the 5-methyl group is excluded by the side chains of Pro 36, Pro 90 and Phe 116 (4). By contrast an exocyclic amino group at the C2 of guanines would result in a steric clash with the main chain oxygen of Glu 111, thus precluding binding. This steric clash also explains the lack of binding seen with xanthine (the deamination product of guanine) (5), which contains an exocyclic oxygen at this position.
Coupling deaminated base recognition to 3′-5′ exonuclease activity
Polymerase stalling in response to uracil and hypoxanthine is a DNA damage avoidance mechanism, designed to avoid copying of these pro-mutagenic bases. With the U+4 structure (4), the DNA is bound in an editing mode away from the polymerization active site and, additionally, the lack of unwinding of the primer strand inhibits proof-reading exonuclease activity (6). Thus, the nucleic acid is stably and inertly bound, presumably awaiting the recruitment of, as yet, unidentified repair proteins. However, stalling does not confer an absolute block to DNA replication and prolonged incubation leads to read through of deaminated bases (6). As the polymerase inches towards the lesion, uracil and hypoxanthine become progressively located three, two and one base ahead of the primer-template junction and this progression results in a profound stimulation of proof-reading exonuclease activity (6). Thereby the slowly, and inappropriately, extending primer is cut back, to reset the deaminated base to the +4 position. Such “idling” cycles of extension and degradation provide an additional barrier to replication beyond the damaged bases, as shown by the increased propensity of exo- mutants to extend beyond uracil and hypoxanthine (6). The structure with hypoxanthine at the + 2 position rationalizes the link between deaminated base binding and activation of the polymerase proof reading exonuclease. The U+4 and H+2 structures are broadly similar with regard to base recognition, the disposition of the polymerase domains and the localization of the bound nucleic acid. There exists, however, one key difference; with U+4 the primer-template is fully base-paired, whereas with H+2 the last two primer bases are single stranded and located in the editing channel. The denaturing seen with H+2 is mediated by the β-hairpin motif, a critical region in family B polymerases for the partition of primer strands between the polymerase and exonuclease active sites (39, 40). In an editing complex seen with the family-B polymerase from bacteriophage RB69, Arg 260, an amino acid found in the β-hairpin, interacts with the primer-template and appears to contribute to the melting of three primer bases and positioning of the resulting single stranded primer region in the editing channel (32). It has been proposed that the equivalent amino acid in the archaeal family-B polymerase from Pyrococcus kodakaraensis KOD1, Arg 247, fulfils a similar role (41). With Tgo-Pol bound to hypoxanthine at +2, Arg 247 pushes into the template strand, between the two bases in the double-stranded region immediately adjacent to the primer-template junction (G5 and A6). This wedge-like insertion pries G5 and A6 apart and unwinds the complementary primer bases, C28 and T27. Comparing the H+2 and U+4 structures shows almost perfect superimposition of the β-hairpin (figure 7) and the entire amino-terminal and exonuclease domains overlap almost completely. In particular the separations between the deaminated base binding pocket, the β-hairpin and the nearest double-stranded base-pair are largely unchanged between the two structures (figure 7). In the case of U+4 this locates the hairpin near single stranded template bases and both Arg 247 and the proximal bases appear disordered. With H+2 the hairpin is close to the double stranded region of the primer-template and Arg 247 behaves differently, becoming ordered and acting as a wedge. Thus the two polymerase domains differentially manipulate and distort the conformation of bound DNA, dependent on the relative positions of the deaminated base and the nearest base-pair, thereby activating exonuclease activity with U/H at +2. With euryarchaeal polymerases from thermococcus and pyrococcus species, sequence alignment shows that amino acid 247 is Arg (21), Met (8, with Pfu-Pol having this residue) or Ser (9) (numbers in parenthesis refer to frequency of occurrence). While the long side chain of Met could act similarly to Arg inserting between bases, it is unclear if the much shorter Ser could fulfill this role. The same alignment shows that the amino acid at position 246, which hydrogen bonds with base A6, is overwhelmingly Asp, which occurs 36 times. There are single instances of Glu and Gln at this location but all three bases are compatible with the function proposed and, in principle, able to hydrogen bond to any standard DNA base at this position. However, other species of euryarchaeal family B polymerases show very little amino acid conservation at these two locations. This may be related to variability in the overall size of the β-hairpin observed between polymerases e.g. from phage RB69 (31) and herpes simplex virus (42) and the tendency for structural rather than amino acid sequence conservation (39). In contrast amino acid 261, which prevents re-hybridization by blocking access of T27 to A6 is functionally well conserved in all euryarchaeal family B polymerases, being either an aromatic or bulky hydrophobic residue: Y(51), F(12), W(12), L(21).
Figure 7.
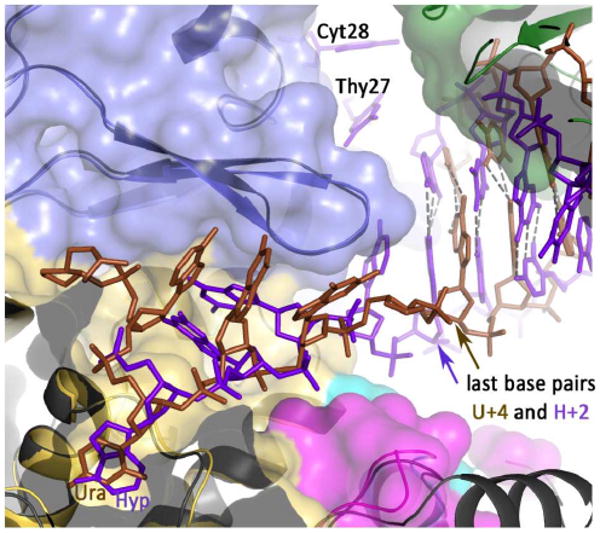
Relative spatial positions of uracil/hypoxanthine, the β-hairpin and the nearest double-stranded base for the U+4 and H+2 structures. The two structures have been superimposed and the space fill for H+2 is shown (thumb domain, green; exonuclease domain, blue; amino-terminal domain, yellow). The uracil-containing DNA is brown and the hypoxanthine-containing DNA blue, with both the deaminated bases labeled. The β-hairpin (β-strand/loop/β-strand) is illustrated for both structures with that of uracil in slightly darker blue. The positions of the base pairs (last base pairs) nearest the deaminated bases and the β-hairpin are arrowed, showing the relative disposition of these three elements are unchanged between he two structures. However, with U+4 the last base-pair contains the 3′ base of the primer. This is not the case with H+2, as the two bases at the 3′ end of the primer (T27 and C28) become single stranded and positioned in the editing channel of the polymerase.
Hydrogen bonding and shape recognition
In order to assess the relative contributions of hydrogen bonds and shape recognition for specific recognition of deaminated bases, use has been made of the uracil and hypoxanthine isosteric analogues illustrated in figure 1. With difluorobenzene some interaction with the polymerase is seen using primer-extensions, binding assays and single turnover dNTP incorporation. However, the preference over controls is only 4-10 fold (binding and single turnovers, respectively), significantly lower than that observed with uracil. In the case of fluorobenzimidazole, none of the assays reveal any indication of positive binding to the polymerase. The stronger binding of diFBz relative to FBzIm mimics the behavior seen with their parent bases, uracil and hypoxanthine, respectively (5, 8). diFBz and FBzIm are shape-matched to uracil and hypoxanthine but cannot participate in polar interactions; therefore, it can be concluded that the hydrogen bonds seen between the polymerase and the deaminated bases play a key role in selectivity. The ability to specifically bind the damaged bases uracil and hypoxanthine, which occur at very low levels in DNA, while simultaneously rejecting the four canonical bases, present in large excess, is demanding. Therefore, it appears likely that the polymerase would make use of every distinguishing feature of the two deaminated bases. It is possible to differentiate between the uracil/thymine and hypoxanthine/guanine pairs using size criteria, as the canonical bases are larger. Indeed rejection of thymine and guanine relies largely on the pocket being shaped to prevent binding of the additional 5-CH3 and 2-NH2 groups on steric grounds. However, it is almost impossible to distinguish the near isosteric bases uracil/cytosine and hypoxanthine/adenine in this manner and for this purpose the polymerase makes use of hydrogen bonding. The critical nature of these interactions accounts for the loss of tight binding when diFBz and FBzIm are used.
DNA polymerases are able to accept non-polar isosteric base analogues with relatively high efficiency during replication, inferring a preference for the production of a correctly shaped base pair, rather than Watson-Crick hydrogen bonds (12-15). However, in this publication isosteres have been used to probe specific recognition of a damaged base by the polymerase, a DNA repair response, rather than DNA synthesis itself. Examples of the use of isosteric bases for investigation of DNA repair include Fpg (20) and AlkA (43), which target 8-oxoguanuine and hypoxanthine respectively. Locating an isostere opposite the damaged base, in double-stranded DNA, usually results in faster excision, as abolition of Watson-Crick base pairs facilitates flipping of the damaged base into the enzyme active site. However, in both cases the isostere does not directly probe recognition of the damaged base by the protein, rather the influence of base-pairing on overall repair. MutY removes a canonical DNA base, adenine, when paired opposite the damaged base 8-oxoguanine. In general, replacing adenine with an isosteric analogue results in better binding by promoting base flipping (18, 44). However, catalysis is severely compromised and a study with adenine isosteres in which the ring nitrogens were gradually replaced, uncovered a critical role for N3 and N7 (20). Recognition of uracil and hypoxanthine by DNA polymerases differs, somewhat, from the repair systems previously analyzed. No catalytic activity is manifested and the polymerase interacts with the deaminated bases in single stranded DNA, removing any influence of complementary bases. Nevertheless, the results found with the polymerase are reminiscent of those seen with MutY; isosteric bases strongly reduce binding in the case of MutY and drastically diminish catalysis with the polymerase. It is clear that base-pairs can be recognized largely by shape and steric factors, as seen with DNA polymerases during replication. However, it remains to be demonstrated whether such a mechanism is adequate for highly specific interaction of a single isolated base with a protein. With archaeal polymerases faithful recognition of deaminated bases is clearly dependent on hydrogen bonding. However, as the enzyme must recognize both uracil and hypoxanthine, extreme steric tailoring, needed to distinguish near isosteric base combinations such as uracil/cytidine and hypoxanthine/adenine, is almost certainly precluded. Therefore, the question of shape recognition of single bases by other proteins remains an open one.
Footnotes
BAC was supported by grants from the European Commission (MRTN-CT-2005-019566) and the UK BBSRC (BB/F00687X/1). TK is a UK BBSRC supported PhD student. ETK acknowledges support from the U.S. National Institutes of Health (GM072705).
Abbreviations: Fpg, formamidopyrimidine (fapy)-DNA glycosylase (also known as 8-oxoguanine DNA glycosylase); diFBz, difluorobenzene; FBzIm, fluorobenzimidazole; U, uracil; H, hypoxanthine; Hex, hexachlorofluorescein; Cy-5, cyanine-5; Pfu-Pol, the family-B DNA polymerase from Pyrococcus furiosus; Tgo-Pol, the family-B DNA polymerase from Thermococcus gorgonarius: PCNA, proliferating cell nuclear antigen.
References
- 1.Greagg MA, Fogg MJ, Panayotou G, Evans SJ, Connolly BA, Pearl LH. A read-ahead function in archaeal DNA polymerases detects promutagenic template-strand uracil. Proc Natl Acad Sci USA. 1999;96:9045–9050. doi: 10.1073/pnas.96.16.9045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fogg MJ, Pearl LH, Connolly BA. Structural basis for uracil recognition by archaeal family B DNA polymerases. Nat Struct Biol. 2002;9:922–927. doi: 10.1038/nsb867. [DOI] [PubMed] [Google Scholar]
- 3.Shuttleworth G, Fogg MJ, Kurpiewski MR, Jen-Jacobson L, Connolly BA. Recognition of the Pro-mutagenic Base Uracil by Family B DNA Polymerases from Archaea. J Mol Biol. 2004;337:621–634. doi: 10.1016/j.jmb.2004.01.021. [DOI] [PubMed] [Google Scholar]
- 4.Firbank SJ, Wardle J, Heslop P, Lewis RJ, Connolly BA. Uracil Recognition in Archaeal DNA Polymerases Captured by X-ray Crystallography. J Mol Biol. 2008;381:529–539. doi: 10.1016/j.jmb.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 5.Gill S, O'Neill R, Lewis RJ, Connolly BA. Interaction of the family-B DNA polymerase from the archaeon Pyrococcus furiosus with deaminated bases. J Mol Biol. 2007;372:855–863. doi: 10.1016/j.jmb.2007.07.015. [DOI] [PubMed] [Google Scholar]
- 6.Russell HJ, Richardson TT, Emptage K, Connolly BA. The 3′-5′ proofreading exonuclease of archaeal family-B DNA polymerase hinders the copying of template strand deaminated bases. Nucleic Acids Research. 2009;37:7603–7611. doi: 10.1093/nar/gkp800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wardle J, Burgers PMJ, Cann IKO, Darley K, Heslop P, Johansson E, Lin LJ, McGlynn P, Sanvoisin J, Stith CM, Connolly BA. Uracil recognition by replicative DNA polymerases is limited to the archaea, not occurring with bacteria and eukarya. Nucleic Acids Res. 2008;36:793–802. doi: 10.1093/nar/gkm1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Emptage K, O'Neill R, Solovyova A, Connolly BA. Interplay between DNA polymerase and proliferating cell nuclear antigen switches off base excision repair of uracil and hypoxanthine during replication in archaea. J Mol Biol. 2008;383:762–771. doi: 10.1016/j.jmb.2008.08.018. [DOI] [PubMed] [Google Scholar]
- 9.Schweitzer BA, Kool ET. Nonpolar aromatic nucleosides as hydrophobic isosteres of DNA nucleosides. J Org Chem. 1994;59:7238–7242. doi: 10.1021/jo00103a013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kool ET. Replacing the nucleobases in DNA with designer molecules. Accounts Chem Res. 2002;35:936–943. doi: 10.1021/ar000183u. [DOI] [PubMed] [Google Scholar]
- 11.Krueger AT, Kool ET. Model systems for understanding DNA base pairing. Curr Opin Chem Biol. 2007;11:588–594. doi: 10.1016/j.cbpa.2007.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim TW, Delaney JC, Essigmann JM, Kool ET. Probing the Active Site Tightness of DNA Polymerase in Sub-Angstrom Increments. Proc Natl Acad Sci USA. 2005;102:15803–15808. doi: 10.1073/pnas.0505113102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kool ET, Morales JC, Guckian KM. Mimicking the structure and function of DNA: Insights into DNA stability and replication. Angew Chem Int Ed. 2000;39:990–1009. doi: 10.1002/(sici)1521-3773(20000317)39:6<990::aid-anie990>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 14.Kool ET. Hydrogen bonding, base stacking, and steric effects in DNA replication. Ann Rev Biophys Biomol Struct. 2001;30:1–22. doi: 10.1146/annurev.biophys.30.1.1. [DOI] [PubMed] [Google Scholar]
- 15.Kool ET, Sintim HO. The difluorotoluene debate - a decade later. Chem Commun. 2006:3665–3675. doi: 10.1039/b605414e. [DOI] [PubMed] [Google Scholar]
- 16.Potapova O, Chan C, DeLucia AM, Helquist SA, Kool ET, Grindley NDF, Joyce CM. DNA polymerase catalysis in the absence of Watson-Crick hydrogen Bonds: analysis by single-turnover kinetics. Biochemistry. 2006;45:890–898. doi: 10.1021/bi051792i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wolfle WT, Washington MT, Kool ET, Spratt TE, Helquist SA, Prakash L, Prakash S. Evidence for a Watson-Crick hydrogen bonding requirement in DNA synthesis by human DNA polymerase kappa. Mol Cell Biol. 2005;25:7137–7143. doi: 10.1128/MCB.25.16.7137-7143.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Irimia A, Eoff RL, Pallan PS, Guengerich FP, Egli M. Structure and activity of Y-class DNA polymerase DPO4 from Sulfolobus solfataricus with templates containing the hydrophobic thymine analog 2,4-difluorotoluene. J Biol Chem. 2007;282:36421–36433. doi: 10.1074/jbc.M707267200. [DOI] [PubMed] [Google Scholar]
- 19.Morales JC, Kool ET. Importance of Terminal Base Pair Hydrogen-Bonding in 3′-End Proofreading by the Klenow Fragment of DNA Polymerase I. Biochemistry. 2000;39:2626–2632. doi: 10.1021/bi992173a. [DOI] [PubMed] [Google Scholar]
- 20.Francis AW, Helquist SA, Kool ET, David SS. Probing the Requirements for Recognition and Catalysis in Fpg and MutY with Nonpolar Adenine Isosteres. J Am Chem Soc. 2003;125:16235–16242. doi: 10.1021/ja0374426. [DOI] [PubMed] [Google Scholar]
- 21.Evans SJ, Fogg MJ, Mamone A, Davis M, Pearl LH, Connolly BA. Improving dideoxynucleotide-triphosphate utilization by the hyper-thermophilic DNA polymerase from Pyrococcus furiosus. Nucleic Acids Res. 2000;28:1059–1066. doi: 10.1093/nar/28.5.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leslie AGW. Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4 + ESF-EAMCB Newsletter on Protein Crystallography. 1992;26 [Google Scholar]
- 23.Evans PR. Data reduction. Proceedings of CCP4 Study Weekend on Data Collection and Processing. 1993:114–122. [Google Scholar]
- 24.CCP4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 25.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Crystallogr. 1997;30:1022–1025. [Google Scholar]
- 26.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 27.Murshudov G, Vagin A, Dodson E. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 28.Kleywegt GJ. Use of non-crystallographic symmetry in protein structure refinement. Acta Crystallogr D. 1996;52:842–857. doi: 10.1107/S0907444995016477. [DOI] [PubMed] [Google Scholar]
- 29.Lai JS, Qu J, Kool ET. Fluorinated DNA bases as probes of electrostatic effects in DNA base stacking. Angew Chem Int Ed. 2003;42:5973–5977. doi: 10.1002/anie.200352531. [DOI] [PubMed] [Google Scholar]
- 30.Seela F, Bourgeois W, Rosemeyer H, Wenzel T. Helv Chim Act. 1996;79:488–498. [Google Scholar]
- 31.Franklin MC, Wang J, Steitz TA. Structure of the replicating complex of a Pol alpha family DNA polymerase. Cell. 2001;105:657–666. doi: 10.1016/s0092-8674(01)00367-1. [DOI] [PubMed] [Google Scholar]
- 32.Shamoo Y, Steitz TA. Building a replisome from interacting pieces: sliding clamp complexed to a peptide from DNA polymerase and a polymerase editing complex. Cell. 1999;99:155–166. doi: 10.1016/s0092-8674(00)81647-5. [DOI] [PubMed] [Google Scholar]
- 33.Hopfner KP, Eichinger A, Engh RA, Laue F, Ankenbauer W, Huber R, Angerer B. Crystal structure of a thermostable type B DNA polymerase from Thermococcus gorgonarius. Proc Natl Acad Sci USA. 1999;96:3600–3605. doi: 10.1073/pnas.96.7.3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beese LS, Steitz TA. Structural basis for the 3′-5′ exonuclease activity of Escherichia coli DNA polymerase I: a two metal ion mechanism. EMBO J. 1991;10:25–33. doi: 10.1002/j.1460-2075.1991.tb07917.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim SH, Kim DU, Kim JK, Kang LW, Cho HS. Crystal structure of Pfu, the high fidelity polymerase from Pyrococcus furiosus. Int J Biol Macromol. 2008;42:356–361. doi: 10.1016/j.ijbiomac.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 36.Carver TE, Jr, Sexton DJ, Benkovic SJ. Dissociation of bacteriophage T4 DNA polymerase and its processivity clamp after completion of Okazaki fragment synthesis. Biochemistry. 1997;36:14409–14417. doi: 10.1021/bi971423p. [DOI] [PubMed] [Google Scholar]
- 37.Pearl LH. Structure and function in the uracil-DNA glycosylase superfamily. Mutation Res. 2000;460:165–181. doi: 10.1016/s0921-8777(00)00025-2. [DOI] [PubMed] [Google Scholar]
- 38.Huffman JL, Sundheim O, Tainer JA. Structural features of DNA glycosylases and AP endonucleases. In: Siede W, Kow YW, Doetsch PW, editors. DNA damage recognition. Taylor and Francis; New York: 2006. pp. 299–321. [Google Scholar]
- 39.Hogg M, Aller P, Konigsberg W, Wallace SS, Doublié Structural and biochemical investigation of the role in proofreading of a β hairpin loop found in the exonuclease domain of a replicative DNA polymerase of the B family. J Biol Chem. 2007;282:1432–1444. doi: 10.1074/jbc.M605675200. [DOI] [PubMed] [Google Scholar]
- 40.Trzenecka A, Pochocka D, Bebenek A. Different behaviors in vivo of mutations in the β hairpin loop of the DNA polymerases of the closely related phages T4 and RB69. J Mol Biol. 2009;289:797–807. doi: 10.1016/j.jmb.2009.04.055. [DOI] [PubMed] [Google Scholar]
- 41.Hashimoto H, Nishioka M, Fujiwara S, Takagi M, Imanaka T, Inoue T, Kai Y. Crystal structure of DNA polymerase from hyperthermophilic archaeon Pyrococcus kodakaraensis KOD1. J Mol Biol. 2001;306:469–477. doi: 10.1006/jmbi.2000.4403. [DOI] [PubMed] [Google Scholar]
- 42.Liu S, Knafels JD, Chang JS, Waszak GA, Baldwin ET, Deibel MR, Thomsen DR, Homa FL, Wells PA, Tory MC, Poorman RA, Gao H, Qiu X, Seddon AP. Crystal structure of the herpes simplex virus 1 DNA polymerase. J Biol Chem. 2006;281:18193–18200. doi: 10.1074/jbc.M602414200. [DOI] [PubMed] [Google Scholar]
- 43.Vallur AC, Feller JA, Abner CW, Tran RK, Bloom LB. Effects of hydrogen bonding within a damaged base pair on the activity of wild type and DNA-intercalating mutants of human alkyladenine DNA glycosylase. J Biol Chem. 2002;277:31673–31678. doi: 10.1074/jbc.M204475200. [DOI] [PubMed] [Google Scholar]
- 44.Chepanoske CL, Langlier CR, Chmiel NH, David SS. Recognition of the nonpolar base 4-methylindole in DNA by the DNA repair adenine glycosylase MutY. Organic Lett. 2000;2:1341–1344. doi: 10.1021/ol005831o. [DOI] [PubMed] [Google Scholar]